p53 Isoforms and Their Implications in Cancer


Abstract
:
1. p53—The Gene, the Protein, and the Isoforms
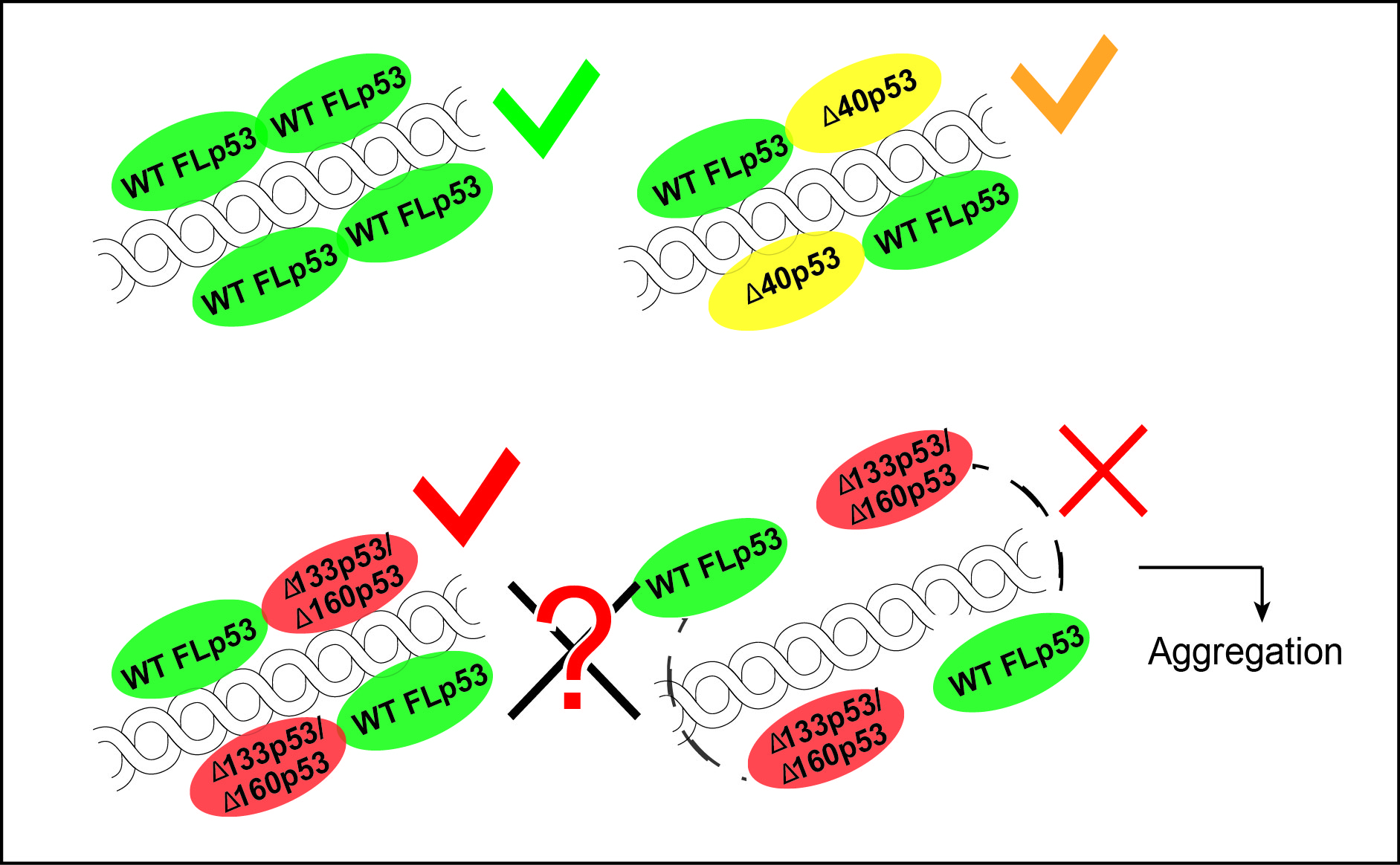
1.1. The TP53 Gene and the Synthesis of the p53 Isoforms
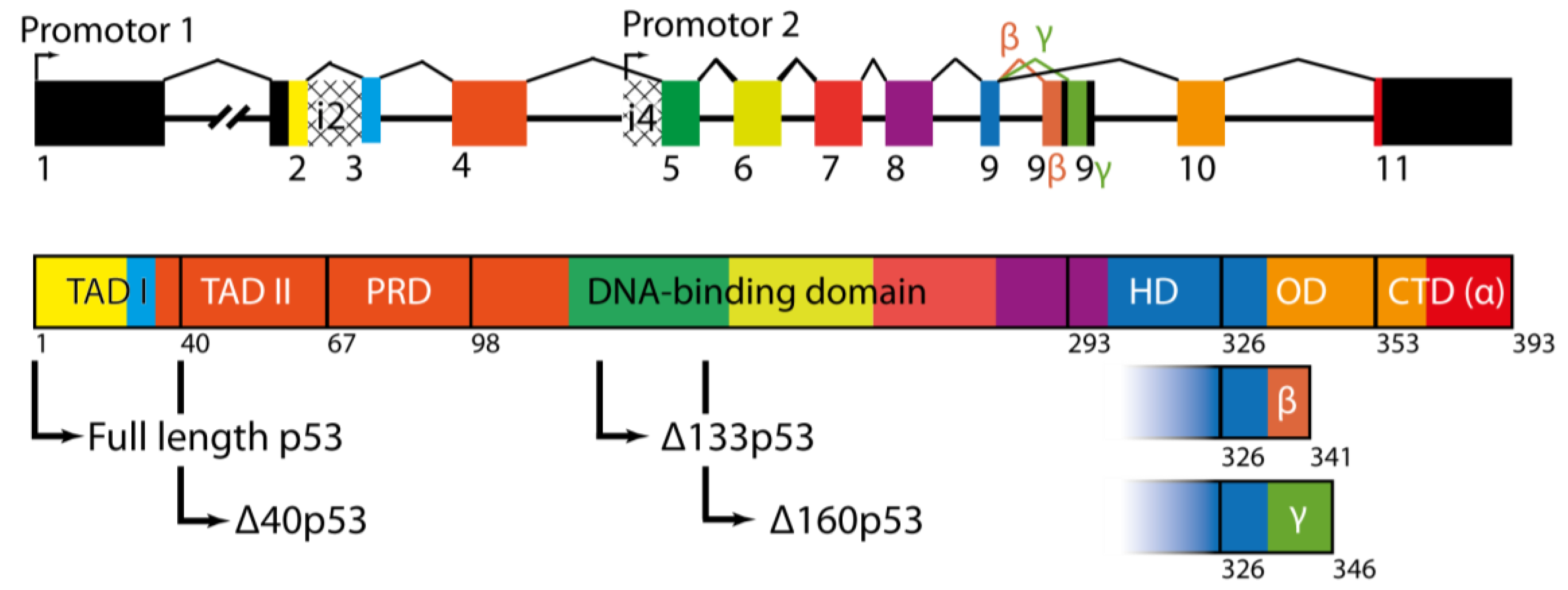
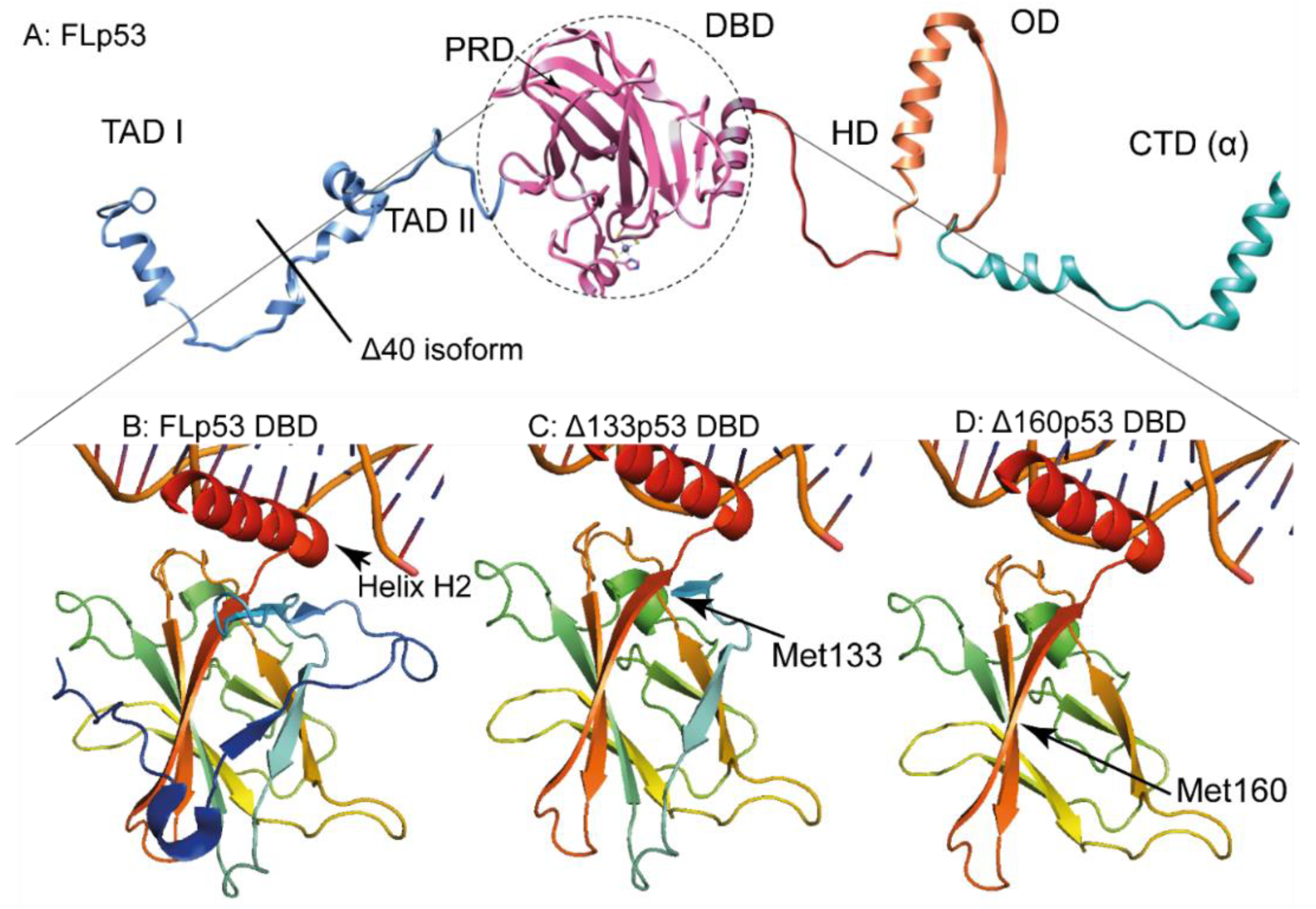
1.2. p53 Protein and Its Different Domains
1.3. p53 Isoforms Are Variants of p53 with N- and C-Terminal Truncations
2. Major N-Terminal Isoforms of p53
2.1. The ∆40p53 Isoform
2.2. The ∆133p53 Isoform
2.3. The ∆160p53 Isoform
3. Disease Perspective of the p53 Isoforms
4. Ways to Inactivate or Modulate the p53 Pathway
4.1. Mutations
4.2. Dominant Negative Effect
4.3. Gain of Function
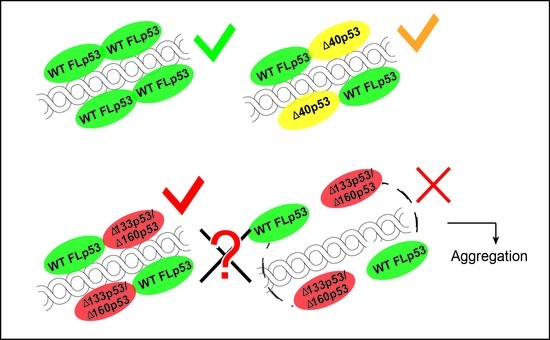
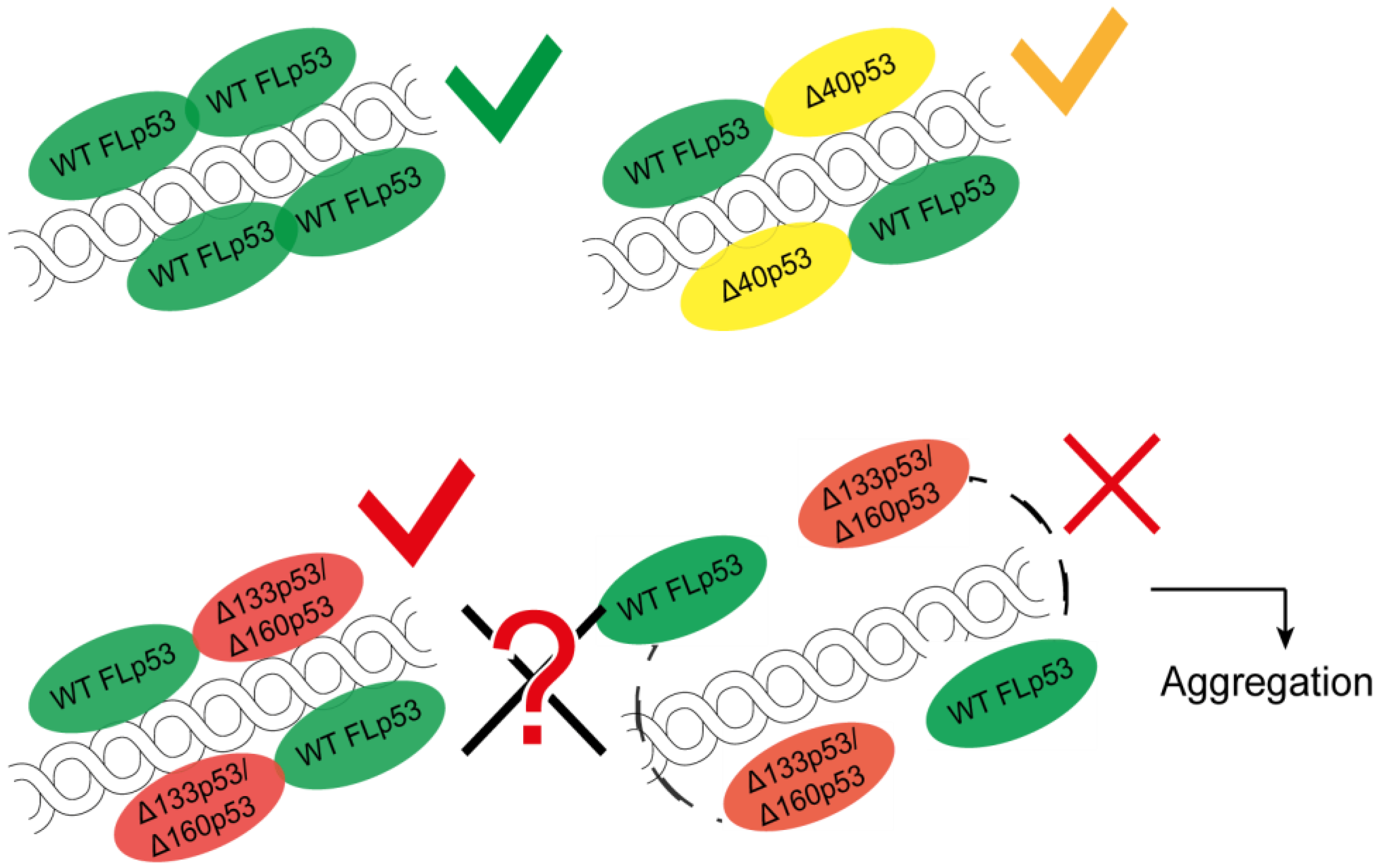
4.4. Aggregation
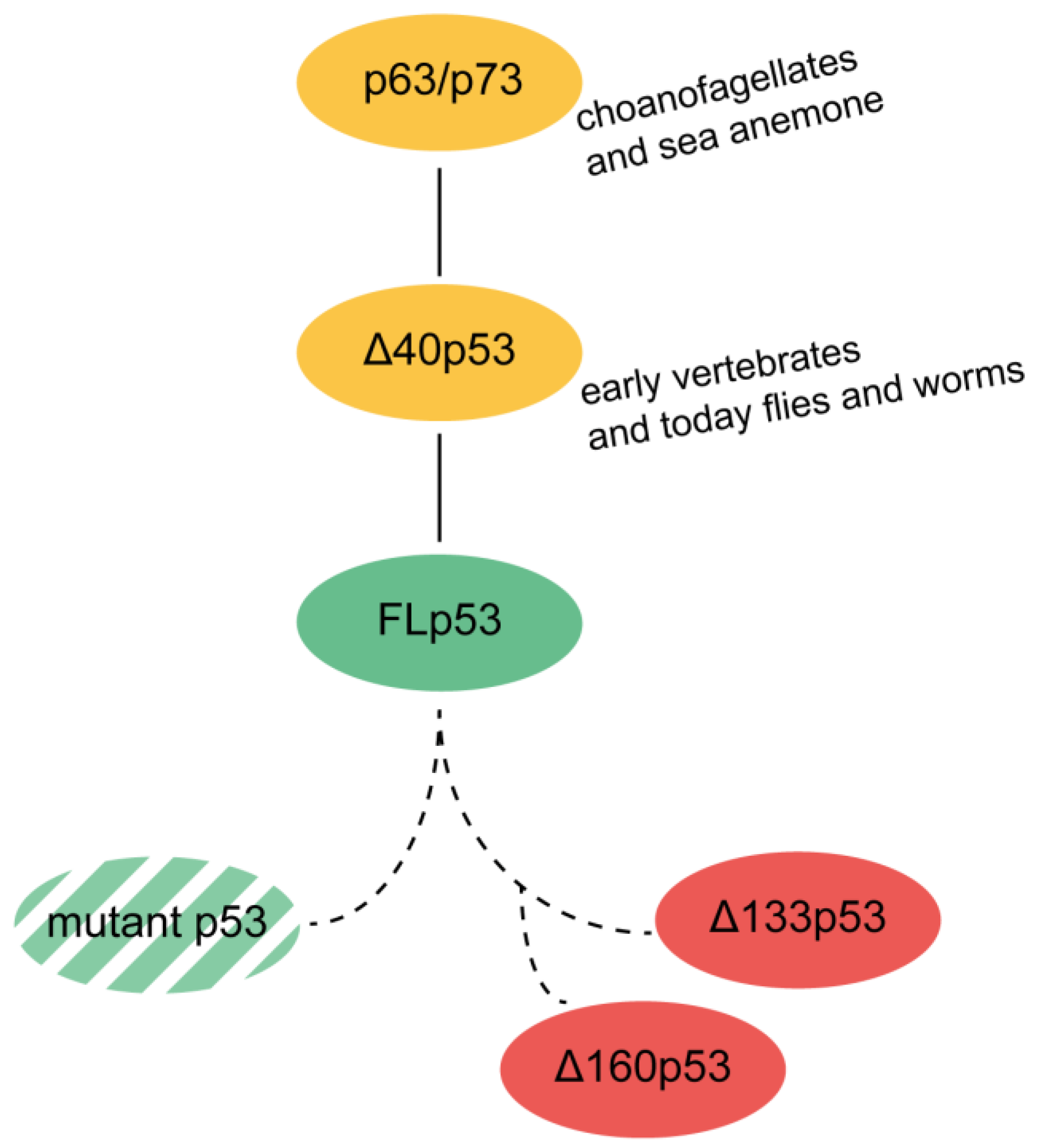
5. Evolutionary Origin
6. Conclusions/Summary/Key Points
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Lane, D.P. p53, guardian of the genome. Nature 1992, 358, 15–16. [Google Scholar] [CrossRef] [PubMed]
- Hainaut, P. TP53: Coordinator of the processes that underlie the hallmarks of cancer. In p53 in the Clinics; Springer New York: New York, NY, USA, 2013; pp. 1–23. ISBN 9781461436768. [Google Scholar]
- Harris, S.L.; Levine, A.J. The p53 pathway: Positive and negative feedback loops. Oncogene 2005, 24, 2899–2908. [Google Scholar] [CrossRef] [PubMed]
- Powell, D.J.; Hrstka, R.; Candeias, M.; Bourougaa, K.; Vojtesek, B.; Fåhraeus, R. Stress-dependent changes in the properties of p53 complexes by the alternative translation product p53/47. Cell Cycle 2008, 7, 950–959. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kitayner, M.; Rozenberg, H.; Kessler, N.; Rabinovich, D.; Shaulov, L.; Haran, T.E.; Shakked, Z. Structural Basis of DNA Recognition by p53 Tetramers. Mol. Cell 2006, 22, 741–753. [Google Scholar] [CrossRef] [PubMed]
- Lahav, G.; Rosenfeld, N.; Sigal, A.; Geva-Zatorsky, N.; Levine, A.J.; Elowitz, M.B.; Alon, U. Dynamics of the p53-Mdm2 feedback loop in individual cells. Nat. Genet. 2004, 36, 147–150. [Google Scholar] [CrossRef] [PubMed]
- Brown, C.J.; Lain, S.; Verma, C.S.; Fersht, A.R.; Lane, D.P. Awakening guardian angels: Drugging the p53 pathway. Nat. Rev. Cancer 2009, 9, 862–873. [Google Scholar] [CrossRef] [PubMed]
- Chen, J. Loss of function of def selectively up-regulates 113p53 expression to arrest expansion growth of digestive organs in zebrafish. Genes Dev. 2005, 19, 2900–2911. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gong, H.; Zhang, Y.; Jiang, K.; Ye, S.; Chen, S.; Zhang, Q.; Peng, J.; Chen, J. p73 coordinates with Δ133p53 to promote DNA double-strand break repair. Cell Death Differ. 2018, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Olivier, M. TP53 Somatic Mutations: Prognostic and Predictive Value in Human Cancers. In p53 in the Clinics; Hainaut, P., Olivier, M., Wiman, K.G., Eds.; Springer New York: New York, NY, USA, 2013; pp. 127–146. [Google Scholar]
- Chen, Y.; Dey, R.; Chen, L. Crystal Structure of the p53 Core Domain Bound to a Full Consensus Site as a Self-Assembled Tetramer. Structure 2010, 18, 246–256. [Google Scholar] [CrossRef] [PubMed]
- Joruiz, S.M.; Bourdon, J.-C. p53 Isoforms: Key Regulators of the Cell Fate Decision. Cold Spring Harb. Perspect. Med. 2016, 6, a026039. [Google Scholar] [CrossRef] [PubMed]
- Khoury, M.P.; Bourdon, J.-C. The isoforms of the p53 protein. Cold Spring Harb. Perspect. Biol. 2010, 2, a000927. [Google Scholar] [CrossRef] [PubMed]
- Candeias, M.M.; Powell, D.J.; Roubalova, E.; Apcher, S.; Bourougaa, K.; Vojtesek, B.; Bruzzoni-Giovanelli, H.; Fåhraeus, R. Expression of p53 and p53/47 are controlled by alternative mechanisms of messenger RNA translation initiation. Oncogene 2006, 25, 6936–6947. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ray, P.S.; Grover, R.; Das, S. Two internal ribosome entry sites mediate the translation of p53 isoforms. EMBO Rep. 2006, 7, 404–410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vousden, K.H.; Lane, D.P. p53 in health and disease. Nat. Rev. Mol. Cell Biol. 2007, 8, 275–283. [Google Scholar] [CrossRef] [PubMed]
- Jeffrey, P.D.; Gorina, S.; Pavletich, N.P. Crystal structure of the tetramerization domain of the p53 tumor suppressor at 1.7 angstroms. Science 1995, 267, 1498–1502. [Google Scholar] [CrossRef] [PubMed]
- Stenger, J.E.; Tegtmeyer, P.; Mayr, G.A.; Reed, M.; Wang, Y.; Wang, P.; Hough, P.V.; Mastrangelo, I.A. p53 oligomerization and DNA looping are linked with transcriptional activation. EMBO J. 1994, 13, 6011–6020. [Google Scholar] [PubMed]
