Proteasome 26S Subunit, non-ATPase 3 (PSMD3) Regulates Breast Cancer by Stabilizing HER2 from Degradation

 ,
,
Abstract
:1. Introduction
2. Results
2.1. PSMD3 Expression Is Coupled with HER2 Expression
2.2. PSMD3 Expression Is Associated with HER2+ Patients, According to the Oncomine and TCGA Databases
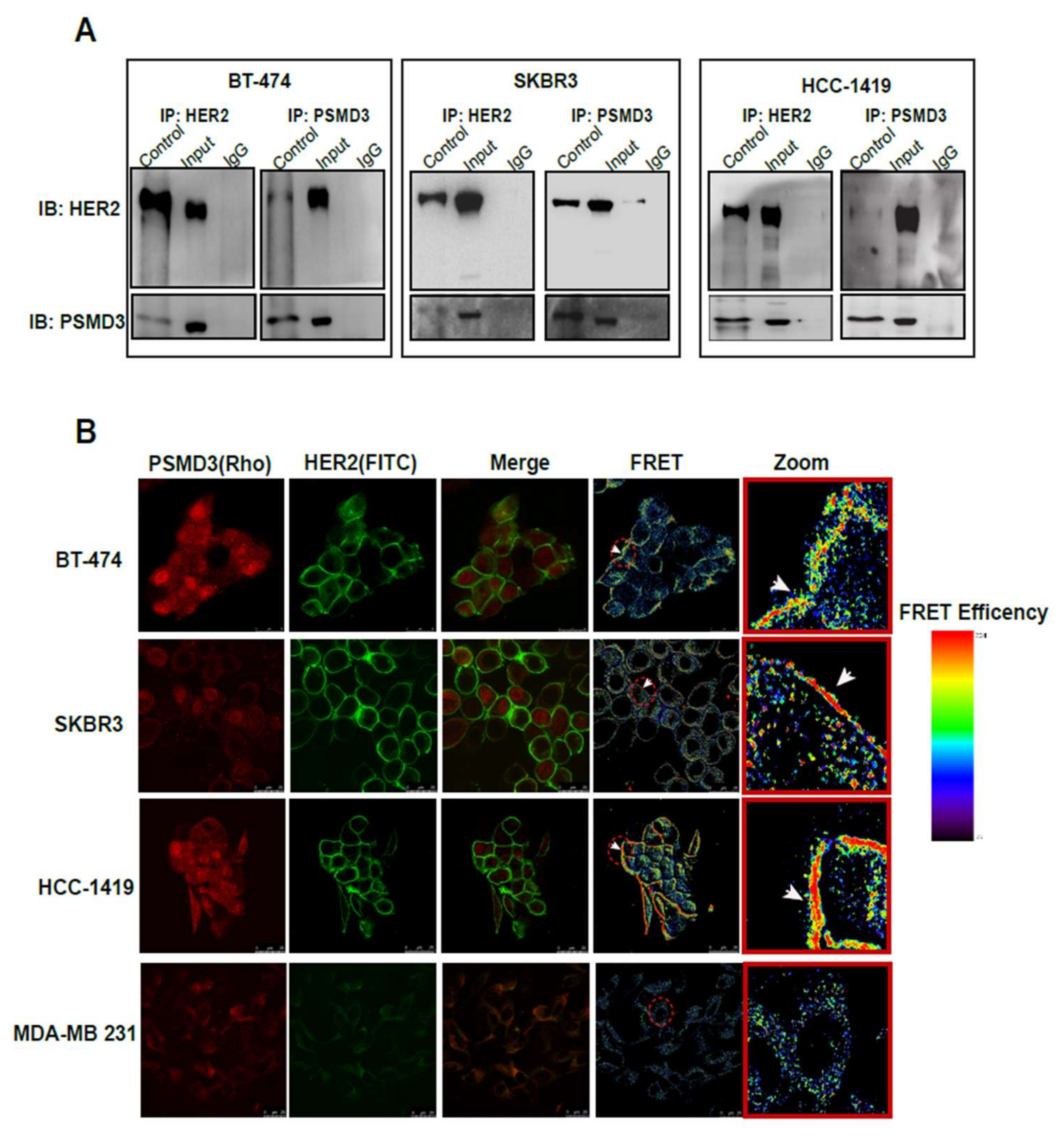
2.3. Direct Interaction Between PSMD3 and HER2 in HER2+ BC Cell Lines
2.4. PSMD3 Stabilizes HER2 at Protein Level
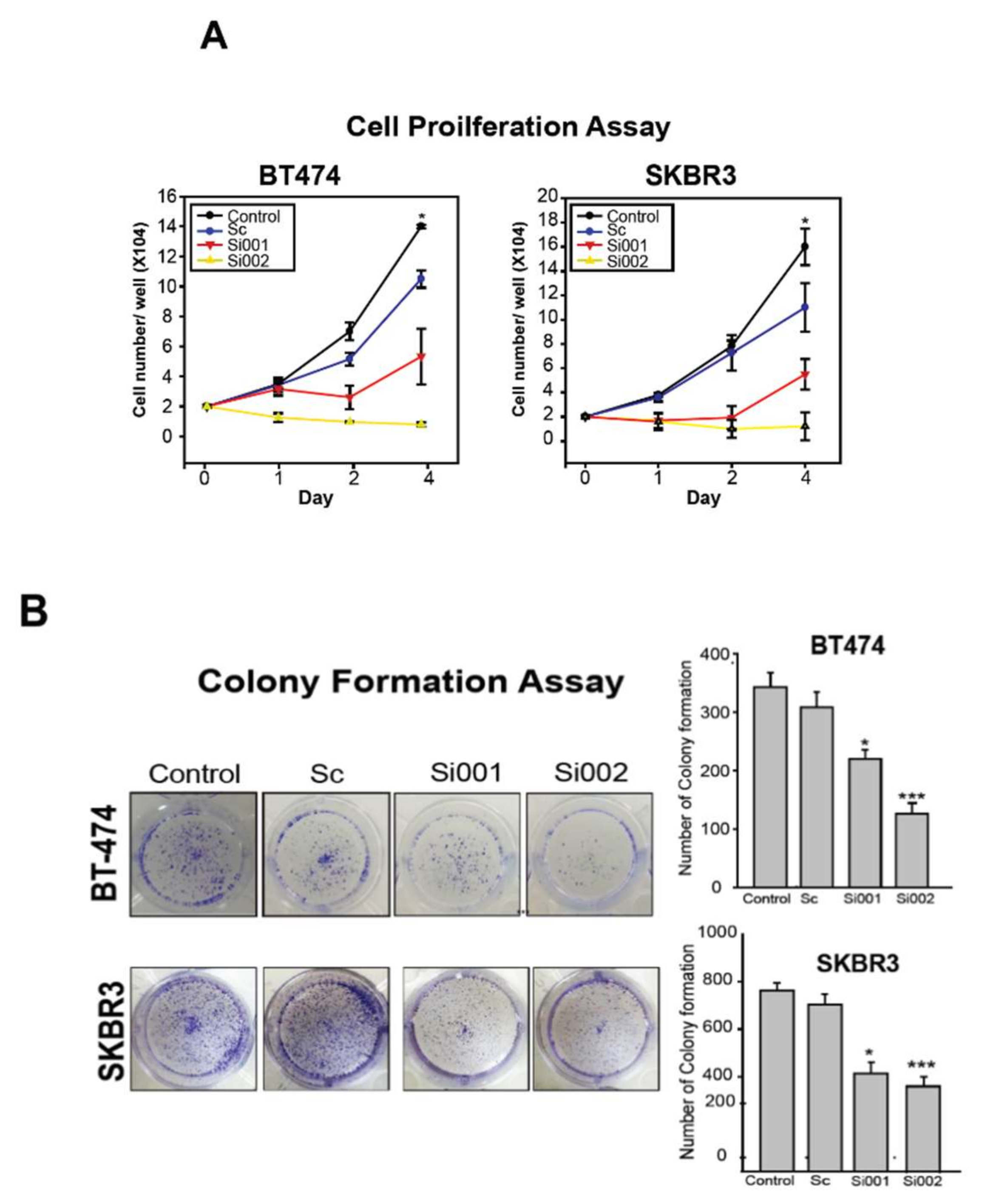
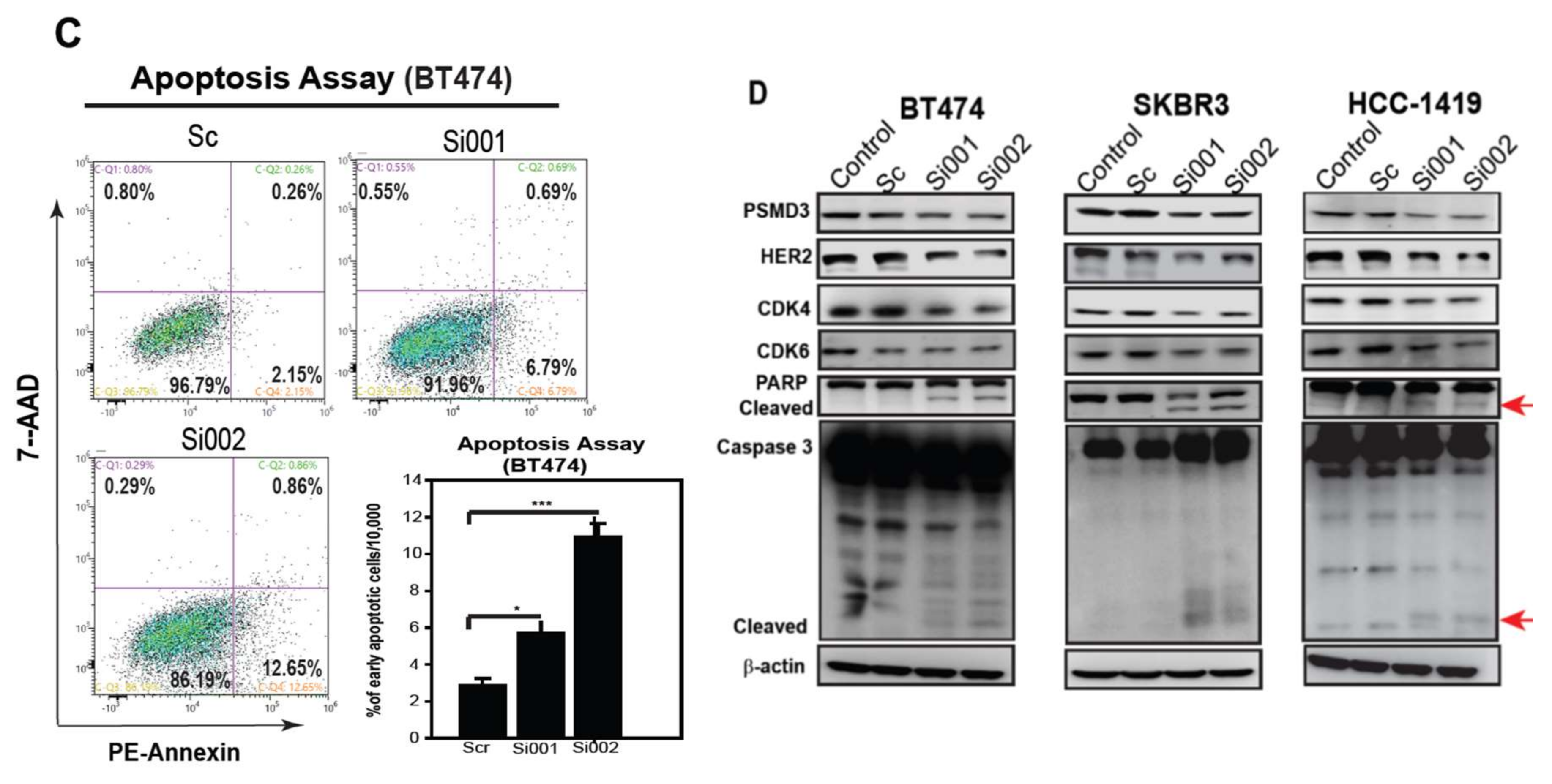
2.5. PSMD3 Silencing Inhibits Cell Proliferation and Induces Cellular Apoptosis
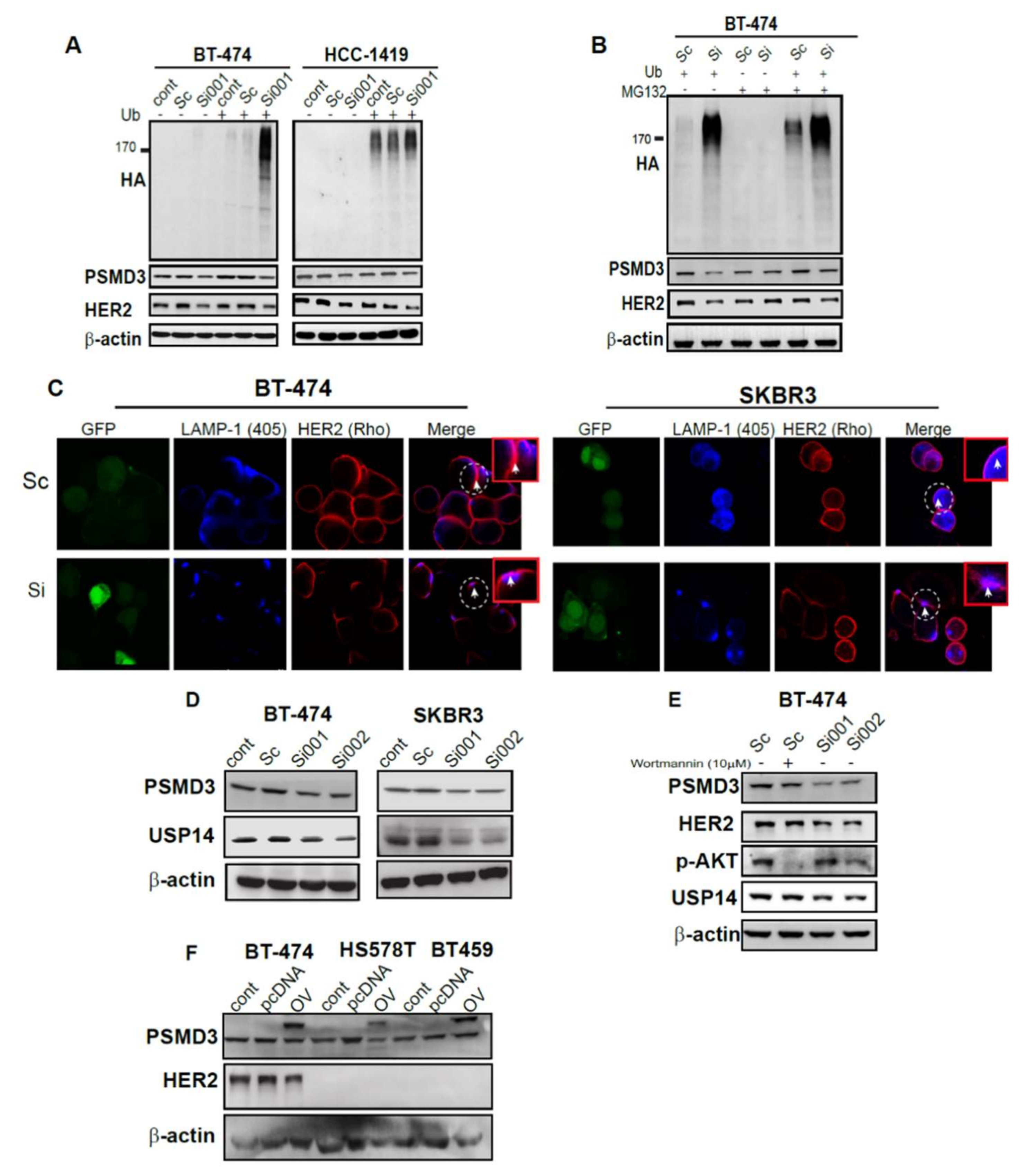
2.6. Silencing of PSMD3 Enhanced Ubiquitination and Lysosomal Process in Correlation with Decrease HER2 Level
2.7. PSMD3 Is Dependent on Proteasome
2.8. Correlation of PSMD3 Expression with Clinicopathological Features in BC
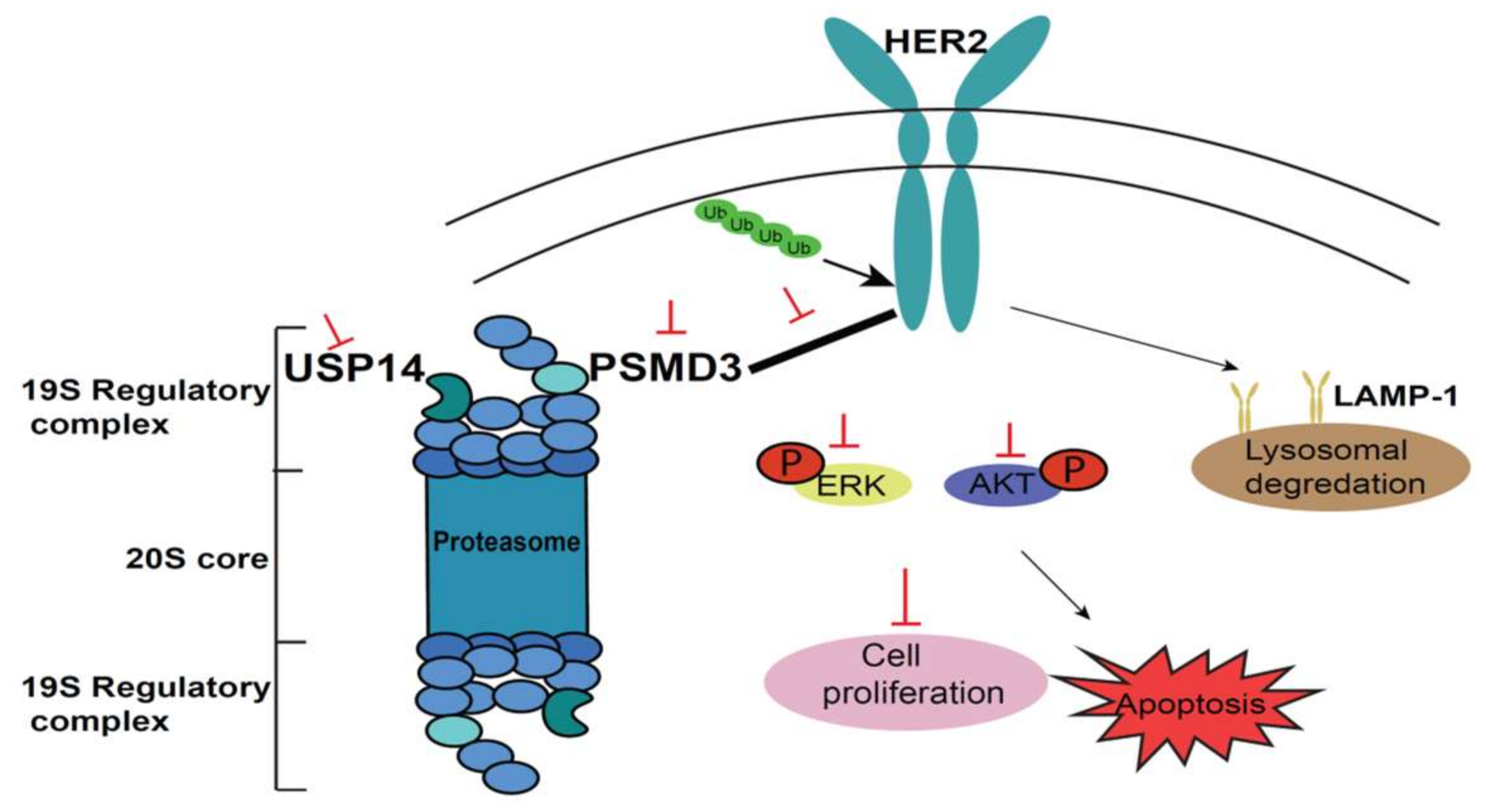
3. Discussions
4. Materials and Methods
4.1. Clinical Patient Samples
4.2. Cell lines and Cell Culture
4.3. RNA Isolation, Conventional PCR, and Real-Time Quantitative PCR
4.4. Oncomine, TCGA and Kaplan-Meier Plotter Data Set Analysis
4.5. Immunohistochemistry (IHC) Staining and Scoring System
4.6. Immunoprecipitation (IP)
4.7. Immunoblotting
4.8. Construction of Plasmids
4.9. Immunofluorescence (IF) Staining and Confocal Microscopy
4.10. Cell Proliferation and Colony Formation Assays
4.11. Apoptotic Assay
4.12. Ubiquitination Assay
4.13. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Torre, L.A.; Bray, F.; Siegel, R.L.; Ferlay, J.; Lortet-Tieulent, J.; Jemal, A. Global cancer statistics, 2012. CA Cancer J. Clin. 2015, 65, 87–108. [Google Scholar] [CrossRef] [PubMed]
- Sorlie, T.; Perou, C.M.; Tibshirani, R.; Aas, T.; Geisler, S.; Johnsen, H.; Hastie, T.; Eisen, M.B.; van de Rijn, M.; Jeffrey, S.S.; et al. Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc. Natl. Acad. Sci. USA 2001, 98, 10869–10874. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Network. Comprehensive molecular portraits of human breast tumours. Nature 2012, 490, 61–70. [Google Scholar] [CrossRef] [PubMed]
