The p53 Pathway in Glioblastoma

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
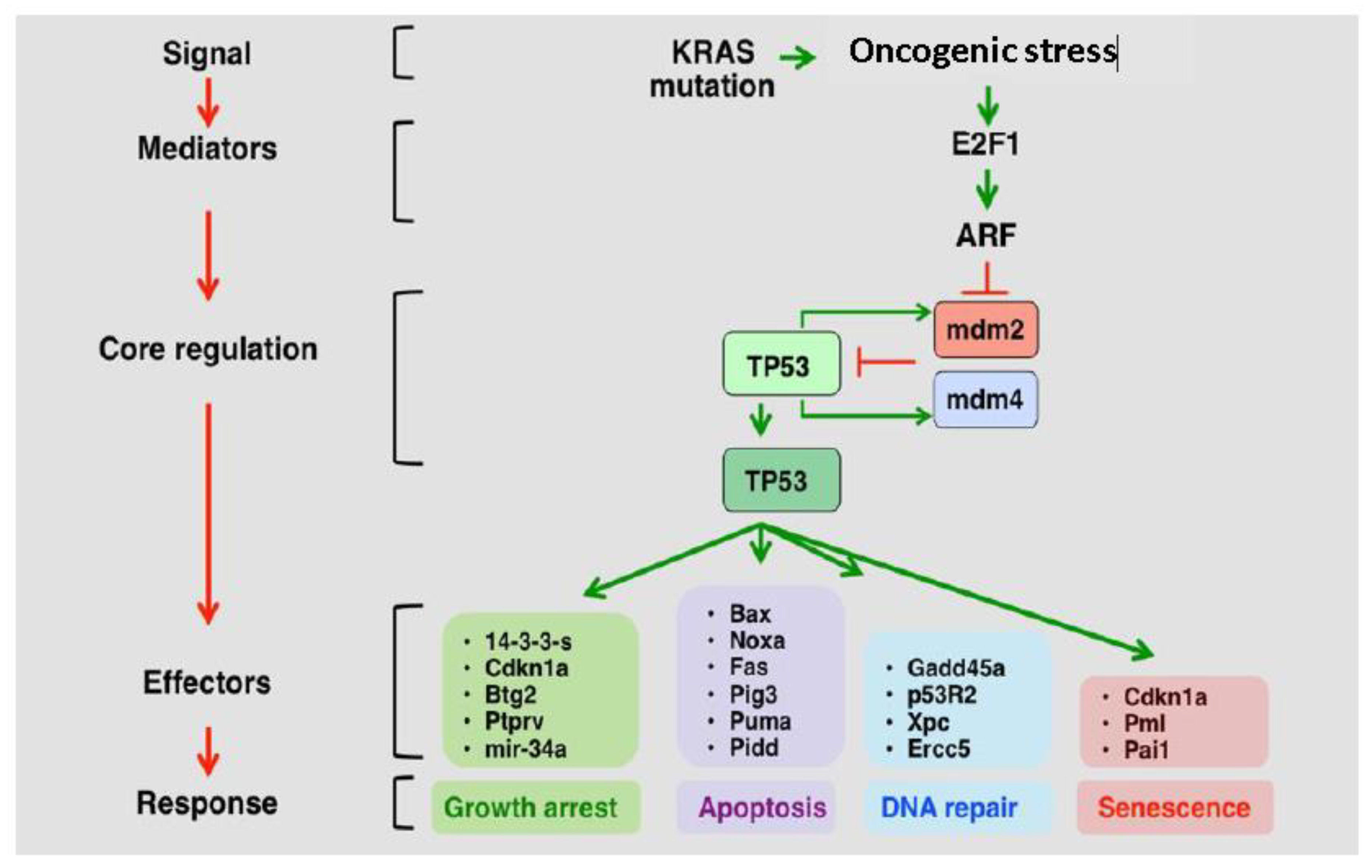
2. P53 Role and Deregulation in GBM
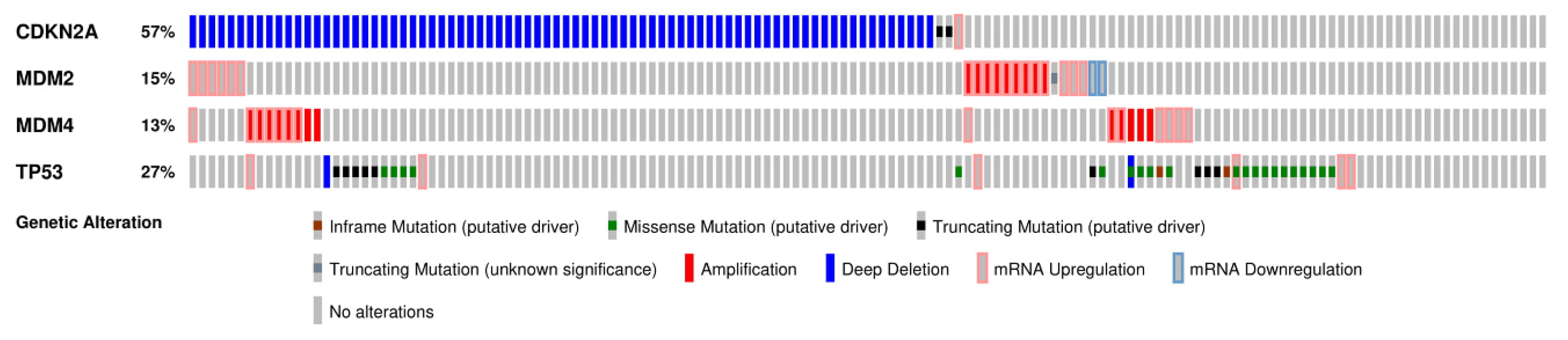
2.1. The p53 Pathway is Frequently Deregulated in GBM
2.2. P53 Is Implicated in GBM Progression.
2.3. CDKN2A/ARF Is the Most Commonly Deregulated Component of the p53 Pathway in GBM
2.4. MDM2 and MDM4 Are Amplified in GBM and Negatively Regulate p53
2.5. The p53 Pathway Is Regulated by Various Non-Coding Elements in GBM
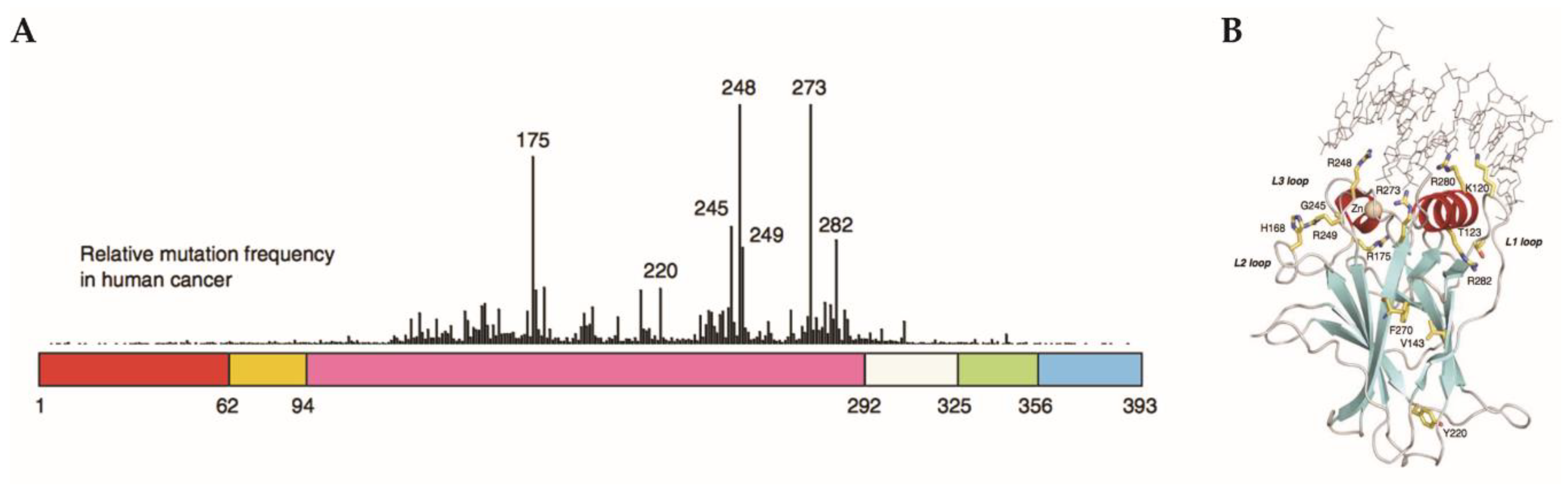
2.6. Gain-of-Function Mut-p53 in Cancer and GBM
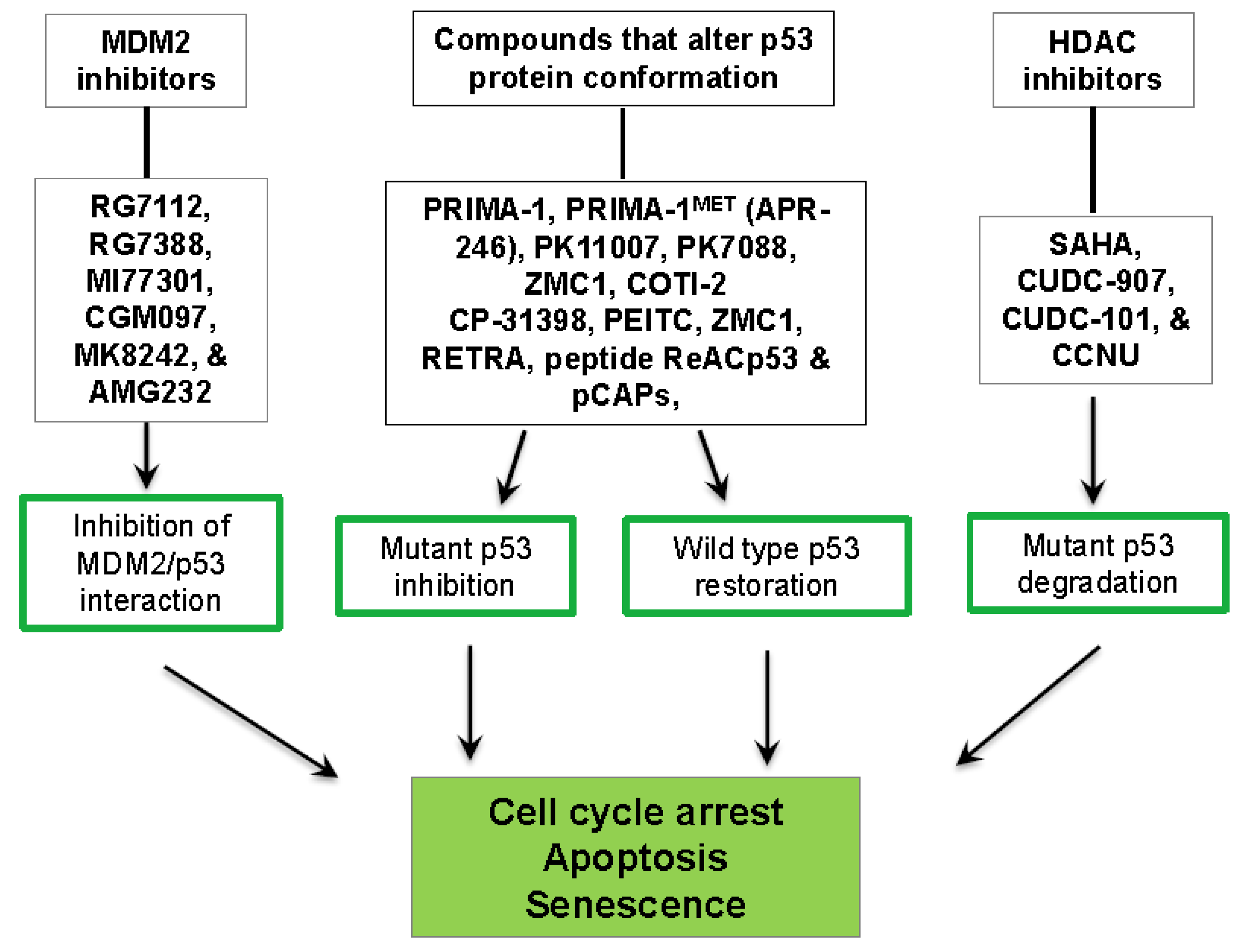
3. P53-Targeted Therapies
3.1. Inhibition of the MDM2/p53 Complex
3.2. Restoration of wt-p53 Conformation and Function
3.3. Degradation of Mut-p53
4. Controversies and Future Perspectives
Funding
Conflicts of Interest
References
- Jeffrey, P.D.; Gorina, S.; Pavletich, N.P. Crystal structure of the tetramerization domain of the p53 tumor suppressor at 1.7 angstroms. Science 1995, 267, 1498–1502. [Google Scholar] [CrossRef] [PubMed]
- Rajagopalan, S.; Huang, F.; Fersht, A.R. Single-Molecule characterization of oligomerization kinetics and equilibria of the tumor suppressor p53. Nucleic Acids Res. 2011, 39, 2294–2303. [Google Scholar] [CrossRef] [PubMed]
- El-Deiry, W.S.; Kern, S.E.; Pietenpol, J.A.; Kinzler, K.W.; Vogelstein, B. Definition of a consensus binding site for p53. Nat. Genet. 1992, 1, 45–49. [Google Scholar] [CrossRef] [PubMed]
- Wei, C.L.; Wu, Q.; Vega, V.B.; Chiu, K.P.; Ng, P.; Zhang, T.; Shahab, A.; Yong, H.C.; Fu, Y.; Weng, Z.; et al. A global map of p53 transcription-factor binding sites in the human genome. Cell 2006, 124, 207–219. [Google Scholar] [CrossRef] [PubMed]
- Wade, M.; Li, Y.C.; Wahl, G.M. MDM2, MDMX and p53 in oncogenesis and cancer therapy. Nat. Rev. Cancer 2013, 13, 83–96. [Google Scholar] [CrossRef] [PubMed]
- Kastenhuber, E.R.; Lowe, S.W. Putting p53 in Context. Cell 2017, 170, 1062–1078. [Google Scholar] [CrossRef] [PubMed]
- Lane, D.P. Cancer. p53, guardian of the genome. Nature 1992, 358, 15–16. [Google Scholar] [CrossRef] [PubMed]
- Mills, K.D. Tumor suppression: Putting p53 in context. Cell Cycle 2013, 12, 3461–3462. [Google Scholar] [CrossRef] [PubMed]
- Bieging, K.T.; Mello, S.S.; Attardi, L.D. Unravelling mechanisms of p53-mediated tumour suppression. Nat. Rev. Cancer 2014, 14, 359–370. [Google Scholar] [CrossRef] [PubMed]
- Schapira, A.H.V. Neurology and Clinical Neuroscience; Mosby Elsevier: Philadelphia PA, USA, 2007; p. 1336. [Google Scholar]
- Gallego, O. Nonsurgical treatment of recurrent glioblastoma. Curr. Oncol. 2015, 22, e273–e281. [Google Scholar] [CrossRef] [PubMed]
- Louis, D.N.; Perry, A.; Reifenberger, G.; von Deimling, A.; Figarella-Branger, D.; Cavenee, W.K.; Ohgaki, H.; Wiestler, O.D.; Kleihues, P.; Ellison, D.W. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: A summary. Acta Neuropathol. 2016, 131, 803–820. [Google Scholar] [CrossRef] [PubMed]
- Kloosterhof, N.K.; Bralten, L.B.; Dubbink, H.J.; French, P.J.; van den Bent, M.J. Isocitrate dehydrogenase-1 mutations: A fundamentally new understanding of diffuse glioma? Lancet Oncol. 2011, 12, 83–91. [Google Scholar] [CrossRef]
- Brennan, C.W.; Verhaak, R.G.; McKenna, A.; Campos, B.; Noushmehr, H.; Salama, S.R.; Zheng, S.; Chakravarty, D.; Sanborn, J.Z.; Berman, S.H.; et al. The somatic genomic landscape of glioblastoma. Cell 2013, 155, 462–477. [Google Scholar] [CrossRef] [PubMed]
- Verhaak, R.G.; Hoadley, K.A.; Purdom, E.; Wang, V.; Qi, Y.; Wilkerson, M.D.; Miller, C.R.; Ding, L.; Golub, T.; Mesirov, J.P.; et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 2010, 17, 98–110. [Google Scholar] [CrossRef] [PubMed]
- Nandeesh, B.N.; Naskar, S.; Shashtri, A.H.; Arivazhagan, A.; Santosh, V. Recurrent Glioblastomas Exhibit Higher Expression of Biomarkers with Stem-like Properties. J. Neurosci. Rural Pract. 2018, 9, 86–91. [Google Scholar] [PubMed]
- Stark, A.M.; Witzel, P.; Strege, R.J.; Hugo, H.H.; Mehdorn, H.M. p53, mdm2, EGFR, and msh2 expression in paired initial and recurrent glioblastoma multiforme. J. Neurol. Neurosurg. Psychiatry 2003, 74, 779–783. [Google Scholar] [CrossRef] [PubMed]
- Wiewrodt, D.; Nagel, G.; Dreimuller, N.; Hundsberger, T.; Perneczky, A.; Kaina, B. MGMT in primary and recurrent human glioblastomas after radiation and chemotherapy and comparison with p53 status and clinical outcome. Int. J. Cancer 2008, 122, 1391–1399. [Google Scholar] [CrossRef] [PubMed]
- Rivlin, N.; Brosh, R.; Oren, M.; Rotter, V. Mutations in the p53 Tumor Suppressor Gene: Important Milestones at the Various Steps of Tumorigenesis. Genes Cancer 2011, 2, 466–474. [Google Scholar] [CrossRef] [PubMed]
