The Presence of Concomitant Mutations Affects the Activity of EGFR Tyrosine Kinase Inhibitors in EGFR-Mutant Non-Small Cell Lung Cancer (NSCLC) Patients

 , , , ,
, , , , 

Abstract
:1. Introduction
2. Results
2.1. Patients’ Characteristics
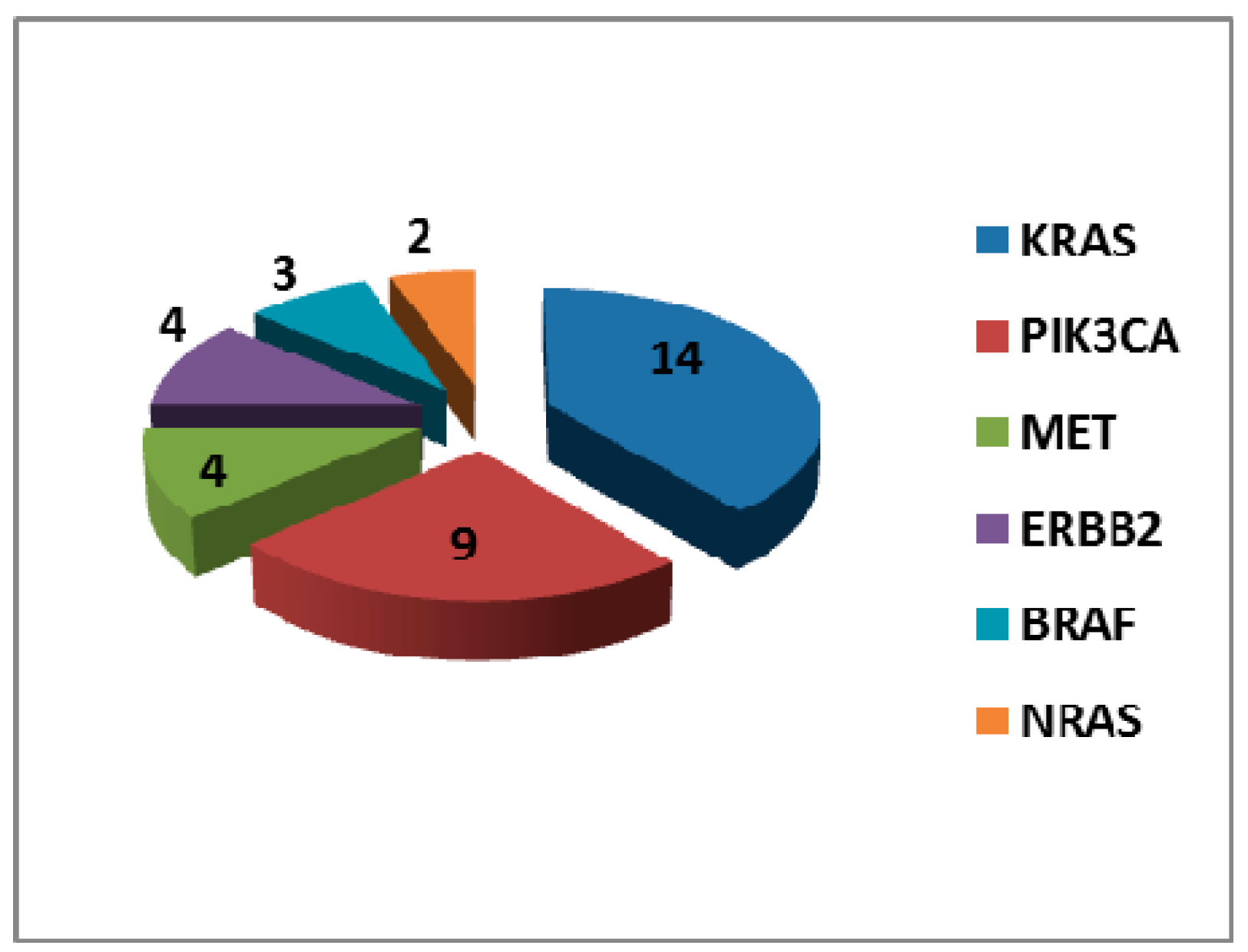
2.2. Mutational Landscape of EGFR Mutant Tumours
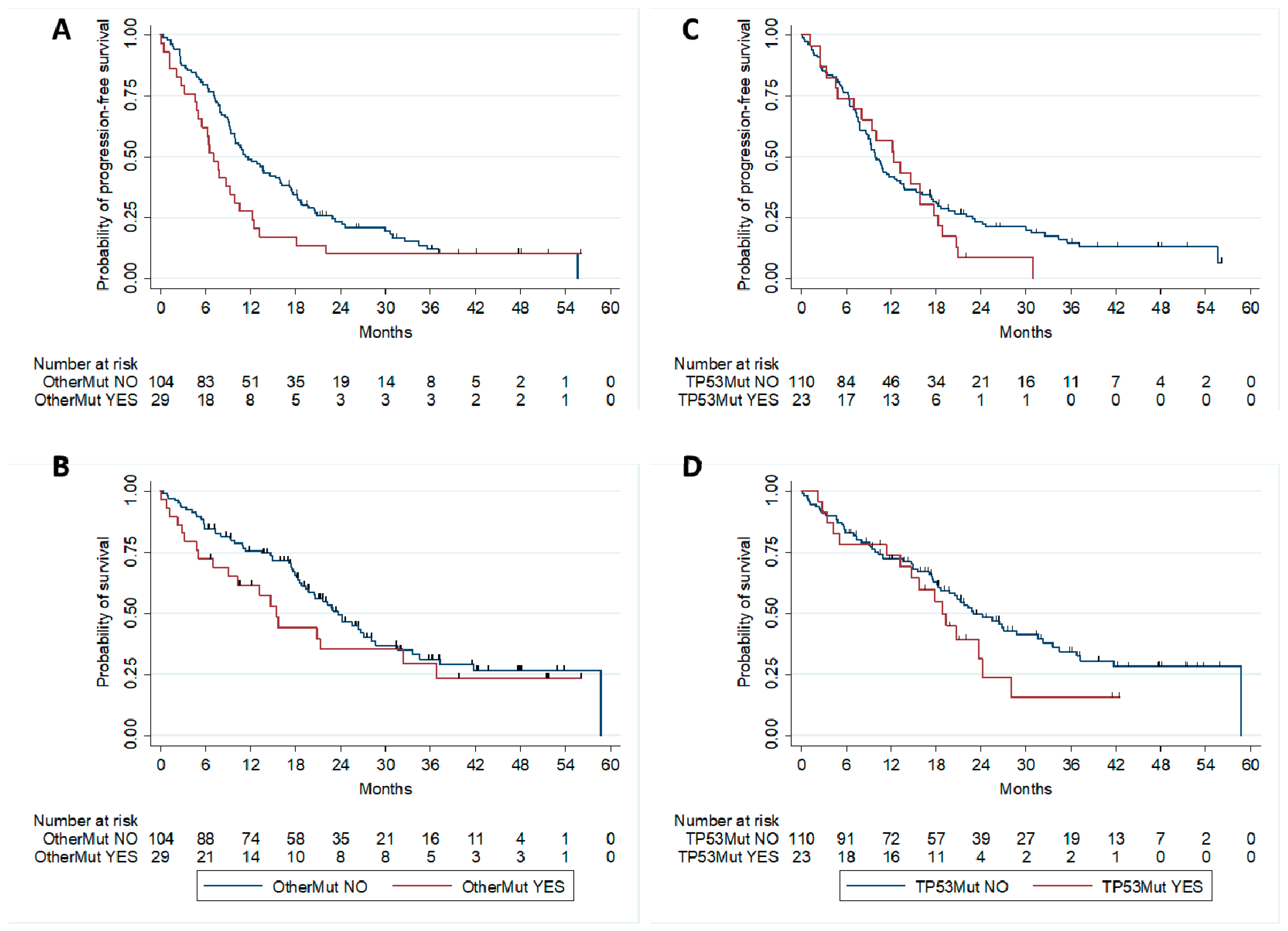
2.3. Correlation with Patients’ Outcome
3. Discussion
4. Materials and Methods
4.1. Study Design
4.2. Mutational Analysis
4.3. Study Treatment and Assessments
4.4. Statistical Analyses
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Rossi, A.; Pasquale, R.; Esposito, C.; Normanno, N. Should epidermal growth factor receptor tyrosine kinase inhibitors be considered ideal drugs for the treatment of selected advanced non-small cell lung cancer patients. Cancer Treat. Rev. 2013, 39, 489–497. [Google Scholar] [CrossRef] [PubMed]
- Camidge, D.R.; Pao, W.; Sequist, L.V. Acquired resistance to TKIs in solid tumours: Learning from lung cancer. Nat. Rev. Clin. Oncol. 2014, 11, 473–481. [Google Scholar] [CrossRef] [PubMed]
- Jamal-Hanjani, M.; Wilson, G.A.; McGranahan, N.; Birkbak, N.J.; Watkins, T.B.K.; Veeriah, S.; Shafi, S.; Jhonson, H.D.; Mitter, R.; Rosenthal, R.; et al. Tracking the evolution of non-small-cell lung cancer. N. Engl. J. Med. 2017, 376, 2109–2121. [Google Scholar] [CrossRef] [PubMed]
- De Luca, A.; Normanno, N. Predictive biomarkers to tyrosine kinase inhibitors for the epidermal growth factor receptor in non-small-cell lung cancer. Curr. Drug. Targets 2010, 11, 851–864. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Li, L.; Zhu, Y.; Huang, C.; Qin, Y.; Liu, H.; Ren-Heidenreich, L.; Shi, B.; Ren, H.; Chu, X.; et al. Coexistence of EGFR with KRAS, or BRAF, or PIK3CA somatic mutations in lung cancer: A comprehensive mutation profiling from 5125 Chinese cohorts. Br. J. Cancer 2014, 110, 2812–2820. [Google Scholar] [CrossRef] [PubMed]
- Scheffler, M.; Bos, M.; Gardizi, M.; Konig, K.; Michels, S.; Fassunke, J.; Heydt, C.; Kunstlinger, H.; Ihle, M.; Veckeroth, F.; et al. PIK3CA mutations in non-small cell lung cancer (NSCLC): Genetic heterogeneity, prognostic impact and incidence of prior malignancies. Oncotarget 2015, 6, 1315–1326. [Google Scholar] [CrossRef] [PubMed]
