Co-Expression of the Epstein-Barr Virus-Encoded Latent Membrane Proteins and the Pathogenesis of Classic Hodgkin Lymphoma


{kind=link}
{kind=link}
{kind=link}
{kind=link}
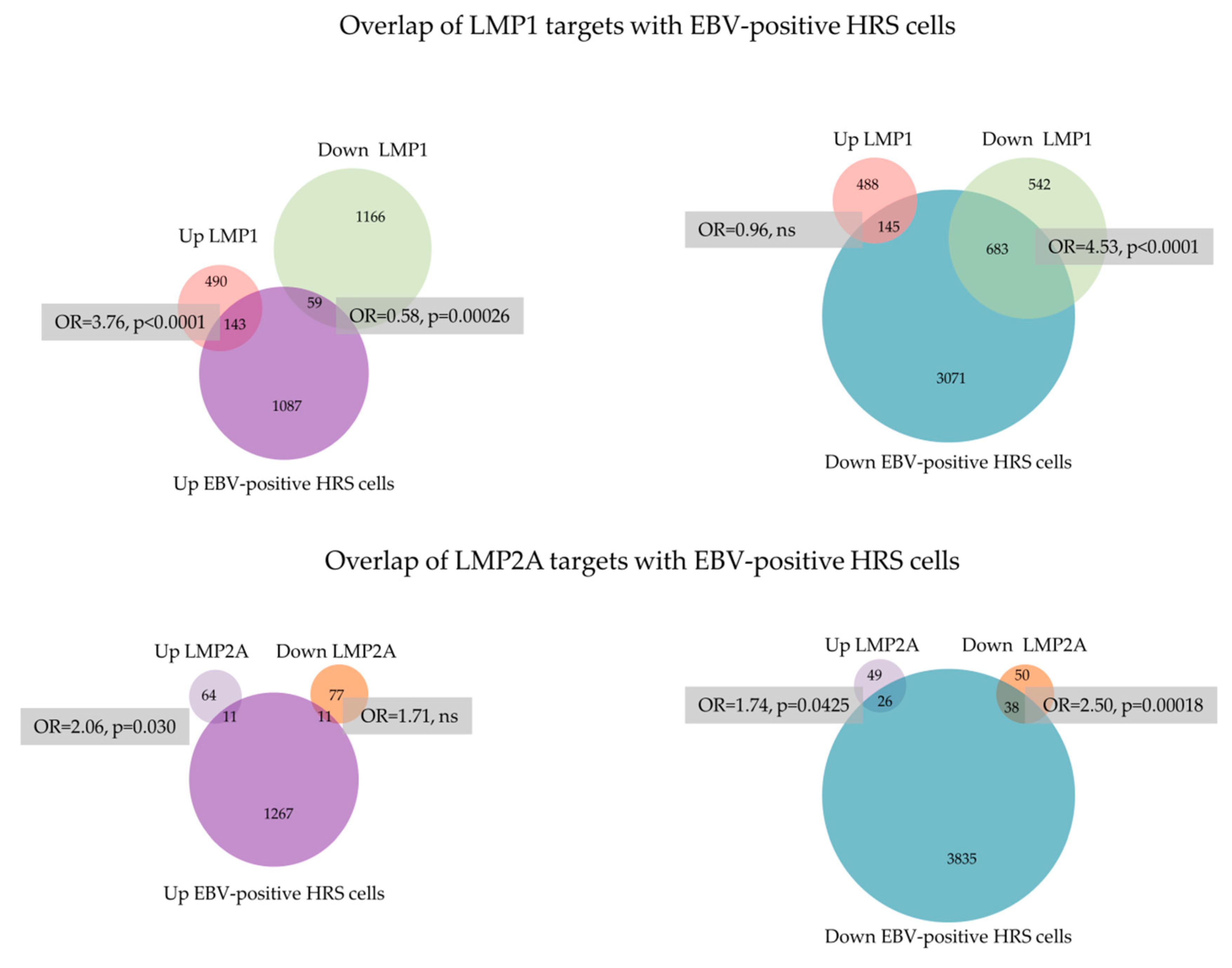{kind=link}
Abstract
1. Epstein-Barr Virus and the Pathogenesis of Classic Hodgkin Lymphoma
2. The Role of EBV Latent Membrane Proteins in Viral Persistence
3. EBV Latent Membrane Proteins and the Pathogenesis of Classic Hodgkin Lymphoma
4. Effects of the Latent Membrane Proteins on the Phenotype of B Cells in Mouse Models
5. Transcriptional Effects of LMP1 and LMP2 in Primary Human Germinal Centre B Cells
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Kuppers, R.; Rajewsky, K.; Zhao, M.; Simons, G.; Laumann, R.; Fischer, R.; Hansmann, M.L. Hodgkin disease: Hodgkin and Reed-Sternberg cells picked from histological sections show clonal immunoglobulin gene rearrangements and appear to be derived from B cells at various stages of development. Proc. Natl. Acad. Sci. USA 1994, 91, 10962–10966. [Google Scholar] [CrossRef] [PubMed]
- Kuppers, R.; Rajewsky, K.; Zhao, M.; Simons, G.; Laumann, R.; Fischer, R.; Hansmann, M.L. Hodgkin’s disease: Clonal Ig gene rearrangements in Hodgkin and Reed-Sternberg cells picked from histological sections. Ann. N. Y. Acad. Sci. 1995, 764, 523–524. [Google Scholar] [CrossRef] [PubMed]
- Kanzler, H.; Kuppers, R.; Hansmann, M.L.; Rajewsky, K. Hodgkin and Reed-Sternberg cells in Hodgkin’s disease represent the outgrowth of a dominant tumor clone derived from (crippled) germinal center B cells. J. Exp. Med. 1996, 184, 1495–1505. [Google Scholar] [CrossRef] [PubMed]
- Kuppers, R.; Kanzler, H.; Hansmann, M.L.; Rajewsky, K. Single cell analysis of Hodgkin/Reed-Sternberg cells. Ann. Oncol. 1996, 7 (Suppl. 4), 27–30. [Google Scholar] [CrossRef]
- Ushmorov, A.; Ritz, O.; Hummel, M.; Leithauser, F.; Moller, P.; Stein, H.; Wirth, T. Epigenetic silencing of the immunoglobulin heavy-chain gene in classical Hodgkin lymphoma-derived cell lines contributes to the loss of immunoglobulin expression. Blood 2004, 104, 3326–3334. [Google Scholar] [CrossRef] [PubMed]
- Hertel, C.B.; Zhou, X.G.; Hamilton-Dutoit, S.J.; Junker, S. Loss of B cell identity correlates with loss of B cell-specific transcription factors in Hodgkin/Reed-Sternberg cells of classical Hodgkin lymphoma. Oncogene 2002, 21, 4908–4920. [Google Scholar] [CrossRef] [PubMed]
- Schwering, I.; Brauninger, A.; Klein, U.; Jungnickel, B.; Tinguely, M.; Diehl, V.; Hansmann, M.L.; Dalla-Favera, R.; Rajewsky, K.; Kuppers, R. Loss of the B-lineage-specific gene expression program in Hodgkin and Reed-Sternberg cells of Hodgkin lymphoma. Blood 2003, 101, 1505–1512. [Google Scholar] [CrossRef] [PubMed]
- Kuppers, R.; Klein, U.; Schwering, I.; Distler, V.; Brauninger, A.; Cattoretti, G.; Tu, Y.; Stolovitzky, G.A.; Califano, A.; Hansmann, M.L.; et al. Identification of Hodgkin and Reed-Sternberg cell-specific genes by gene expression profiling. J. Clin. Investig. 2003, 111, 529–537. [Google Scholar] [CrossRef] [PubMed]
- Tiacci, E.; Doring, C.; Brune, V.; van Noesel, C.J.; Klapper, W.; Mechtersheimer, G.; Falini, B.; Kuppers, R.; Hansmann, M.L. Analyzing primary Hodgkin and Reed-Sternberg cells to capture the molecular and cellular pathogenesis of classical Hodgkin lymphoma. Blood 2012, 120, 4609–4620. [Google Scholar] [CrossRef] [PubMed]
- Steidl, C.; Diepstra, A.; Lee, T.; Chan, F.C.; Farinha, P.; Tan, K.; Telenius, A.; Barclay, L.; Shah, S.P.; Connors, J.M.; et al. Gene expression profiling of microdissected Hodgkin Reed-Sternberg cells correlates with treatment outcome in classical Hodgkin lymphoma. Blood 2012, 120, 3530–3540. [Google Scholar] [CrossRef] [PubMed]
- Renne, C.; Martin-Subero, J.I.; Eickernjager, M.; Hansmann, M.L.; Kuppers, R.; Siebert, R.; Brauninger, A. Aberrant expression of ID2, a suppressor of B-cell-specific gene expression, in Hodgkin’s lymphoma. Am. J. Pathol. 2006, 169, 655–664. [Google Scholar] [CrossRef] [PubMed]
- Bohle, V.; Doring, C.; Hansmann, M.L.; Kuppers, R. Role of early B-cell factor 1 (EBF1) in Hodgkin lymphoma. Leukemia 2013, 27, 671–679. [Google Scholar] [CrossRef] [PubMed]