- Iqbal, N.; Iqbal, N. Human Epidermal Growth Factor Receptor 2 (HER2) in Cancers: Overexpression and Therapeutic Implications. Mol. Biol. Int. 2014, 2014, 852748. [Google Scholar] [CrossRef] [PubMed]
- Arteaga, C.L.; Engelman, J.A. ERBB receptors: From oncogene discovery to basic science to mechanism-based cancer therapeutics. Cancer Cell 2014, 25, 282–303. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.J.; Seo, A.N.; Kim, E.J.; Jang, M.H.; Kim, Y.J.; Kim, J.H.; Kim, S.W.; Ryu, H.S.; Park, I.A.; Im, S.A.; et al. Prognostic and predictive values of EGFR overexpression and EGFR copy number alteration in HER2-positive breast cancer. Br. J. Cancer 2015, 112, 103–111. [Google Scholar] [CrossRef] [PubMed]
- Smith, I.; Procter, M.; Gelber, R.D.; Guillaume, S.; Feyereislova, A.; Dowsett, M.; Goldhirsch, A.; Untch, M.; Mariani, G.; Baselga, J.; et al. 2-year follow-up of trastuzumab after adjuvant chemotherapy in HER2-positive breast cancer: A randomised controlled trial. Lancet 2007, 369, 29–36. [Google Scholar] [CrossRef]
- Piccart-Gebhart, M.J.; Procter, M.; Leyland-Jones, B.; Goldhirsch, A.; Untch, M.; Smith, I.; Gianni, L.; Baselga, J.; Bell, R.; Jackisch, C.; et al. Trastuzumab after adjuvant chemotherapy in HER2-positive breast cancer. N. Engl. J. Med. 2005, 353, 1659–1672. [Google Scholar] [CrossRef] [PubMed]
- Cameron, D.; Piccart-Gebhart, M.J.; Gelber, R.D.; Procter, M.; Goldhirsch, A.; de Azambuja, E.; Castro, G., Jr.; Untch, M.; Smith, I.; Gianni, L.; et al. 11 years, follow-up of trastuzumab after adjuvant chemotherapy in HER2-positive early breast cancer: Final analysis of the HERceptin Adjuvant (HERA) trial. Lancet 2017, 389, 1195–1205. [Google Scholar] [CrossRef]
- Veeraraghavan, J.; De Angelis, C.; Reis-Filho, J.S.; Pascual, T.; Prat, A.; Rimawi, M.F.; Osborne, C.K.; Schiff, R. De-escalation of treatment in HER2-positive breast cancer: Determinants of response and mechanisms of resistance. Breast 2017, 34 (Suppl. 1), S19–S26. [Google Scholar] [CrossRef]