- Bria, E.; Pilotto, S.; Amato, E.; Fassan, M.; Novello, S.; Peretti, U.; Vavala, T.; Kingspergher, S.; Righi, L.; Santo, A.; et al. Molecular heterogeneity assessment by next-generation sequencing and response to gefitinib of EGFR mutant advanced lung adenocarcinoma. Oncotarget 2015, 6, 12783–12795. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ju, L.; Han, M.; Zhao, C.; Li, X. EGFR, KRAS and ROS1 variants coexist in a lung adenocarcinoma patient. Lung Cancer 2016, 95, 94–97. [Google Scholar] [CrossRef] [PubMed]
- Lim, S.M.; Kim, H.R.; Cho, E.K.; Min, Y.J.; Ahn, J.S.; Ahn, M.J.; Park, K.; Cho, B.C.; Lee, J.H.; Jeong, H.C.; et al. Targeted sequencing identifies genetic alterations that confer primary resistance to EGFR tyrosine kinase inhibitor (Korean Lung Cancer Consortium). Oncotarget 2016, 7, 36311–36320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, T.; Lee, B.; Choi, Y.L.; Han, J.; Ahn, M.J.; Um, S.W. Non-small cell lung cancer with concomitant EGFR, KRAS, and ALK mutation: Clinicopathologic features of 12 cases. J. Pathol. Transl. Med. 2016, 50, 197–203. [Google Scholar] [CrossRef] [PubMed]
- Choughule, A.; Sharma, R.; Trivedi, V.; Thavamani, A.; Noronha, V.; Joshi, A.; Desai, S.; Chandrani, P.; Sundaram, P.; Utture, S.; et al. Coexistence of KRAS mutation with mutant but not wild-type EGFR predicts response to tyrosine-kinase inhibitors in human lung cancer. Br. J. Cancer 2014, 111, 2203–2204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tiseo, M.; Bersanelli, M.; Perrone, F.; Tamborini, E.; Settanni, G.; Busico, A.; Rossi, G.; Ardizzoni, A.; Pelosi, G. Different clinical effects upon separate inhibition of coexisting EGFR and PI3KCA mutations in a lung adenocarcinoma patient. Lung Cancer 2015, 87, 204–206. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.A.; Jordan, E.; Ni, A.; Feldman, D.; Rodriguez, C.; Kim, H.R.; Kris, G.M.; Solit, B.D.; Berger, F.M.; Ladanyi, M.; et al. Concurrent genetic alterations identified by next-generation sequencing in pre-treatment, metastatic EGFR-mutant lung cancers. J. Clin. Oncol. 2016, 34, 9053. [Google Scholar] [CrossRef]
- Canale, M.; Petracci, E.; Delmonte, A.; Chiadini, E.; Dazzi, C.; Papi, M.; Cappeli, L.; Casanova, C.; De Luigi, N.; Mariotti, N.; et al. Impact of TP53 mutations on outcome in EGFR-mutated patients treated with first-line tyrosine kinase inhibitors. Clin. Cancer Res. 2017, 23, 2195–2202. [Google Scholar] [CrossRef] [PubMed]
