Differentiation Therapy Targeting the β-Catenin/CBP Interaction in Pancreatic Cancer

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
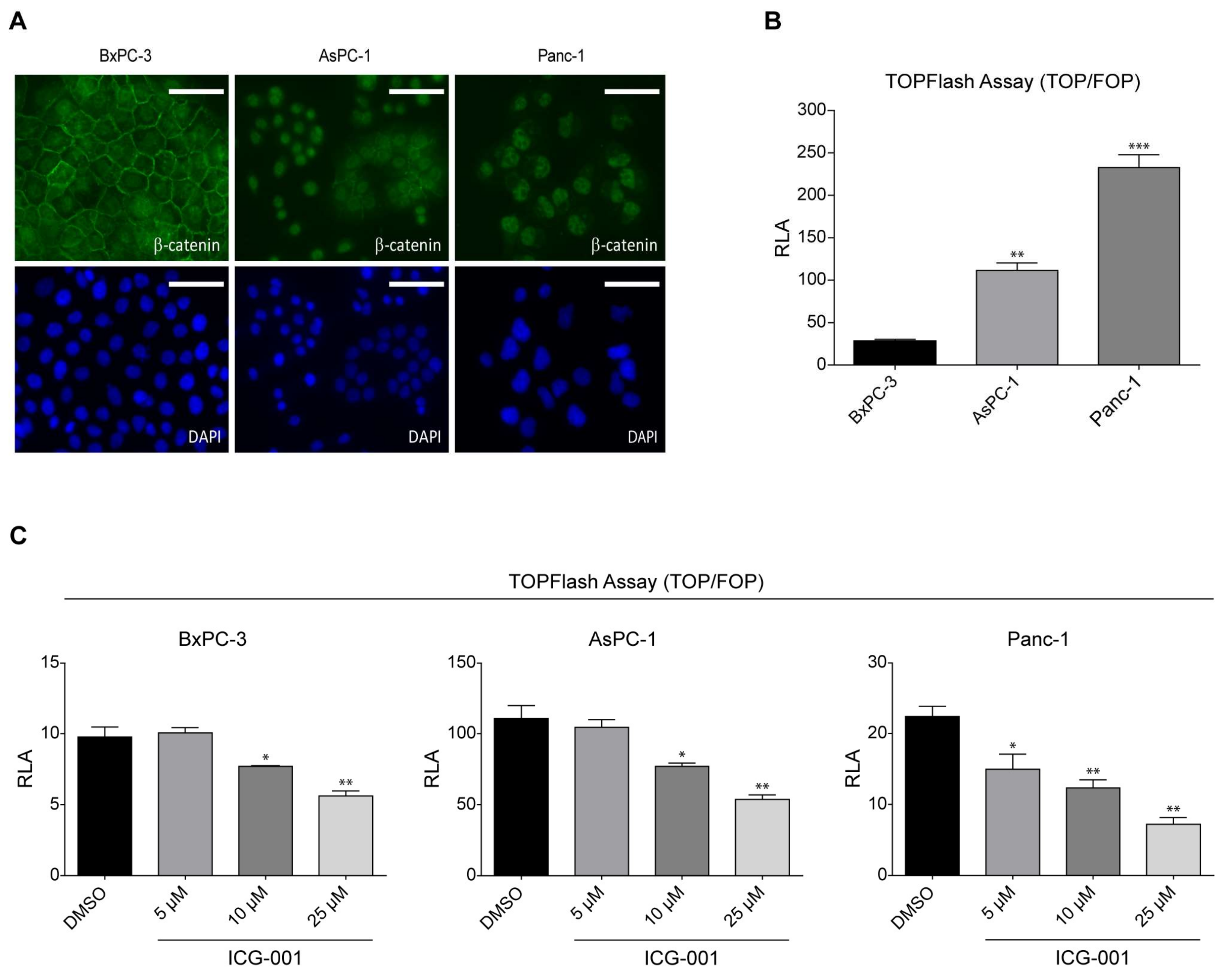
2.1. K-Ras Mutations Increase Wnt/β-Catenin Signaling in Pancreatic Cancer Cells
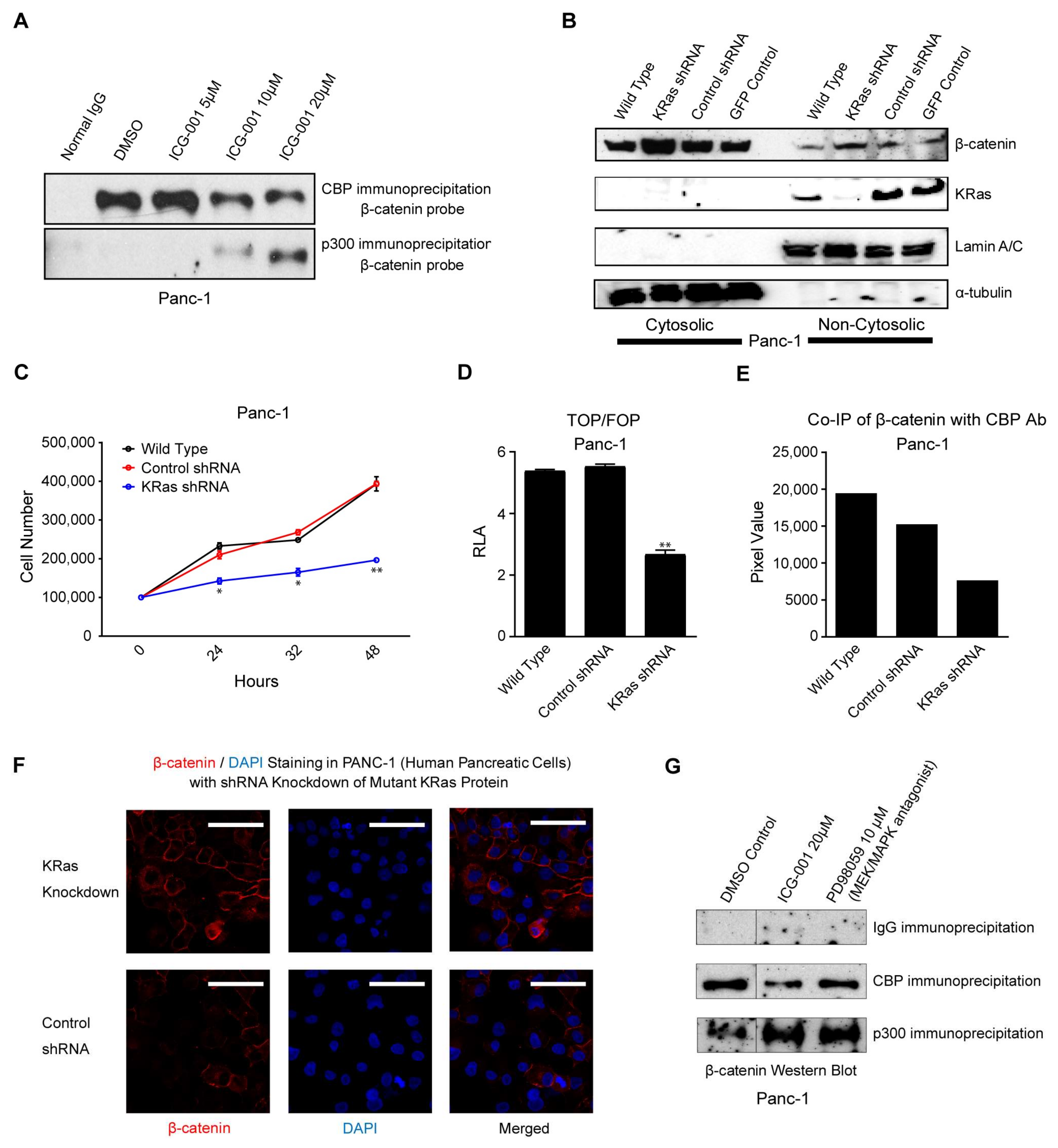
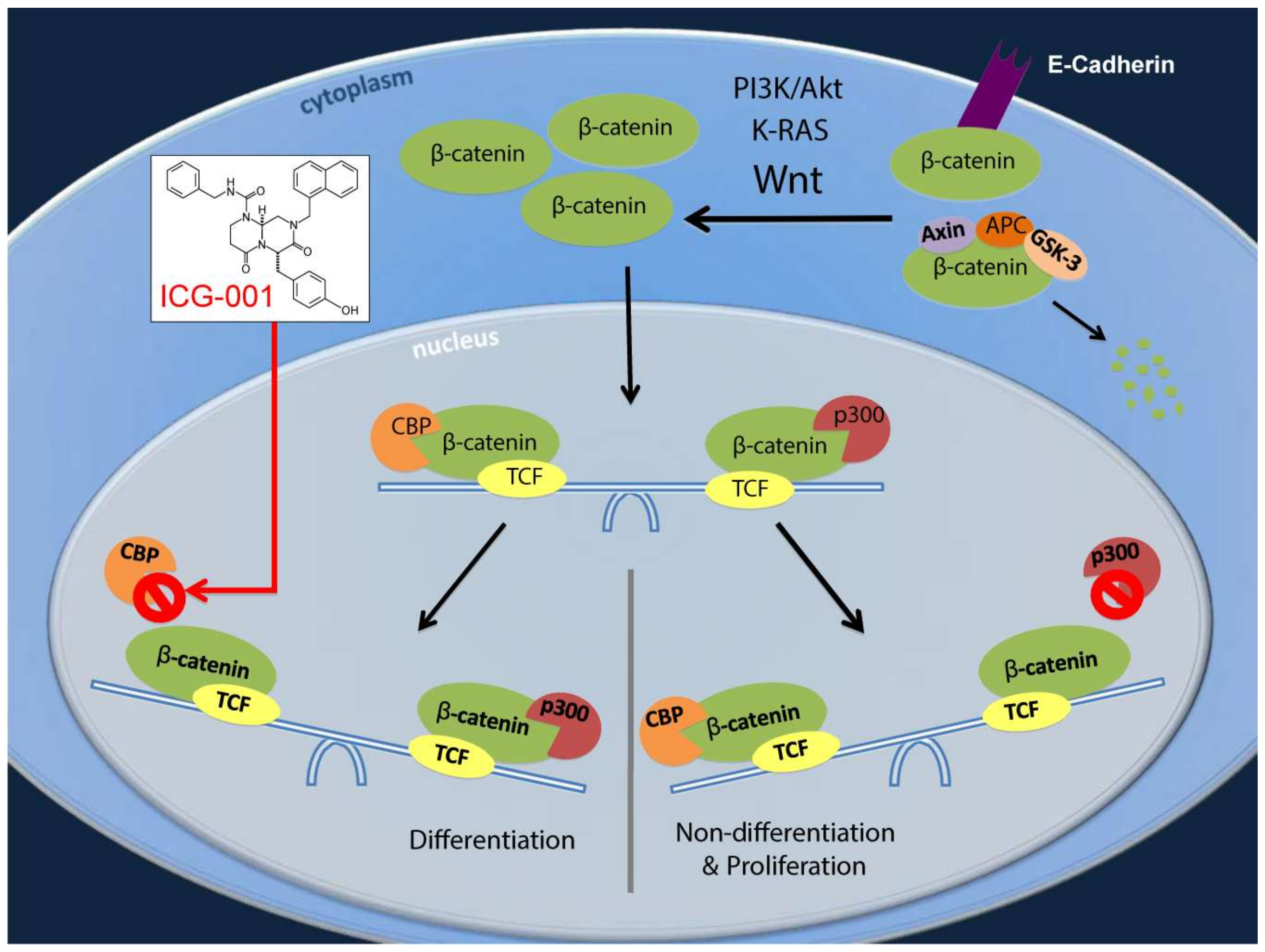
2.2. ICG-001 Effects a Change in β-Catenin Coactivator Usage in Pancreatic Cancer Cells
2.3. K-Ras Activation Increases the CBP/β-Catenin Interaction
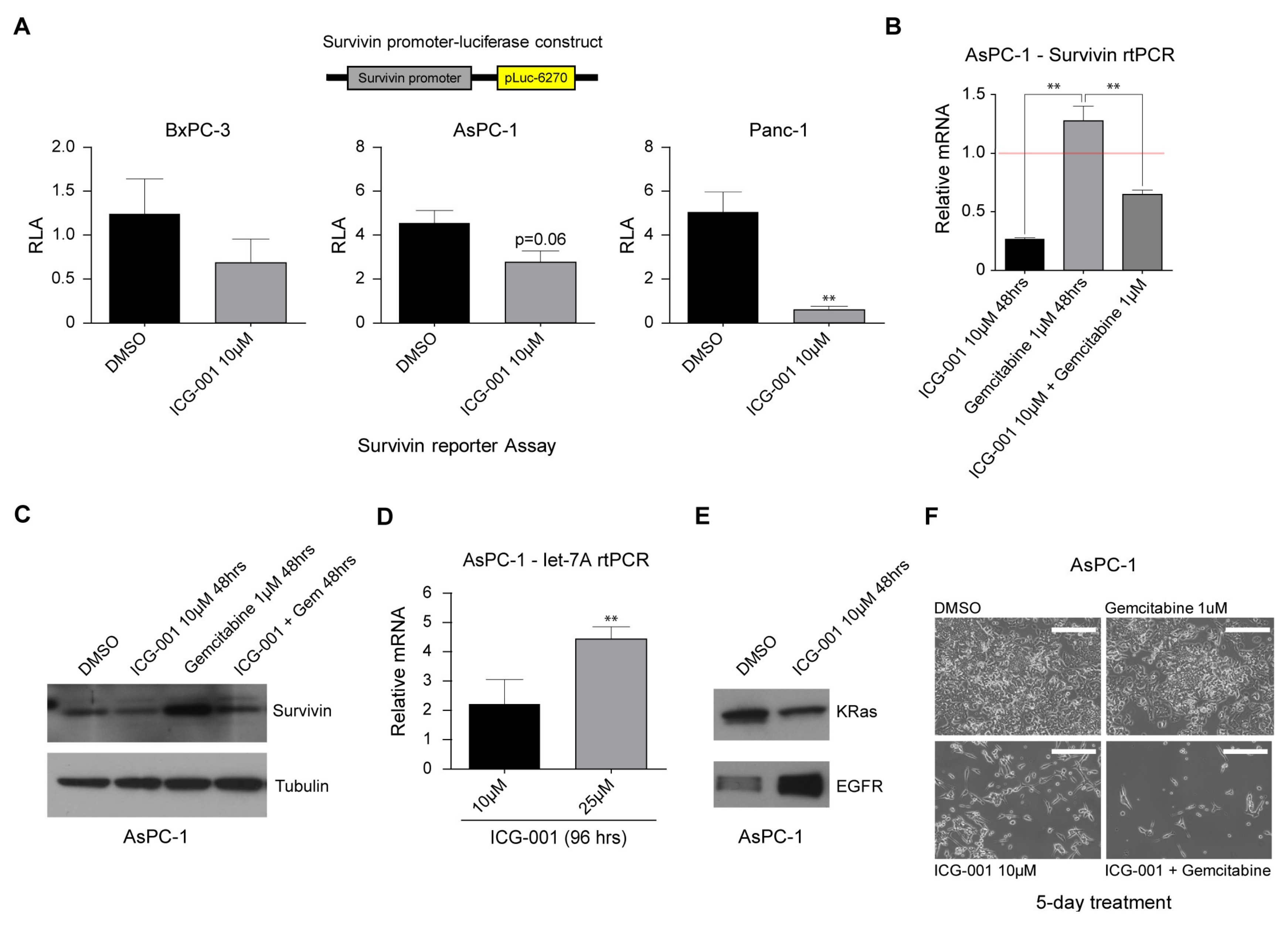
2.4. Decreasing CBP/β-Catenin Interaction While Increasing p300/β-Catenin Interaction Results in Reduced Expression of Genes Associated with Poor Prognosis
2.5. ICG-001 Induces let-7 miRNA Expression and Suppresses K-Ras Protein Levels while Increasing EGFR Protein Levels in Pancreatic Cancer Cells
2.6. ICG-001 Effectively Sensitizes Pancreatic Cancer Cells to Conventional Chemotherapy
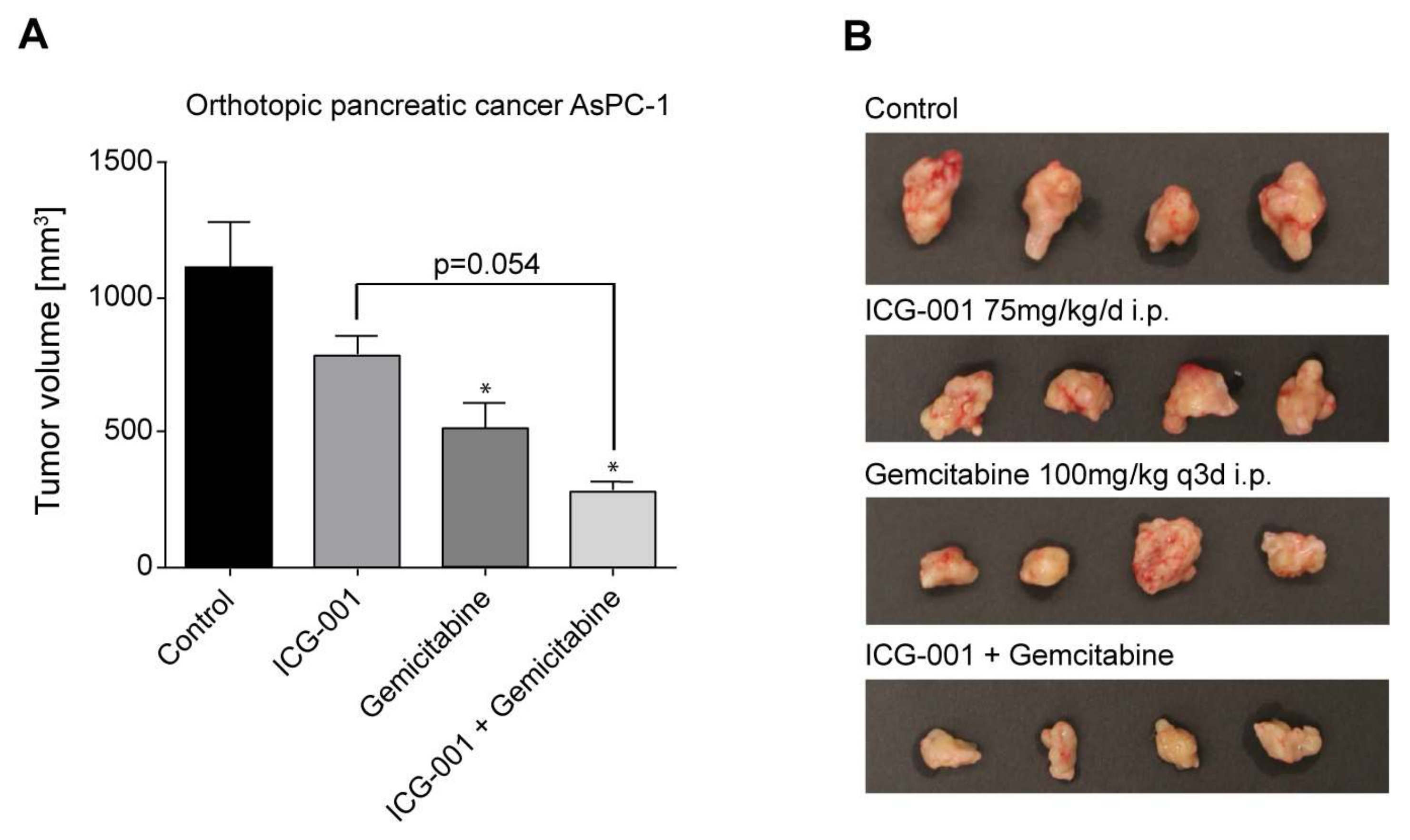
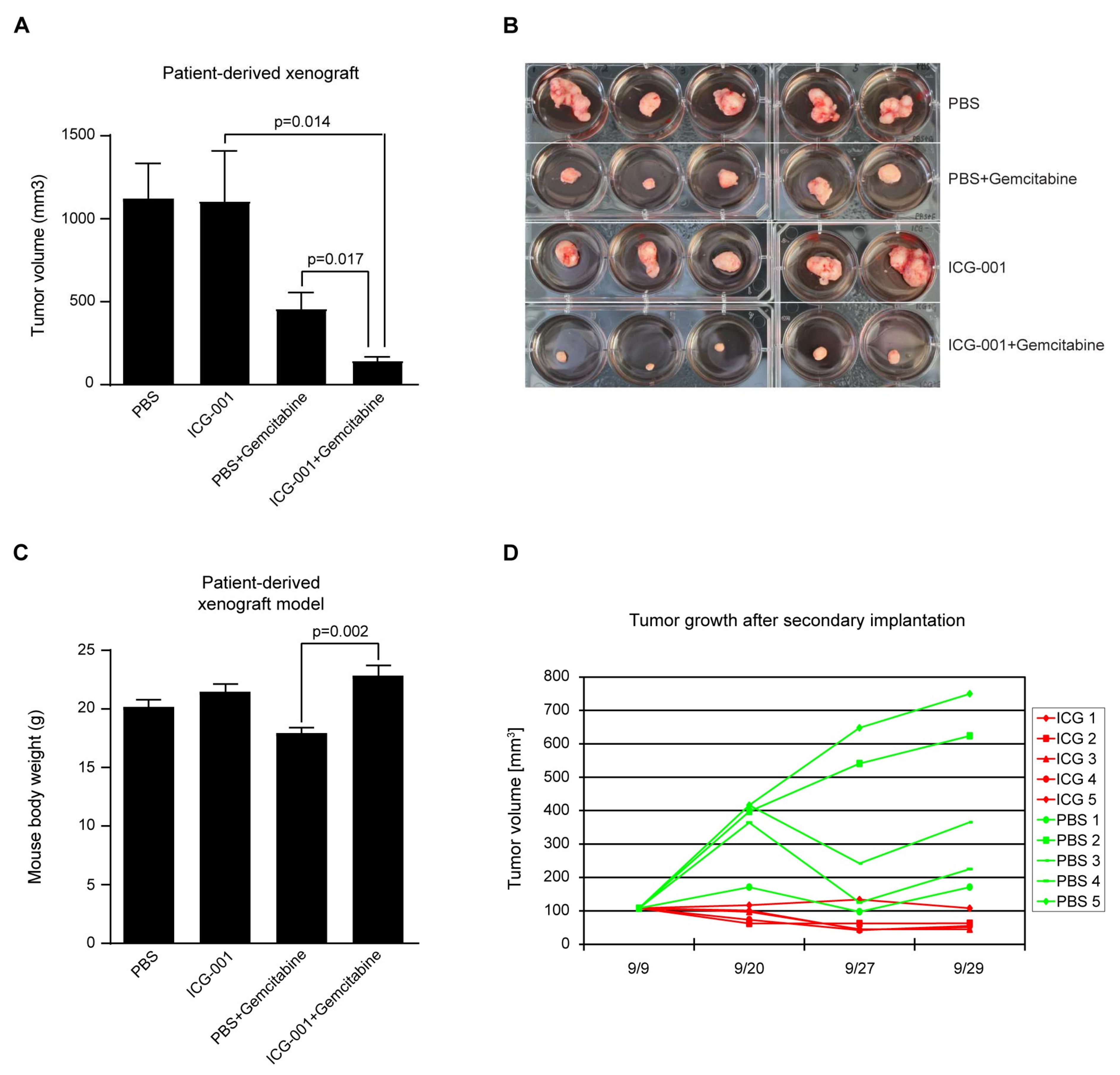
2.7. Efficacy of CBP/Catenin Antagonists in In Vivo Models
3. Discussion
4. Materials and Methods
4.1. Cancer Cell Lines
4.2. Pharmacologic Agents
4.3. Orthotopic Pancreatic Cancer in Nude Mice
4.4. Human Patient-Derived Xenograft Model of Pancreatic Ductal Adenocarcinoma (PDAC)
4.5. Antibodies
4.6. Immunofluorescence
4.7. TOPFlash/FOPFlash and Survivin Reporter Assays
4.8. Co-Immunoprecipitation
4.9. Lentiviral shRNA Knockdown of K-Ras
4.10. Western Blot
4.11. Reverse Transcription Quantitative Polymerase Chain Reaction (RT-qPCR) Analysis
4.12. Data Analysis
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Werner, J.; Combs, S.E.; Springfeld, C.; Hartwig, W.; Hackert, T.; Büchler, M.W. Advanced-stage pancreatic cancer: Therapy options. Nat. Rev. Clin. Oncol. 2013, 10, 323–333. [Google Scholar] [CrossRef] [PubMed]
- Abraham, S.C.; Wu, T.T.; Klimstra, D.S.; Finn, L.S.; Lee, J.H.; Yeo, C.J.; Cameron, J.L.; Hruban, R.H. Distinctive molecular genetic alterations in sporadic and familial adenomatous polyposis-associated pancreatoblastomas: Frequent alterations in the APC/β-catenin pathway and chromosome 11p. Am. J. Pathol. 2001, 159, 1619–1627. [Google Scholar] [CrossRef]
