Translocation Breakpoints Preferentially Occur in Euchromatin and Acrocentric Chromosomes



Abstract
:1. Introduction
2. Results
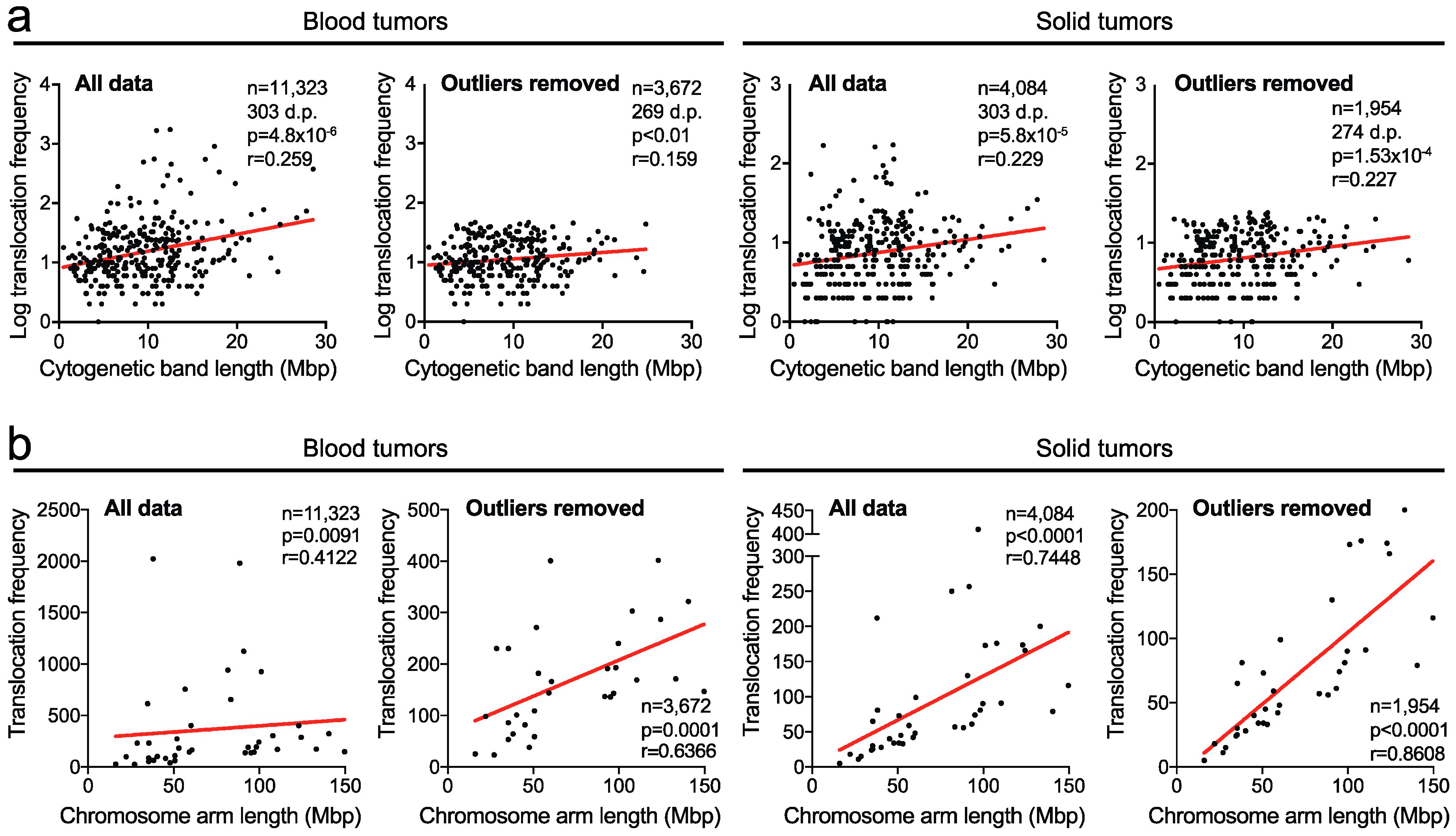
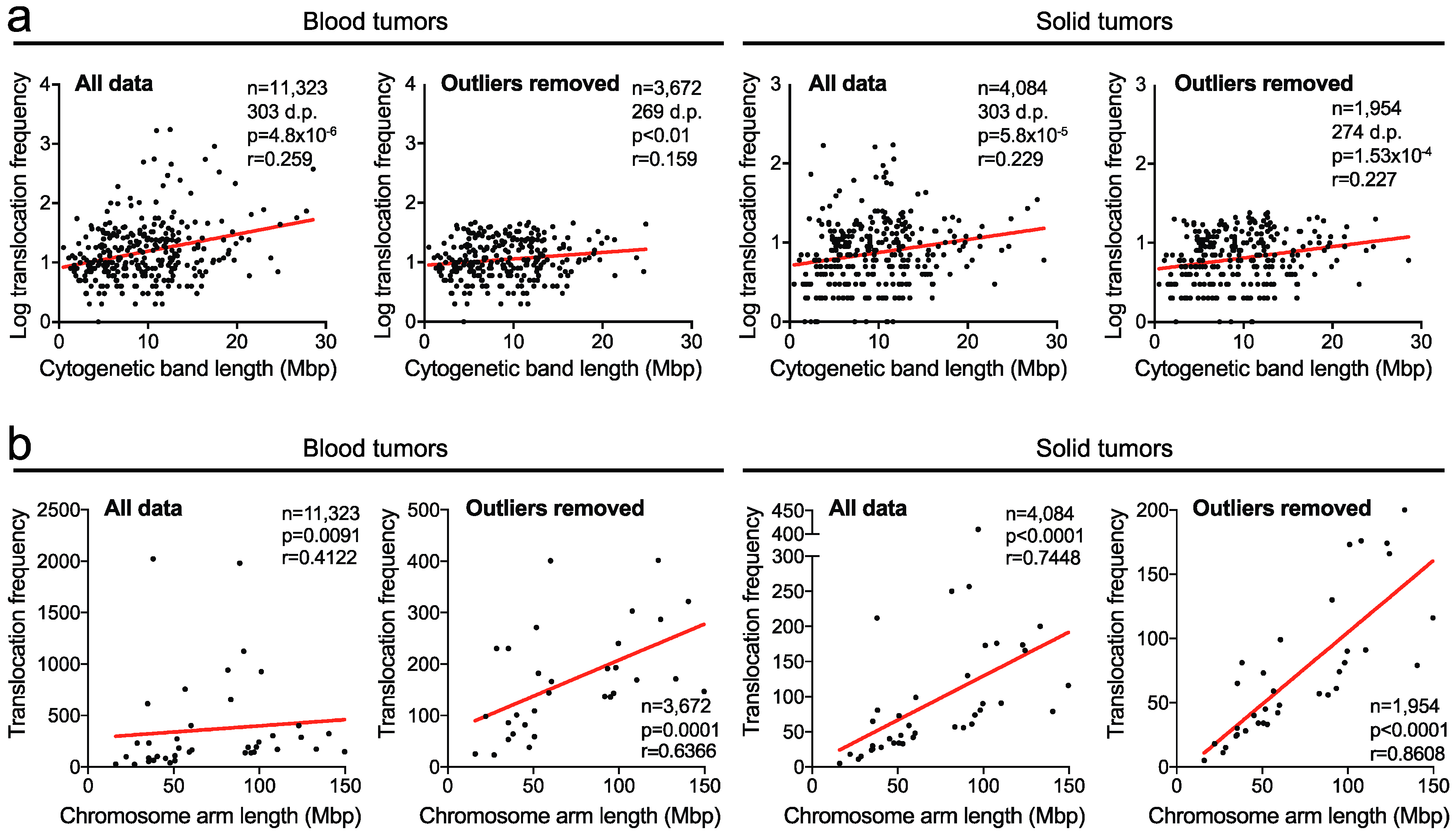
2.1. Translocation Breakpoints Preferentially Occur in Longer Cytogenetic Chromosome Bands
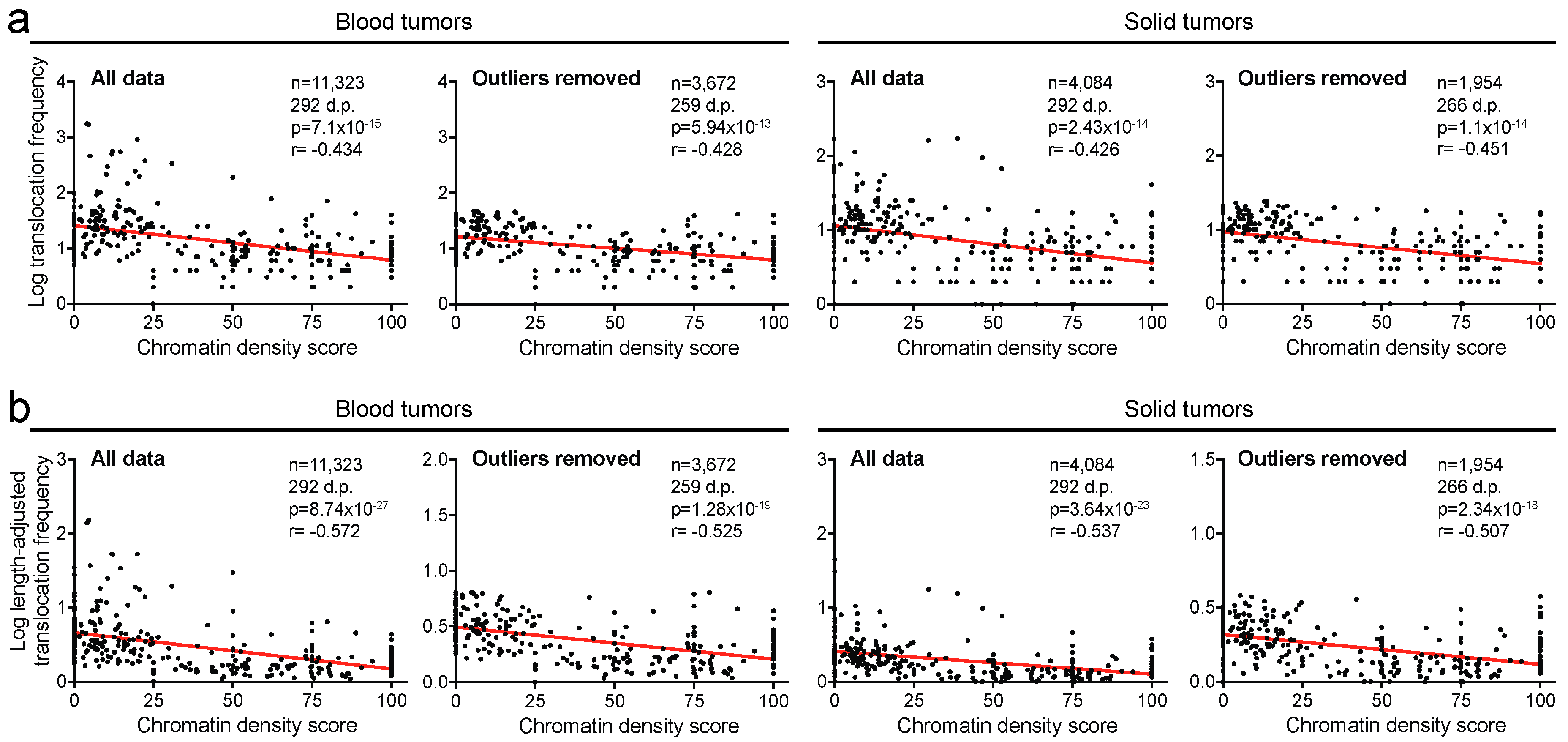
2.2. Translocation Breakpoints Preferentially Occur in Open Chromatin
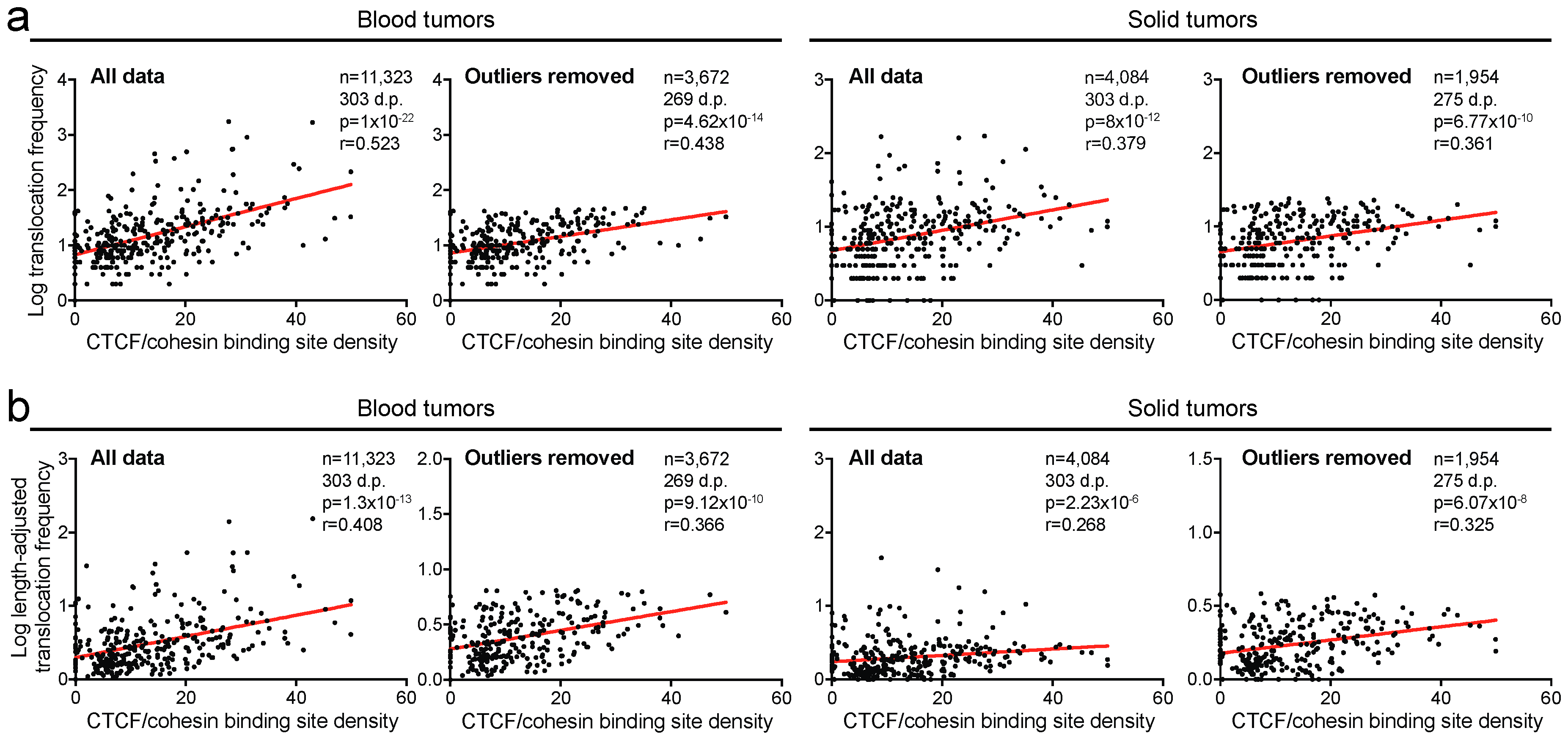
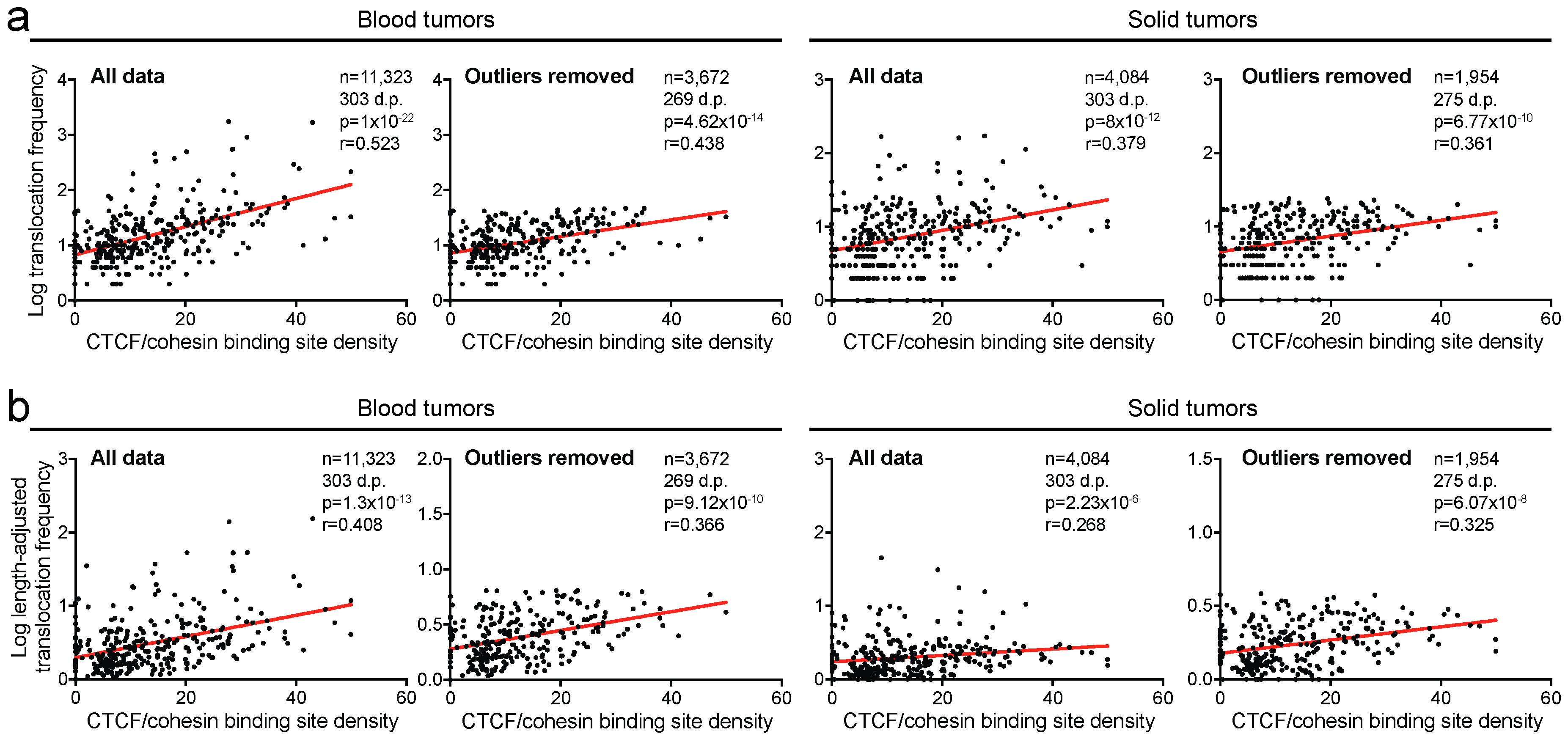
2.3. Translocation Breakpoints Preferentially Occur in Regions Rich in CTCF/Cohesin Binding Sites
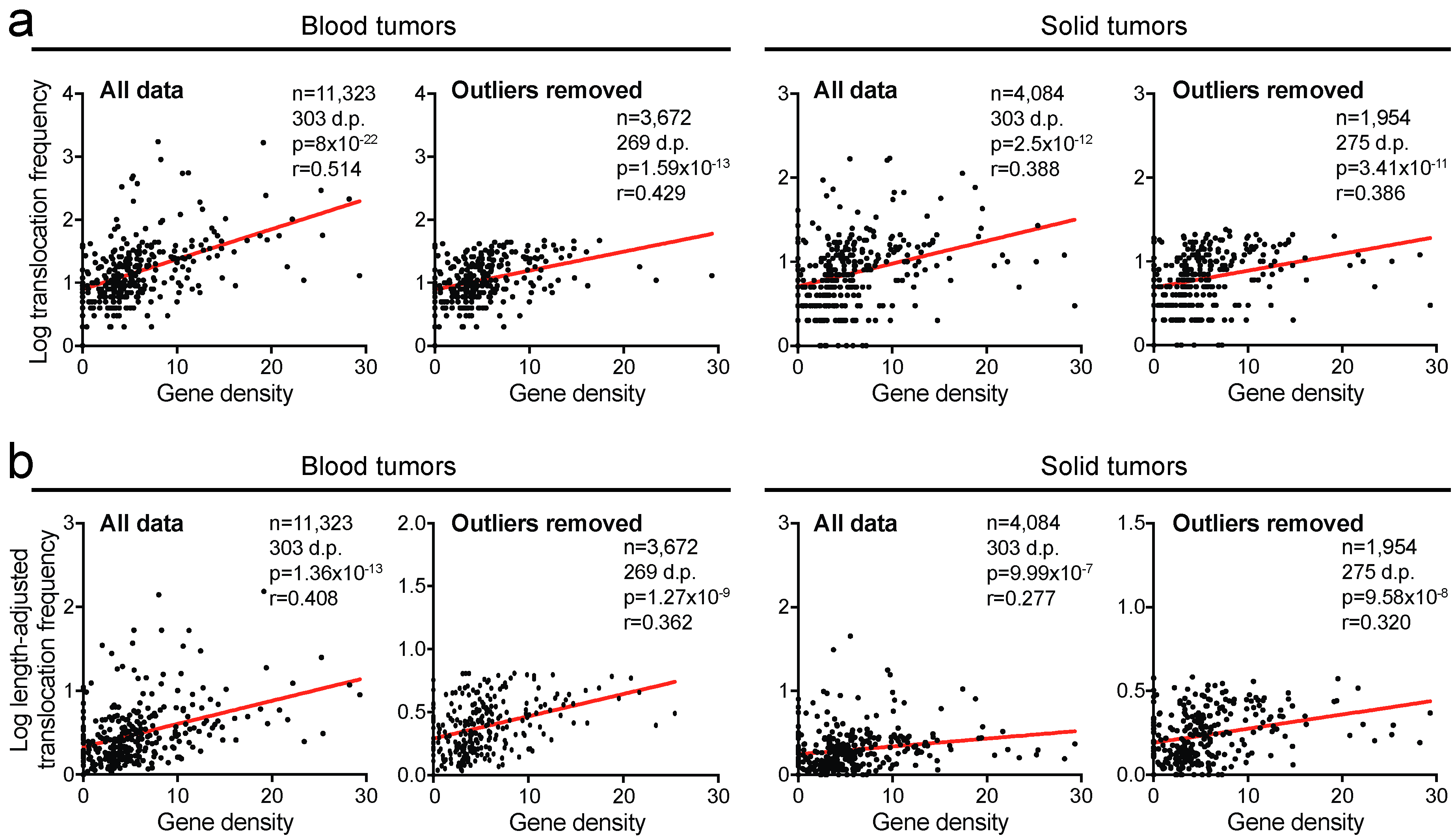
2.4. Translocation Breakpoints Preferentially Occur in Gene-Rich Regions
2.5. Chromatin Density is the Primary Predictor for Translocation Breakpoints
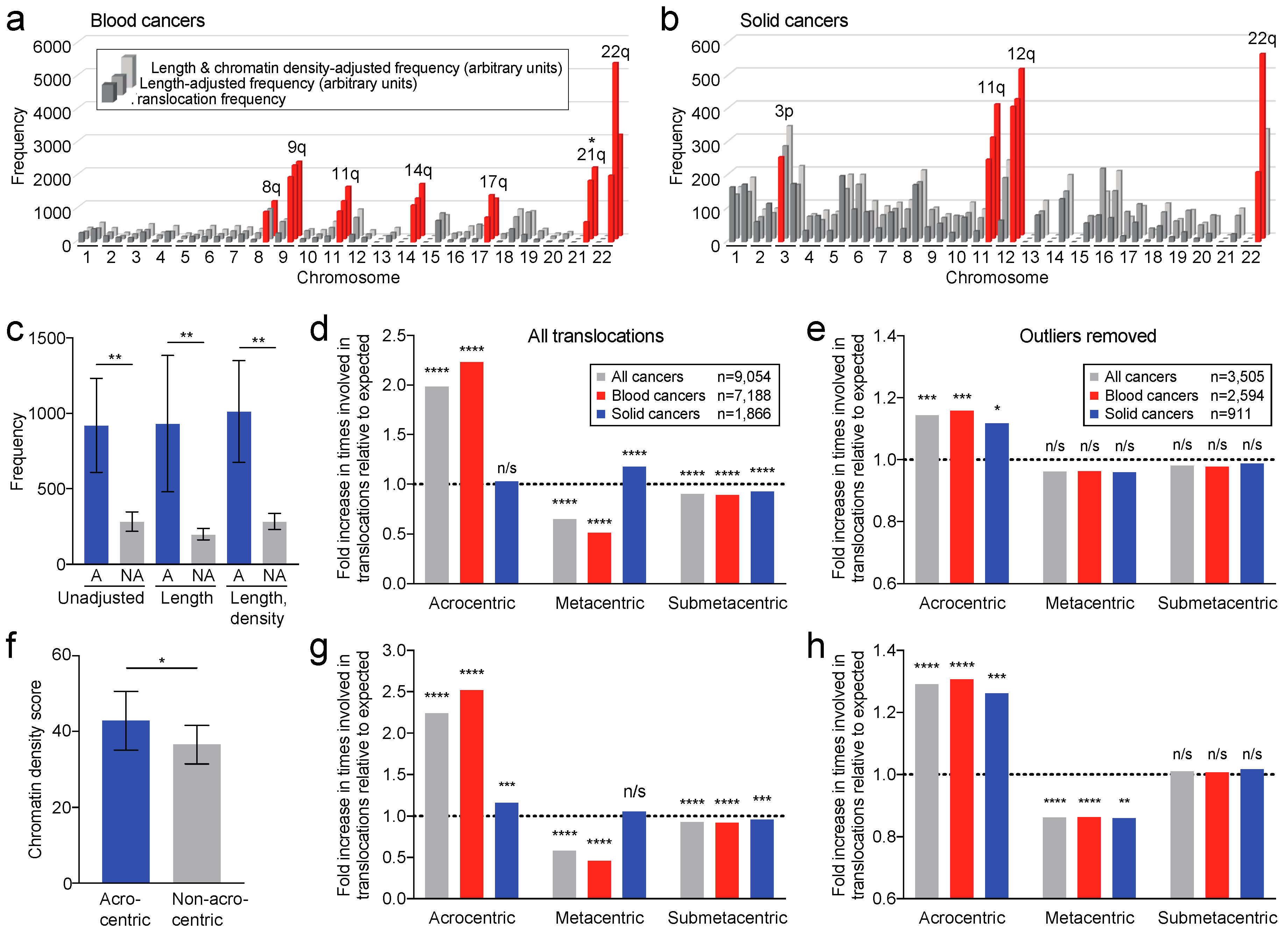
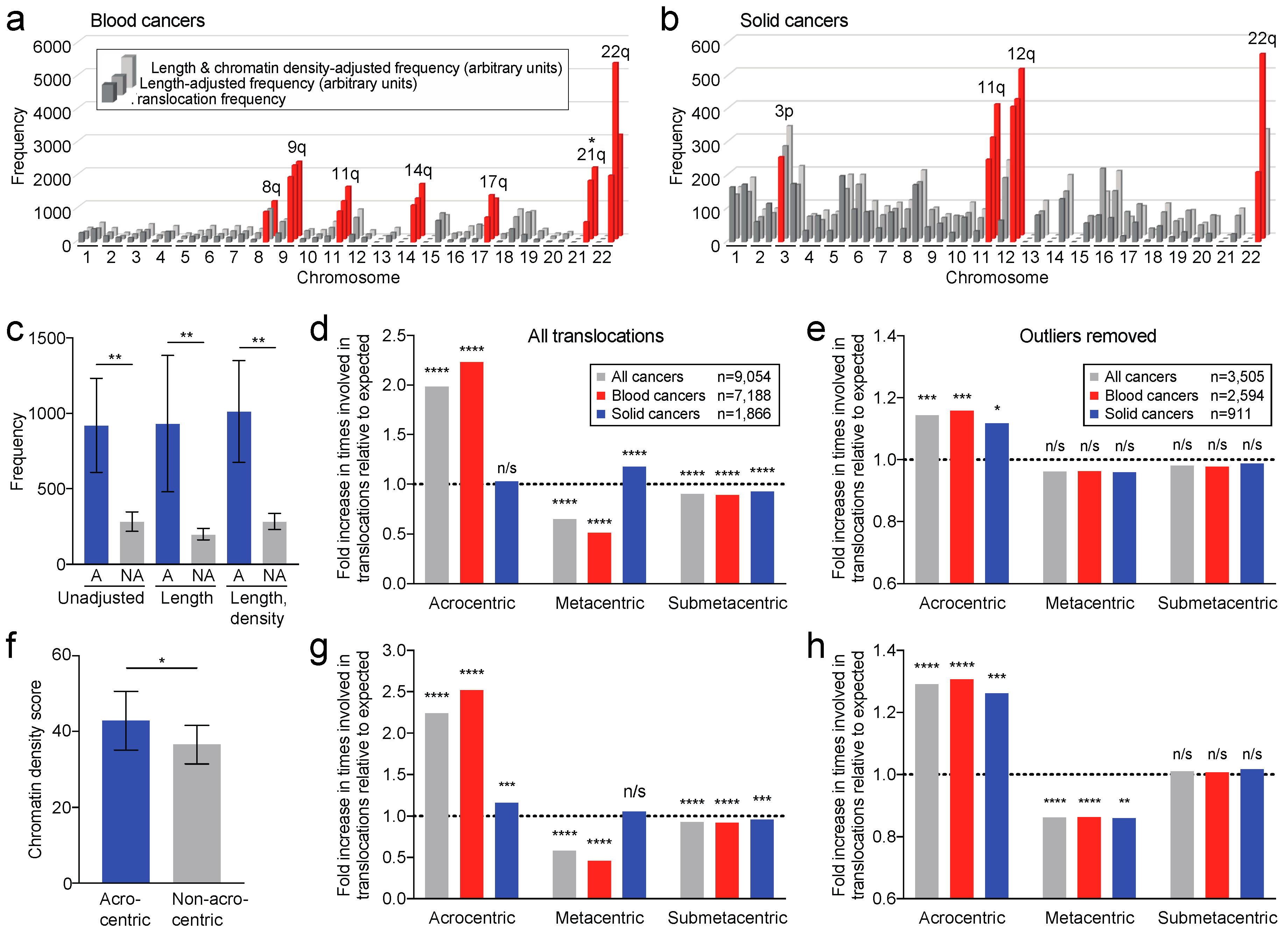
2.6. Translocation Breakpoints Preferentially Occur in Acrocentric Chromosome Arms
2.7. Translocation Breakpoints Preferentially Occur in Acrocentric Chromosomes
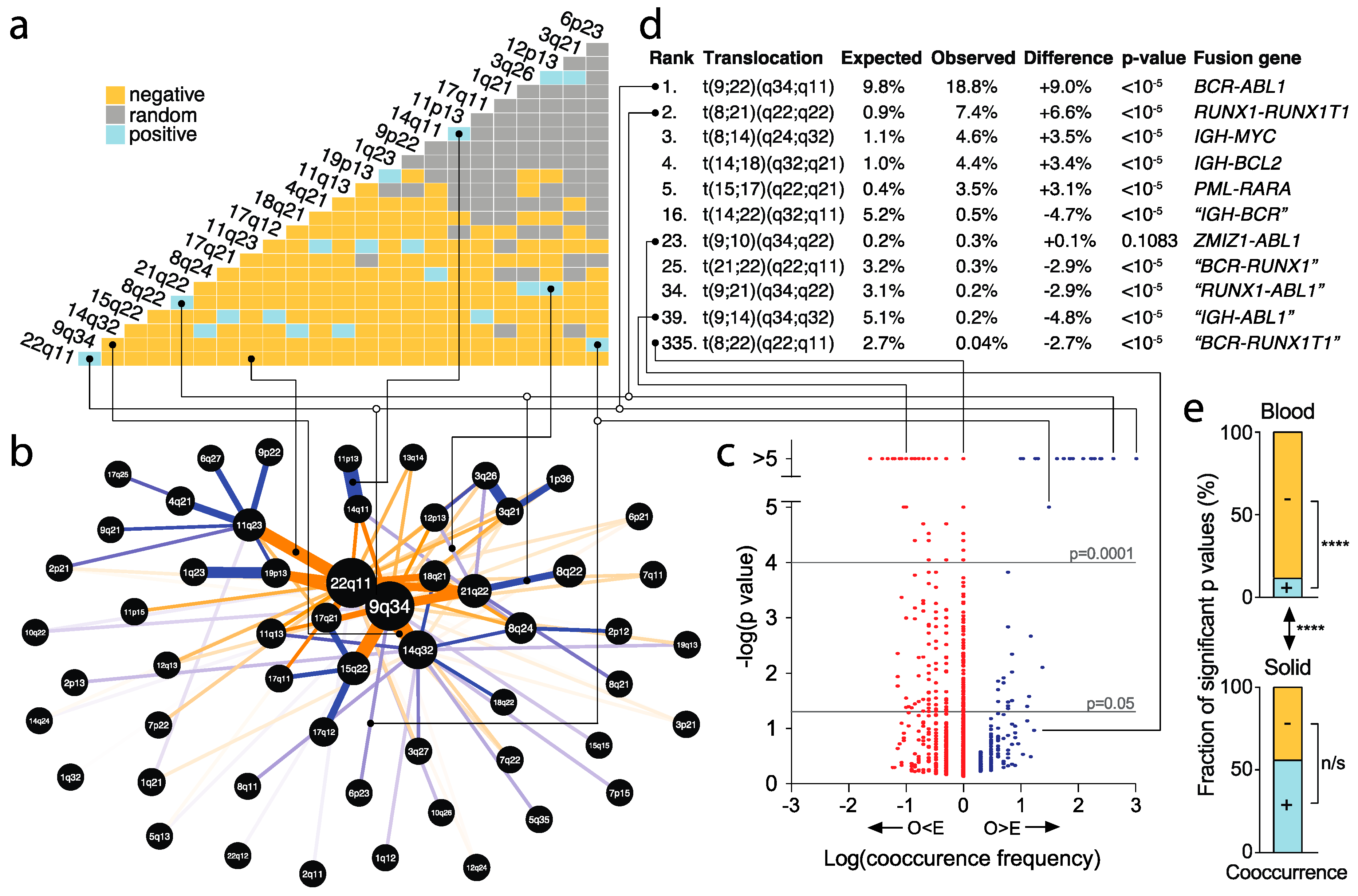
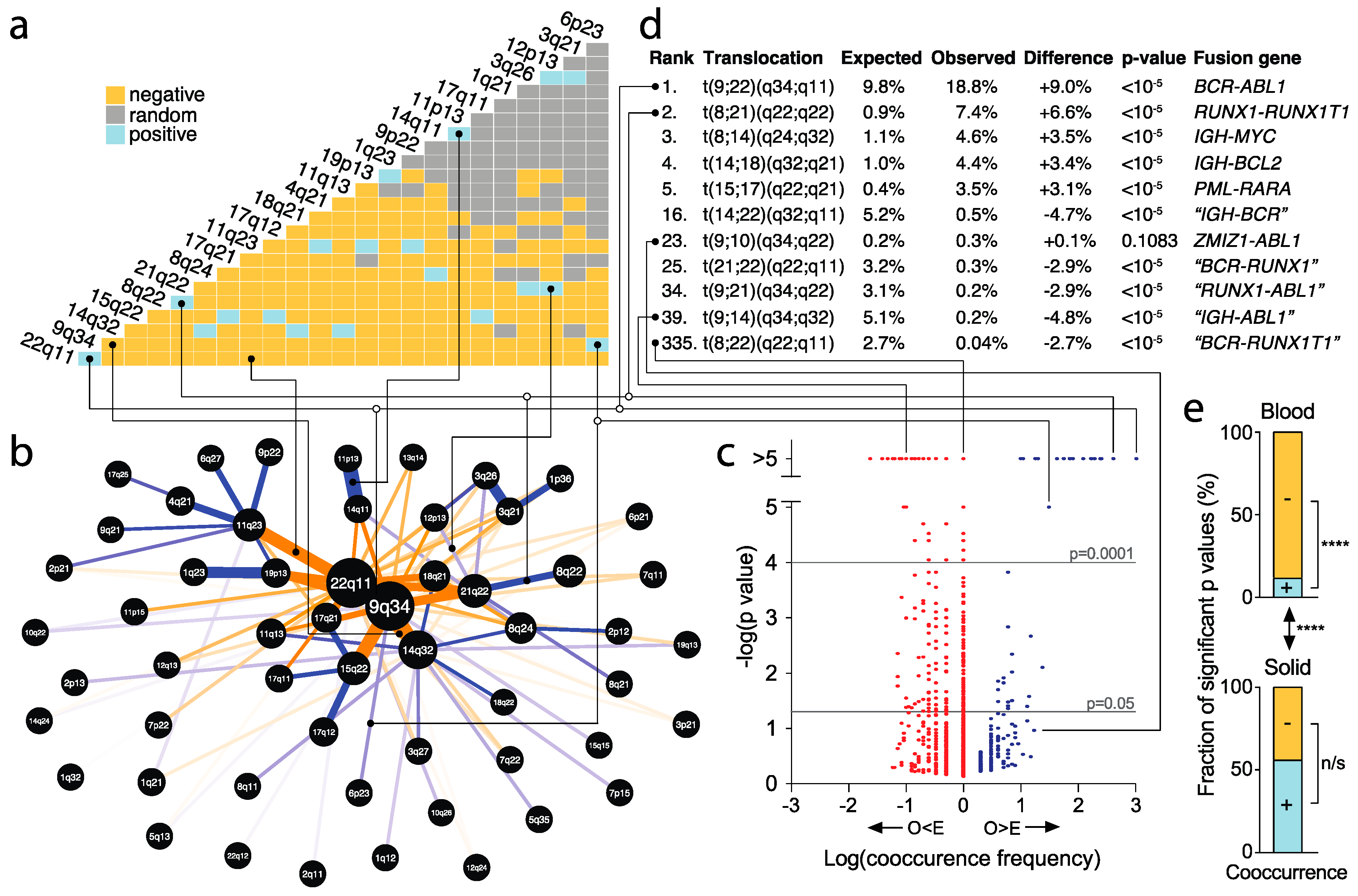
2.8. Identification of Significantly Recurrent and Underrepresented Translocations
3. Discussion
4. Materials and Methods
4.1. Karyotype Selection and Translocation Frequencies
4.2. Definitions of Parameters
4.3. Statistical Analyses
4.3.1. Data Distributions, Outliers and Linear Regression
4.3.2. Multiple Regression Analysis
4.3.3. Chromosome Type Analyses
4.3.4. Probabilistic Co-Occurrence Modeling
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Negrini, S.; Gorgoulis, V.G.; Halazonetis, T.D. Genomic instability—An evolving hallmark of cancer. Nat. Rev. Mol. Cell Biol. 2010, 11, 220–228. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, K.; Hirota, T. Chromosomal instability: A common feature and a therapeutic target of cancer. Biochim. Biophys. Acta 2016, 1866, 64–75. [Google Scholar] [CrossRef] [PubMed]
- Habermann, J.K.; Doering, J.; Hautaniemi, S.; Roblick, U.J.; Bundgen, N.K.; Nicorici, D.; Kronenwett, U.; Rathnagiriswaran, S.; Mettu, R.K.; Ma, Y.; et al. The gene expression signature of genomic instability in breast cancer is an independent predictor of clinical outcome. Int. J. Cancer 2009, 124, 1552–1564. [Google Scholar] [CrossRef] [PubMed]