- Labbe, C.; Korpanty, G.; Tomasini, P.; Doherty, M.; Mascaux, C.; Jao, K.; Pitcher, B.; Pintilie, M.; Leighle, B.N.; Feld, R.; et al. Prognostic and predictive effects of TP53 mutation in patients with EGFR-mutated non-small cell lung cancer (NSCLC). J. Clin. Oncol. 2016, 34, 11585. [Google Scholar] [CrossRef]
- Roeper, J.; Netchaeva, M.; Lueers, A.C.; Regina, P.; Sriba, D.; Willborn, K.; Stropiep, U.; Hallas, C.; Falk, M.; Tiemann, M.; et al. P53 Non-disruptive mutation is a negative Predictive factor for OS and PFS in EGFR M+ NSCLC treated with TKI. In Proceedings of the World Conference on Lung Cancer, Vienna, Austria, 11 October 2016; p. ID5879. [Google Scholar]
- Elamin, Y.Y.; Rinsurongkawong, W.; Tran, H.T.; Gold, K.A.; Lewis, J.; Roarty, E.; Futreal, A.; Zhang, J.; Heymach, J. The impact of genomic landscape of EGFR Mutant NSCLC on response to targeted and immune therapy. J. Thorac. Oncol. 2016, 12, S423–S424. [Google Scholar] [CrossRef]
- Alizadeh, A.A.; Aranda, V.; Bardelli, A.; Blanpain, C.; Bock, C.; Borowski, C.; Caldas, C.; Califano, A.; Doherty, M.; Elsner, M.; et al. Toward understanding and exploiting tumor heterogeneity. Nat. Med. 2015, 21, 846–853. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McGranahan, N.; Favero, F.; De Bruin, E.C.; Birkbak, N.J.; Szallasi, Z.; Swanton, C. Clonal status of actionable driver events and the timing of mutational processes in cancer evolution. Sci. Transl. Med. 2015, 7, 283ra54. [Google Scholar] [CrossRef] [PubMed]
- Normanno, N.; Rachiglio, A.M.; Lambiase, M.; Martinelli, E.; Fenizia, F.; Esposito, C.; Roma, C.; Troiani, T.; Rizzi, D.; Tatangelo, F.; et al. Heterogeneity of KRAS, NRAS, BRAF and PIK3CA mutations in metastatic colorectal cancer and potential effects on therapy in the CAPRI GOIM trial. Ann. Oncol. 2015, 26, 1710–1714. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andor, N.; Graham, T.A.; Jansen, M.; Xia, L.C.; Aktipis, C.A.; Petritsch, C.; Ji, P.H.; Maley, C.C. Pan-cancer analysis of the extent and consequences of intratumor heterogeneity. Nat Med. 2016, 22, 105–113. [Google Scholar] [CrossRef] [PubMed]
- Spigel, D.R.; Schrock, A.B.; Fabrizio, D.; Frampton, G.M.; Sun, J.; He, J.; Johnson, L.M.; Bauer, M.T.; Kalemkerian, P.G.; Raez, E.L.; et al. Total mutation burden (TMB) in lung cancer (LC) and relationship with response to PD-1/PD-L1 targeted therapies. J.Clin. Oncol. 2016, 34, 9017. [Google Scholar] [CrossRef]