- Abraham, S.C.; Wu, T.T.; Hruban, R.H.; Lee, J.H.; Yeo, C.J.; Conlon, K.; Brennan, M.; Cameron, J.L.; Klimstra, D.S. Genetic and immunohistochemical analysis of pancreatic acinar cell carcinoma: Frequent allelic loss on chromosome 11p and alterations in the APC/β-catenin pathway. Am. J. Pathol. 2002, 160, 953–962. [Google Scholar] [CrossRef]
- Abraham, S.C.; Klimstra, D.S.; Wilentz, R.E.; Yeo, C.J.; Conlon, K.; Brennan, M.; Cameron, J.L.; Wu, T.T.; Hruban, R.H. Solid-pseudopapillary tumors of the pancreas are genetically distinct from pancreatic ductal adenocarcinomas and almost always harbor β-catenin mutations. Am. J. Pathol. 2002, 160, 1361–1369. [Google Scholar] [CrossRef]
- Zeng, G.; Germinaro, M.; Micsenyi, A.; Monga, N.K.; Bell, A.; Sood, A.; Malhotra, V.; Sood, N.; Midda, V.; Monga, D.K.; et al. Aberrant Wnt/β-catenin signaling in pancreatic adenocarcinoma. Neoplasia 2006, 8, 279–289. [Google Scholar] [CrossRef] [PubMed]
- Pasca di Magliano, M.; Biankin, A.V.; Heiser, P.W.; Cano, D.A.; Gutierrez, P.J.; Deramaudt, T.; Segara, D.; Dawson, A.C.; Kench, J.G.; Henshall, S.M.; et al. Common activation of canonical Wnt signaling in pancreatic adenocarcinoma. PLoS ONE 2007, 2, e1155. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Gou, S.M.; Wang, C.Y.; Wu, H.S.; Xiong, J.X.; Zhou, F. Pancreas duodenal homeobox-1 expression and significance in pancreatic cancer. World J. Gastroenterol. 2007, 13, 2615–2618. [Google Scholar] [CrossRef] [PubMed]
- Brabletz, T.; Jung, A.; Spaderna, S.; Hlubek, F.; Kirchner, T. Opinion: Migrating cancer stem cells—An integrated concept of malignant tumour progression. Nat. Rev. Cancer 2005, 5, 744–749. [Google Scholar] [CrossRef] [PubMed]
- Peitzsch, C.; Tyutyunnykova, A.; Pantel, K.; Dubrovska, A. Cancer stem cells: The root of tumor recurrence and metastases. Semin. Cancer Biol. 2017, 44, 10–24. [Google Scholar] [CrossRef] [PubMed]