- Wang, Y.C.; Morrison, G.; Gillihan, R.; Guo, J.; Ward, R.M.; Fu, X.; Botero, M.F.; Healy, N.A.; Hilsenbeck, S.G.; Phillips, G.L.; et al. Different mechanisms for resistance to trastuzumab versus lapatinib in HER2-positive breast cancers—Role of estrogen receptor and HER2 reactivation. Breast Cancer Res. 2011, 13, R121. [Google Scholar] [CrossRef] [PubMed]
- Collins, G.A.; Goldberg, A.L. The Logic of the 26S Proteasome. Cell 2017, 169, 792–806. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.H.; Zhai, B.; Gygi, S.P.; Goldberg, A.L. mTOR inhibition activates overall protein degradation by the ubiquitin proteasome system as well as by autophagy. Proc. Natl. Acad. Sci. USA 2015, 112, 15790–15797. [Google Scholar] [CrossRef] [PubMed]
- Clague, M.J.; Urbe, S. Ubiquitin: Same molecule, different degradation pathways. Cell 2010, 143, 682–685. [Google Scholar] [CrossRef] [PubMed]
- Clague, M.J.; Heride, C.; Urbe, S. The demographics of the ubiquitin system. Trends Cell Biol. 2015, 25, 417–426. [Google Scholar] [CrossRef] [PubMed]
- Ciechanover, A. Intracellular protein degradation: From a vague idea through the lysosome and the ubiquitin-proteasome system and onto human diseases and drug targeting. Bioorg. Med. Chem. 2013, 21, 3400–3410. [Google Scholar] [CrossRef] [PubMed]
- Vilchez, D.; Saez, I.; Dillin, A. The role of protein clearance mechanisms in organismal ageing and age-related diseases. Nat. Commun. 2014, 5, 5659. [Google Scholar] [CrossRef]
- Reyes-Turcu, F.E.; Ventii, K.H.; Wilkinson, K.D. Regulation and cellular roles of ubiquitin-specific deubiquitinating enzymes. Annu. Rev. Biochem. 2009, 78, 363–397. [Google Scholar] [CrossRef] [PubMed]
- Fraile, J.M.; Quesada, V.; Rodriguez, D.; Freije, J.M.; Lopez-Otin, C. Deubiquitinases in cancer: New functions and therapeutic options. Oncogene 2012, 31, 2373–2388. [Google Scholar] [CrossRef]
- Verma, R.; Aravind, L.; Oania, R.; McDonald, W.H.; Yates, J.R., 3rd; Koonin, E.V.; Deshaies, R.J. Role of Rpn11 metalloprotease in deubiquitination and degradation by the 26S proteasome. Science 2002, 298, 611–615. [Google Scholar] [CrossRef]
- Borodovsky, A.; Kessler, B.M.; Casagrande, R.; Overkleeft, H.S.; Wilkinson, K.D.; Ploegh, H.L. A novel active site-directed probe specific for deubiquitylating enzymes reveals proteasome association of USP14. EMBO J. 2001, 20, 5187–5196. [Google Scholar] [CrossRef]