- Thompson, L.L.; Jeusset, L.M.; Lepage, C.C.; McManus, K.J. Evolving therapeutic strategies to exploit chromosome instability in cancer. Cancers 2017, 9, 151. [Google Scholar] [CrossRef] [PubMed]
- Duijf, P.H.; Schultz, N.; Benezra, R. Cancer cells preferentially lose small chromosomes. Int. J. Cancer 2013, 132, 2316–2326. [Google Scholar] [CrossRef] [PubMed]
- Duijf, P.H.; Benezra, R. The cancer biology of whole-chromosome instability. Oncogene 2013, 32, 4727–4736. [Google Scholar] [CrossRef] [PubMed]
- Van Jaarsveld, R.H.; Kops, G.J. Difference makers: Chromosomal instability versus aneuploidy in cancer. Trends Cancer 2016, 2, 561–571. [Google Scholar] [CrossRef] [PubMed]
- Ghadimi, B.M.; Sackett, D.L.; Difilippantonio, M.J.; Schrock, E.; Neumann, T.; Jauho, A.; Auer, G.; Ried, T. Centrosome amplification and instability occurs exclusively in aneuploid, but not in diploid colorectal cancer cell lines, and correlates with numerical chromosomal aberrations. Genes Chromosomes Cancer 2000, 27, 183–190. [Google Scholar] [CrossRef]
- Vaidyanathan, S.; Cato, K.; Tang, L.; Pavey, S.; Haass, N.K.; Gabrielli, B.G.; Duijf, P.H. In vivo overexpression of emi1 promotes chromosome instability and tumorigenesis. Oncogene 2016, 35, 5446–5455. [Google Scholar] [CrossRef] [PubMed]
- Thangavelu, P.U.; Lin, C.Y.; Vaidyanathan, S.; Nguyen, T.H.M.; Dray, E.; Duijf, P.H.G. Overexpression of the E2F target gene CENPI promotes chromosome instability and predicts poor prognosis in estrogen receptor-positive breast cancer. Oncotarget 2017, 8, 62167–62182. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J. Oncogenic chromosomal translocations and human cancer (review). Oncol. Rep. 2013, 30, 2011–2019. [Google Scholar] [CrossRef] [PubMed]
- Nowell, P.C. Discovery of the Philadelphia chromosome: A personal perspective. J. Clin. Invest. 2007, 117, 2033–2035. [Google Scholar] [CrossRef] [PubMed]
- Chaganti, S.R.; Chen, W.; Parsa, N.; Offit, K.; Louie, D.C.; Dalla-Favera, R.; Chaganti, R.S. Involvement of BCL6 in chromosomal aberrations affecting band 3q27 in B-cell non-Hodgkin lymphoma. Genes Chromosomes Cancer 1998, 23, 323–327. [Google Scholar] [CrossRef]
- Kim, S.K.; Park, Y.K. Ewing sarcoma: A chronicle of molecular pathogenesis. Hum. Pathol. 2016, 55, 91–100. [Google Scholar] [CrossRef] [PubMed]
- Nambiar, M.; Kari, V.; Raghavan, S.C. Chromosomal translocations in cancer. Biochim. Biophys. Acta 2008, 1786, 139–152. [Google Scholar] [CrossRef] [PubMed]