- Jackson, P.; Mastrangelo, I.; Reed, M.; Tegtmeyer, P.; Yardley, G.; Barrett, J. Synergistic transcriptional activation of the MCK promoter by p53: Tetramers link separated DNA response elements by DNA looping. Oncogene 1998, 16, 283–292. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; An, S.S.A. Role of p53 isoforms and aggregations in cancer. Medicine (Baltimore) 2016, 95, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Candau, R.; Scolnick, D.M.; Darpino, P.; Ying, C.Y.; Halazonetis, T.D.; Berger, S.L. Two tandem and independent sub-activation domains in the amino terminus of p53 require the adaptor complex for activity. Oncogene 1997, 15, 807–816. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brady, C.A.; Jiang, D.; Mello, S.S.; Johnson, T.M.; Jarvis, L.A.; Kozak, M.M.; Broz, D.K.; Basak, S.; Park, E.J.; McLaughlin, M.E.; et al. Distinct p53 Transcriptional Programs Dictate Acute DNA-Damage Responses and Tumor Suppression. Cell 2011, 145, 571–583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dawson, R.; Müller, L.; Dehner, A.; Klein, C.; Kessler, H.; Buchner, J. The N-terminal Domain of p53 is Natively Unfolded. J. Mol. Biol. 2003, 332, 1131–1141. [Google Scholar] [CrossRef] [PubMed]
- Ferreon, J.C.; Lee, C.W.; Arai, M.; Martinez-Yamout, M.A.; Dyson, H.J.; Wright, P.E. Cooperative regulation of p53 by modulation of ternary complex formation with CBP/p300 and HDM2. Proc. Nat. Acad. Sci. USA 2009, 106, 6591–6596. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, J.; Kim, D.H.; Lee, S.W.; Choi, K.Y.; Sung, Y.C. Transactivation ability of p53 transcriptional activation domain is directly related to the binding affinity to TATA-binding protein. J. Biol. Chem. 1995, 270, 25014–25019. [Google Scholar] [CrossRef] [PubMed]
- Di Lello, P.; Jenkins, L.M.M.; Jones, T.N.; Nguyen, B.D.; Hara, T.; Yamaguchi, H.; Dikeakos, J.D.; Appella, E.; Legault, P.; Omichinski, J.G. Structure of the Tfb1/p53 Complex: Insights into the Interaction between the p62/Tfb1 Subunit of TFIIH and the Activation Domain of p53. Mol. Cell 2006, 22, 731–740. [Google Scholar] [CrossRef] [PubMed]
- Walker, K.K.; Levine, A.J. Identification of a novel p53 functional domain that is necessary for efficient growth suppression. Proc. Nat. Acad. Sci. USA 1996, 93, 15335–15340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, H.; Chen, J.K.; Feng, S.; Dalgarno, D.C.; Brauer, A.W.; Schrelber, S.L. Structural basis for the binding of proline-rich peptides to SH3 domains. Cell 1994, 76, 933–945. [Google Scholar] [CrossRef]
- Venot, C.; Maratrat, M.; Dureuil, C.; Conseiller, E.; Bracco, L.; Debussche, L. The requirement for the p53 proline-rich functional domain for mediation of apoptosis is correlated with specific PIG3 gene transactivation and with transcriptional repression. EMBO J. 1998, 17, 4668–4679. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riley, T.; Sontag, E.; Chen, P.; Levine, A. Transcriptional control of human p53-regulated genes. Nat. Rev. Mol. Cell Biol. 2008, 9, 402–412. [Google Scholar] [CrossRef] [PubMed]
- Cho, Y.; Gorina, S.; Jeffrey, P.; Pavletich, N. Crystal structure of a p53 tumor suppressor-DNA complex: Understanding tumorigenic mutations. Science 1994, 265, 346–355. [Google Scholar] [CrossRef] [PubMed]
- Melero, R.; Rajagopalan, S.; Lázaro, M.; Joerger, A.C.; Brandt, T.; Veprintsev, D.B.; Lasso, G.; Gil, D.; Scheres, S.H.W.; Carazo, J.M.; et al. Electron microscopy studies on the quaternary structure of p53 reveal different binding modes for p53 tetramers in complex with DNA. Proc. Natl. Acad. Sci. USA 2011, 108, 557–562. [Google Scholar] [CrossRef] [PubMed]
- D’Abramo, M.; Bešker, N.; Desideri, A.; Levine, A.J.; Melino, G.; Chillemi, G. The p53 tetramer shows an induced-fit interaction of the C-terminal domain with the DNA-binding domain. Oncogene 2016, 35, 3272–3281. [Google Scholar] [CrossRef] [PubMed]
- Riley, K.J.-L.; Maher, L.J. p53 RNA interactions: New clues in an old mystery. RNA 2007, 13, 1825–1833. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chillemi, G.; Kehrloesser, S.; Bernassola, F.; Desideri, A.; Dötsch, V.; Levine, A.J.; Melino, G. Structural Evolution and Dynamics of the p53 Proteins. Cold Spring Harb. Perspect. Med. 2017, 7, a028308. [Google Scholar] [CrossRef] [PubMed]
- Bell, S.; Klein, C.; Müller, L.; Hansen, S.; Buchner, J. p53 contains large unstructured regions in its native state. J. Mol. Biol. 2002, 322, 917–927. [Google Scholar] [CrossRef]