- Ishii, N.; Maier, D.; Merlo, A.; Tada, M.; Sawamura, Y.; Diserens, A.C.; Van Meir, E.G. Frequent co-alterations of TP53, p16/CDKN2A, p14ARF, PTEN tumor suppressor genes in human glioma cell lines. Brain Pathol. 1999, 9, 469–479. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.J.; Huang, M.S.; Hong, C.Y.; Tse, V.; Silverberg, G.D.; Hsiao, M. Comparisons of tumor suppressor p53, p21, and p16 gene therapy effects on glioblastoma tumorigenicity in situ. Biochem. Biophys. Res. Commun. 2001, 287, 173–180. [Google Scholar] [CrossRef] [PubMed]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal. 2013, 6, pl1. [Google Scholar] [CrossRef] [PubMed]
- Leroy, B.; Fournier, J.L.; Ishioka, C.; Monti, P.; Inga, A.; Fronza, G.; Soussi, T. The TP53 website: An integrative resource centre for the TP53 mutation database and TP53 mutant analysis. Nucleic Acids Res. 2013, 41, D962–D969. [Google Scholar] [CrossRef] [PubMed]
- Tsuchiya, K. Functional restoration of tumor suppressor p53 alters susceptibility of glioblastoma cells to irradiation—Analysis using a cell line containing a temperature-sensitive mutant. Hokkaido Igaku Zasshi 2000, 75, 265–274. [Google Scholar] [PubMed]
- Lin, T.; Chao, C.; Saito, S.; Mazur, S.J.; Murphy, M.E.; Appella, E.; Xu, Y. p53 induces differentiation of mouse embryonic stem cells by suppressing Nanog expression. Nat. Cell Biol. 2005, 7, 165–171. [Google Scholar] [CrossRef] [PubMed]
- England, B.; Huang, T.; Karsy, M. Current understanding of the role and targeting of tumor suppressor p53 in glioblastoma multiforme. Tumour Biol. 2013, 34, 2063–2074. [Google Scholar] [CrossRef] [PubMed]
- Zheng, H.; Ying, H.; Yan, H.; Kimmelman, A.C.; Hiller, D.J.; Chen, A.J.; Perry, S.R.; Tonon, G.; Chu, G.C.; Ding, Z.; et al. p53 and Pten control neural and glioma stem/progenitor cell renewal and differentiation. Nature 2008, 455, 1129–1133. [Google Scholar] [CrossRef] [PubMed]
- Krex, D.; Mohr, B.; Appelt, H.; Schackert, H.K.; Schackert, G. Genetic analysis of a multifocal glioblastoma multiforme: A suitable tool to gain new aspects in glioma development. Neurosurgery 2003, 53, 1377–1384. [Google Scholar] [CrossRef] [PubMed]
- Djuzenova, C.S.; Fiedler, V.; Memmel, S.; Katzer, A.; Hartmann, S.; Krohne, G.; Zimmermann, H.; Scholz, C.J.; Polat, B.; Flentje, M.; et al. Actin cytoskeleton organization, cell surface modification and invasion rate of 5 glioblastoma cell lines differing in PTEN and p53 status. Exp. Cell Res. 2015, 330, 346–357. [Google Scholar] [CrossRef] [PubMed]
- Park, C.M.; Park, M.J.; Kwak, H.J.; Moon, S.I.; Yoo, D.H.; Lee, H.C.; Park, I.C.; Rhee, C.H.; Hong, S.I. Induction of p53-mediated apoptosis and recovery of chemosensitivity through p53 transduction in human glioblastoma cells by cisplatin. Int. J. Oncol. 2006, 28, 119–125. [Google Scholar] [CrossRef] [PubMed]
- Petitjean, A.; Achatz, M.I.; Borresen-Dale, A.L.; Hainaut, P.; Olivier, M. TP53 mutations in human cancers: Functional selection and impact on cancer prognosis and outcomes. Oncogene 2007, 26, 2157–2165. [Google Scholar] [CrossRef] [PubMed]
- Rich, J.N.; Hans, C.; Jones, B.; Iversen, E.S.; McLendon, R.E.; Rasheed, B.K.; Dobra, A.; Dressman, H.K.; Bigner, D.D.; Nevins, J.R.; et al. Gene expression profiling and genetic markers in glioblastoma survival. Cancer Res. 2005, 65, 4051–4058. [Google Scholar] [CrossRef] [PubMed]
- Kraus, J.A.; Glesmann, N.; Beck, M.; Krex, D.; Klockgether, T.; Schackert, G.; Schlegel, U. Molecular analysis of the PTEN, TP53 and CDKN2A tumor suppressor genes in long-term survivors of glioblastoma multiforme. J. Neurooncol. 2000, 48, 89–94. [Google Scholar] [CrossRef] [PubMed]
- Zadeh, M.D.; Amini, R.; Firoozray, M.; Derakhshandeh-Peykar, P. Frequent homozygous deletion of p16/CDKN2A gene in malignant gliomas of Iranian patients. Pak. J. Biol. Sci. 2007, 10, 4246–4250. [Google Scholar] [PubMed]
- Zerrouqi, A.; Pyrzynska, B.; Febbraio, M.; Brat, D.J.; Van Meir, E.G. P14ARF inhibits human glioblastoma-induced angiogenesis by upregulating the expression of TIMP3. J. Clin. Investig. 2012, 122, 1283–1295. [Google Scholar] [CrossRef] [PubMed]
- Rickert, C.H.; Riemenschneider, M.J.; Schachenmayr, W.; Richter, H.P.; Bockhorn, J.; Reifenberger, G.; Paulus, W. Glioblastoma with adipocyte-like tumor cell differentiation—Histological and molecular features of a rare differentiation pattern. Brain Pathol. 2009, 19, 431–438. [Google Scholar] [CrossRef] [PubMed]
- Ghimenti, C.; Fiano, V.; Chiado-Piat, L.; Chio, A.; Cavalla, P.; Schiffer, D. Deregulation of the p14ARF/Mdm2/p53 pathway and G1/S transition in two glioblastoma sets. J. Neurooncol. 2003, 61, 95–102. [Google Scholar] [CrossRef] [PubMed]