- Mahanic, C.S.; Budhavarapu, V.; Graves, J.D.; Li, G.; Lin, W.C. Regulation of E2 Promoter Binding Factor 1 (E2F1) Transcriptional Activity through a Deubiquitinating Enzyme, UCH37. J. Biol. Chem. 2015, 290, 26508–26522. [Google Scholar] [CrossRef]
- Melvin, A.T.; Woss, G.S.; Park, J.H.; Waters, M.L.; Allbritton, N.L. Measuring Activity in the Ubiquitin-Proteasome System: From Large Scale Discoveries to Single Cells Analysis. Cell Biochem. Biophys. 2013, 67, 75–89. [Google Scholar] [CrossRef]
- Mocciaro, A.; Rape, M. Emerging regulatory mechanisms in ubiquitin-dependent cell cycle control. J. Cell Sci. 2012, 125, 255–263. [Google Scholar] [CrossRef] [PubMed]
- Tu, Y.; Chen, C.; Pan, J.; Xu, J.; Zhou, Z.G.; Wang, C.Y. The Ubiquitin Proteasome Pathway (UPP) in the regulation of cell cycle control and DNA damage repair and its implication in tumorigenesis. Int. J. Clin. Exp. Pathol. 2012, 5, 726–738. [Google Scholar]
- Gadhave, K.; Bolshette, N.; Ahire, A.; Pardeshi, R.; Thakur, K.; Trandafir, C.; Istrate, A.; Ahmed, S.; Lahkar, M.; Muresanu, D.F.; et al. The ubiquitin proteasomal system: A potential target for the management of Alzheimer’s disease. J. Cell. Mol. Med. 2016, 20, 1392–1407. [Google Scholar] [CrossRef]
- Li, Y.; Huang, J.; Zeng, B.; Yang, D.; Sun, J.; Yin, X.; Lu, M.; Qiu, Z.; Peng, W.; Xiang, T.; et al. PSMD2 regulates breast cancer cell proliferation and cell cycle progression by modulating p21 and p27 proteasomal degradation. Cancer Lett. 2018, 430, 109–122. [Google Scholar] [CrossRef] [PubMed]
- Matsuyama, Y.; Suzuki, M.; Arima, C.; Huang, Q.M.; Tomida, S.; Takeuchi, T.; Sugiyama, R.; Itoh, Y.; Yatabe, Y.; Goto, H.; et al. Proteasomal Non-catalytic Subunit PSMD2 as a Potential Therapeutic Target in Association With Various Clinicopathologic Features in Lung Adenocarcinomas. Mol. Carcinog. 2011, 50, 301–309. [Google Scholar] [CrossRef]