- Joerger, A.C.; Fersht, A.R. Structural Biology of the Tumor Suppressor p53. Annu. Rev. Biochem. 2008, 77, 557–582. [Google Scholar] [CrossRef] [PubMed]
- Kastenhuber, E.R.; Lowe, S.W. Putting p53 in Context. Cell 2017, 170, 1062–1078. [Google Scholar] [CrossRef] [PubMed]
- Bourdon, J.-C.; Fernandes, K.; Murray-Zmijewski, F.; Liu, G.; Diot, A.; Xirodimas, D.P.; Saville, M.K.; Lane, D.P. p53 isoforms can regulate p53 transcriptional activity. Genes Dev. 2005, 19, 2122–2137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anensen, N.; Oyan, A.M.; Bourdon, J.C.; Kalland, K.H.; Bruserud, O.; Gjertsen, B.T. A distinct p53 protein isoform signature reflects the onset of induction chemotherapy for acute myeloid leukemia. Clin. Cancer Res. 2006, 12, 3985–3992. [Google Scholar] [CrossRef] [PubMed]
- Matlashewski, G.; Lamb, P.; Pim, D.; Peacock, J.; Crawford, L.; Benchimol, S. Isolation and characterization of a human p53 cDNA clone: Expression of the human p53 gene. EMBO J. 1984, 3, 3257–3262. [Google Scholar] [PubMed]
- Wolf, D.; Harris, N.; Goldfinger, N.; Rotter, V. Isolation of a full-length mouse cDNA clone coding for an immunologically distinct p53 molecule. Mol. Cell. Biol. 1985, 5, 127–132. [Google Scholar] [CrossRef] [PubMed]
- Hafsi, H.; Santos-Silva, D.; Courtois-Cox, S.; Hainaut, P. Effects of Δ40p53, an isoform of p53 lacking the N-terminus, on transactivation capacity of the tumor suppressor protein p53. BMC Cancer 2013, 13, 134. [Google Scholar] [CrossRef] [PubMed]
- Slatter, T.L.; Hung, N.; Bowie, S.; Campbell, H.; Rubio, C.; Speidel, D.; Wilson, M.; Baird, M.; Royds, J.A.; Braithwaite, A.W. Δ122p53, a mouse model of Δ133p53α, enhances the tumor-suppressor activities of an attenuated p53 mutant. Cell Death Dis. 2015, 6, e1783. [Google Scholar] [CrossRef] [PubMed]
- Courtois, S.; Verhaegh, G.; North, S.; Luciani, M.-G.; Lassus, P.; Hibner, U.; Oren, M.; Hainaut, P. ΔN-p53, a natural isoform of p53 lacking the first transactivation domain, counteracts growth suppression by wild-type p53. Oncogene 2002, 21, 6722–6728. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharathchandra, A.; Katoch, A.; Das, S. IRES mediated translational regulation of p53 isoforms. Wiley Interdiscip. Rev. RNA 2014, 5, 131–139. [Google Scholar] [CrossRef] [PubMed]
- Ohki, R.; Kawase, T.; Ohta, T.; Ichikawa, H.; Taya, Y. Dissecting functional roles of p53 N-terminal transactivation domains by microarray expression analysis. Cancer Sci. 2007, 98, 189–200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, W.M.; Siu, W.Y.; Lau, A.; Poon, R.Y.C. How Many Mutant p53 Molecules Are Needed To Inactivate a Tetramer? Mol. Cell. Biol. 2004, 24, 3536–3551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weinberg, R.L.; Veprintsev, D.B.; Fersht, A.R. Cooperative binding of tetrameric p53 to DNA. J. Mol. Biol. 2004, 341, 1145–1159. [Google Scholar] [CrossRef] [PubMed]
- Maier, B.; Gluba, W.; Bernier, B.; Turner, T.; Mohammad, K.; Guise, T.; Sutherland, A.; Thorner, M.; Scrable, H. Modulation of mammalian life span by the short isoform of p53. Genes Dev. 2004, 18, 306–319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horikawa, I.; Park, K.; Isogaya, K.; Hiyoshi, Y.; Li, H.; Anami, K.; Robles, A.I.; Mondal, A.M.; Fujita, K.; Serrano, M.; et al. Δ133P53 Represses P53-Inducible Senescence Genes and Enhances the Generation of Human Induced Pluripotent Stem Cells. Cell Death Differ. 2017, 24, 1017–1028. [Google Scholar] [CrossRef] [PubMed]
- Bernard, H.; Garmy-Susini, B.; Ainaoui, N.; Van Den Berghe, L.; Peurichard, A.; Javerzat, S.; Bikfalvi, A.; Lane, D.P.; Bourdon, J.C.; Prats, A.-C. The p53 isoform, Δ133p53α, stimulates angiogenesis and tumour progression. Oncogene 2013, 32, 2150–2160. [Google Scholar] [CrossRef] [PubMed]
- Xie, N.; Chen, M.; Dai, R.; Zhang, Y.; Zhao, H.; Song, Z.; Zhang, L.; Li, Z.; Feng, Y.; Gao, H.; et al. SRSF1 promotes vascular smooth muscle cell proliferation through a Δ133p53/EGR1/KLF5 pathway. Nat. Commun. 2017, 8, 16016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campbell, H.; Fleming, N.; Roth, I.; Mehta, S.; Wiles, A.; Williams, G.; Vennin, C.; Arsic, N.; Parkin, A.; Pajic, M.; et al. ∆133p53 isoform promotes tumour invasion and metastasis via interleukin-6 activation of JAK-STAT and RhoA-ROCK signalling. Nat. Commun. 2018, 9, 254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.; Ng, S.M.; Chang, C.; Zhang, Z.; Bourdon, J.-C.; Lane, D.P.; Peng, J. p53 isoform delta113p53 is a p53 target gene that antagonizes p53 apoptotic activity via BclxL activation in zebrafish. Genes Dev. 2009, 23, 278–290. [Google Scholar] [CrossRef] [PubMed]