- Fulci, G.; Labuhn, M.; Maier, D.; Lachat, Y.; Hausmann, O.; Hegi, M.E.; Janzer, R.C.; Merlo, A.; Van Meir, E.G. p53 gene mutation and ink4a-arf deletion appear to be two mutually exclusive events in human glioblastoma. Oncogene 2000, 19, 3816–3822. [Google Scholar] [CrossRef] [PubMed]
- Biernat, W.; Debiec-Rychter, M.; Liberski, P.P. Mutations of TP53, amplification of EGFR, MDM2 and CDK4, and deletions of CDKN2A in malignant astrocytomas. Pol. J. Pathol. 1998, 49, 267–271. [Google Scholar] [PubMed]
- Crespo, I.; Vital, A.L.; Gonzalez-Tablas, M.; Patino Mdel, C.; Otero, A.; Lopes, M.C.; de Oliveira, C.; Domingues, P.; Orfao, A.; Tabernero, M.D. Molecular and Genomic Alterations in Glioblastoma Multiforme. Am. J. Pathol. 2015, 185, 1820–1833. [Google Scholar] [CrossRef] [PubMed]
- Nobusawa, S.; Lachuer, J.; Wierinckx, A.; Kim, Y.H.; Huang, J.; Legras, C.; Kleihues, P.; Ohgaki, H. Intratumoral patterns of genomic imbalance in glioblastomas. Brain Pathol. 2010, 20, 936–944. [Google Scholar] [PubMed]
- Rao, S.K.; Edwards, J.; Joshi, A.D.; Siu, I.M.; Riggins, G.J. A survey of glioblastoma genomic amplifications and deletions. J. Neurooncol. 2010, 96, 169–179. [Google Scholar] [CrossRef] [PubMed]
- He, J.; Reifenberger, G.; Liu, L.; Collins, V.P.; James, C.D. Analysis of glioma cell lines for amplification and overexpression of MDM2. Genes Chromosomes Cancer 1994, 11, 91–96. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Cai, S.; Bailey, B.J.; Reza Saadatzadeh, M.; Ding, J.; Tonsing-Carter, E.; Georgiadis, T.M.; Zachary Gunter, T.; Long, E.C.; Minto, R.E.; et al. Combination therapy in a xenograft model of glioblastoma: Enhancement of the antitumor activity of temozolomide by an MDM2 antagonist. J. Neurosurg. 2017, 126, 446–459. [Google Scholar] [CrossRef] [PubMed]
- Rolle, K. miRNA Multiplayers in glioma. From bench to bedside. Acta Biochim. Pol. 2015, 62, 353–365. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Yin, F.; Zhang, J.; Wicha, M.S.; Chang, A.E.; Fan, W.; Chen, L.; Fan, M.; Li, Q. Regulatory roles of miRNA in the human neural stem cell transformation to glioma stem cells. J. Cell Biochem. 2014, 115, 1368–1380. [Google Scholar] [CrossRef] [PubMed]
- Lavorgna, G.; Vago, R.; Sarmini, M.; Montorsi, F.; Salonia, A.; Bellone, M. Long non-coding RNAs as novel therapeutic targets in cancer. Pharmacol. Res. 2016, 110, 131–138. [Google Scholar] [CrossRef] [PubMed]
- Ruvkun, G. Molecular biology. Glimpses of a tiny RNA world. Science 2001, 294, 797–799. [Google Scholar] [CrossRef] [PubMed]
- Lee, R.C.; Feinbaum, R.L.; Ambros, V. The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell 1993, 75, 843–854. [Google Scholar] [CrossRef]
- Li, Y.; Guessous, F.; Zhang, Y.; Dipierro, C.; Kefas, B.; Johnson, E.; Marcinkiewicz, L.; Jiang, J.; Yang, Y.; Schmittgen, T.D.; et al. MicroRNA-34a inhibits glioblastoma growth by targeting multiple oncogenes. Cancer Res. 2009, 69, 7569–7576. [Google Scholar] [CrossRef] [PubMed]
- Wu, N.; Lin, X.; Zhao, X.; Zheng, L.; Xiao, L.; Liu, J.; Ge, L.; Cao, S. MiR-125b acts as an oncogene in glioblastoma cells and inhibits cell apoptosis through p53 and p38MAPK-independent pathways. Br. J. Cancer 2013, 109, 2853–2863. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.H.; Wu, S.Q.; Zhang, Y.D. Downregulation of miR-124 promotes the growth and invasiveness of glioblastoma cells involving upregulation of PPP1R13L. Int. J. Mol. Med. 2013, 32, 101–107. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Liu, Y.; Granberg, K.J.; Wang, Q.; Moore, L.M.; Ji, P.; Gumin, J.; Sulman, E.P.; Calin, G.A.; Haapasalo, H.; et al. Two mature products of MIR-491 coordinate to suppress key cancer hallmarks in glioblastoma. Oncogene 2015, 34, 1619–1628. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Yang, B.B. Stress response of glioblastoma cells mediated by miR-17-5p targeting PTEN and the passenger strand miR-17-3p targeting MDM2. Oncotarget 2012, 3, 1653–1668. [Google Scholar] [CrossRef] [PubMed]
- Mattick, J.S.; Rinn, J.L. Discovery and annotation of long noncoding RNAs. Nat. Struct. Mol. Biol. 2015, 22, 5–7. [Google Scholar] [CrossRef] [PubMed]
- He, Z.; Wang, Y.; Huang, G.; Wang, Q.; Zhao, D.; Chen, L. The lncRNA UCA1 interacts with miR-182 to modulate glioma proliferation and migration by targeting iASPP. Arch. Biochem. Biophys. 2017, 623–624, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Peng, R.; Liu, Q.; Liu, D.; Du, P.; Yuan, J.; Peng, G.; Liao, Y. The lncRNA H19 interacts with miR-140 to modulate glioma growth by targeting iASPP. Arch. Biochem. Biophys. 2016, 610, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Dittmer, D.; Pati, S.; Zambetti, G.; Chu, S.; Teresky, A.K.; Moore, M.; Finlay, C.; Levine, A.J. Gain of function mutations in p53. Nat. Genet. 1993, 4, 42–46. [Google Scholar] [CrossRef] [PubMed]