- Reya, T.; Clevers, H. Wnt signalling in stem cells and cancer. Nature 2005, 434, 843–850. [Google Scholar] [CrossRef] [PubMed]
- Thomas, P.D.; Kahn, M. Kat3 coactivators in somatic stem cells and cancer stem cells: Biological roles, evolution, and pharmacologic manipulation. Cell Biol. Toxicol. 2016, 32, 61–81. [Google Scholar] [CrossRef] [PubMed]
- Huang, M.E.; Ye, Y.C.; Chen, S.R.; Chai, J.R.; Lu, J.X.; Zhoa, L.; Gu, L.J.; Wang, Z.Y. Use of all-trans retinoic acid in the treatment of acute promyelocytic leukemia. Blood 1988, 72, 567–572. [Google Scholar] [PubMed]
- Nowak, D.; Stewart, D.; Koeffler, H.P. Differentiation therapy of leukemia: 3 Decades of development. Blood 2009, 113, 3655–3665. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.M.; Ma, H.; Oehler, V.G.; Gang, E.J.; Nguyen, C.; Masiello, D.; Liu, H.; Zhao, Y.; Radich, J.; Kahn, M. The gamma catenin/CBP complex maintains survivin transcription in β-catenin deficient/depleted cancer cells. Curr. Cancer Drug Targets 2011, 11, 213–225. [Google Scholar] [CrossRef] [PubMed]
- Gang, E.J.; Hsieh, Y.T.; Pham, J.; Zhao, Y.; Nguyen, C.; Huantes, S.; Park, E.; Naing, K.; Klemm, L.; Swaminathan, S.; et al. Small-molecule inhibition of CBP/catenin interactions eliminates drug-resistant clones in acute lymphoblastic leukemia. Oncogene 2014, 33, 2169–2178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wend, P.; Fang, L.; Zhu, Q.; Schipper, J.H.; Loddenkemper, C.; Kosel, F.; Brinkmann, V.; Eckert, K.; Hindersin, S.; Holland, J.D.; et al. Wnt/β-catenin signalling induces mll to create epigenetic changes in salivary gland tumours. EMBO J. 2013, 32, 1977–1989. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Masiello, D.; McMillian, M.; Nguyen, C.; Wu, Y.; Melendez, E.; Smbatyan, G.; Kida, A.; He, Y.; Teo, J.L.; et al. CBP/catenin antagonist safely eliminates drug-resistant leukemia-initiating cells. Oncogene 2016, 35, 3705–3717. [Google Scholar] [CrossRef] [PubMed]