- Aoubala, M.; Murray-Zmijewski, F.; Khoury, M.P.; Fernandes, K.; Perrier, S.; Bernard, H.; Prats, A.-C.; Lane, D.P.; Bourdon, J.-C. p53 directly transactivates Δ133p53α, regulating cell fate outcome in response to DNA damage. Cell Death Differ. 2011, 18, 248–258. [Google Scholar] [CrossRef] [PubMed]
- Kovachev, P.S.; Banerjee, D.; Rangel, L.P.; Eriksson, J.; Pedrote, M.M.; Martins-Dinis, M.M.D.C.; Edwards, K.; Cordeiro, Y.; Silva, J.L.; Sanyal, S. Distinct modulatory role of RNA in the aggregation of the tumor suppressor protein p53 core domain. J. Biol. Chem. 2017, 292, 9345–9357. [Google Scholar] [CrossRef] [PubMed]
- Khoury, M.P.; Bourdon, J.-C. p53 Isoforms: An Intracellular Microprocessor? Genes Cancer 2011, 2, 453–465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gong, L.; Gong, H.; Pan, X.; Chang, C.; Ou, Z.; Ye, S.; Yin, L.; Yang, L.; Tao, T.; Zhang, Z.; et al. p53 isoform Δ113p53/Δ133p53 promotes DNA double-strand break repair to protect cell from death and senescence in response to DNA damage. Cell Res. 2015, 25, 351–369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mondal, A.M.; Horikawa, I.; Pine, S.R.; Fujita, K.; Morgan, K.M.; Vera, E.; Mazur, S.J.; Appella, E.; Vojtesek, B.; Blasco, M.A.; et al. P53 isoforms regulate aging- and tumor-associated replicative senescence in T lymphocytes. J. Clin. Investig. 2013, 123, 5247–5257. [Google Scholar] [CrossRef] [PubMed]
- Marcel, V.; Perrier, S.; Aoubala, M.; Ageorges, S.; Groves, M.J.; Diot, A.; Fernandes, K.; Tauro, S.; Bourdon, J.C. D160p53 is a novel N-terminal p53 isoform encoded by D133p53 transcript. FEBS Lett. 2010, 584, 4463–4468. [Google Scholar] [CrossRef] [PubMed]
- Marcel, V.; Dichtel-Danjoy, M.-L.; Sagne, C.; Hafsi, H.; Ma, D.; Ortiz-Cuaran, S.; Olivier, M.; Hall, J.; Mollereau, B.; Hainaut, P.; et al. Biological functions of p53 isoforms through evolution: Lessons from animal and cellular models. Cell Death Differ. 2011, 18, 1815–1824. [Google Scholar] [CrossRef] [PubMed]
- Candeias, M.M.; Hagiwara, M.; Matsuda, M. Cancer-specific mutations in p53 induce the translation of Δ160p53 promoting tumorigenesis. EMBO Rep. 2016, 17, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Bourdon, J.-C.; Khoury, M.P.; Diot, A.; Baker, L.; Fernandes, K.; Aoubala, M.; Quinlan, P.; Purdie, C.A.; Jordan, L.B.; Prats, A.-C.; et al. p53 mutant breast cancer patients expressing p53g have as good a prognosis as wild-type p53 breast cancer patients. Breast Cancer Res. 2011, 13, R7. [Google Scholar] [CrossRef] [PubMed]
- Surget, S.; Khoury, M.P.; Bourdon, J. Uncovering the role of p53 splice variants in human malignancy: A clinical perspective. OncoTargets Ther. 2013, 7, 57–68. [Google Scholar] [CrossRef]
- Petitjean, A.; Achatz, M.; Borresen-dale, A.; Hainaut, P.; Olivier, M. TP53 mutations in human cancers: Functional selection and impact on cancer prognosis and outcomes. Oncogene 2007, 26, 2157–2165. [Google Scholar] [CrossRef] [PubMed]
- Bouaoun, L.; Sonkin, D.; Ardin, M.; Hollstein, M.; Byrnes, G.; Zavadil, J.; Olivier, M. TP53 Variations in Human Cancers: New Lessons from the IARC TP53 Database and Genomics Data. Hum. Mutat. 2016, 37, 865–876. [Google Scholar] [CrossRef] [PubMed]
- Vogelstein, B.; Lane, D.; Levine, A.J. Surfing the p53 network. Nature 2000, 408, 307–310. [Google Scholar] [CrossRef] [PubMed]
- Soria, C.; Estermann, F.E.; Espantman, K.C.; O’Shea, C.C. Heterochromatin silencing of p53 target genes by a small viral protein. Nature 2010, 466, 1076–1081. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Collot-Teixeira, S.; Bass, J.; Denis, F.; Ranger-Rogez, S. Human tumor suppressor p53 and DNA viruses. Rev. Med. Virol. 2004, 14, 301–319. [Google Scholar] [CrossRef] [PubMed]
- Wei, J.; Noto, J.; Zaika, E.; Romero-Gallo, J.; Correa, P.; El-Rifai, W.; Peek, R.M.; Zaika, A. Pathogenic bacterium Helicobacter pylori alters the expression profile of p53 protein isoforms and p53 response to cellular stresses. Proc. Nat. Acad. Sci. USA 2012, 109, E2543–E2550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ishimaru, D.; Andrade, L.R.; Teixeira, L.S.P.; Quesado, P.A.; Maiolino, L.M.; Lopez, P.M.; Cordeiro, Y.; Costa, L.T.; Heckl, W.M.; Weissmüller, G.; et al. Fibrillar aggregates of the tumor suppressor p53 core domain. Biochemistry 2003, 42, 9022–9027. [Google Scholar] [CrossRef] [PubMed]