- Lang, G.A.; Iwakuma, T.; Suh, Y.A.; Liu, G.; Rao, V.A.; Parant, J.M.; Valentin-Vega, Y.A.; Terzian, T.; Caldwell, L.C.; Strong, L.C.; et al. Gain of function of a p53 hot spot mutation in a mouse model of Li-Fraumeni syndrome. Cell 2004, 119, 861–872. [Google Scholar] [CrossRef] [PubMed]
- Walker, D.R.; Bond, J.P.; Tarone, R.E.; Harris, C.C.; Makalowski, W.; Boguski, M.S.; Greenblatt, M.S. Evolutionary conservation and somatic mutation hotspot maps of p53: Correlation with p53 protein structural and functional features. Oncogene 1999, 18, 211–218. [Google Scholar] [CrossRef] [PubMed]
- Cho, Y.; Gorina, S.; Jeffrey, P.D.; Pavletich, N.P. Crystal structure of a p53 tumor suppressor-DNA complex: Understanding tumorigenic mutations. Science 1994, 265, 346–355. [Google Scholar] [CrossRef] [PubMed]
- Galia, A.; Calogero, A.E.; Condorelli, R.; Fraggetta, F.; La Corte, A.; Ridolfo, F.; Bosco, P.; Castiglione, R.; Salemi, M. PARP-1 protein expression in glioblastoma multiforme. Eur. J. Histochem. 2012, 56, e9. [Google Scholar] [CrossRef] [PubMed]
- Harris, S.L.; Levine, A.J. The p53 pathway: Positive and negative feedback loops. Oncogene 2005, 24, 2899–2908. [Google Scholar] [CrossRef] [PubMed]
- Lukashchuk, N.; Vousden, K.H. Ubiquitination and degradation of mutant p53. Mol. Cell Biol. 2007, 27, 8284–8295. [Google Scholar] [CrossRef] [PubMed]
- Zheng, T.; Wang, J.; Zhao, Y.; Zhang, C.; Lin, M.; Wang, X.; Yu, H.; Liu, L.; Feng, Z.; Hu, W. Spliced MDM2 isoforms promote mutant p53 accumulation and gain-of-function in tumorigenesis. Nat. Commun. 2013, 4, 2996. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Marchenko, N.D.; Schulz, R.; Fischer, V.; Velasco-Hernandez, T.; Talos, F.; Moll, U.M. Functional inactivation of endogenous MDM2 and CHIP by HSP90 causes aberrant stabilization of mutant p53 in human cancer cells. Mol. Cancer Res. 2011, 9, 577–588. [Google Scholar] [CrossRef] [PubMed]
- Muller, P.A.; Caswell, P.T.; Doyle, B.; Iwanicki, M.P.; Tan, E.H.; Karim, S.; Lukashchuk, N.; Gillespie, D.A.; Ludwig, R.L.; Gosselin, P.; et al. Mutant p53 drives invasion by promoting integrin recycling. Cell 2009, 139, 1327–1341. [Google Scholar] [CrossRef] [PubMed]
- Blandino, G.; Deppert, W.; Hainaut, P.; Levine, A.; Lozano, G.; Olivier, M.; Rotter, V.; Wiman, K.; Oren, M. Mutant p53 protein, master regulator of human malignancies: A report on the Fifth Mutant p53 Workshop. Cell Death Differ. 2012, 19, 180–183. [Google Scholar] [CrossRef] [PubMed]
- Freed-Pastor, W.A.; Mizuno, H.; Zhao, X.; Langerod, A.; Moon, S.H.; Rodriguez-Barrueco, R.; Barsotti, A.; Chicas, A.; Li, W.; Polotskaia, A.; et al. Mutant p53 disrupts mammary tissue architecture via the mevalonate pathway. Cell 2012, 148, 244–258. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Liu, J.; Liang, Y.; Wu, R.; Zhao, Y.; Hong, X.; Lin, M.; Yu, H.; Liu, L.; Levine, A.J.; et al. Tumour-associated mutant p53 drives the Warburg effect. Nat. Commun. 2013, 4, 2935. [Google Scholar] [CrossRef] [PubMed]
- Muller, P.A.; Vousden, K.H.; Norman, J.C. p53 and its mutants in tumor cell migration and invasion. J. Cell Biol. 2011, 192, 209–218. [Google Scholar] [CrossRef] [PubMed]
- Yue, X.; Zhao, Y.; Xu, Y.; Zheng, M.; Feng, Z.; Hu, W. Mutant p53 in Cancer: Accumulation, Gain-of-Function, and Therapy. J. Mol. Biol. 2017, 429, 1595–1606. [Google Scholar] [CrossRef] [PubMed]
- Di Agostino, S.; Strano, S.; Emiliozzi, V.; Zerbini, V.; Mottolese, M.; Sacchi, A.; Blandino, G.; Piaggio, G. Gain of function of mutant p53: The mutant p53/NF-Y protein complex reveals an aberrant transcriptional mechanism of cell cycle regulation. Cancer Cell 2006, 10, 191–202. [Google Scholar] [CrossRef] [PubMed]
- Dupont, S.; Mamidi, A.; Cordenonsi, M.; Montagner, M.; Zacchigna, L.; Adorno, M.; Martello, G.; Stinchfield, M.J.; Soligo, S.; Morsut, L.; et al. FAM/USP9x, a deubiquitinating enzyme essential for TGFbeta signaling, controls Smad4 monoubiquitination. Cell 2009, 136, 123–135. [Google Scholar] [CrossRef] [PubMed]
- Sampath, J.; Sun, D.; Kidd, V.J.; Grenet, J.; Gandhi, A.; Shapiro, L.H.; Wang, Q.; Zambetti, G.P.; Schuetz, J.D. Mutant p53 cooperates with ETS and selectively up-regulates human MDR1 not MRP1. J. Biol. Chem. 2001, 276, 39359–39367. [Google Scholar] [CrossRef] [PubMed]
- Stambolsky, P.; Tabach, Y.; Fontemaggi, G.; Weisz, L.; Maor-Aloni, R.; Siegfried, Z.; Shiff, I.; Kogan, I.; Shay, M.; Kalo, E.; et al. Modulation of the vitamin D3 response by cancer-associated mutant p53. Cancer Cell 2010, 17, 273–285. [Google Scholar] [CrossRef] [PubMed]
- Brosh, R.; Rotter, V. When mutants gain new powers: News from the mutant p53 field. Nat. Rev. Cancer 2009, 9, 701–713. [Google Scholar] [CrossRef] [PubMed]