- Mathas, S.; Janz, M.; Hummel, F.; Hummel, M.; Wollert-Wulf, B.; Lusatis, S.; Anagnostopoulos, I.; Lietz, A.; Sigvardsson, M.; Jundt, F.; et al. Intrinsic inhibition of transcription factor E2A by HLH proteins ABF-1 and Id2 mediates reprogramming of neoplastic B cells in Hodgkin lymphoma. Nat. Immunol. 2006, 7, 207–215. [Google Scholar] [CrossRef] [PubMed]
- Bargou, R.C.; Leng, C.; Krappmann, D.; Emmerich, F.; Mapara, M.Y.; Bommert, K.; Royer, H.D.; Scheidereit, C.; Dorken, B. High-level nuclear NF-kappa B and Oct-2 is a common feature of cultured Hodgkin/Reed-Sternberg cells. Blood 1996, 87, 4340–4347. [Google Scholar] [PubMed]
- Carbone, A.; Gloghini, A.; Gattei, V.; Aldinucci, D.; Degan, M.; De Paoli, P.; Zagonel, V.; Pinto, A. Expression of functional CD40 antigen on Reed-Sternberg cells and Hodgkin’s disease cell lines. Blood 1995, 85, 780–789. [Google Scholar] [PubMed]
- Fiumara, P.; Snell, V.; Li, Y.; Mukhopadhyay, A.; Younes, M.; Gillenwater, A.M.; Cabanillas, F.; Aggarwal, B.B.; Younes, A. Functional expression of receptor activator of nuclear factor kappaB in Hodgkin disease cell lines. Blood 2001, 98, 2784–2790. [Google Scholar] [CrossRef] [PubMed]
- Horie, R.; Watanabe, T.; Morishita, Y.; Ito, K.; Ishida, T.; Kanegae, Y.; Saito, I.; Higashihara, M.; Mori, S.; Kadin, M.E.; et al. Ligand-independent signaling by overexpressed CD30 drives NF-kappaB activation in Hodgkin-Reed-Sternberg cells. Oncogene 2002, 21, 2493–2503. [Google Scholar] [CrossRef] [PubMed]
- Chiu, A.; Xu, W.; He, B.; Dillon, S.R.; Gross, J.A.; Sievers, E.; Qiao, X.; Santini, P.; Hyjek, E.; Lee, J.W.; et al. Hodgkin lymphoma cells express TACI and BCMA receptors and generate survival and proliferation signals in response to BAFF and APRIL. Blood 2007, 109, 729–739. [Google Scholar] [CrossRef] [PubMed]
- Carbone, A.; Gloghini, A.; Gruss, H.J.; Pinto, A. CD40 ligand is constitutively expressed in a subset of T cell lymphomas and on the microenvironmental reactive T cells of follicular lymphomas and Hodgkin’s disease. Am. J. Pathol. 1995, 147, 912–922. [Google Scholar] [PubMed]
- Pinto, A.; Aldinucci, D.; Gloghini, A.; Zagonel, V.; Degan, M.; Perin, V.; Todesco, M.; De Iuliis, A.; Improta, S.; Sacco, C.; et al. The role of eosinophils in the pathobiology of Hodgkin’s disease. Ann. Oncol. 1997, 8 (Suppl. 2), 89–96. [Google Scholar] [CrossRef]
- Barth, T.F.; Martin-Subero, J.I.; Joos, S.; Menz, C.K.; Hasel, C.; Mechtersheimer, G.; Parwaresch, R.M.; Lichter, P.; Siebert, R.; Mooller, P. Gains of 2p involving the REL locus correlate with nuclear c-Rel protein accumulation in neoplastic cells of classical Hodgkin lymphoma. Blood 2003, 101, 3681–3686. [Google Scholar] [CrossRef] [PubMed]
- Joos, S.; Granzow, M.; Holtgreve-Grez, H.; Siebert, R.; Harder, L.; Martin-Subero, J.I.; Wolf, J.; Adamowicz, M.; Barth, T.F.; Lichter, P.; et al. Hodgkin’s lymphoma cell lines are characterized by frequent aberrations on chromosomes 2p and 9p including REL and JAK2. Int. J. Cancer 2003, 103, 489–495. [Google Scholar] [CrossRef] [PubMed]
- Martin-Subero, J.I.; Gesk, S.; Harder, L.; Sonoki, T.; Tucker, P.W.; Schlegelberger, B.; Grote, W.; Novo, F.J.; Calasanz, M.J.; Hansmann, M.L.; et al. Recurrent involvement of the REL and BCL11A loci in classical Hodgkin lymphoma. Blood 2002, 99, 1474–1477. [Google Scholar] [CrossRef] [PubMed]
- Cabannes, E.; Khan, G.; Aillet, F.; Jarrett, R.F.; Hay, R.T. Mutations in the IkBa gene in Hodgkin’s disease suggest a tumour suppressor role for IkappaBalpha. Oncogene 1999, 18, 3063–3070. [Google Scholar] [CrossRef] [PubMed]
- Emmerich, F.; Meiser, M.; Hummel, M.; Demel, G.; Foss, H.D.; Jundt, F.; Mathas, S.; Krappmann, D.; Scheidereit, C.; Stein, H.; et al. Overexpression of I kappa B alpha without inhibition of NF-kappaB activity and mutations in the I kappa B alpha gene in Reed-Sternberg cells. Blood 1999, 94, 3129–3134. [Google Scholar] [PubMed]
- Jungnickel, B.; Staratschek-Jox, A.; Brauninger, A.; Spieker, T.; Wolf, J.; Diehl, V.; Hansmann, M.L.; Rajewsky, K.; Kuppers, R. Clonal deleterious mutations in the IkappaBalpha gene in the malignant cells in Hodgkin’s lymphoma. J. Exp. Med. 2000, 191, 395–402. [Google Scholar] [CrossRef] [PubMed]
- Emmerich, F.; Theurich, S.; Hummel, M.; Haeffker, A.; Vry, M.S.; Dohner, K.; Bommert, K.; Stein, H.; Dorken, B. Inactivating I kappa B epsilon mutations in Hodgkin/Reed-Sternberg cells. J. Pathol. 2003, 201, 413–420. [Google Scholar] [CrossRef] [PubMed]