- Janssen, K.P.; Alberici, P.; Fsihi, H.; Gaspar, C.; Breukel, C.; Franken, P.; Rosty, C.; Abal, M.; El Marjou, F.; Smits, R.; et al. APC and oncogenic KRAS are synergistic in enhancing Wnt signaling in intestinal tumor formation and progression. Gastroenterology 2006, 131, 1096–1109. [Google Scholar] [CrossRef] [PubMed]
- Sansom, O.J.; Meniel, V.; Wilkins, J.A.; Cole, A.M.; Oien, K.A.; Marsh, V.; Jamieson, T.J.; Guerra, C.; Ashton, G.H.; Barbacid, M.; et al. Loss of APC allows phenotypic manifestation of the transforming properties of an endogenous K-ras oncogene in vivo. Proc. Natl. Acad. Sci. USA 2006, 103, 14122–14127. [Google Scholar] [CrossRef] [PubMed]
- Harada, N.; Oshima, H.; Katoh, M.; Tamai, Y.; Oshima, M.; Taketo, M.M. Hepatocarcinogenesis in mice with β-catenin and Ha-ras gene mutations. Cancer Res. 2004, 64, 48–54. [Google Scholar] [CrossRef] [PubMed]
- Araki, Y.; Okamura, S.; Hussain, S.P.; Nagashima, M.; He, P.; Shiseki, M.; Miura, K.; Harris, C.C. Regulation of cyclooxygenase-2 expression by the Wnt and RAS pathways. Cancer Res. 2003, 63, 728–734. [Google Scholar] [PubMed]
- Kerkhoff, E.; Houben, R.; Löffler, S.; Troppmair, J.; Lee, J.E.; Rapp, U.R. Regulation of c-myc expression by Ras/Raf signalling. Oncogene 1998, 16, 211–216. [Google Scholar] [CrossRef] [PubMed]
- Masckauchán, T.N.; Shawber, C.J.; Funahashi, Y.; Li, C.M.; Kitajewski, J. Wnt/β-catenin signaling induces proliferation, survival and interleukin-8 in human endothelial cells. Angiogenesis 2005, 8, 43–51. [Google Scholar] [CrossRef] [PubMed]
- Cui, J.; Jiang, W.; Wang, S.; Wang, L.; Xie, K. Role of Wnt/β-catenin signaling in drug resistance of pancreatic cancer. Curr. Pharm. Des. 2012, 18, 2464–2471. [Google Scholar] [CrossRef] [PubMed]
- Emami, K.H.; Nguyen, C.; Ma, H.; Kim, D.H.; Jeong, K.W.; Eguchi, M.; Moon, R.T.; Teo, J.L.; Oh, S.W.; Kim, H.Y.; et al. A small molecule inhibitor of β-catenin/creb-binding protein transcription [corrected]. Proc. Natl. Acad. Sci. USA 2004, 101, 12682–12687. [Google Scholar] [CrossRef] [PubMed]