- Hong, S.; Gao, F.; Fu, S.; Wang, Y.; Fang, W.; Huang, Y.; Zhang, L. Concomitant genetic alterations with response to treatment and epidermal growth factor receptor tyrosine Kinase inhibitors in patients with EGFR-Mutant advanced non-small cell lung cancer. JAMA Oncol. 2018, 4, 739–742. [Google Scholar] [CrossRef] [PubMed]
- Jakobsen, J.N.; Santoni-Rugiu, E.; Grauslund, M.; Melchior, L.; Sorensen, J.B. Concomitant driver mutations in advanced EGFR-mutated non-small-cell lung cancer and their impact on erlotinib treatment. Oncotarget 2018, 9, 26195–26208. [Google Scholar] [CrossRef] [PubMed]
- Ohashi, K.; Sequist, L.V.; Arcila, M.E.; Moran, T.; Chmielecki, J.; Lin, Y.L.; Pan, Y.; Wang, L.; de Stanchina, E.; Shein, K.; et al. Lung cancers with acquired resistance to EGFR inhibitors occasionally harbor BRAF gene mutations but lack mutations in KRAS, NRAS, or MEK1. Proc. Natl. Acad. Sci. USA 2012, 109, E2127–E2133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Del Re, M.; Tiseo, M.; Bordi, P.; D’Incecco, A.; Camerini, A.; Petrini, I.; Lucchesi, M.; Inno, A.; Spada, D.; Vasile, E.; et al. Contribution of KRAS mutations and c.2369C > T (p.T790M) EGFR to acquired resistance to EGFR-TKIs in EGFR mutant NSCLC: A study on circulating tumor DNA. Oncotarget 2017, 8, 13611–13619. [Google Scholar] [CrossRef] [PubMed]
- Chabon, J.J.; Simmons, A.D.; Lovejoy, A.F.; Esfahani, M.S.; Newman, A.M.; Haringsma, H.J.; Kurtz, M.D.; Stehr, H.; Schere, F.; Karlovich, A.C.; et al. Circulating tumour DNA profiling reveals heterogeneity of EGFR inhibitor resistance mechanisms in lung cancer patients. Nat Commun. 2016, 7, 11815. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, H.A.; Arcila, M.E.; Hellmann, M.D.; Kris, M.G.; Ladanyi, M.; Riely, G.J. Poor response to erlotinib in patients with tumors containing baseline EGFR T790M mutations found by routine clinical molecular testing. Ann. Oncol. 2014, 25, 423–428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maheswaran, S.; Sequist, L.V.; Nagrath, S.; Ulkus, L.; Brannigan, B.; Collura, C.V.; Inserra, E.; Diederich, S.; Iafrate, J.; Bell, W.D.; et al. Detection of mutations in EGFR in circulating lung-cancer cells. N. Engl. J. Med. 2008, 359, 366–377. [Google Scholar] [CrossRef] [PubMed]