- Ano Bom, A.P.D.D.; Rangel, L.P.; Costa, D.C.F.F.; de Oliveira, G.A.P.P.; Sanches, D.; Braga, C.A.; Gava, L.M.; Ramos, C.H.I.I.; Cepeda, A.O.T.T.; Stumbo, A.C.; et al. Mutant p53 Aggregates into Prion-like Amyloid Oligomers and Fibrils. J. Biol. Chem. 2012, 287, 28152–28162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Terzian, T.; Lozano, G. Mutant p53-Driven Tumorigenesis. In p53 in the Clinics; Hainaut, P., Olivier, M., Wiman, K.G., Eds.; Springer New York: New York, NY, USA, 2013; pp. 77–93. [Google Scholar]
- Freed-Pastor, W.A.; Prives, C. Mutant p53: One name, many proteins. Genes Dev. 2012, 26, 1268–1286. [Google Scholar] [CrossRef] [PubMed]
- Olivier, M.; Eeles, R.; Hollstein, M.; Khan, M.A.; Harris, C.C.; Hainaut, P. The IARC TP53 database: New online mutation analysis and recommendations to users. Hum. Mutat. 2002, 19, 607–614. [Google Scholar] [CrossRef] [PubMed]
- Bullock, A.N.; Henckel, J.; Fersht, A.R. Quantitative analysis of residual folding and DNA binding in mutant p53 core domain: Definition of mutant states for rescue in cancer therapy. Oncogene 2000, 19, 1245–1256. [Google Scholar] [CrossRef] [PubMed]
- Kern, S.E.; Kinzler, K.W.; Baker, S.J.; Nigro, J.M.; Rotter, V.; Levine, A.J.; Friedman, P.; Prives, C.; Vogelstein, B. Mutant p53 proteins bind DNA abnormally in vitro. Oncogene 1991, 6, 131–136. [Google Scholar] [PubMed]
- Langerød, A.; Olivier, M.; Børresen-Dale, A.-L. Assessing TP53 Status in Human Tumors: Lessons from Breast Cancer. In p53 in the Clinics; Hainaut, P., Olivier, M., Wiman, K.G., Eds.; Springer New York: New York, NY, USA, 2013; pp. 147–165. ISBN 9781461436768. [Google Scholar]
- Milner, J.; Medcalf, E.A. Cotranslation of activated mutant p53 with wild type drives the wild-type p53 protein into the mutant conformation. Cell 1991, 65, 765–774. [Google Scholar] [CrossRef]
- Kim, M.P.; Lozano, G. Mutant p53 partners in crime. Cell Death Differ. 2017, 25, 161–168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Terzian, T.; Suh, Y.A.; Iwakuma, T.; Post, S.M.; Neumann, M.; Lang, G.A.; Van Pelt, C.S.; Lozano, G. The inherent instability of mutant p53 is alleviated by Mdm2 or p16 INK4a loss. Genes Dev. 2008, 22, 1337–1344. [Google Scholar] [CrossRef] [PubMed]
- Johnstone, R.W.; Ruefli, A.A.; Lowe, S.W. Apoptosis: A link between cancer genetics and chemotherapy. Cell 2002, 108, 153–164. [Google Scholar] [CrossRef]
- Xu, J.; Reumers, J.; Couceiro, J.R.; De Smet, F.; Gallardo, R.; Rudyak, S.; Cornelis, A.; Rozenski, J.; Zwolinska, A.; Marine, J.-C.; et al. Gain of function of mutant p53 by coaggregation with multiple tumor suppressors. Nat. Chem. Biol. 2011, 7, 285–295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ko, L.J.; Prives, C. p53: Puzzle and paradigm. Genes Dev. 1996, 10, 1054–1072. [Google Scholar] [CrossRef] [PubMed]
- Brosh, R.; Rotter, V. When mutants gain new powers: News from the mutant p53 field. Nat. Rev. Cancer 2009, 9, 701–713. [Google Scholar] [CrossRef] [PubMed]
- Willis, A.; Jung, E.J.; Wakefield, T.; Chen, X. Mutant p53 exerts a dominant negative effect by preventing wild-type p53 from binding to the promoter of its target genes. Oncogene 2004, 23, 2330–2338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Billant, O.; Léon, A.; Le Guellec, S.; Friocourt, G.; Blondel, M.; Voisset, C. The dominant-negative interplay between p53, p63 and p73: A family affair. Oncotarget 2016, 7, 69549–69564. [Google Scholar] [CrossRef] [PubMed]
- Levine, A.J.; Momand, J.; Finlay, C.A. The p53 tumour suppressor gene. Nature 1991, 351, 453–456. [Google Scholar] [CrossRef] [PubMed]
- Pusch, J.; Votteler, M.; Göhler, S.; Engl, J.; Hampel, M.; Walles, H.; Schenke-Layland, K. The physiological performance of a three-dimensional model that mimics the microenvironment of the small intestine. Biomaterials 2011, 32, 7469–7478. [Google Scholar] [CrossRef] [PubMed]
- Aschauer, L.; Muller, P.A.J. Novel targets and interaction partners of mutant p53 Gain-Of-Function. Biochem. Soc. Trans. 2016, 44, 460–466. [Google Scholar] [CrossRef] [PubMed]
- Muller, P.A.J.; Vousden, K.H. P53 Mutations in Cancer. Nat. Cell Biol. 2013, 15, 2–8. [Google Scholar] [CrossRef] [PubMed]
- Yao, Y.; Dai, W. Genomic instability and cancer. J. Carcinog. Mutagen. 2014, 5, 165. [Google Scholar] [CrossRef]
- Shen, Z. Genomic instability and cancer: An introduction. J. Mol. Cell Biol. 2011, 3, 1–3. [Google Scholar] [CrossRef] [PubMed]