- Lake, A.; Shield, L.A.; Cordano, P.; Chui, D.T.; Osborne, J.; Crae, S.; Wilson, K.S.; Tosi, S.; Knight, S.J.; Gesk, S.; et al. Mutations of NFKBIA, encoding IkappaB alpha, are a recurrent finding in classical Hodgkin lymphoma but are not a unifying feature of non-EBV-associated cases. Int. J. Cancer 2009, 125, 1334–1342. [Google Scholar] [CrossRef] [PubMed]
- Ranuncolo, S.M.; Pittaluga, S.; Evbuomwan, M.O.; Jaffe, E.S.; Lewis, B.A. Hodgkin lymphoma requires stabilized NIK and constitutive RelB expression for survival. Blood 2012, 120, 3756–3763. [Google Scholar] [CrossRef] [PubMed]
- Otto, C.; Giefing, M.; Massow, A.; Vater, I.; Gesk, S.; Schlesner, M.; Richter, J.; Klapper, W.; Hansmann, M.L.; Siebert, R.; et al. Genetic lesions of the TRAF3 and MAP3K14 genes in classical Hodgkin lymphoma. Br. J. Haematol. 2012, 157, 702–708. [Google Scholar] [CrossRef] [PubMed]
- Schmitz, R.; Hansmann, M.L.; Bohle, V.; Martin-Subero, J.I.; Hartmann, S.; Mechtersheimer, G.; Klapper, W.; Vater, I.; Giefing, M.; Gesk, S.; et al. TNFAIP3 (A20) is a tumor suppressor gene in Hodgkin lymphoma and primary mediastinal B cell lymphoma. J. Exp. Med. 2009, 206, 981–989. [Google Scholar] [CrossRef] [PubMed]
- Hinz, M.; Lemke, P.; Anagnostopoulos, I.; Hacker, C.; Krappmann, D.; Mathas, S.; Dorken, B.; Zenke, M.; Stein, H.; Scheidereit, C. Nuclear factor kappaB-dependent gene expression profiling of Hodgkin’s disease tumor cells, pathogenetic significance, and link to constitutive signal transducer and activator of transcription 5a activity. J. Exp. Med. 2002, 196, 605–617. [Google Scholar] [PubMed]
- Skinnider, B.F.; Elia, A.J.; Gascoyne, R.D.; Patterson, B.; Trumper, L.; Kapp, U.; Mak, T.W. Signal transducer and activator of transcription 6 is frequently activated in Hodgkin and Reed-Sternberg cells of Hodgkin lymphoma. Blood 2002, 99, 618–626. [Google Scholar] [CrossRef] [PubMed]
- Kube, D.; Holtick, U.; Vockerodt, M.; Ahmadi, T.; Haier, B.; Behrmann, I.; Heinrich, P.C.; Diehl, V.; Tesch, H. STAT3 is constitutively activated in Hodgkin cell lines. Blood 2001, 98, 762–770. [Google Scholar] [CrossRef] [PubMed]
- Joos, S.; Kupper, M.; Ohl, S.; von Bonin, F.; Mechtersheimer, G.; Bentz, M.; Marynen, P.; Moller, P.; Pfreundschuh, M.; Trumper, L.; et al. Genomic imbalances including amplification of the tyrosine kinase gene JAK2 in CD30+ Hodgkin cells. Cancer Res. 2000, 60, 549–552. [Google Scholar] [PubMed]
- Weniger, M.A.; Melzner, I.; Menz, C.K.; Wegener, S.; Bucur, A.J.; Dorsch, K.; Mattfeldt, T.; Barth, T.F.; Moller, P. Mutations of the tumor suppressor gene SOCS-1 in classical Hodgkin lymphoma are frequent and associated with nuclear phospho-STAT5 accumulation. Oncogene 2006, 25, 2679–2684. [Google Scholar] [CrossRef] [PubMed]
- Gunawardana, J.; Chan, F.C.; Telenius, A.; Woolcock, B.; Kridel, R.; Tan, K.L.; Ben-Neriah, S.; Mottok, A.; Lim, R.S.; Boyle, M.; et al. Recurrent somatic mutations of PTPN1 in primary mediastinal B cell lymphoma and Hodgkin lymphoma. Nat. Genet. 2014, 46, 329–335. [Google Scholar] [CrossRef] [PubMed]
- Glaser, S.L.; Lin, R.J.; Stewart, S.L.; Ambinder, R.F.; Jarrett, R.F.; Brousset, P.; Pallesen, G.; Gulley, M.L.; Khan, G.; O’Grady, J.; et al. Epstein-Barr virus-associated Hodgkin’s disease: Epidemiologic characteristics in international data. Int. J. Cancer 1997, 70, 375–382. [Google Scholar] [CrossRef]
- Weiss, L.M.; Movahed, L.A.; Warnke, R.A.; Sklar, J. Detection of Epstein-Barr viral genomes in Reed-Sternberg cells of Hodgkin’s disease. N. Engl. J. Med. 1989, 320, 502–506. [Google Scholar] [CrossRef] [PubMed]
- Flavell, K.J.; Biddulph, J.P.; Powell, J.E.; Parkes, S.E.; Redfern, D.; Weinreb, M.; Nelson, P.; Mann, J.R.; Young, L.S.; Murray, P.G. South Asian ethnicity and material deprivation increase the risk of Epstein-Barr virus infection in childhood Hodgkin’s disease. Br. J. Cancer 2001, 85, 350–356. [Google Scholar] [CrossRef] [PubMed]
- Chang, K.L.; Albujar, P.F.; Chen, Y.Y.; Johnson, R.M.; Weiss, L.M. High prevalence of Epstein-Barr virus in the Reed-Sternberg cells of Hodgkin’s disease occurring in Peru. Blood 1993, 81, 496–501. [Google Scholar] [PubMed]
- Weinreb, M.; Day, P.J.; Niggli, F.; Green, E.K.; Nyong’o, A.O.; Othieno-Abinya, N.A.; Riyat, M.S.; Raafat, F.; Mann, J.R. The consistent association between Epstein-Barr virus and Hodgkin’s disease in children in Kenya. Blood 1996, 87, 3828–3836. [Google Scholar] [PubMed]