- Su, K.Y.; Chen, H.Y.; Li, K.C.; Kuo, M.L.; Yang, J.C.H.; Chan, W.K.; Ho, B.C.; Chang, G.C.; Shih, J.Y.; Yu, S.L.; et al. Pretreatment epidermal growth factor receptor (EGFR) T790M mutation predicts shorter EGFR tyrosine kinase inhibitor response duration in patients with non-small-cell lung cancer. J. Clin. Oncol. 2012, 30, 433–440. [Google Scholar] [CrossRef] [PubMed]
- Costa, C.; Molina, M.A.; Drozdowskyj, A.; Gimenez-Capitan, A.; Bertran-Alamillo, J.; Karachaliou, N.; Geravis, R.; Massuti, B.; Wei, J.; Moran, T.; et al. The impact of EGFR T790M mutations and BIM mRNA expression on outcome in patients with EGFR-mutant NSCLC treated with erlotinib or chemotherapy in the randomized phase III EURTAC trial. Clin. Cancer Res. 2014, 20, 2001–2010. [Google Scholar] [CrossRef] [PubMed]
- Morabito, A.; Costanzo, R.; Rachiglio, A.M.; Pasquale, R.; Sandomenico, C.; Franco, R.; Montanino, A.; De Lutio, E.; Rocco, G.; Normanno, N. Activity of gefitinib in a non–small-cell lung cancer patient with both activating and resistance EGFR mutations. J. Thorac. Oncol. 2013, 8, e59–e60. [Google Scholar] [CrossRef] [PubMed]
- Tops, B.B.; Normanno, N.; Kurth, H.; Amato, E.; Mafficini, A.; Rieber, N.; Le Corre, D.; Rachinglio, A.M.; Reiman, A.; Sheli, O.; et al. Development of a semi-conductor sequencing-based panel for genotyping of colon and lung cancer by the Onconetwork consortium. BMC Cancer. 2015, 15, 26. [Google Scholar] [CrossRef] [PubMed]
- Takagi, Y.; Osada, H.; Kuroishi, T.; Mitsudomi, T.; Kondo, M.; Niimi, T.; Saji, S.; Gazdar, A.F.; Takahashi, T.; Minna, J.D.; et al. p53 mutations in non-small-cell lung cancers occurring in individuals without a past history of active smoking. Br. J. Cancer 1998, 77, 1568–1572. [Google Scholar] [CrossRef] [PubMed]
- Husgafvel-Pursiainen, K.; Boffetta, P.; Kannio, A.; Nyberg, F.; Pershagen, G.; Mukeria, A.; Constantinescu, V.; Fortes, C.; Benhamou, S. p53 Mutations and exposure to environmental tobacco smoke in a multicenter study on lung cancer. Cancer Res. 2000, 60, 2906–2911. [Google Scholar] [PubMed]
- Vahakangas, K.H.; Bennett, W.P.; Castren, K.; Welsh, J.A.; Khan, M.A.; Blomeke, B.; Alavanja, C.R.M.; Harris, C.C. p53 and K-ras mutations in lung cancers from former and never-smoking women. Cancer Res. 2001, 61, 4350–4356. [Google Scholar] [PubMed]



{kind=link}
{kind=link}
| Characteristics | All (N = 133) | Pts without Other Mutations (N = 104) | Pts with Other Mutations (N = 29) | p-Value |
|---|---|---|---|---|
| Age, median (range) | 71 (41–92) | 71 (41–92) | 69 (42–84) | 0.31 * |