- Kahn, M. Can we safely target the Wnt pathway? Nat. Rev. Drug Discov. 2014, 13, 513–532. [Google Scholar] [CrossRef] [PubMed]
- Hasegawa, K.; Yasuda, S.Y.; Teo, J.L.; Nguyen, C.; McMillan, M.; Hsieh, C.L.; Suemori, H.; Nakatsuji, N.; Yamamoto, M.; Miyabayashi, T.; et al. Wnt signaling orchestration with a small molecule dyrk inhibitor provides long-term xeno-free human pluripotent cell expansion. Stem Cells Transl. Med. 2012, 1, 18–28. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.R.; Scehnet, J.S.; Ley, E.J.; Singh, J.; Krasnoperov, V.; Liu, R.; Manchanda, P.K.; Ladner, R.D.; Hawes, D.; Weaver, F.A.; et al. Preferential induction of EphB4 over EphB2 and its implication in colorectal cancer progression. Cancer Res. 2009, 69, 3736–3745. [Google Scholar] [CrossRef] [PubMed]
- Miyabayashi, T.; Teo, J.L.; Yamamoto, M.; McMillan, M.; Nguyen, C.; Kahn, M. Wnt/β-catenin/CBP signaling maintains long-term murine embryonic stem cell pluripotency. Proc. Natl. Acad. Sci. USA 2007, 104, 5668–5673. [Google Scholar] [CrossRef] [PubMed]
- Velculescu, V.E.; Madden, S.L.; Zhang, L.; Lash, A.E.; Yu, J.; Rago, C.; Lal, A.; Wang, C.J.; Beaudry, G.A.; Ciriello, K.M.; et al. Analysis of human transcriptomes. Nat. Genet. 1999, 23, 387–388. [Google Scholar] [CrossRef] [PubMed]
- Ma, H.; Nguyen, C.; Lee, K.S.; Kahn, M. Differential roles for the coactivators CBP and p300 on TCF/β-catenin-mediated survivin gene expression. Oncogene 2005, 24, 3619–3631. [Google Scholar] [CrossRef] [PubMed]
- Henderson, W.R.; Chi, E.Y.; Ye, X.; Nguyen, C.; Tien, Y.T.; Zhou, B.; Borok, Z.; Knight, D.A.; Kahn, M. Inhibition of Wnt/β-catenin/creb binding protein (CBP) signaling reverses pulmonary fibrosis. Proc. Natl. Acad. Sci. USA 2010, 107, 14309–14314. [Google Scholar] [CrossRef] [PubMed]
- Guo, Q.; Chen, Y.; Zhang, B.; Kang, M.; Xie, Q.; Wu, Y. Potentiation of the effect of gemcitabine by emodin in pancreatic cancer is associated with survivin inhibition. Biochem. Pharmacol. 2009, 77, 1674–1683. [Google Scholar] [CrossRef] [PubMed]