- Yaswen, P.; MacKenzie, K.L.; Keith, W.N.; Hentosh, P.; Rodier, F.; Zhu, J.; Firestone, G.L.; Matheu, A.; Carnero, A.; Bilsland, A.; et al. Therapeutic targeting of replicative immortality. Semin. Cancer Biol. 2015, 35, S104–S128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nishida, N.; Yano, H.; Nishida, T.; Kamura, T.; Kojiro, M. Angiogenesis in cancer. Vasc. Health Risk Manag. 2006, 2, 213–219. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, M.L. Deregulation of Energy Metabolism as a Cause and Consequence of Oncogenic Process: Review of Literature. Anat. Physiol. 2016, 6, 1–5. [Google Scholar] [CrossRef]
- Oren, M.; Rotter, V. Mutant p53 gain-of-function in cancer. Cold Spring Harb. Perspect. Biol. 2010, 2, a001107. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, S.; Salot, S.; Sengupta, S.; Navalkar, A.; Ghosh, D.; Jacob, R.; Das, S.; Kumar, R.; Jha, N.N.; Sahay, S.; et al. P53 Amyloid Formation Leading To Its Loss of Function: Implications in Cancer Pathogenesis. Cell Death Differ. 2017, 24, 1784–1798. [Google Scholar] [CrossRef] [PubMed]
- Silva, J.L.; Cino, E.A.; Soares, I.N.; Ferreira, V.F.; de Oliveira, G.A.P. Targeting the Prion-like Aggregation of Mutant p53 to Combat Cancer. Acc. Chem. Res. 2018, 51, 181–190. [Google Scholar] [CrossRef] [PubMed]
- Kraiss, S.; Quaiser, A.; Oren, M.; Montenarh, M. Oligomerization of oncoprotein p53. J. Virol. 1988, 62, 4737–4744. [Google Scholar] [PubMed]
- Costa, D.C.F.; de Oliveira, G.A.P.; Cino, E.A.; Soares, I.N.; Rangel, L.P.; Silva, J.L. Aggregation and Prion-Like Properties of Misfolded Tumor Suppressors: Is Cancer a Prion Disease? Cold Spring Harb. Perspect. Biol. 2016, 8, a023614. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rangel, L.P.; Costa, D.C.; Vieira, T.C.; Silva, J.L. The aggregation of mutant p53 produces prion-like properties in cancer. Prion 2014, 8, 75–84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lasagna-Reeves, C.A.; Clos, A.L.; Castillo-Carranza, D.; Sengupta, U.; Guerrero-Muñoz, M.; Kelly, B.; Wagner, R.; Kayed, R. Dual role of p53 amyloid formation in cancer; loss of function and gain of toxicity. Biochem. Biophys. Res. Commun. 2013, 430, 963–968. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Fersht, A.R. Propagation of aggregated p53: Cross-reaction and coaggregation vs. seeding. Proc. Natl. Acad. Sci. USA 2015, 112, 2443–2448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rigacci, S.; Bucciantini, M.; Relini, A.; Pesce, A.; Gliozzi, A.; Berti, A.; Stefani, M. The (1–63) Region of the p53 Transactivation Domain Aggregates In Vitro into Cytotoxic Amyloid Assemblies. Biophys. J. 2008, 94, 3635–3646. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Higashimoto, Y.; Asanomi, Y.; Takakusagi, S.; Lewis, M.S.; Uosaki, K.; Durell, S.R.; Anderson, C.W.; Appella, E.; Sakaguchi, K. Unfolding, Aggregation, and Amyloid Formation by the Tetramerization Domain from Mutant p53 Associated with Lung Cancer. Biochemistry 2006, 45, 1608–1619. [Google Scholar] [CrossRef] [PubMed]
- Lei, J.; Qi, R.; Wei, G.; Nussinov, R.; Ma, B. Self-aggregation and coaggregation of the p53 core fragment with its aggregation gatekeeper variant. Phys. Chem. Chem. Phys. 2016, 18, 8098–8107. [Google Scholar] [CrossRef] [PubMed]
- Bullock, A.N.; Henckel, J.; DeDecker, B.S.; Johnson, C.M.; Nikolova, P.V.; Proctor, M.R.; Lane, D.P.; Fersht, A.R. Thermodynamic stability of wild-type and mutant p53 core domain. Proc. Natl. Acad. Sci. USA 1997, 94, 14338–14342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silva, J.L.; Gallo, C.V.D.M.; Costa, D.C.F.; Rangel, L.P. Prion-like aggregation of mutant p53 in cancer. Trends Biochem. Sci. 2014, 39, 260–267. [Google Scholar] [CrossRef] [PubMed]
- Strano, S.; Rossi, M.; Fontemaggi, G.; Munarriz, E.; Soddu, S.; Sacchi, A.; Blandino, G. From p63 to p53 across p73. FEBS Lett. 2001, 490, 163–170. [Google Scholar] [CrossRef] [Green Version]
- Strano, S.; Fontemaggi, G.; Costanzo, A.; Rizzo, M.G.; Monti, O.; Baccarini, A.; Del Sal, G.; Levrero, M.; Sacchi, A.; Oren, M.; et al. Physical interaction with human tumor-derived p53 mutants inhibits p63 activities. J. Biol. Chem. 2002, 277, 18817–18826. [Google Scholar] [CrossRef] [PubMed]
- Fujita, K.; Mondal, A.M.; Horikawa, I.; Nguyen, G.H.; Kumamoto, K.; Sohn, J.J.; Bowman, E.D.; Mathe, E.A.; Schetter, A.J.; Pine, S.R.; et al. P53 Isoforms Δ133P53 and P53B Are Endogenous Regulators of Replicative Cellular Senescence. Nat. Cell Biol. 2009, 11, 1135–1142. [Google Scholar] [CrossRef] [PubMed]
- Song, W.; Huo, S.; Lü, J.; Liu, Z.; Fang, X.; Jin, X.; Yuan, M. Expression of p53 isoforms in renal cell carcinoma. Chin. Med. J. 2009, 122, 921–926. [Google Scholar] [CrossRef] [PubMed]