- Anagnostopoulos, I.; Herbst, H.; Niedobitek, G.; Stein, H. Demonstration of monoclonal EBV genomes in Hodgkin’s disease and Ki-1-positive anaplastic large cell lymphoma by combined Southern blot and in situ hybridization. Blood 1989, 74, 810–816. [Google Scholar] [PubMed]
- Coates, P.J.; Slavin, G.; D’Ardenne, A.J. Persistence of Epstein-Barr virus in Reed-Sternberg cells throughout the course of Hodgkin’s disease. J. Pathol. 1991, 164, 291–297. [Google Scholar] [CrossRef] [PubMed]
- Bräuninger, A.; Schmitz, R.; Bechtel, D.; Renné, C.; Hansmann, M.-L.; Küppers, R. Molecular biology of Hodgkin’s and Reed/Sternberg cells in Hodgkin’s lymphoma. Int. J. Cancer 2006, 118, 1853–1861. [Google Scholar] [CrossRef] [PubMed]
- Chaganti, S.; Bell, A.I.; Pastor, N.B.; Milner, A.E.; Drayson, M.; Gordon, J.; Rickinson, A.B. Epstein-Barr virus infection in vitro can rescue germinal center B cells with inactivated immunoglobulin genes. Blood 2005, 106, 4249–4252. [Google Scholar] [CrossRef] [PubMed]
- Bechtel, D.; Kurth, J.J.; Unkel, C.; Küppers, R. Transformation of BCR-deficient germinal-center B cells by EBV supports a major role of the virus in the pathogenesis of Hodgkin and posttransplantation lymphomas. Blood 2005, 106, 4345–4350. [Google Scholar] [CrossRef] [PubMed]
- Mancao, C.; Altmann, M.; Jungnickel, B.; Hammerschmidt, W. Rescue of “crippled” germinal center B cells from apoptosis by Epstein-Barr virus. Blood 2005, 106, 4339–4343. [Google Scholar] [CrossRef] [PubMed]
- Nalesnik, M.A.; Randhawa, P.; Demetris, A.J.; Casavilla, A.; Fung, J.J.; Locker, J. Lymphoma resembling Hodgkin disease after posttransplant lymphoproliferative disorder in a liver transplant recipient. Cancer 1993, 72, 2568–2573. [Google Scholar] [CrossRef]
- Bierman, P.J.; Vose, J.M.; Langnas, A.N.; Rifkin, R.M.; Hauke, R.J.; Smir, B.N.; Greiner, T.C. Hodgkin’s disease following solid organ transplantation. Ann. Oncol. 1996, 7, 265–270. [Google Scholar] [CrossRef] [PubMed]
- Rowlings, P.A.; Curtis, R.E.; Passweg, J.R.; Deeg, H.J.; Socie, G.; Travis, L.B.; Kingma, D.W.; Jaffe, E.S.; Sobocinski, K.A.; Horowitz, M.M. Increased incidence of Hodgkin’s disease after allogeneic bone marrow transplantation. J. Clin. Oncol. 1999, 17, 3122–3127. [Google Scholar] [CrossRef] [PubMed]
- Cohen, J.I.; Kimura, H.; Nakamura, S.; Ko, Y.H.; Jaffe, E.S. Epstein-Barr virus-associated lymphoproliferative disease in non-immunocompromised hosts: A status report and summary of an international meeting, 8–9 September 2008. Ann. Oncol. 2009, 20, 1472–1482. [Google Scholar] [CrossRef] [PubMed]
- Anderton, E.; Yee, J.; Smith, P.; Crook, T.; White, R.E.; Allday, M.J. Two Epstein–Barr virus (EBV) oncoproteins cooperate to repress expression of the proapoptotic tumour-suppressor Bim: Clues to the pathogenesis of Burkitt’s lymphoma. Oncogene 2007, 27, 421–433. [Google Scholar] [CrossRef] [PubMed]
- Kilger, E.; Kieser, A.; Baumann, M.; Hammerschmidt, W. Epstein-Barr virus-mediated B-cell proliferation is dependent upon latent membrane protein 1, which simulates an activated CD40 receptor. EMBO J. 1998, 17, 1700–1709. [Google Scholar] [CrossRef] [PubMed]
- Bornkamm, G.W.; Hudewentz, J.; Freese, U.K.; Zimber, U. Deletion of the nontransforming Epstein-Barr virus strain P3HR-1 causes fusion of the large internal repeat to the DSL region. J. Virol. 1982, 43, 952–968. [Google Scholar] [PubMed]
- Wysokenski, D.A.; Yates, J.L. Multiple EBNA1-binding sites are required to form an EBNA1-dependent enhancer and to activate a minimal replicative origin within oriP of Epstein-Barr virus. J. Virol. 1989, 63, 2657–2666. [Google Scholar] [PubMed]
- Lupton, S.; Levine, A.J. Mapping Genetic Elements of Epstein-Barr Virus That Facilitate Extrachromosomal Persistence of Epstein-Barr Virus-Derived Plasmids in Human-Cells. Mol. Cell. Biol. 1985, 5, 2533–2542. [Google Scholar] [CrossRef] [PubMed]
- Gahn TA, S.B. An EBNA1-dependent enhancer acts from a distance of 10 kilobase pairs to increase expression of the Epstein-Barr virus LMP gene. J. Virol. 1995, 69, 2633–2636. [Google Scholar] [PubMed]
- Reisman, D.; Sugden, B. trans activation of an Epstein-Barr viral transcriptional enhancer by the Epstein-Barr viral nuclear antigen 1. Mol. Cell. Biol. 1986, 6, 3838–3846. [Google Scholar] [CrossRef] [PubMed]