- Lin, P.L.; Chang, J.T.; Wu, D.W.; Huang, C.C.; Lee, H. Cytoplasmic localization of Nrf2 promotes colorectal cancer with more aggressive tumors via upregulation of PSMD4. Free Radic. Biol. Med. 2016, 95, 121–132. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.M.; Lin, P.L.; Wu, D.W.; Wang, L.; Huang, C.C.; Lee, H. PSMD4 is a novel therapeutic target in chemoresistant colorectal cancer activated by cytoplasmic localization of Nrf2. Oncotarget 2018, 9, 26342–26352. [Google Scholar] [CrossRef] [PubMed]
- Marx, C.; Yau, C.; Banwait, S.; Zhou, Y.; Scott, G.K.; Hann, B.; Park, J.W.; Benz, C.C. Proteasome-regulated ERBB2 and estrogen receptor pathways in breast cancer. Mol. Pharmacol. 2007, 71, 1525–1534. [Google Scholar] [CrossRef] [PubMed]
- Fagerberg, L.; Hallstrom, B.M.; Oksvold, P.; Kampf, C.; Djureinovic, D.; Odeberg, J.; Habuka, M.; Tahmasebpoor, S.; Danielsson, A.; Edlund, K.; et al. Analysis of the human tissue-specific expression by genome-wide integration of transcriptomics and antibody-based proteomics. Mol. Cell. Proteom. 2014, 13, 397–406. [Google Scholar] [CrossRef]
- Zheng, J.S.; Arnett, D.K.; Parnell, L.D.; Lee, Y.C.; Ma, Y.; Smith, C.E.; Richardson, K.; Li, D.; Borecki, I.B.; Ordovas, J.M.; et al. Genetic variants at PSMD3 interact with dietary fat and carbohydrate to modulate insulin resistance. J. Nutr. 2013, 143, 354–361. [Google Scholar] [CrossRef]
- Iio, E.; Matsuura, K.; Nishida, N.; Maekawa, S.; Enomoto, N.; Nakagawa, M.; Sakamoto, N.; Yatsuhashi, H.; Kurosaki, M.; Izumi, N.; et al. Genome-wide association study identifies a PSMD3 variant associated with neutropenia in interferon-based therapy for chronic hepatitis C. Hum. Genet. 2015, 134, 279–289. [Google Scholar] [CrossRef]
- Kotoula, V.; Bobos, M.; Alexopoulou, Z.; Papadimitriou, C.; Papadopoulou, K.; Charalambous, E.; Tsolaki, E.; Xepapadakis, G.; Nicolaou, I.; Papaspirou, I.; et al. Adjusting breast cancer patient prognosis with non-HER2-gene patterns on chromosome 17. PLoS ONE 2014, 9, e103707. [Google Scholar] [CrossRef]
- Sahlberg, K.K.; Hongisto, V.; Edgren, H.; Makela, R.; Hellstrom, K.; Due, E.U.; Vollan, H.K.M.; Sahlberg, N.; Wolf, M.; Borresen-Dale, A.L.; et al. The HER2 amplicon includes several genes required for the growth and survival of HER2 positive breast cancer cells. Mol. Oncol. 2013, 7, 392–401. [Google Scholar] [CrossRef]