- Salmon, J.M.; Bots, M.; Vidacs, E.; Stanley, K.L.; Atadja, P.; Zuber, J.; Johnstone, R.W. Combining the differentiating effect of panobinostat with the apoptotic effect of arsenic trioxide leads to significant survival benefit in a model of t(8;21) acute myeloid leukemia. Clin. Epigenetics 2015, 7, 2. [Google Scholar] [CrossRef] [PubMed]
- Guo, G.; Kang, Q.; Zhu, X.; Chen, Q.; Wang, X.; Chen, Y.; Ouyang, J.; Zhang, L.; Tan, H.; Chen, R.; et al. A long noncoding RNA critically regulates Bcr-Abl-mediated cellular transformation by acting as a competitive endogenous RNA. Oncogene 2015, 34, 1768–1779. [Google Scholar] [CrossRef] [PubMed]
- Chapman, J.R.; Taylor, M.R.; Boulton, S.J. Playing the end game: DNA double-strand break repair pathway choice. Mol. Cell 2012, 47, 497–510. [Google Scholar] [CrossRef] [PubMed]
- Janssen, A.; van der Burg, M.; Szuhai, K.; Kops, G.J.; Medema, R.H. Chromosome segregation errors as a cause of DNA damage and structural chromosome aberrations. Science 2011, 333, 1895–1898. [Google Scholar] [CrossRef] [PubMed]
- Weiler, K.S.; Wakimoto, B.T. Heterochromatin and gene expression in drosophila. Annu. Rev. Genet. 1995, 29, 577–605. [Google Scholar] [CrossRef] [PubMed]
- Schatz, D.G.; Baltimore, D. Uncovering the V(D)J recombinase. Cell 2004, 116, S103–S108. [Google Scholar] [CrossRef]
- Fugmann, S.D.; Lee, A.I.; Shockett, P.E.; Villey, I.J.; Schatz, D.G. The RAG proteins and V(D)J recombination: Complexes, ends, and transposition. Annu. Rev. Immunol. 2000, 18, 495–527. [Google Scholar] [CrossRef] [PubMed]
- Lieber, M.R. The mechanism of double-strand DNA break repair by the nonhomologous DNA end-joining pathway. Annu. Rev. Biochem. 2010, 79, 181–211. [Google Scholar] [CrossRef] [PubMed]
- Raghavan, S.C.; Kirsch, I.R.; Lieber, M.R. Analysis of the V(D)J recombination efficiency at lymphoid chromosomal translocation breakpoints. J. Biol. Chem. 2001, 276, 29126–29133. [Google Scholar] [CrossRef] [PubMed]
- Marculescu, R.; Le, T.; Simon, P.; Jaeger, U.; Nadel, B. V(D)J-mediated translocations in lymphoid neoplasms: A functional assessment of genomic instability by cryptic sites. J. Exp. Med. 2002, 195, 85–98. [Google Scholar] [CrossRef] [PubMed]