- Finlay, C.A.; Hinds, P.W.; Levine, A.J. The p53 proto-oncogene can act as a suppressor of transformation. Cell 1989, 57, 1083–1093. [Google Scholar] [CrossRef]
- Gaiddon, C.; Lokshin, M.; Ahn, J.; Zhang, T.; Prives, C. A subset of tumor-derived mutant forms of p53 down-regulate p63 and p73 through a direct interaction with the p53 core domain. Mol. Cell Biol. 2001, 21, 1874–1887. [Google Scholar] [CrossRef] [PubMed]
- Vogiatzi, F.; Brandt, D.T.; Schneikert, J.; Fuchs, J.; Grikscheit, K.; Wanzel, M.; Pavlakis, E.; Charles, J.P.; Timofeev, O.; Nist, A.; et al. Mutant p53 promotes tumor progression and metastasis by the endoplasmic reticulum UDPase ENTPD5. Proc. Natl. Acad. Sci. USA 2016, 113, E8433–E8442. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.A.; Menendez, D.; Resnick, M.A.; Anderson, C.W. Mutant TP53 posttranslational modifications: Challenges and opportunities. Hum. Mutat. 2014, 35, 738–755. [Google Scholar] [CrossRef] [PubMed]
- Suh, Y.A.; Post, S.M.; Elizondo-Fraire, A.C.; Maccio, D.R.; Jackson, J.G.; El-Naggar, A.K.; Van Pelt, C.; Terzian, T.; Lozano, G. Multiple stress signals activate mutant p53 in vivo. Cancer Res. 2011, 71, 7168–7175. [Google Scholar] [CrossRef] [PubMed]
- Nagpal, J.; Jamoona, A.; Gulati, N.D.; Mohan, A.; Braun, A.; Murali, R.; Jhanwar-Uniyal, M. Revisiting the role of p53 in primary and secondary glioblastomas. Anticancer Res. 2006, 26, 4633–4639. [Google Scholar] [PubMed]
- Kraus, J.A.; Wenghoefer, M.; Glesmann, N.; Mohr, S.; Beck, M.; Schmidt, M.C.; Schroder, R.; Berweiler, U.; Roggendorf, W.; Diete, S.; et al. TP53 gene mutations, nuclear p53 accumulation, expression of Waf/p21, Bcl-2, and CD95 (APO-1/Fas) proteins are not prognostic factors in de novo glioblastoma multiforme. J. Neurooncol. 2001, 52, 263–272. [Google Scholar] [CrossRef] [PubMed]
- Ohgaki, H. Genetic pathways to glioblastomas. Neuropathology 2005, 25, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, K.; Sato, K.; Biernat, W.; Tachibana, O.; von Ammon, K.; Ogata, N.; Yonekawa, Y.; Kleihues, P.; Ohgaki, H. Incidence and timing of p53 mutations during astrocytoma progression in patients with multiple biopsies. Clin. Cancer Res. 1997, 3, 523–530. [Google Scholar] [PubMed]
- Peraud, A.; Kreth, F.W.; Wiestler, O.D.; Kleihues, P.; Reulen, H.J. Prognostic impact of TP53 mutations and P53 protein overexpression in supratentorial WHO grade II astrocytomas and oligoastrocytomas. Clin. Cancer Res. 2002, 8, 1117–1124. [Google Scholar] [PubMed]
- Okamoto, Y.; Di Patre, P.L.; Burkhard, C.; Horstmann, S.; Jourde, B.; Fahey, M.; Schuler, D.; Probst-Hensch, N.M.; Yasargil, M.G.; Yonekawa, Y.; et al. Population-based study on incidence, survival rates, and genetic alterations of low-grade diffuse astrocytomas and oligodendrogliomas. Acta Neuropathol. 2004, 108, 49–56. [Google Scholar] [CrossRef] [PubMed]
- Green, D.R.; Kroemer, G. Cytoplasmic functions of the tumour suppressor p53. Nature 2009, 458, 1127–1130. [Google Scholar] [CrossRef] [PubMed]
- Martin, S.; Janouskova, H.; Dontenwill, M. Integrins and p53 pathways in glioblastoma resistance to temozolomide. Front. Oncol. 2012, 2, 157. [Google Scholar] [CrossRef] [PubMed]
- Pathania, M.; De Jay, N.; Maestro, N.; Harutyunyan, A.S.; Nitarska, J.; Pahlavan, P.; Henderson, S.; Mikael, L.G.; Richard-Londt, A.; Zhang, Y.; et al. H3.3(K27M) Cooperates with Trp53 Loss and PDGFRA Gain in Mouse Embryonic Neural Progenitor Cells to Induce Invasive High-Grade Gliomas. Cancer Cell 2017, 32, 684–700. [Google Scholar] [CrossRef] [PubMed]
- Hesselager, G.; Uhrbom, L.; Westermark, B.; Nister, M. Complementary effects of platelet-derived growth factor autocrine stimulation and p53 or Ink4a-Arf deletion in a mouse glioma model. Cancer Res. 2003, 63, 4305–4309. [Google Scholar] [PubMed]
- Zalcenstein, A.; Stambolsky, P.; Weisz, L.; Muller, M.; Wallach, D.; Goncharov, T.M.; Krammer, P.H.; Rotter, V.; Oren, M. Mutant p53 gain of function: Repression of CD95(Fas/APO-1) gene expression by tumor-associated p53 mutants. Oncogene 2003, 22, 5667–5676. [Google Scholar] [CrossRef] [PubMed]
- Kleber, S.; Sancho-Martinez, I.; Wiestler, B.; Beisel, A.; Gieffers, C.; Hill, O.; Thiemann, M.; Mueller, W.; Sykora, J.; Kuhn, A.; et al. Yes and PI3K bind CD95 to signal invasion of glioblastoma. Cancer Cell 2008, 13, 235–248. [Google Scholar] [CrossRef] [PubMed]
- Wisniewski, P.; Ellert-Miklaszewska, A.; Kwiatkowska, A.; Kaminska, B. Non-apoptotic Fas signaling regulates invasiveness of glioma cells and modulates MMP-2 activity via NFkappaB-TIMP-2 pathway. Cell Signal. 2010, 22, 212–220. [Google Scholar] [CrossRef] [PubMed]
- Sano, T.; Lin, H.; Chen, X.; Langford, L.A.; Koul, D.; Bondy, M.L.; Hess, K.R.; Myers, J.N.; Hong, Y.K.; Yung, W.K.; et al. Differential expression of MMAC/PTEN in glioblastoma multiforme: Relationship to localization and prognosis. Cancer Res. 1999, 59, 1820–1824. [Google Scholar] [PubMed]