| Gender, n (%) Male Female | 41 (31) 92 (69) | 32 (31) 72 (69) | 9 (31) 20 (69) | 0.98 § |
| Smoking habits, n (%) Never smoker Ever smoker Unknown | 81 (61) 51 (38) 1 (1) | 3 (61) 40 (39) 1 (<1) | 18 (62) 11 (38) - | 0.93 § |
| EGFR mutation type, n (%) Exon 19 del p.L858R Other | 83 (62) 39 (29) 11 (8) | 66 (63) 28 (27) 10 (10) | 17 (59) 11 (38) 1 (3) | 0.36 § |
| 1st line EGFR TKI Gefitinib Erlotinib Afatinib | 114 (86) 11 (8) 8 (6) | 91 (87) 8 (8) 5 (5) | 23 (79) 3 (10) 3 (10) | 0.47 § |
| PatientID | EGFR | KRAS | PFS | BR | ||
|---|---|---|---|---|---|---|
| NGS (VAF) | ddPCR (VAF) | NGS (VAF) | ddPCR (VAF) | |||
| 1512# | p.E746_A750del (40%) | - | p.Gly12Cys (3%) | Codon 12/13 mutation (1.93%) | 13.19 | PR |
| 1616# | p.E746_A750del (58.9%) | - | p.Gly13Asp (11.8%) | Codon 12/13 mutation (10%) | 12.3 | PR |
| 3426# | mutation not detected | Ex19 del (2.5%) | p.Gly12Asp (38%) | Codon 12/13 mutation (33%) | 4.83 | SD |
| 3981# | mutation not detected | Ex19 del (1.2%) | p.Gly12Cys (15%) | Codon 12/13 mutation (12%) | 2.7 | PD |
| 4733# | p.E746_A750 > DP (2.6%) | - | p.Gly12Ala (10.7%) | Codon 12/13 mutation (9.3%) | 0.43 | PD |
| 4840# | mutation not detected | Ex19 del (0,5%) 1 | p.Gly13Cys (10.1%) | Codon 12/13 mutation (0.4%) 1 | 2.14 | PD |
| 4990# | mutation not detected | Ex19 del (0.7%) | p.Gly13Cys (28%) | Codon 12/13 mutation (24%) | 1.18 | PD |
| 5074# | p.E746_A750del (12.8%); p.L858R (16.9%) | - | p.Gly12Cys (3.3%) | Codon 12/13 mutation (0.13%) | 3.26 | PD |
| 5374# | mutation not detected | Ex19 del (1.8%) | p.Gly12Cys (13.4%) | Codon 12/13 mutation (11%) | 4.64 | PR |
| 6541# | p.E746_A750del (47.2%) | - | p.Gly13Asp (12.7%) | Codon 12/13 mutation (11.3%) | 0.06 | NE |
| 6545# | p.L858R (75%) | - | p.Ala59Thr (6.2%) | Tissue and plasma not available | 9.87 | SD |
| 6548# | p.L858R (55.9%) | - | p.Gln61His (3.3%) | p.Gln61His (0.43%) | 6.48 | PD |
| 7567# | p.L858R (35.4%); p.T790M (0.9%) | - | p.Gly12Cys (9.2%) | Codon 12/13 mutation (8.7%) | 12.43 | PR |
| 7964# | p.E746_A750del (56.4%) | - | p.Ala146Thr (2%) | p.Ala146Thr (0.4%) | 51.58 | CR |
| No Other Mutation (n = 104) | Any Other Mutation (n = 29) | KRAS MUT (n = 14) | NRAS MUT (n = 2) | BRAF MUT (n = 3) | PIK3CA MUT (n = 9) | ERBB2 MUT (n = 4) | MET MUT (n = 4) | |
|---|---|---|---|---|---|---|---|---|
| Objective Response | ||||||||