- Rooney, S.; Alt, F.W.; Lombard, D.; Whitlow, S.; Eckersdorff, M.; Fleming, J.; Fugmann, S.; Ferguson, D.O.; Schatz, D.G.; Sekiguchi, J. Defective DNA repair and increased genomic instability in artemis-deficient murine cells. J. Exp. Med. 2003, 197, 553–565. [Google Scholar] [CrossRef] [PubMed]
- Guo, C.; Yoon, H.S.; Franklin, A.; Jain, S.; Ebert, A.; Cheng, H.L.; Hansen, E.; Despo, O.; Bossen, C.; Vettermann, C.; et al. CTCF-binding elements mediate control of V(D)J recombination. Nature 2011, 477, 424–430. [Google Scholar] [CrossRef] [PubMed]
- Ong, C.T.; Corces, V.G. CTCF: An architectural protein bridging genome topology and function. Nat. Rev. Genet. 2014, 15, 234–246. [Google Scholar] [CrossRef] [PubMed]
- Parelho, V.; Hadjur, S.; Spivakov, M.; Leleu, M.; Sauer, S.; Gregson, H.C.; Jarmuz, A.; Canzonetta, C.; Webster, Z.; Nesterova, T.; et al. Cohesins functionally associate with CTCF on mammalian chromosome arms. Cell 2008, 132, 422–433. [Google Scholar] [CrossRef] [PubMed]
- Rubio, E.D.; Reiss, D.J.; Welcsh, P.L.; Disteche, C.M.; Filippova, G.N.; Baliga, N.S.; Aebersold, R.; Ranish, J.A.; Krumm, A. CTCF physically links cohesin to chromatin. Proc. Natl. Acad. Sci. USA 2008, 105, 8309–8314. [Google Scholar] [CrossRef] [PubMed]
- Wendt, K.S.; Yoshida, K.; Itoh, T.; Bando, M.; Koch, B.; Schirghuber, E.; Tsutsumi, S.; Nagae, G.; Ishihara, K.; Mishiro, T.; et al. Cohesin mediates transcriptional insulation by CCCTC-binding factor. Nature 2008, 451, 796–801. [Google Scholar] [CrossRef] [PubMed]
- Herold, M.; Bartkuhn, M.; Renkawitz, R. CTCF: Insights into insulator function during development. Development 2012, 139, 1045–1057. [Google Scholar] [CrossRef] [PubMed]
- Howarth, K.D.; Blood, K.A.; Ng, B.L.; Beavis, J.C.; Chua, Y.; Cooke, S.L.; Raby, S.; Ichimura, K.; Collins, V.P.; Carter, N.P.; et al. Array painting reveals a high frequency of balanced translocations in breast cancer cell lines that break in cancer-relevant genes. Oncogene 2008, 27, 3345–3359. [Google Scholar] [CrossRef] [PubMed]
- Bates, S.E. Classical cytogenetics: Karyotyping techniques. Methods Mol. Biol. 2011, 767, 177–190. [Google Scholar] [PubMed]
- Ozery-Flato, M.; Linhart, C.; Trakhtenbrot, L.; Izraeli, S.; Shamir, R. Large-scale analysis of chromosomal aberrations in cancer karyotypes reveals two distinct paths to aneuploidy. Genome Biol. 2011, 12, R61. [Google Scholar] [CrossRef] [PubMed]