- Boldrup, L.; Bourdon, J.C.; Coates, P.J.; Sjöström, B.; Nylander, K. Expression of p53 isoforms in squamous cell carcinoma of the head and neck. Eur. J. Cancer 2007, 43, 617–623. [Google Scholar] [CrossRef] [PubMed]
- Avery-Kiejda, K.A.; Zhang, X.D.; Adams, L.J.; Scott, R.J.; Vojtesek, B.; Lane, D.P.; Hersey, P. Small Molecular Weight Variants of p53 Are Expressed in Human Melanoma Cells and Are Induced by the DNA-Damaging Agent Cisplatin. Clin. Cancer Res. 2008, 14, 1659–1668. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bischof, K.; Knappskog, S.; Stefansson, I.; McCormack, E.M.; Trovik, J.; Werner, H.M.J.; Woie, K.; Gjertsen, B.T.; Bjorge, L. High expression of the p53 isoform γ is associated with reduced progression-free survival in uterine serous carcinoma. BMC Cancer 2018, 18, 684. [Google Scholar] [CrossRef] [PubMed]
- Ota, A.; Nakao, H.; Sawada, Y.; Karnan, S.; Wahiduzzaman, M.; Inoue, T.; Kobayashi, Y.; Yamamoto, T.; Ishii, N.; Ohashi, T.; et al. Δ40p53α suppresses tumor cell proliferation and induces cellular senescence in hepatocellular carcinoma cells. J. Cell Sci. 2017, 130, 614–625. [Google Scholar] [CrossRef] [PubMed]
- Hofstetter, G.; Berger, A.; Berger, R.; Zorić, A.; Braicu, E.I.; Reimer, D.; Fiegl, H.; Marth, C.; Zeimet, A.G.; Ulmer, H.; et al. The N-Terminally Truncated p53 Isoform Δ40p53 Influences Prognosis in Mucinous Ovarian Cancer. Int. J. Gynecol. Cancer 2012, 22, 372–379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takahashi, R.; Giannini, C.; Sarkaria, J.N.; Schroeder, M.; Rogers, J.; Mastroeni, D.; Scrable, H. P53 isoform profiling in glioblastoma and injured brain. Oncogene 2013, 32, 3165–3174. [Google Scholar] [CrossRef] [PubMed]
- Hofstetter, G.; Berger, A.; Schuster, E.; Wolf, A.; Hager, G.; Vergote, I.; Cadron, I.; Sehouli, J.; Braicu, E.I.; Mahner, S.; et al. Δ133p53 is an independent prognostic marker in p53 mutant advanced serous ovarian cancer. Br. J. Cancer 2011, 105, 1593–1599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marcel, V.; Palmero, E.I.; Falagan-Lotsch, P.; Martel-Planche, G.; Ashton-Prolla, P.; Olivier, M.; Brentani, R.R.; Hainaut, P.; Achatz, M.I. TP53 PIN3 and MDM2 SNP309 polymorphisms as genetic modifiers in the Li-Fraumeni syndrome: Impact on age at first diagnosis. J. Med. Genet. 2009, 46, 766–772. [Google Scholar] [CrossRef] [PubMed]
- Nutthasirikul, N.; Limpaiboon, T.; Leelayuwat, C.; Patrakitkomjorn, S.; Jearanaikoon, P. Ratio disruption of the ∆133p53 and TAp53 isoform equilibrium correlates with poor clinical outcome in intrahepatic cholangiocarcinoma. Int. J. Oncol. 2013, 42, 1181–1188. [Google Scholar] [CrossRef] [PubMed]
- Gadea, G.; Arsic, N.; Fernandes, K.; Diot, A.; Joruiz, S.M.; Abdallah, S.; Meuray, V.; Vinot, S.; Anguille, C.; Remenyi, J.; et al. TP53 drives invasion through expression of its Δ133p53β variant. Elife 2016, 5. [Google Scholar] [CrossRef] [PubMed]
- Arsic, N.; Gadea, G.; Lagerqvist, E.L.; Bußon, M.; Cahuzac, N.; Brock, C.; Hollande, F.; Gire, V.; Pannequin, J.; Roux, P. The p53 isoform Δ133p53β promotes cancer stem cell potential. Stem Cell Rep. 2015, 4, 531–540. [Google Scholar] [CrossRef] [PubMed]
- Nedelcu, A.M.; Tan, C. Early diversification and complex evolutionary history of the p53 tumor suppressor gene family. Dev. Genes Evol. 2007, 217, 801–806. [Google Scholar] [CrossRef] [PubMed]
- Pankow, S.; Bamberger, C. The p53 Tumor Suppressor-Like Protein nvp63 Mediates Selective Germ Cell Death in the Sea Anemone Nematostella vectensis. PLoS ONE 2007, 2, e782. [Google Scholar] [CrossRef] [PubMed]
- Levine, A.J.; Tomasini, R.; McKeon, F.D.; Mak, T.W.; Melino, G. The p53 family: Guardians of maternal reproduction. Nat. Rev. Mol. Cell Biol. 2011, 12, 259–265. [Google Scholar] [CrossRef] [PubMed]
- Donehower, L.A.; Levine, A.J. p53, Cancer, and Longevity. In Molecular Biology of Aging; Guarente, L., Partridge, L., Wallace, D.C., Eds.; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, New York, NY, USA, 2008; pp. 127–152. ISBN 9780879698249. [Google Scholar]
- Tucci, P. Caloric restriction: Is mammalian life extension linked to p53? Aging (Albany. NY) 2012, 4, 525–534. [Google Scholar] [CrossRef] [PubMed]
- Rovinski, B.; Benchimol, S. Immortalization of rat embryo fibroblasts by the cellular p53 oncogene. Oncogene 1988, 2, 445–452. [Google Scholar] [PubMed]
- Lavigueur, A.; Maltby, V.; Mock, D.; Rossant, J.; Pawson, T.; Bernstein, A. High incidence of lung, bone, and lymphoid tumors in transgenic mice overexpressing mutant alleles of the p53 oncogene. Mol. Cell. Biol. 1989, 9, 3982–3991. [Google Scholar] [CrossRef] [PubMed]