- Vikhanskaya, F.; Lee, M.K.; Mazzoletti, M.; Broggini, M.; Sabapathy, K. Cancer-derived p53 mutants suppress p53-target gene expression--potential mechanism for gain of function of mutant p53. Nucleic. Acids Res. 2007, 35, 2093–2104. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.; Liang, Y.; Zhu, H.; Zhang, J.; Zhong, X. R280T mutation of p53 gene promotes proliferation of human glioma cells through GSK-3beta/PTEN pathway. Neurosci. Lett. 2012, 529, 60–65. [Google Scholar] [CrossRef] [PubMed]
- Mendrysa, S.M.; Ghassemifar, S.; Malek, R. p53 in the CNS: Perspectives on Development, Stem Cells, and Cancer. Genes Cancer 2011, 2, 431–442. [Google Scholar] [CrossRef] [PubMed]
- Burness, M.L.; Sipkins, D.A. The stem cell niche in health and malignancy. Semin. Cancer Biol. 2010, 20, 107–115. [Google Scholar] [CrossRef] [PubMed]
- Flores, E.R.; Sengupta, S.; Miller, J.B.; Newman, J.J.; Bronson, R.; Crowley, D.; Yang, A.; McKeon, F.; Jacks, T. Tumor predisposition in mice mutant for p63 and p73: Evidence for broader tumor suppressor functions for the p53 family. Cancer Cell 2005, 7, 363–373. [Google Scholar] [CrossRef] [PubMed]
- Moll, U.M.; Erster, S.; Zaika, A. p53, p63 and p73—Solos, alliances and feuds among family members. Biochim. Biophys. Acta 2001, 1552, 47–59. [Google Scholar] [CrossRef]
- Palani, M.; Devan, S.; Arunkumar, R.; Vanisree, A.J. Frequency variations in the methylated pattern of p73/p21 genes and chromosomal aberrations correlating with different grades of glioma among south Indian population. Med. Oncol. 2011, 28 (Suppl 1), S445–S452. [Google Scholar] [CrossRef] [PubMed]
- Ham, S.W.; Jeon, H.Y.; Jin, X.; Kim, E.J.; Kim, J.K.; Shin, Y.J.; Lee, Y.; Kim, S.H.; Lee, S.Y.; Seo, S.; et al. TP53 gain-of-function mutation promotes inflammation in glioblastoma. Cell Death Differ. 2018. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Guessous, F.; Kwon, S.; Kumar, M.; Ibidapo, O.; Fuller, L.; Johnson, E.; Lal, B.; Hussaini, I.; Bao, Y.; et al. PTEN has tumor-promoting properties in the setting of gain-of-function p53 mutations. Cancer Res. 2008, 68, 1723–1731. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Zhang, Y.; Tang, Y.; Butler, N.; Kim, J.; Guessous, F.; Schiff, D.; Mandell, J.; Abounader, R. A novel PTEN/mutant p53/c-Myc/Bcl-XL axis mediates context-dependent oncogenic effects of PTEN with implications for cancer prognosis and therapy. Neoplasia 2013, 15, 952–965. [Google Scholar] [CrossRef] [PubMed]
- Pfister, N.T.; Fomin, V.; Regunath, K.; Zhou, J.Y.; Zhou, W.; Silwal-Pandit, L.; Freed-Pastor, W.A.; Laptenko, O.; Neo, S.P.; Bargonetti, J.; et al. Mutant p53 cooperates with the SWI/SNF chromatin remodeling complex to regulate VEGFR2 in breast cancer cells. Genes Dev. 2015, 29, 1298–1315. [Google Scholar] [CrossRef] [PubMed]
- Brazdova, M.; Quante, T.; Togel, L.; Walter, K.; Loscher, C.; Tichy, V.; Cincarova, L.; Deppert, W.; Tolstonog, G.V. Modulation of gene expression in U251 glioblastoma cells by binding of mutant p53 R273H to intronic and intergenic sequences. Nucleic Acids Res. 2009, 37, 1486–1500. [Google Scholar] [CrossRef] [PubMed]
- Quante, T.; Otto, B.; Brazdova, M.; Kejnovska, I.; Deppert, W.; Tolstonog, G.V. Mutant p53 is a transcriptional co-factor that binds to G-rich regulatory regions of active genes and generates transcriptional plasticity. Cell Cycle 2012, 11, 3290–3303. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Sammons, M.A.; Donahue, G.; Dou, Z.; Vedadi, M.; Getlik, M.; Barsyte-Lovejoy, D.; Al-awar, R.; Katona, B.W.; Shilatifard, A.; et al. Gain-of-function p53 mutants co-opt chromatin pathways to drive cancer growth. Nature 2015, 525, 206–211. [Google Scholar] [CrossRef] [PubMed]
- Joerger, A.C.; Fersht, A.R. Structural biology of the tumor suppressor p53. Annu. Rev. Biochem. 2008, 77, 557–582. [Google Scholar] [CrossRef] [PubMed]
- Muller, P.A.; Vousden, K.H. Mutant p53 in cancer: New functions and therapeutic opportunities. Cancer Cell 2014, 25, 304–317. [Google Scholar] [CrossRef] [PubMed]
- Vu, B.; Wovkulich, P.; Pizzolato, G.; Lovey, A.; Ding, Q.; Jiang, N.; Liu, J.J.; Zhao, C.; Glenn, K.; Wen, Y.; et al. Discovery of RG7112: A Small-Molecule MDM2 Inhibitor in Clinical Development. ACS Med. Chem. Lett. 2013, 4, 466–469. [Google Scholar] [CrossRef] [PubMed]
- Canon, J.; Osgood, T.; Olson, S.H.; Saiki, A.Y.; Robertson, R.; Yu, D.; Eksterowicz, J.; Ye, Q.; Jin, L.; Chen, A.; et al. The MDM2 Inhibitor AMG 232 Demonstrates Robust Antitumor Efficacy and Potentiates the Activity of p53-Inducing Cytotoxic Agents. Mol. Cancer Ther. 2015, 14, 649–658. [Google Scholar] [CrossRef] [PubMed]