- Motulsky, H.J.; Brown, R.E. Detecting outliers when fitting data with nonlinear regression—a new method based on robust nonlinear regression and the false discovery rate. BMC Bioinformatics 2006, 7, 123. [Google Scholar] [CrossRef] [PubMed]
- Straughen, J.K.; Sipahi, L.; Uddin, M.; Misra, D.P.; Misra, V.K. Racial differences in IGF1 methylation and birth weight. Clin. Epigenetics 2015, 7, 47. [Google Scholar] [CrossRef] [PubMed]
- Huret, J.L.; Ahmad, M.; Arsaban, M.; Bernheim, A.; Cigna, J.; Desangles, F.; Guignard, J.C.; Jacquemot-Perbal, M.C.; Labarussias, M.; Leberre, V.; et al. Atlas of genetics and cytogenetics in oncology and haematology in 2013. Nucleic Acids Res. 2013, 41, D920–D924. [Google Scholar] [CrossRef] [PubMed]
- Veech, J.A. A probabilistic model for analysing species co-occurrence. Global Ecol. Biogeogr. 2013, 22, 252–260. [Google Scholar] [CrossRef]
- Tognon, C.; Knezevich, S.R.; Huntsman, D.; Roskelley, C.D.; Melnyk, N.; Mathers, J.A.; Becker, L.; Carneiro, F.; MacPherson, N.; Horsman, D.; et al. Expression of the ETV6-NTRK3 gene fusion as a primary event in human secretory breast carcinoma. Cancer Cell 2002, 2, 367–376. [Google Scholar] [CrossRef]
- Soda, M.; Choi, Y.L.; Enomoto, M.; Takada, S.; Yamashita, Y.; Ishikawa, S.; Fujiwara, S.; Watanabe, H.; Kurashina, K.; Hatanaka, H.; et al. Identification of the transforming EML4-ALK fusion gene in non-small-cell lung cancer. Nature 2007, 448, 561–566. [Google Scholar] [CrossRef] [PubMed]
- Iarovaia, O.V.; Rubtsov, M.; Ioudinkova, E.; Tsfasman, T.; Razin, S.V.; Vassetzky, Y.S. Dynamics of double strand breaks and chromosomal translocations. Mol. Cancer 2014, 13, 249. [Google Scholar] [CrossRef] [PubMed]
- Mills, K.D.; Ferguson, D.O.; Alt, F.W. The role of DNA breaks in genomic instability and tumorigenesis. Immunol. Rev. 2003, 194, 77–95. [Google Scholar] [CrossRef] [PubMed]
- Aguilera, A. The connection between transcription and genomic instability. EMBO J. 2002, 21, 195–201. [Google Scholar] [CrossRef] [PubMed]
- Klein, I.A.; Resch, W.; Jankovic, M.; Oliveira, T.; Yamane, A.; Nakahashi, H.; Di Virgilio, M.; Bothmer, A.; Nussenzweig, A.; Robbiani, D.F.; et al. Translocation-capture sequencing reveals the extent and nature of chromosomal rearrangements in b lymphocytes. Cell 2011, 147, 95–106. [Google Scholar] [CrossRef] [PubMed]