- Pang, Y.; Ball, G.; Rakha, E.; Powe, D.; Caldas, C.; Ellis, I.; Green, A. SOX11 and PSMD3 Expression in HER2 Positive Breast Cancer. J. Pathol. 2010, 222, S3. [Google Scholar] [CrossRef]
- Fararjeh, A.S.; Tu, S.H.; Chen, L.C.; Liu, Y.R.; Lin, Y.K.; Chang, H.L.; Chang, H.W.; Wu, C.H.; Hwang-Verslues, W.W.; Ho, Y.S. The impact of the effectiveness of GATA3 as a prognostic factor in breast cancer. Hum. Pathol. 2018, 80, 219–230. [Google Scholar] [CrossRef]
- Lin, J.H.; Tu, S.H.; Chen, L.C.; Huang, C.C.; Chang, H.L.; Cheng, T.C.; Chang, H.W.; Wu, C.H.; Wu, H.C.; Ho, Y.S. Oestrogen receptor-regulated glutathione S-transferase mu 3 expression attenuates hydrogen peroxide-induced cytotoxicity, which confers tamoxifen resistance on breast cancer cells. Breast Cancer Res. Treat. 2018, 172, 45–59. [Google Scholar] [CrossRef]
- Li, Y.; Huang, J.; Sun, J.; Xiang, S.; Yang, D.; Ying, X.; Lu, M.; Li, H.; Ren, G. The transcription levels and prognostic values of seven proteasome alpha subunits in human cancers. Oncotarget 2017, 8, 4501–4519. [Google Scholar] [CrossRef] [PubMed]
- Fejzo, M.S.; Anderson, L.; Chen, H.W.; Guandique, E.; Kalous, O.; Conklin, D.; Slamon, D.J. Proteasome ubiquitin receptor PSMD4 is an amplification target in breast cancer and may predict sensitivity to PARPi. Genes Chromosomes Cancer 2017, 56, 589–597. [Google Scholar] [CrossRef]
- Bader, M.; Steller, H. Regulation of cell death by the ubiquitin-proteasome system. Curr. Opin. Cell Biol. 2009, 21, 878–884. [Google Scholar] [CrossRef]
- Canton, I.; Battaglia, G. Endocytosis at the nanoscale. Chem. Soc. Rev. 2012, 41, 2718–2739. [Google Scholar] [CrossRef]
- Lerdrup, M.; Bruun, S.; Grandal, M.V.; Roepstorff, K.; Kristensen, M.M.; Hommelgaard, A.M.; van Deurs, B. Endocytic down-regulation of ErbB2 is stimulated by cleavage of its C-terminus. Mol. Biol. Cell 2007, 18, 3656–3666. [Google Scholar] [CrossRef] [PubMed]
- Bertelsen, V.; Stang, E. The Mysterious Ways of ErbB2/HER2 Trafficking. Membranes 2014, 4, 424–446. [Google Scholar] [CrossRef]