- Her, N.G.; Oh, J.W.; Oh, Y.J.; Han, S.; Cho, H.J.; Lee, Y.; Ryu, G.H.; Nam, D.H. Potent effect of the MDM2 inhibitor AMG232 on suppression of glioblastoma stem cells. Cell Death Dis. 2018, 9, 792. [Google Scholar] [CrossRef] [PubMed]
- Verreault, M.; Schmitt, C.; Goldwirt, L.; Pelton, K.; Haidar, S.; Levasseur, C.; Guehennec, J.; Knoff, D.; Labussiere, M.; Marie, Y.; et al. Preclinical Efficacy of the MDM2 Inhibitor RG7112 in MDM2-Amplified and TP53 Wild-type Glioblastomas. Clin. Cancer Res. 2016, 22, 1185–1196. [Google Scholar] [CrossRef] [PubMed]
- Daniele, S.; Taliani, S.; Da Pozzo, E.; Giacomelli, C.; Costa, B.; Trincavelli, M.L.; Rossi, L.; La Pietra, V.; Barresi, E.; Carotenuto, A.; et al. Apoptosis therapy in cancer: The first single-molecule co-activating p53 and the translocator protein in glioblastoma. Sci. Rep. 2014, 4, 4749. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, B.D.; Pietenpol, J.A. Targeting mutant p53 in human tumors. J. Clin. Oncol. 2012, 30, 3648–3650. [Google Scholar] [CrossRef] [PubMed]
- Maslon, M.M.; Hupp, T.R. Drug discovery and mutant p53. Trends Cell Biol. 2010, 20, 542–555. [Google Scholar] [CrossRef] [PubMed]
- Wiman, K.G. Pharmacological reactivation of mutant p53: From protein structure to the cancer patient. Oncogene 2010, 29, 4245–4252. [Google Scholar] [CrossRef] [PubMed]
- Weinmann, L.; Wischhusen, J.; Demma, M.J.; Naumann, U.; Roth, P.; Dasmahapatra, B.; Weller, M. A novel p53 rescue compound induces p53-dependent growth arrest and sensitises glioma cells to Apo2L/TRAIL-induced apoptosis. Cell Death Differ. 2008, 15, 718–729. [Google Scholar] [CrossRef] [PubMed]
- Wischhusen, J.; Naumann, U.; Ohgaki, H.; Rastinejad, F.; Weller, M. CP-31398, a novel p53-stabilizing agent, induces p53-dependent and p53-independent glioma cell death. Oncogene 2003, 22, 8233–8245. [Google Scholar] [CrossRef] [PubMed]
- Bykov, V.J.; Issaeva, N.; Shilov, A.; Hultcrantz, M.; Pugacheva, E.; Chumakov, P.; Bergman, J.; Wiman, K.G.; Selivanova, G. Restoration of the tumor suppressor function to mutant p53 by a low-molecular-weight compound. Nat. Med. 2002, 8, 282–288. [Google Scholar] [CrossRef] [PubMed]
- Lambert, J.M.; Gorzov, P.; Veprintsev, D.B.; Soderqvist, M.; Segerback, D.; Bergman, J.; Fersht, A.R.; Hainaut, P.; Wiman, K.G.; Bykov, V.J. PRIMA-1 reactivates mutant p53 by covalent binding to the core domain. Cancer Cell 2009, 15, 376–388. [Google Scholar] [CrossRef] [PubMed]
- Nahi, H.; Lehmann, S.; Mollgard, L.; Bengtzen, S.; Selivanova, G.; Wiman, K.G.; Paul, C.; Merup, M. Effects of PRIMA-1 on chronic lymphocytic leukaemia cells with and without hemizygous p53 deletion. Br. J. Haematol. 2004, 127, 285–291. [Google Scholar] [CrossRef] [PubMed]
- Nahi, H.; Merup, M.; Lehmann, S.; Bengtzen, S.; Mollgard, L.; Selivanova, G.; Wiman, K.G.; Paul, C. PRIMA-1 induces apoptosis in acute myeloid leukaemia cells with p53 gene deletion. Br. J. Haematol. 2006, 132, 230–236. [Google Scholar] [CrossRef] [PubMed]
- Patyka, M.; Sharifi, Z.; Petrecca, K.; Mansure, J.; Jean-Claude, B.; Sabri, S. Sensitivity to PRIMA-1MET is associated with decreased MGMT in human glioblastoma cells and glioblastoma stem cells irrespective of p53 status. Oncotarget 2016, 7, 60245–60269. [Google Scholar] [CrossRef] [PubMed]
- Bykov, V.J.; Zache, N.; Stridh, H.; Westman, J.; Bergman, J.; Selivanova, G.; Wiman, K.G. PRIMA-1(MET) synergizes with cisplatin to induce tumor cell apoptosis. Oncogene 2005, 24, 3484–3491. [Google Scholar] [CrossRef] [PubMed]
- Zache, N.; Lambert, J.M.; Wiman, K.G.; Bykov, V.J. PRIMA-1MET inhibits growth of mouse tumors carrying mutant p53. Cell Oncol. 2008, 30, 411–418. [Google Scholar] [PubMed]
- Lehmann, S.; Bykov, V.J.; Ali, D.; Andren, O.; Cherif, H.; Tidefelt, U.; Uggla, B.; Yachnin, J.; Juliusson, G.; Moshfegh, A.; et al. Targeting p53 in vivo: A first-in-human study with p53-targeting compound APR-246 in refractory hematologic malignancies and prostate cancer. J. Clin. Oncol. 2012, 30, 3633–3639. [Google Scholar] [CrossRef] [PubMed]
- Duffy, M.J.; Synnott, N.C.; Crown, J. Mutant p53 as a target for cancer treatment. Eur. J. Cancer 2017, 83, 258–265. [Google Scholar] [CrossRef] [PubMed]
- Aggarwal, M.; Saxena, R.; Sinclair, E.; Fu, Y.; Jacobs, A.; Dyba, M.; Wang, X.; Cruz, I.; Berry, D.; Kallakury, B.; et al. Reactivation of mutant p53 by a dietary-related compound phenethyl isothiocyanate inhibits tumor growth. Cell Death Differ. 2016, 23, 1615–1627. [Google Scholar] [CrossRef] [PubMed]
- Soragni, A.; Janzen, D.M.; Johnson, L.M.; Lindgren, A.G.; Thai-Quynh Nguyen, A.; Tiourin, E.; Soriaga, A.B.; Lu, J.; Jiang, L.; Faull, K.F.; et al. A Designed Inhibitor of p53 Aggregation Rescues p53 Tumor Suppression in Ovarian Carcinomas. Cancer Cell 2016, 29, 90–103. [Google Scholar] [CrossRef] [PubMed]