- Chiarle, R.; Zhang, Y.; Frock, R.L.; Lewis, S.M.; Molinie, B.; Ho, Y.J.; Myers, D.R.; Choi, V.W.; Compagno, M.; Malkin, D.J.; et al. Genome-wide translocation sequencing reveals mechanisms of chromosome breaks and rearrangements in B cells. Cell 2011, 147, 107–119. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Gostissa, M.; Hildebrand, D.G.; Becker, M.S.; Boboila, C.; Chiarle, R.; Lewis, S.; Alt, F.W. The role of mechanistic factors in promoting chromosomal translocations found in lymphoid and other cancers. Adv. Immunol. 2010, 106, 93–133. [Google Scholar] [PubMed]
- Roix, J.J.; McQueen, P.G.; Munson, P.J.; Parada, L.A.; Misteli, T. Spatial proximity of translocation-prone gene loci in human lymphomas. Nat. Genet. 2003, 34, 287–291. [Google Scholar] [CrossRef] [PubMed]
- Neves, H.; Ramos, C.; da Silva, M.G.; Parreira, A.; Parreira, L. The nuclear topography of ABL, BCR, PML, and RARα genes: Evidence for gene proximity in specific phases of the cell cycle and stages of hematopoietic differentiation. Blood 1999, 93, 1197–1207. [Google Scholar] [PubMed]
- Osborne, C.S.; Chakalova, L.; Mitchell, J.A.; Horton, A.; Wood, A.L.; Bolland, D.J.; Corcoran, A.E.; Fraser, P. Myc dynamically and preferentially relocates to a transcription factory occupied by Igh. PLoS Biol. 2007, 5, e192. [Google Scholar] [CrossRef] [PubMed]
- Schvartzman, J.M.; Duijf, P.H.; Sotillo, R.; Coker, C.; Benezra, R. Mad2 is a critical mediator of the chromosome instability observed upon Rb and p53 pathway inhibition. Cancer Cell 2011, 19, 701–714. [Google Scholar] [CrossRef] [PubMed]
- Cremer, T.; Cremer, M. Chromosome territories. Cold Spring Harb Perspect Biol. 2010, 2, a003889. [Google Scholar] [CrossRef] [PubMed]
- Bolzer, A.; Kreth, G.; Solovei, I.; Koehler, D.; Saracoglu, K.; Fauth, C.; Muller, S.; Eils, R.; Cremer, C.; Speicher, M.R.; et al. Three-dimensional maps of all chromosomes in human male fibroblast nuclei and prometaphase rosettes. PLoS Biol. 2005, 3, e157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henderson, A.S.; Warburton, D.; Atwood, K.C. Location of ribosomal DNA in the human chromosome complement. Proc. Natl. Acad. Sci. USA 1972, 69, 3394–3398. [Google Scholar] [CrossRef] [PubMed]