- Mowat, M.; Cheng, A.; Kimura, N.; Bernstein, A.; Benchimol, S. Rearrangements of the cellular p53 gene in erythroleukaemic cells transformed by Friend virus. Nature 1985, 314, 633–636. [Google Scholar] [CrossRef] [PubMed]
- Hofstetter, G.; Berger, A.; Fiegl, H.; Slade, N.; Zori, A.; Holzer, B.; Schuster, E.; Mobus, V.J.; Reimer, D.; Daxenbichler, G.; et al. Alternative splicing of p53 and p73: The novel p53 splice variant p53 is an independent prognostic marker in ovarian cancer. Oncogene 2010, 29, 1997–2004. [Google Scholar] [CrossRef] [PubMed]
- Rant, U.; Arinaga, K.; Fujita, S.; Yokoyama, N.; Abstreiter, G.; Tornow, M. Dynamic electrical switching of DNA layers on a metal surface. Nano Lett. 2004, 4, 2441–2445. [Google Scholar] [CrossRef]
- Silva, J.L.; Rangel, L.P.; Costa, D.C.F.; Cordeiro, Y.; De Moura Gallo, C.V. Expanding the prion concept to cancer biology: Dominant-negative effect of aggregates of mutant p53 tumour suppressor. Biosci. Rep. 2013, 33, 593–603. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| p53 Isoforms with Description | Discovered and Reported Cellular Effects | Cancers with Appearance or Overexpression | |
|---|---|---|---|
| FLp53β | Lacks 52 amino acids from the C-terminus of FLp53; devoid of OD and CTD | Promotes senescence [113] loss of expression in breast cancer [39] | Colorectal cancer [52] Renal cell carcinoma [114] Acute myeloid leukemia (AML) [40] Loss of expression in breast cancer [39] Squamous cell carcinoma [115] Expressed only melanoma, not in melanocytes [116] |
| FLp53γ | 47 amino acid truncated from the C-terminus; devoid of an OD or CTD | Cytotoxic [58] Patients with p53γ have an overall good survival rate, along with a low cancer reoccurrence, and without p53γ a particularly poor prognosis [58] | Breast cancer (good prognosis) [64] Acute myeloid leukemia (AML) (overexpression after chemotherapy followed by higher overall survival) [40] Shorter progression-free survival in uterine serous carcinoma [117] |
| Δ40p53 | N-terminal truncated p53, starts from Met 40; lacks TAD I | Induces cell death [46] Prolongs pluripotency [46] Decreases pancreatic-β-cell proliferation and regulates glucose homeostasis [46] tumor suppression [118], and balance with tissue regeneration [50] regulates folding, oligomerization, and post-translational modification of p53 complexes [4] | Mucinous ovarian cancer [119] glioblastoma multiforme [120] Serous ovarian cancer [121] Expressed in melanoma, but not in melanocytes; and improved survival in ovarian cancer [116] Li-Fraumeni syndrome [122] |
| Δ133p53α | Lacks 132 amino acids from the N-terminus; lacks TAD I, TAD II, the proline-rich region (PRD), and part of the DBD | Pro-survival factor [9] Inhibits senescence [113], p53 mediated apoptosis [39], and p53 transcriptional activity [58] Extends cellular replicative life spans in human fibroblasts [113] Delays the onset of replicative senescence [60] May rescue embryogenic cells from radiation induced apoptosis [55] | Colorectal cancer [52] Serous ovarian cancer [121] Overexpression in primary breast cancer [39] Cholangiocarcinoma with shortened overall survival [123] |
| Δ133p53β | Lacks 132 amino acids from NTD including TAD I, TAD II, PRD, parts of DBD. Is also devoid of OD, and CTD as other β forms | Promotes epithelial–mesenchymal transition in breast cancer cells [124] Fosters cancer stem cell potential [125] | Enhances cancer cell stemness in breast cancer [125] Expressed in melanoma cell lines [116] Increased breast cancer pervasiveness [124] |
| Δ160p53 | Truncated at N-terminus, starts with Met 160; lacks TAD I, TAD II, PRD, and parts of the DBD | Bears pro-oncogenic traits [63] can induce mutant-like phenotypes [63] probably widely involved in oncogenic mutant p53 gain of functions [63] | Unknown to date |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vieler, M.; Sanyal, S. p53 Isoforms and Their Implications in Cancer. Cancers 2018, 10, 288. https://doi.org/10.3390/cancers10090288
Vieler M, Sanyal S. p53 Isoforms and Their Implications in Cancer. Cancers. 2018; 10(9):288. https://doi.org/10.3390/cancers10090288
Chicago/Turabian StyleVieler, Maximilian, and Suparna Sanyal. 2018. "p53 Isoforms and Their Implications in Cancer" Cancers 10, no. 9: 288. https://doi.org/10.3390/cancers10090288
APA StyleVieler, M., & Sanyal, S. (2018). p53 Isoforms and Their Implications in Cancer. Cancers, 10(9), 288. https://doi.org/10.3390/cancers10090288