- Visus, C.; Wang, Y.; Lozano-Leon, A.; Ferris, R.L.; Silver, S.; Szczepanski, M.J.; Brand, R.E.; Ferrone, C.R.; Whiteside, T.L.; Ferrone, S.; et al. Targeting aldh(bright) human carcinoma-initiating cells with aldh1a1-specific cd8+ t cells. Clin. Cancer Res. 2011, 17, 6174–6184. [Google Scholar] [CrossRef] [PubMed]
- Teo, J.L.; Ma, H.; Nguyen, C.; Lam, C.; Kahn, M. Specific inhibition of CBP/β-catenin interaction rescues defects in neuronal differentiation caused by a presenilin-1 mutation. Proc. Natl. Acad. Sci. USA 2005, 102, 12171–12176. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; VandenBoom, T.G.; Kong, D.; Wang, Z.; Ali, S.; Philip, P.A.; Sarkar, F.H. Up-regulation of miR-200 and let-7 by natural agents leads to the reversal of epithelial-to-mesenchymal transition in gemcitabine-resistant pancreatic cancer cells. Cancer Res. 2009, 69, 6704–6712. [Google Scholar] [CrossRef] [PubMed]
- Melton, C.; Judson, R.L.; Blelloch, R. Opposing microrna families regulate self-renewal in mouse embryonic stem cells. Nature 2010, 463, 621–626. [Google Scholar] [CrossRef] [PubMed]
- Bruns, C.J.; Harbison, M.T.; Kuniyasu, H.; Eue, I.; Fidler, I.J. In vivo selection and characterization of metastatic variants from human pancreatic adenocarcinoma by using orthotopic implantation in nude mice. Neoplasia 1999, 1, 50–62. [Google Scholar] [CrossRef] [PubMed]
- Burris, H.A.; Moore, M.J.; Andersen, J.; Green, M.R.; Rothenberg, M.L.; Modiano, M.R.; Cripps, M.C.; Portenoy, R.K.; Storniolo, A.M.; Tarassoff, P.; et al. Improvements in survival and clinical benefit with gemcitabine as first-line therapy for patients with advanced pancreas cancer: A randomized trial. J. Clin. Oncol. 1997, 15, 2403–2413. [Google Scholar] [CrossRef] [PubMed]
- Burris, H.A. Recent updates on the role of chemotherapy in pancreatic cancer. Semin. Oncol. 2005, 32, S1–S3. [Google Scholar] [CrossRef] [PubMed]
- Moore, M.J.; Goldstein, D.; Hamm, J.; Figer, A.; Hecht, J.R.; Gallinger, S.; Au, H.J.; Murawa, P.; Walde, D.; Wolff, R.A.; et al. Erlotinib plus gemcitabine compared with gemcitabine alone in patients with advanced pancreatic cancer: A phase III trial of the national cancer institute of Canada clinical trials group. J. Clin. Oncol. 2007, 25, 1960–1966. [Google Scholar] [CrossRef] [PubMed]