| Responder, N (%) | 71 (68.3%) | 17 (58.6%) | 6 (42.9%) | 2 (100%) | 0 | 7 (77.8%) | 1 (25.0%) | 4 (100.0%) |
| Non responder, n (%) | 33 (31.7%) | 12 (41.4%) | 8 (57.1%) | 0 | 3 (100.0%) | 2 (22.2%) | 3 (75.0%) | 0 |
| PFS, months (95% CI) | 11.3 (9.4–15.9) | 7.0 (4.8–9.9) | 4.6 (1.2–12.3) | NA * | 3.3 (0.4–NR) | 8.7 (5.5–NR) | 3.3 (1.2–NR) | 6.4 (6.2–NR) |
| OS, months (95% CI) | 23.7 (19.4–28. 1) | 15.5 (7.0–32.4) | 5.1 (1.2–20.8) | NA * | 3.3 (0.8–NR) | 36.8 (9.1–NR) | 3.3 (2.2–NR) | 32.4 (10.3–NR) |
| Variable | HR | 95% CI | P |
|---|---|---|---|
| Other mutations | 1.63 | 1.04–2.58 | 0.03 |
| Sex | 0.98 | 0.6–1.63 | 0.97 |
| Age | 1 | 0.98–1.02 | 0.70 |
| Ever smoker | 1.22 | 0.76–1.95 | 0.41 |
| T790M | 1.06 | 0.53–2.13 | 0.86 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rachiglio, A.M.; Fenizia, F.; Piccirillo, M.C.; Galetta, D.; Crinò, L.; Vincenzi, B.; Barletta, E.; Pinto, C.; Ferraù, F.; Lambiase, M.; et al. The Presence of Concomitant Mutations Affects the Activity of EGFR Tyrosine Kinase Inhibitors in EGFR-Mutant Non-Small Cell Lung Cancer (NSCLC) Patients. Cancers 2019, 11, 341. https://doi.org/10.3390/cancers11030341
Rachiglio AM, Fenizia F, Piccirillo MC, Galetta D, Crinò L, Vincenzi B, Barletta E, Pinto C, Ferraù F, Lambiase M, et al. The Presence of Concomitant Mutations Affects the Activity of EGFR Tyrosine Kinase Inhibitors in EGFR-Mutant Non-Small Cell Lung Cancer (NSCLC) Patients. Cancers. 2019; 11(3):341. https://doi.org/10.3390/cancers11030341
Chicago/Turabian StyleRachiglio, Anna Maria, Francesca Fenizia, Maria Carmela Piccirillo, Domenico Galetta, Lucio Crinò, Bruno Vincenzi, Emiddio Barletta, Carmine Pinto, Francesco Ferraù, Matilde Lambiase, and et al. 2019. "The Presence of Concomitant Mutations Affects the Activity of EGFR Tyrosine Kinase Inhibitors in EGFR-Mutant Non-Small Cell Lung Cancer (NSCLC) Patients" Cancers 11, no. 3: 341. https://doi.org/10.3390/cancers11030341
APA StyleRachiglio, A. M., Fenizia, F., Piccirillo, M. C., Galetta, D., Crinò, L., Vincenzi, B., Barletta, E., Pinto, C., Ferraù, F., Lambiase, M., Montanino, A., Roma, C., Ludovini, V., Montagna, E. S., De Luca, A., Rocco, G., Botti, G., Perrone, F., Morabito, A., & Normanno, N. (2019). The Presence of Concomitant Mutations Affects the Activity of EGFR Tyrosine Kinase Inhibitors in EGFR-Mutant Non-Small Cell Lung Cancer (NSCLC) Patients. Cancers, 11(3), 341. https://doi.org/10.3390/cancers11030341