- Sugden, B.; Warren, N. A Promoter of Epstein-Barr Virus That Can Function during Latent Infection Can Be Transactivated by Ebna-1, a Viral Protein Required for Viral-DNA Replication during Latent Infection. J. Virol. 1989, 63, 2644–2649. [Google Scholar] [PubMed]
- Flavell, J.R.; Baumforth, K.R.; Wood, V.H.; Davies, G.L.; Wei, W.; Reynolds, G.M.; Morgan, S.; Boyce, A.; Kelly, G.L.; Young, L.S.; et al. Down-regulation of the TGF-beta target gene, PTPRK, by the Epstein-Barr virus encoded EBNA1 contributes to the growth and survival of Hodgkin lymphoma cells. Blood 2008, 111, 292–301. [Google Scholar] [CrossRef] [PubMed]
- Wood, V.H.; O’Neil, J.D.; Wei, W.; Stewart, S.E.; Dawson, C.W.; Young, L.S. Epstein-Barr virus-encoded EBNA1 regulates cellular gene transcription and modulates the STAT1 and TGFbeta signaling pathways. Oncogene 2007, 26, 4135–4147. [Google Scholar] [CrossRef] [PubMed]
- Rowe, M.; Lear, A.L.; Croom-Carter, D.; Davies, A.H.; Rickinson, A.B. Three pathways of Epstein-Barr virus gene activation from EBNA1-positive latency in B lymphocytes. J. Virol. 1992, 66, 122–131. [Google Scholar] [PubMed]
- Deacon, E.M.; Pallesen, G.; Niedobitek, G.; Crocker, J.; Brooks, L.; Rickinson, A.B.; Young, L.S. Epstein-Barr virus and Hodgkin’s disease: Transcriptional analysis of virus latency in the malignant cells. J. Exp. Med. 1993, 177, 339–349. [Google Scholar] [CrossRef] [PubMed]
- Grasser, F.A.; Murray, P.G.; Kremmer, E.; Klein, K.; Remberger, K.; Feiden, W.; Reynolds, G.; Niedobitek, G.; Young, L.S.; Mueller-Lantzsch, N. Monoclonal antibodies directed against the Epstein-Barr virus-encoded nuclear antigen 1 (EBNA1): Immunohistologic detection of EBNA1 in the malignant cells of Hodgkin’s disease. Blood 1994, 84, 3792–3798. [Google Scholar] [PubMed]
- Murray, P.G.; Young, L.S.; Rowe, M.; Crocker, J. Immunohistochemical demonstration of the Epstein-Barr virus-encoded latent membrane protein in paraffin sections of Hodgkin’s disease. J. Pathol. 1992, 166, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Babcock, G.J.; Decker, L.L.; Volk, M.; Thorley-Lawson, D.A. EBV persistence in memory B cells in vivo. Immunity 1998, 9, 395–404. [Google Scholar] [CrossRef]
- Babcock, G.J.; Hochberg, D.; Thorley-Lawson, D.A. The expression pattern of Epstein-Barr virus latent genes in vivo is dependent upon the differentiation stage of the infected B cell. Immunity 2000, 13, 497–506. [Google Scholar] [CrossRef]
- Eliopoulos, A.G.; Dawson, C.W.; Mosialos, G.; Floettmann, J.E.; Rowe, M.; Armitage, R.J.; Dawson, J.; Zapata, J.M.; Kerr, D.J.; Wakelam, M.J.; et al. CD40-induced growth inhibition in epithelial cells is mimicked by Epstein-Barr Virus-encoded LMP1: Involvement of TRAF3 as a common mediator. Oncogene 1996, 13, 2243–2254. [Google Scholar] [PubMed]
- Gires, O.; Zimber-Strobl, U.; Gonnella, R.; Ueffing, M.; Marschall, G.; Zeidler, R.; Pich, D.; Hammerschmidt, W. Latent membrane protein 1 of Epstein-Barr virus mimics a constitutively active receptor molecule. EMBO J. 1997, 16, 6131–6140. [Google Scholar] [CrossRef] [PubMed]
- Merchant, M.; Swart, R.; Katzman, R.B.; Ikeda, M.; Ikeda, A.; Longnecker, R.; Dykstra, M.L.; Pierce, S.K. The effects of the Epstein-Barr virus latent membrane protein 2A on B cell function. Int. Rev. Immunol. 2001, 20, 805–835. [Google Scholar] [CrossRef] [PubMed]
- Caldwell, R.G.; Wilson, J.B.; Anderson, S.J.; Longnecker, R. Epstein-Barr virus LMP2A drives B cell development and survival in the absence of normal B cell receptor signals. Immunity 1998, 9, 405–411. [Google Scholar] [CrossRef]
- Laichalk, L.L.; Thorley-Lawson, D.A. Terminal differentiation into plasma cells initiates the replicative cycle of Epstein-Barr virus in vivo. J. Virol. 2005, 79, 1296–1307. [Google Scholar] [CrossRef] [PubMed]
- Vrzalikova, K.; Vockerodt, M.; Leonard, S.; Bell, A.; Wei, W.; Schrader, A.; Wright, K.L.; Kube, D.; Rowe, M.; Woodman, C.B.; et al. Down-regulation of BLIMP1α by the EBV oncogene, LMP-1, disrupts the plasma cell differentiation program and prevents viral replication in B cells: Implications for the pathogenesis of EBV-associated B-cell lymphomas. Blood 2011, 117, 5907–5917. [Google Scholar] [CrossRef] [PubMed]