- Lee, H.; Dam, D.H.; Ha, J.W.; Yue, J.; Odom, T.W. Enhanced Human Epidermal Growth Factor Receptor 2 Degradation in Breast Cancer Cells by Lysosome-Targeting Gold Nanoconstructs. ACS Nano 2015, 9, 9859–9867. [Google Scholar] [CrossRef] [PubMed]
- Sorokin, A.V.; Kim, E.R.; Ovchinnikov, L.P. Proteasome system of protein degradation and processing. Biochemistry 2009, 74, 1411–1442. [Google Scholar] [CrossRef] [PubMed]
- Komada, M. Controlling receptor downregulation by ubiquitination and deubiquitination. Curr. Drug Discov. Technol. 2008, 5, 78–84. [Google Scholar] [CrossRef]
- Ramakrishna, S.; Suresh, B.; Baek, K.H. The role of deubiquitinating enzymes in apoptosis. Cell. Mol. Life Sci. 2011, 68, 15–26. [Google Scholar] [CrossRef]
- Wilson, S.M.; Bhattacharyya, B.; Rachel, R.A.; Coppola, V.; Tessarollo, L.; Householder, D.B.; Fletcher, C.F.; Miller, R.J.; Copeland, N.G.; Jenkins, N.A. Synaptic defects in ataxia mice result from a mutation in Usp14, encoding a ubiquitin-specific protease. Nat. Genet. 2002, 32, 420–425. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wang, J.; Zhong, J.; Deng, Y.; Xi, Q.; He, S.; Yang, S.; Jiang, L.; Huang, M.; Tang, C.; et al. Ubiquitin-specific protease 14 (USP14) regulates cellular proliferation and apoptosis in epithelial ovarian cancer. Med. Oncol. 2015, 32, 379. [Google Scholar] [CrossRef] [PubMed]
- Huang, G.; Li, L.M.; Zhou, W.P. USP14 activation promotes tumor progression in hepatocellular carcinoma. Oncol. Rep. 2015, 34, 2917–2924. [Google Scholar] [CrossRef] [PubMed]
- Liao, Y.; Liu, N.; Hua, X.; Cai, J.; Xia, X.; Wang, X.; Huang, H.; Liu, J. Proteasome-associated deubiquitinase ubiquitin-specific protease 14 regulates prostate cancer proliferation by deubiquitinating and stabilizing androgen receptor. Cell Death Dis. 2017, 8, e2585. [Google Scholar] [CrossRef]
- Liao, Y.; Xia, X.; Liu, N.; Cai, J.; Guo, Z.; Li, Y.; Jiang, L.; Dou, Q.P.; Tang, D.; Huang, H.; et al. Growth arrest and apoptosis induction in androgen receptor-positive human breast cancer cells by inhibition of USP14-mediated androgen receptor deubiquitination. Oncogene 2018, 37, 1896–1910. [Google Scholar] [CrossRef] [PubMed]