- Van Heesch, S.; Simonis, M.; van Roosmalen, M.J.; Pillalamarri, V.; Brand, H.; Kuijk, E.W.; de Luca, K.L.; Lansu, N.; Braat, A.K.; Menelaou, A.; et al. Genomic and functional overlap between somatic and germline chromosomal rearrangements. Cell Rep. 2014, 9, 2001–2010. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petersen, M.B.; Adelsberger, P.A.; Schinzel, A.A.; Binkert, F.; Hinkel, G.K.; Antonarakis, S.E. Down syndrome due to de novo robertsonian translocation t(14q;21q): DNA polymorphism analysis suggests that the origin of the extra 21q is maternal. Am. J. Hum. Genet. 1991, 49, 529–536. [Google Scholar] [PubMed]
- De Klein, A.; van Kessel, A.G.; Grosveld, G.; Bartram, C.R.; Hagemeijer, A.; Bootsma, D.; Spurr, N.K.; Heisterkamp, N.; Groffen, J.; Stephenson, J.R. A cellular oncogene is translocated to the philadelphia chromosome in chronic myelocytic leukaemia. Nature 1982, 300, 765–767. [Google Scholar] [CrossRef] [PubMed]
- Rabbitts, T.H.; Boehm, T. Structural and functional chimerism results from chromosomal translocation in lymphoid tumors. Adv. Immunol. 1991, 50, 119–146. [Google Scholar] [PubMed]
- Borrow, J.; Goddard, A.D.; Sheer, D.; Solomon, E. Molecular analysis of acute promyelocytic leukemia breakpoint cluster region on chromosome 17. Science 1990, 249, 1577–1580. [Google Scholar] [CrossRef] [PubMed]
- Kowarz, E.; Dingermann, T.; Marschalek, R. Do non-genomically encoded fusion transcripts cause recurrent chromosomal translocations? Cancers 2012, 4, 1036–1049. [Google Scholar] [CrossRef] [PubMed]
- Mitelman, F.; Johansson, B.; Mertens, F.E. Mitelman Database of Chromosome Aberrations and Gene Fusions in Cancer. Available online: http://cgap.nci.nih.gov/Chromosomes/Mitelman (accessed on 27 October 2017).
- Thangavelu, P.U.; Krenacs, T.; Dray, E.; Duijf, P.H. In epithelial cancers, aberrant COL17A1 promoter methylation predicts its misexpression and increased invasion. Clin. Epigenetics 2016, 8, 120. [Google Scholar] [CrossRef] [PubMed]
- Vaidyanathan, S.; Thangavelu, P.U.; Duijf, P.H. Overexpression of ran gtpase components regulating nuclear export, but not mitotic spindle assembly, marks chromosome instability and poor prognosis in breast cancer. Target Oncol. 2016, 11, 677–686. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Model | Blood Cancers | Solid Cancers | ||||||
|---|---|---|---|---|---|---|---|---|
| Non-Adjusted | ||||||||
| Variable | B | Standard Error | t Value | p Value | B | Standard Error | t Value | p Value |
| Length | 0.0280 (0.0168) | 0.0046 (0.0038) | 5.97 (4.39) | 6.6 × 10−9 (1.6 × 10−5) | 0.0171 (0.0164) | 0.0043 (0.0034) | 3.96 (4.80) | 9.4 × 10−5 (2.74 × 10−6) |