- Minamitani, T.; Yasui, T.; Ma, Y.; Zhou, H.; Okuzaki, D.; Tsai, C.Y.; Sakakibara, S.; Gewurz, B.E.; Kieff, E.; Kikutani, H. Evasion of affinity-based selection in germinal centers by Epstein-Barr virus LMP2A. Proc. Natl. Acad. Sci. USA 2015, 112, 11612–11617. [Google Scholar] [CrossRef] [PubMed]
- Minamitani, T.; Ma, Y.; Zhou, H.; Kida, H.; Tsai, C.Y.; Obana, M.; Okuzaki, D.; Fujio, Y.; Kumanogoh, A.; Zhao, B.; et, al. Mouse model of Epstein-Barr virus LMP1- and LMP2A-driven germinal center B-cell lymphoproliferative disease. Proc. Natl. Acad. Sci. USA 2017, 114, 4751–4756. [Google Scholar] [CrossRef] [PubMed]
- Ma, S.D.; Tsai, M.H.; Romero-Masters, J.C.; Ranheim, E.A.; Huebner, S.M.; Bristol, J.; Delecluse, H.J.; Kenney, S.C. LMP1 and LMP2A collaborate to promote Epstein-Barr virus (EBV)-induced B cell lymphomas in a cord blood-humanized mouse model but are not essential. J. Virol. 2017, 91. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Liebowitz, D.; Kieff, E. An EBV membrane protein expressed in immortalized lymphocytes transforms established rodent cells. Cell 1985, 43, 831–840. [Google Scholar] [CrossRef]
- Bargou, R.C.; Emmerich, F.; Krappmann, D.; Bommert, K.; Mapara, M.Y.; Arnold, W.; Royer, H.D.; Grinstein, E.; Greiner, A.; Scheidereit, C.; et al. Constitutive nuclear factor-kappaB-relA activation is required for proliferation and survival of Hodgkin’s disease tumor cells. J. Clin. Invest. 1997, 100, 2961–2969. [Google Scholar] [CrossRef] [PubMed]
- Dutton, A.; Reynolds, G.M.; Dawson, C.W.; Young, L.S.; Murray, P.G. Constitutive activation of phosphatidyl-inositide 3 kinase contributes to the survival of Hodgkin’s lymphoma cells through a mechanism involving Akt kinase and mTOR. J. Pathol. 2005, 205, 498–506. [Google Scholar] [CrossRef] [PubMed]
- Holtick, U.; Vockerodt, M.; Pinkert, D.; Schoof, N.; Stürzenhofecker, B.; Kussebi, N.; Lauber, K.; Wesselborg, S.; Löffler, D.; Horn, F.; et al. STAT3 is essential for Hodgkin lymphoma cell proliferation and is a target of tyrphostin AG17 which confers sensitization for apoptosis. Leukemia 2005, 19, 936–944. [Google Scholar] [CrossRef] [PubMed]
- Kieser, A.; Kilger, E.; Gires, O.; Ueffing, M.; Kolch, W.; Hammerschmidt, W. Epstein-Barr virus latent membrane protein-1 triggers AP-1 activity via the c-Jun N-terminal kinase cascade. EMBO J. 1997, 16, 6478–6485. [Google Scholar] [CrossRef] [PubMed]
- Eliopoulos, A.G.; Young, L.S. Activation of the cJun N-terminal kinase (JNK) pathway by the Epstein-Barr virus-encoded latent membrane protein 1 (LMP1). Oncogene 1998, 16, 1731–1742. [Google Scholar] [CrossRef] [PubMed]
- Laherty, C.D.; Hu, H.M.; Opipari, A.W.; Wang, F.; Dixit, V.M. The Epstein-Barr virus LMP1 gene product induces A20 zinc finger protein expression by activating nuclear factor kappa B. J. Biol. Chem. 1992, 267, 24157–24160. [Google Scholar] [PubMed]
- Huen, D.S.; Henderson, S.A.; Croom-Carter, D.; Rowe, M. The Epstein-Barr virus latent membrane protein-1 (LMP1) mediates activation of NF-kappa B and cell surface phenotype via two effector regions in its carboxy-terminal cytoplasmic domain. Oncogene 1995, 10, 549–560. [Google Scholar] [PubMed]
- Gires, O.; Kohlhuber, F.; Kilger, E.; Baumann, M.; Kieser, A.; Kaiser, C.; Zeidler, R.; Scheffer, B.; Ueffing, M.; Hammerschmidt, W. Latent membrane protein 1 of Epstein-Barr virus interacts with JAK3 and activates STAT proteins. EMBO J. 1999, 18, 3064–3073. [Google Scholar] [CrossRef] [PubMed]
- Vockerodt, M.; Morgan, S.L.; Kuo, M.; Wei, W.; Chukwuma, M.B.; Arrand, J.R.; Kube, D.; Gordon, J.; Young, L.S.; Woodman, C.B.; et al. The Epstein-Barr virus oncoprotein, latent membrane protein-1, reprograms germinal centre B cells towards a Hodgkin’s Reed-Sternberg-like phenotype. J. Pathol. 2008, 216, 83–92. [Google Scholar] [CrossRef] [PubMed]
- Henderson, S.; Rowe, M.; Gregory, C.; Croom-Carter, D.; Wang, F.; Longnecker, R.; Kieff, E.; Rickinson, A. Induction of bcl-2 expression by Epstein-Barr virus latent membrane protein 1 protects infected B cells from programmed cell death. Cell 1991, 65, 1107–1115. [Google Scholar] [CrossRef]
- Finke, J.; Fritzen, R.; Ternes, P.; Trivedi, P.; Bross, K.J.; Lange, W.; Mertelsmann, R.; Dolken, G. Expression of bcl-2 in Burkitt’s lymphoma cell lines: Induction by latent Epstein-Barr virus genes. Blood 1992, 80, 459–469. [Google Scholar] [PubMed]