- Tal, P.; Eizenberger, S.; Cohen, E.; Goldfinger, N.; Pietrokovski, S.; Oren, M.; Rotter, V. Cancer therapeutic approach based on conformational stabilization of mutant p53 protein by small peptides. Oncotarget 2016, 7, 11817–11837. [Google Scholar] [CrossRef] [PubMed]
- Joerger, A.C.; Fersht, A.R. The p53 Pathway: Origins, Inactivation in Cancer, and Emerging Therapeutic Approaches. Annu. Rev. Biochem. 2016, 85, 375–404. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Vazquez, A.; Levine, A.J.; Carpizo, D.R. Allele-specific p53 mutant reactivation. Cancer Cell 2012, 21, 614–625. [Google Scholar] [CrossRef] [PubMed]
- Pal, S.; Kozono, D.; Yang, X.; Fendler, W.; Fitts, W.; Ni, J.; Alberta, J.A.; Zhao, J.; Liu, K.X.; Bian, J.; et al. Dual HDAC and PI3K inhibition abrogates NFkappaB- and FOXM1-mediated DNA damage response to radiosensitize pediatric high-grade gliomas. Cancer Res. 2018, 78, 4007–4021. [Google Scholar] [CrossRef] [PubMed]
- Staberg, M.; Michaelsen, S.R.; Rasmussen, R.D.; Villingshoj, M.; Poulsen, H.S.; Hamerlik, P. Inhibition of histone deacetylases sensitizes glioblastoma cells to lomustine. Cell Oncol. 2017, 40, 21–32. [Google Scholar] [CrossRef] [PubMed]
- Liffers, K.; Kolbe, K.; Westphal, M.; Lamszus, K.; Schulte, A. Histone Deacetylase Inhibitors Resensitize EGFR/EGFRvIII-Overexpressing, Erlotinib-Resistant Glioblastoma Cells to Tyrosine Kinase Inhibition. Target Oncol. 2016, 11, 29–40. [Google Scholar] [CrossRef] [PubMed]
- Singh, M.M.; Johnson, B.; Venkatarayan, A.; Flores, E.R.; Zhang, J.; Su, X.; Barton, M.; Lang, F.; Chandra, J. Preclinical activity of combined HDAC and KDM1A inhibition in glioblastoma. Neuro Oncol. 2015, 17, 1463–1473. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Marchenko, N.D.; Moll, U.M. SAHA shows preferential cytotoxicity in mutant p53 cancer cells by destabilizing mutant p53 through inhibition of the HDAC6-Hsp90 chaperone axis. Cell Death Differ. 2011, 18, 1904–1913. [Google Scholar] [CrossRef] [PubMed]
- Kitange, G.J.; Mladek, A.C.; Carlson, B.L.; Schroeder, M.A.; Pokorny, J.L.; Cen, L.; Decker, P.A.; Wu, W.; Lomberk, G.A.; Gupta, S.K.; et al. Inhibition of histone deacetylation potentiates the evolution of acquired temozolomide resistance linked to MGMT upregulation in glioblastoma xenografts. Clin. Cancer Res. 2012, 18, 4070–4079. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.F.; Zhao, Z.J.; Liu, J.J.; Yang, X.H.; Gao, Y.; Zhao, S.; Shi, S.; Huang, K.Q.; Zheng, H.C. SAHA and/or MG132 reverse the aggressive phenotypes of glioma cells: An in vitro and vivo study. Oncotarget 2017, 8, 3156–3169. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.A.; Kwak, P.A.; Park, C.K.; Wang, K.C.; Phi, J.H.; Lee, J.Y.; Lee, C.S.; Lee, J.H.; Kim, S.K. A novel histone deacetylase inhibitor, CKD5, has potent anti-cancer effects in glioblastoma. Oncotarget 2017, 8, 9123–9133. [Google Scholar] [CrossRef] [PubMed]
- Rasmussen, R.D.; Gajjar, M.K.; Jensen, K.E.; Hamerlik, P. Enhanced efficacy of combined HDAC and PARP targeting in glioblastoma. Mol. Oncol. 2016, 10, 751–763. [Google Scholar] [CrossRef] [PubMed]
- Yan, W.; Liu, S.; Xu, E.; Zhang, J.; Zhang, Y.; Chen, X.; Chen, X. Histone deacetylase inhibitors suppress mutant p53 transcription via histone deacetylase 8. Oncogene 2013, 32, 599–609. [Google Scholar] [CrossRef] [PubMed]
- Wakimoto, H.; Tanaka, S.; Curry, W.T.; Loebel, F.; Zhao, D.; Tateishi, K.; Chen, J.; Klofas, L.K.; Lelic, N.; Kim, J.C.; et al. Targetable signaling pathway mutations are associated with malignant phenotype in IDH-mutant gliomas. Clin. Cancer Res. 2014, 20, 2898–2909. [Google Scholar] [CrossRef] [PubMed]




© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, Y.; Dube, C.; Gibert, M.; Cruickshanks, N.; Wang, B.; Coughlan, M.; Yang, Y.; Setiady, I.; Deveau, C.; Saoud, K.; et al. The p53 Pathway in Glioblastoma. Cancers 2018, 10, 297. https://doi.org/10.3390/cancers10090297
Zhang Y, Dube C, Gibert M, Cruickshanks N, Wang B, Coughlan M, Yang Y, Setiady I, Deveau C, Saoud K, et al. The p53 Pathway in Glioblastoma. Cancers. 2018; 10(9):297. https://doi.org/10.3390/cancers10090297
Chicago/Turabian StyleZhang, Ying, Collin Dube, Myron Gibert, Nichola Cruickshanks, Baomin Wang, Maeve Coughlan, Yanzhi Yang, Initha Setiady, Ciana Deveau, Karim Saoud, and et al. 2018. "The p53 Pathway in Glioblastoma" Cancers 10, no. 9: 297. https://doi.org/10.3390/cancers10090297
APA StyleZhang, Y., Dube, C., Gibert, M., Cruickshanks, N., Wang, B., Coughlan, M., Yang, Y., Setiady, I., Deveau, C., Saoud, K., Grello, C., Oxford, M., Yuan, F., & Abounader, R. (2018). The p53 Pathway in Glioblastoma. Cancers, 10(9), 297. https://doi.org/10.3390/cancers10090297