- Xu, D.; Shan, B.; Lee, B.H.; Zhu, K.; Zhang, T.; Sun, H.; Liu, M.; Shi, L.; Liang, W.; Qian, L.; et al. Phosphorylation and activation of ubiquitin-specific protease-14 by Akt regulates the ubiquitin-proteasome system. Elife 2015, 4, e10510. [Google Scholar] [CrossRef] [PubMed]
- Cui, X.; Jing, X.; Yi, Q.; Long, C.; Tian, J.; Zhu, J. Clinicopathological and prognostic significance of SDC1 overexpression in breast cancer. Oncotarget 2017, 8, 111444–111455. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameters | Categories | PSMD3 Low mRNA(n) | PSMD3 High mRNA(n) | p-Value |
|---|---|---|---|---|
| HER2 status | Positive | 52 | 133 | 0.000 *** |
| Negative | 164 | 163 | ||
| NA = 700 | ||||
| ER status | Positive | 151 | 205 | 0.874 |
| Negative | 65 | 91 | ||
| †NA = 700 | ||||
| TNBC | TNBC | 49 | 49 | 0.082 |
| Non-TNBC | 167 | 247 | ||
| †NA = 700 | ||||
| Tumor size | T1 | 173 | 135 | 0.000 *** |
| T2 | 340 | 366 | ||
| T3 | 75 | 75 | ||
| T4 | 6 | 30 | ||
| †NA = 12 | ||||
| Node | N0 | 288 | 273 | 0.787 |
| N1 | 203 | 213 | ||
| N2 | 63 | 69 | ||
| N3 | 53 | 50 | ||
| Stage | I | 120 | 89 | 0.036 * |
| II | 343 | 348 | ||
| III | 130 | 148 | ||
| IV | 13 | 21 | ||
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fararjeh, A.S.; Chen, L.-C.; Ho, Y.-S.; Cheng, T.-C.; Liu, Y.-R.; Chang, H.-L.; Chang, H.-W.; Wu, C.-H.; Tu, S.-H. Proteasome 26S Subunit, non-ATPase 3 (PSMD3) Regulates Breast Cancer by Stabilizing HER2 from Degradation. Cancers 2019, 11, 527. https://doi.org/10.3390/cancers11040527
Fararjeh AS, Chen L-C, Ho Y-S, Cheng T-C, Liu Y-R, Chang H-L, Chang H-W, Wu C-H, Tu S-H. Proteasome 26S Subunit, non-ATPase 3 (PSMD3) Regulates Breast Cancer by Stabilizing HER2 from Degradation. Cancers. 2019; 11(4):527. https://doi.org/10.3390/cancers11040527
Chicago/Turabian StyleFararjeh, Abdulfattah Salah, Li-Ching Chen, Yuan-Soon Ho, Tzu-Chun Cheng, Yun-Ru Liu, Hang-Lung Chang, Hui-Wen Chang, Chih-Hsiung Wu, and Shih-Hsin Tu. 2019. "Proteasome 26S Subunit, non-ATPase 3 (PSMD3) Regulates Breast Cancer by Stabilizing HER2 from Degradation" Cancers 11, no. 4: 527. https://doi.org/10.3390/cancers11040527