- Wang, S.; Rowe, M.; Lundgren, E. Expression of the Epstein Barr virus transforming protein LMP1 causes a rapid and transient stimulation of the Bcl-2 homologue Mcl-1 levels in B-cell lines. Cancer Res. 1996, 56, 4610–4613. [Google Scholar] [PubMed]
- Caldwell, R.G.; Brown, R.C.; Longnecker, R. Epstein-Barr virus LMP2A-induced B-cell survival in two unique classes of EmuLMP2A transgenic mice. J. Virol. 2000, 74, 1101–1113. [Google Scholar] [CrossRef] [PubMed]
- Fukuda, M.; Longnecker, R. Epstein-Barr virus latent membrane protein 2A mediates transformation through constitutive activation of the Ras/PI3-K/Akt Pathway. J. Virol. 2007, 81, 9299–9306. [Google Scholar] [CrossRef] [PubMed]
- Moody, C.A.; Scott, R.S.; Amirghahari, N.; Nathan, C.O.; Young, L.S.; Dawson, C.W.; Sixbey, J.W. Modulation of the cell growth regulator mTOR by Epstein-Barr virus-encoded LMP2A. J. Virol. 2005, 79, 5499–5506. [Google Scholar] [CrossRef] [PubMed]
- Longnecker, R.; Miller, C.L.; Miao, X.Q.; Marchini, A.; Kieff, E. The only domain which distinguishes Epstein-Barr virus latent membrane protein 2A (LMP2A) from LMP2B is dispensable for lymphocyte infection and growth transformation in vitro; LMP2A is therefore nonessential. J. Virol. 1992, 66, 6461–6469. [Google Scholar] [PubMed]
- Rochford, R.; Miller, C.L.; Cannon, M.J.; Izumi, K.M.; Kieff, E.; Longnecker, R. In vivo growth of Epstein-Barr virus transformed B cells with mutations in latent membrane protein 2 (LMP2). Arch. Virol. 1997, 142, 707–720. [Google Scholar] [CrossRef] [PubMed]
- Mancao, C.; Hammerschmidt, W. Epstein-Barr virus latent membrane protein 2A is a B-cell receptor mimic and essential for B-cell survival. Blood 2007, 110, 3715–3721. [Google Scholar] [CrossRef] [PubMed]
- Portis, T.; Dyck, P.; Longnecker, R. Epstein-Barr Virus (EBV) LMP2A induces alterations in gene transcription similar to those observed in Reed-Sternberg cells of Hodgkin lymphoma. Blood 2003, 102, 4166–4178. [Google Scholar] [CrossRef] [PubMed]
- Vockerodt, M.; Wei, W.; Nagy, E.; Prouzova, Z.; Schrader, A.; Kube, D.; Rowe, M.; Woodman, C.B.; Murray, P.G. Suppression of the LMP2A target gene, EGR-1, protects Hodgkin’s lymphoma cells from entry to the EBV lytic cycle. J. Pathol. 2013, 230, 399–409. [Google Scholar] [CrossRef] [PubMed]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Systematic and integrative analysis of large gene lists using DAVID Bioinformatics Resources. Nat. Protoc. 2009, 4, 44–57. [Google Scholar] [CrossRef] [PubMed]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Bioinformatics enrichment tools: Paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009, 37, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Uchida, J.; Yasui, T.; Takaoka-Shichijo, Y.; Muraoka, M.; Kulwichit, W.; Raab-Traub, N.; Kikutani, H. Mimicry of CD40 signals by Epstein-Barr virus LMP1 in B lymphocyte responses. Science 1999, 286, 300–303. [Google Scholar] [CrossRef] [PubMed]
- Stunz, L.L.; Busch, L.K.; Munroe, M.E.; Sigmund, C.D.; Tygrett, L.T.; Waldschmidt, T.J.; Bishop, G.A. Expression of the cytoplasmic tail of LMP1 in mice induces hyperactivation of B lymphocytes and disordered lymphoid architecture. Immunity 2004, 21, 255–266. [Google Scholar] [CrossRef] [PubMed]
- Rastelli, J.; Homig-Holzel, C.; Seagal, J.; Muller, W.; Hermann, A.C.; Rajewsky, K.; Zimber-Strobl, U. LMP1 signaling can replace CD40 signaling in B cells in vivo and has unique features of inducing class-switch recombination to IgG1. Blood 2008, 111, 1448–1455. [Google Scholar] [CrossRef] [PubMed]
- Homig-Holzel, C.; Hojer, C.; Rastelli, J.; Casola, S.; Strobl, L.J.; Muller, W.; Quintanilla-Martinez, L.; Gewies, A.; Ruland, J.; Rajewsky, K.; et al. Constitutive CD40 signaling in B cells selectively activates the noncanonical NF-kappaB pathway and promotes lymphomagenesis. J. Exp. Med. 2008, 205, 1317–1329. [Google Scholar] [CrossRef] [PubMed]
- Kulwichit, W.; Edwards, R.H.; Davenport, E.M.; Baskar, J.F.; Godfrey, V.; Raab-Traub, N. Expression of the Epstein-Barr virus latent membrane protein 1 induces B cell lymphoma in transgenic mice. Proc. Natl. Acad. Sci. USA 1998, 95, 11963–11968. [Google Scholar] [CrossRef] [PubMed]
- Swanson-Mungerson, M.; Bultema, R.; Longnecker, R. Epstein-Barr virus LMP2A enhances B-cell responses in vivo and in vitro. J. Virol. 2006, 80, 6764–6770. [Google Scholar] [CrossRef] [PubMed]