| Chromatin Density Score | −0.0041 (−0.0034) | 0.0008 (0.0006) | −5.20 (−5.74) | 3.7 × 10−7 (2.57 × 10−8) | −0.0047 (−0.0044) | 0.0007 (0.0006) | −6.34 (−7.56) | 8.9 × 10−10 (6.91 × 10−13) |
| CTCF/Cohesin Density | 0.0116 (0.0060) | 0.0039 (0.0031) | 2.93 (1.97) | 0.003 (0.0504) | 1.77 × 10−5 (−0.0005) | 0.0036 (0.0029) | 0.005 (−0.19) | 0.996 (0.849) |
| Gene Density | 0.0146 (0.0108) | 0.0076 (0.0066) | 1.91 (1.62) | 0.056 (0.107) | 0.0114 (0.0066) | 0.0070 (0.0058) | 1.63 (1.14) | 0.105 (0.255) |
| Model Summary | N = 292, R2 = 0.40, adjusted R2 = 0.39, p < 0.001 (N = 259, R2 = 0.32, adjusted R2 = 0.31, p < 0.001) | N = 292, R2 = 0.24, adjusted R2 = 0.23, p < 0.001 (N = 265, R2 = 0.29, adjusted R2 = 0.28, p < 0.001) | ||||||
| Length-Adjusted | ||||||||
| Variable | B | Standard Error | t Value | p Value | B | Standard Error | t Value | p Value |
| Chromatin Density Score | −0.0033 (−0.0021) | 0.0006 (0.0003) | −5.50 (−5.79) | 8.5 × 10−8 (2.08 × 10−8) | −0.0031 (−0.0018) | 0.0004 (0.0003) | −7.11 (−6.34) | 9.4 × 10−12 (1 × 10−9) |
| CTCF/Cohesin Density | 0.0065 (0.0027) | 0.0030 (0.0018) | 2.15 (1.46) | 0.032 (0.147) | 0.0029 (0.0004) | 0.0022 (0.0014) | −1.31 (0.28) | 0.191 (0.779) |
| Gene Density | 0.0102 (0.0073) | 0.0059 (0.0039) | 1.74 (1.90) | 0.083 (0.058) | 0.0062 (0.0025) | 0.0043 (0.0027) | 1.45 (0.91) | 0.149 (0.364) |
| Model Summary | N = 292, R2 = 0.293, adjusted R2 = 0.286, p < 0.001 (N = 257, R2 = 0.302, adjusted R2 = 0.293, p < 0.001) | N = 292, R2 = 0.187, adjusted R2 = 0.179, p < 0.001 (N = 266, R2 = 0.223, adjusted R2 = 0.215, p < 0.001) | ||||||
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lin, C.-Y.; Shukla, A.; Grady, J.P.; Fink, J.L.; Dray, E.; Duijf, P.H.G. Translocation Breakpoints Preferentially Occur in Euchromatin and Acrocentric Chromosomes. Cancers 2018, 10, 13. https://doi.org/10.3390/cancers10010013
Lin C-Y, Shukla A, Grady JP, Fink JL, Dray E, Duijf PHG. Translocation Breakpoints Preferentially Occur in Euchromatin and Acrocentric Chromosomes. Cancers. 2018; 10(1):13. https://doi.org/10.3390/cancers10010013
Chicago/Turabian StyleLin, Cheng-Yu, Ankit Shukla, John P. Grady, J. Lynn Fink, Eloise Dray, and Pascal H.G. Duijf. 2018. "Translocation Breakpoints Preferentially Occur in Euchromatin and Acrocentric Chromosomes" Cancers 10, no. 1: 13. https://doi.org/10.3390/cancers10010013