- Von Hoff, D.D.; Ervin, T.; Arena, F.P.; Chiorean, E.G.; Infante, J.; Moore, M.; Seay, T.; Tjulandin, S.A.; Ma, W.W.; Saleh, M.N.; et al. Increased survival in pancreatic cancer with nab-paclitaxel plus gemcitabine. N. Engl. J. Med. 2013, 369, 1691–1703. [Google Scholar] [CrossRef] [PubMed]
- Bryant, K.L.; Mancias, J.D.; Kimmelman, A.C.; Der, C.J. Kras: Feeding pancreatic cancer proliferation. Trends Biochem. Sci. 2014, 39, 91–100. [Google Scholar] [CrossRef] [PubMed]
- Hezel, A.F.; Kimmelman, A.C.; Stanger, B.Z.; Bardeesy, N.; Depinho, R.A. Genetics and biology of pancreatic ductal adenocarcinoma. Genes Dev. 2006, 20, 1218–1249. [Google Scholar] [CrossRef] [PubMed]
- Dergham, S.T.; Dugan, M.C.; Sarkar, F.H.; Vaitkevicius, V.K. Molecular alterations associated with improved survival in pancreatic cancer patients treated with radiation or chemotherapy. J. Hepatobiliary Pancreat. Surg. 1998, 5, 269–272. [Google Scholar] [CrossRef] [PubMed]
- Rieger, M.E.; Zhou, B.; Solomon, N.; Sunohara, M.; Li, C.; Nguyen, C.; Liu, Y.; Pan, J.H.; Minoo, P.; Crandall, E.D.; et al. P300/β-catenin interactions regulate adult progenitor cell differentiation downstream of WNT5a/protein kinase c (PKC). J. Biol. Chem. 2016, 291, 6569–6582. [Google Scholar] [CrossRef] [PubMed]
- Garg, H.; Suri, P.; Gupta, J.C.; Talwar, G.P.; Dubey, S. Survivin: A unique target for tumor therapy. Cancer Cell Int. 2016, 16, 49. [Google Scholar] [CrossRef] [PubMed]
- Arensman, M.D.; Telesca, D.; Lay, A.R.; Kershaw, K.M.; Wu, N.; Donahue, T.R.; Dawson, D.W. The creb-binding protein inhibitor ICG-001 suppresses pancreatic cancer growth. Mol. Cancer Ther. 2014, 13, 2303–2314. [Google Scholar] [CrossRef] [PubMed]
- Cairns, J. Mutation selection and the natural history of cancer. Nature 1975, 255, 197–200. [Google Scholar] [CrossRef] [PubMed]