- Weniger, M.A.; Tiacci, E.; Schneider, S.; Arnolds, J.; Ruschenbaum, S.; Duppach, J.; Seifert, M.; Doring, C.; Hansmann, M.L.; Kuppers, R. Human CD30+ B cells represent a unique subset related to Hodgkin lymphoma cells. J. Clin. Invest. 2018, 128, 2996–3007. [Google Scholar] [CrossRef] [PubMed]
- Vrazo, A.C.; Chauchard, M.; Raab-Traub, N.; Longnecker, R. Epstein-Barr virus LMP2A reduces hyperactivation induced by LMP1 to restore normal B cell phenotype in transgenic mice. PLoS Pathog. 2012, 8, e1002662. [Google Scholar] [CrossRef] [PubMed]
- Miller, C.L.; Longnecker, R.; Kieff, E. Epstein-Barr virus latent membrane protein 2A blocks calcium mobilization in B lymphocytes. J. Virol. 1993, 67, 3087–3094. [Google Scholar] [PubMed]
- Guasparri, I.; Bubman, D.; Cesarman, E. EBV LMP2A affects LMP1-mediated NF-kappaB signaling and survival of lymphoma cells by regulating TRAF2 expression. Blood 2008, 111, 3813–3820. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Kracker, S.; Yasuda, T.; Casola, S.; Vanneman, M.; Homig-Holzel, C.; Wang, Z.; Derudder, E.; Li, S.; Chakraborty, T.; et al. Immune surveillance and therapy of lymphomas driven by Epstein-Barr virus protein LMP1 in a mouse model. Cell 2012, 148, 739–751. [Google Scholar] [CrossRef] [PubMed]
- Wirtz, T.; Weber, T.; Kracker, S.; Sommermann, T.; Rajewsky, K.; Yasuda, T. Mouse model for acute Epstein-Barr virus infection. Proc. Natl. Acad. Sci. USA 2016, 113, 13821–13826. [Google Scholar] [CrossRef] [PubMed]
- Shair, K.H.; Raab-Traub, N. Transcriptome changes induced by Epstein-Barr virus LMP1 and LMP2A in transgenic lymphocytes and lymphoma. MBio 2012, 3. [Google Scholar] [CrossRef] [PubMed]
- Ma, S.D.; Xu, X.; Plowshay, J.; Ranheim, E.A.; Burlingham, W.J.; Jensen, J.L.; Asimakopoulos, F.; Tang, W.; Gulley, M.L.; Cesarman, E.; et al. LMP1-deficient Epstein-Barr virus mutant requires T cells for lymphomagenesis. J. Clin. Invest. 2015, 125, 304–315. [Google Scholar] [CrossRef] [PubMed]
- Park, G.B.; Kim, Y.S.; Lee, H.K.; Cho, D.H.; Kim, D.; Hur, D.Y. CD80 (B7.1) and CD86 (B7.2) induce EBV-transformed B cell apoptosis through the Fas/FasL pathway. Int. J. Oncol. 2013, 43, 1531–1540. [Google Scholar] [CrossRef] [PubMed]
- Steidl, C.; Telenius, A.; Shah, S.P.; Farinha, P.; Barclay, L.; Boyle, M.; Connors, J.M.; Horsman, D.E.; Gascoyne, R.D. Genome-wide copy number analysis of Hodgkin Reed-Sternberg cells identifies recurrent imbalances with correlations to treatment outcome. Blood 2010, 116, 418–427. [Google Scholar] [CrossRef] [PubMed]
- Dawson, C.W.; George, J.H.; Blake, S.M.; Longnecker, R.; Young, L.S. The Epstein-Barr virus encoded latent membrane protein 2A augments signaling from latent membrane protein 1. Virology 2001, 289, 192–207. [Google Scholar] [CrossRef] [PubMed]
- Shair, K.H.; Bendt, K.M.; Edwards, R.H.; Nielsen, J.N.; Moore, D.T.; Raab-Traub, N. Epstein-Barr virus-encoded latent membrane protein 1 (LMP1) and LMP2A function cooperatively to promote carcinoma development in a mouse carcinogenesis model. J. Virol. 2012, 86, 5352–5365. [Google Scholar] [CrossRef] [PubMed]





© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vrzalikova, K.; Ibrahim, M.; Nagy, E.; Vockerodt, M.; Perry, T.; Wei, W.; Woodman, C.; Murray, P. Co-Expression of the Epstein-Barr Virus-Encoded Latent Membrane Proteins and the Pathogenesis of Classic Hodgkin Lymphoma. Cancers 2018, 10, 285. https://doi.org/10.3390/cancers10090285
Vrzalikova K, Ibrahim M, Nagy E, Vockerodt M, Perry T, Wei W, Woodman C, Murray P. Co-Expression of the Epstein-Barr Virus-Encoded Latent Membrane Proteins and the Pathogenesis of Classic Hodgkin Lymphoma. Cancers. 2018; 10(9):285. https://doi.org/10.3390/cancers10090285
Chicago/Turabian StyleVrzalikova, Katerina, Maha Ibrahim, Eszter Nagy, Martina Vockerodt, Tracey Perry, Wenbin Wei, Ciaran Woodman, and Paul Murray. 2018. "Co-Expression of the Epstein-Barr Virus-Encoded Latent Membrane Proteins and the Pathogenesis of Classic Hodgkin Lymphoma" Cancers 10, no. 9: 285. https://doi.org/10.3390/cancers10090285
APA StyleVrzalikova, K., Ibrahim, M., Nagy, E., Vockerodt, M., Perry, T., Wei, W., Woodman, C., & Murray, P. (2018). Co-Expression of the Epstein-Barr Virus-Encoded Latent Membrane Proteins and the Pathogenesis of Classic Hodgkin Lymphoma. Cancers, 10(9), 285. https://doi.org/10.3390/cancers10090285