- Bajaj, J.; Zimdahl, B.; Reya, T. Fearful symmetry: Subversion of asymmetric division in cancer development and progression. Cancer Res. 2015, 75, 792–797. [Google Scholar] [CrossRef] [PubMed]
- El-Khoueiry, A.B.; Ning, Y.; Yang, D.; Cole, S.; Kahn, M.; Zoghbi, M.; Berg, J.; Fujimori, M.; Inada, T.; et al. A phase I first-in-human study of PRI-724 in patients (pts) with advanced solid tumors. J. Clin. Oncol. 2013, 31, 2501. [Google Scholar]
- Ko, A.; Chiorean, E.; Kwak, E.; Lenz, H.-J.; Nadler, P.; Wood, D.; Fujimori, M.; Inada, T.; Kouji, H.; McWilliams, R. Final results of a phase Ib dose-escalation study of PRI-724, a CBP/β-catenin modulator, plus gemcitabine (GEM) in patients with advanced pancreatic adenocarcinoma (APC) as second-line therapy after FOLFIRINOX or FOLFOX. J. Clin. Oncol. 2016, 34, e15721. [Google Scholar]






© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Manegold, P.; Lai, K.K.Y.; Wu, Y.; Teo, J.-L.; Lenz, H.-J.; Genyk, Y.S.; Pandol, S.J.; Wu, K.; Lin, D.P.; Chen, Y.; et al. Differentiation Therapy Targeting the β-Catenin/CBP Interaction in Pancreatic Cancer. Cancers 2018, 10, 95. https://doi.org/10.3390/cancers10040095
Manegold P, Lai KKY, Wu Y, Teo J-L, Lenz H-J, Genyk YS, Pandol SJ, Wu K, Lin DP, Chen Y, et al. Differentiation Therapy Targeting the β-Catenin/CBP Interaction in Pancreatic Cancer. Cancers. 2018; 10(4):95. https://doi.org/10.3390/cancers10040095
Chicago/Turabian StyleManegold, Philipp, Keane K. Y. Lai, Yongfeng Wu, Jia-Ling Teo, Heinz-Josef Lenz, Yuri S. Genyk, Stephen J. Pandol, Kaijin Wu, David P. Lin, Yibu Chen, and et al. 2018. "Differentiation Therapy Targeting the β-Catenin/CBP Interaction in Pancreatic Cancer" Cancers 10, no. 4: 95. https://doi.org/10.3390/cancers10040095
APA StyleManegold, P., Lai, K. K. Y., Wu, Y., Teo, J.-L., Lenz, H.-J., Genyk, Y. S., Pandol, S. J., Wu, K., Lin, D. P., Chen, Y., Nguyen, C., Zhao, Y., & Kahn, M. (2018). Differentiation Therapy Targeting the β-Catenin/CBP Interaction in Pancreatic Cancer. Cancers, 10(4), 95. https://doi.org/10.3390/cancers10040095