Single-Cell Mechanophenotyping in Microfluidics to Evaluate Behavior of U87 Glioma Cells



Abstract
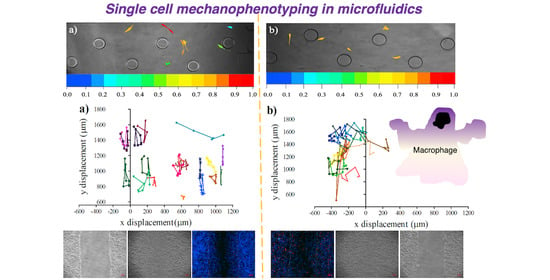
{kind=link}
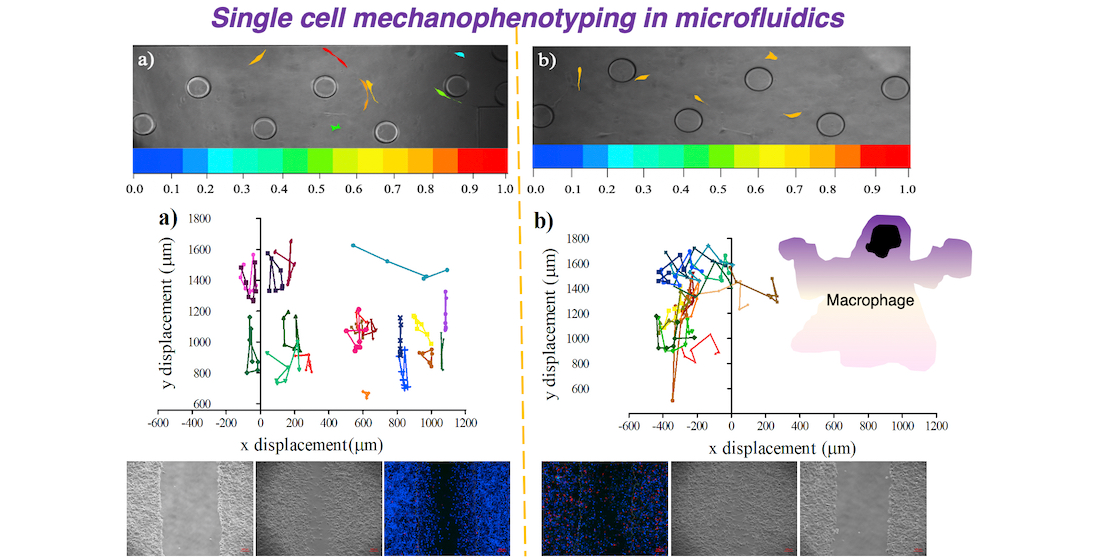
{kind=link}
{kind=link}
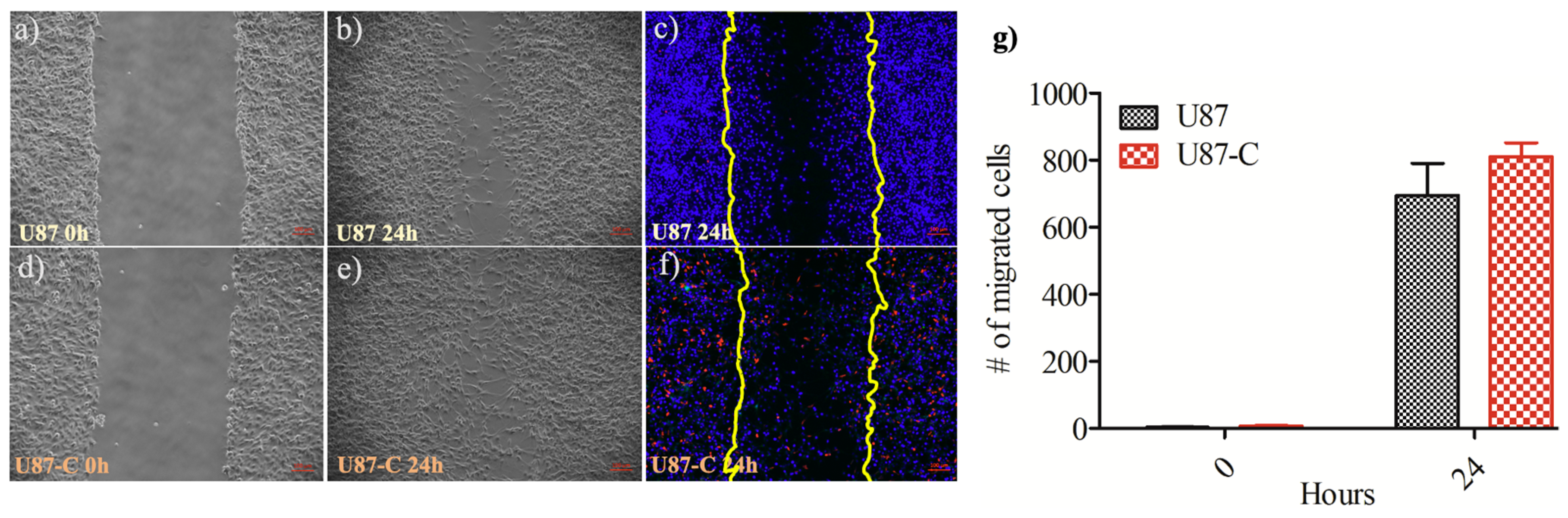{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Cell Culture
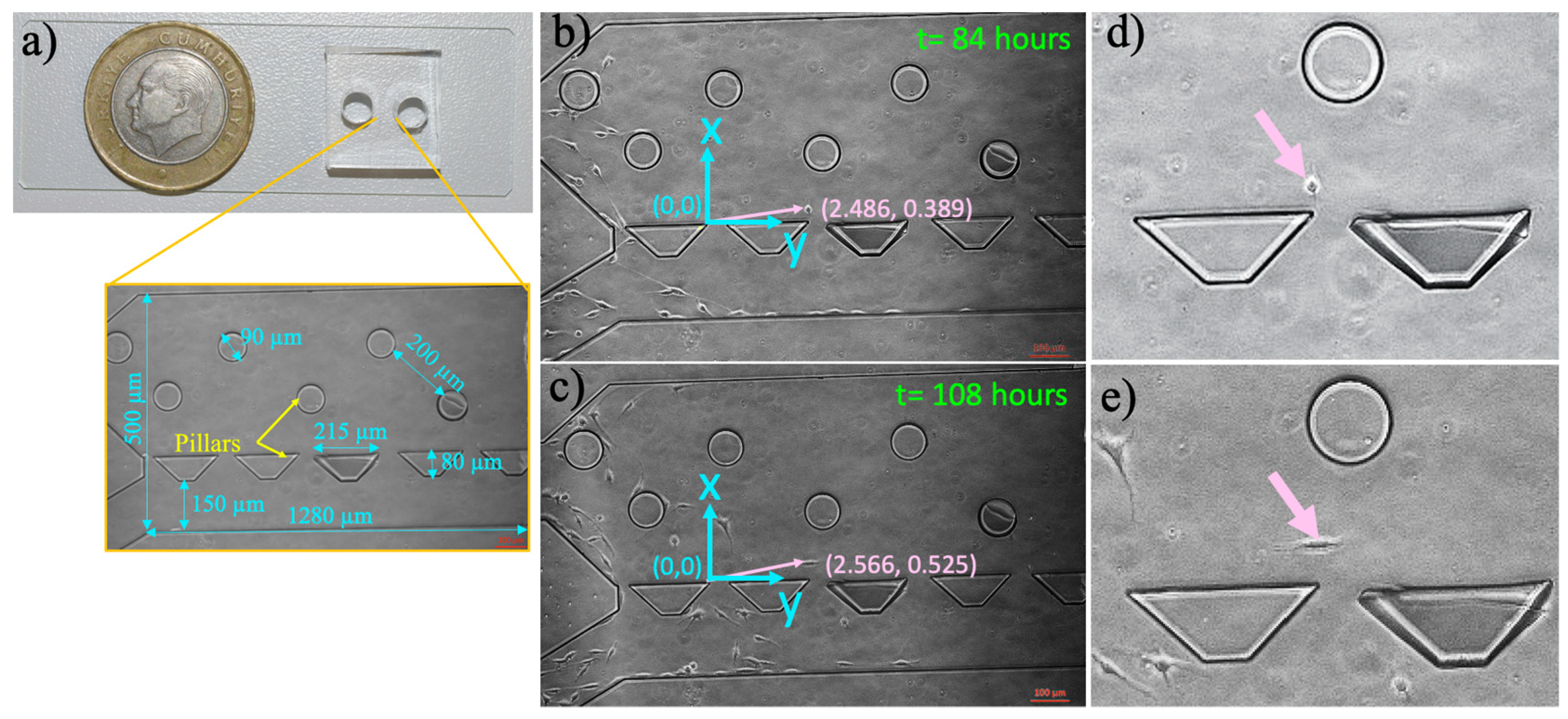
2.2. Microfluidic Chip Fabrication
2.3. Microfluidic Chip Preparation and Culturing Cells in the Microfluidic Device
2.4. Cell Growth in a 12-Well Cell Culture Plate
2.5. Cell Migration by Wound Healing in a 12-Well Cell Culture Plate
2.6. Measurement of Single-Cell Migration in the Microfluidic Device
2.7. Immunohistochemistry
3. Results
3.1. Influence of Conditioned Medium on U87 Proliferation in 12-Well Plate and Microfluidic Device
3.2. Influence of Conditional Medium on U87 Cell Migration by Wound Healing Assay
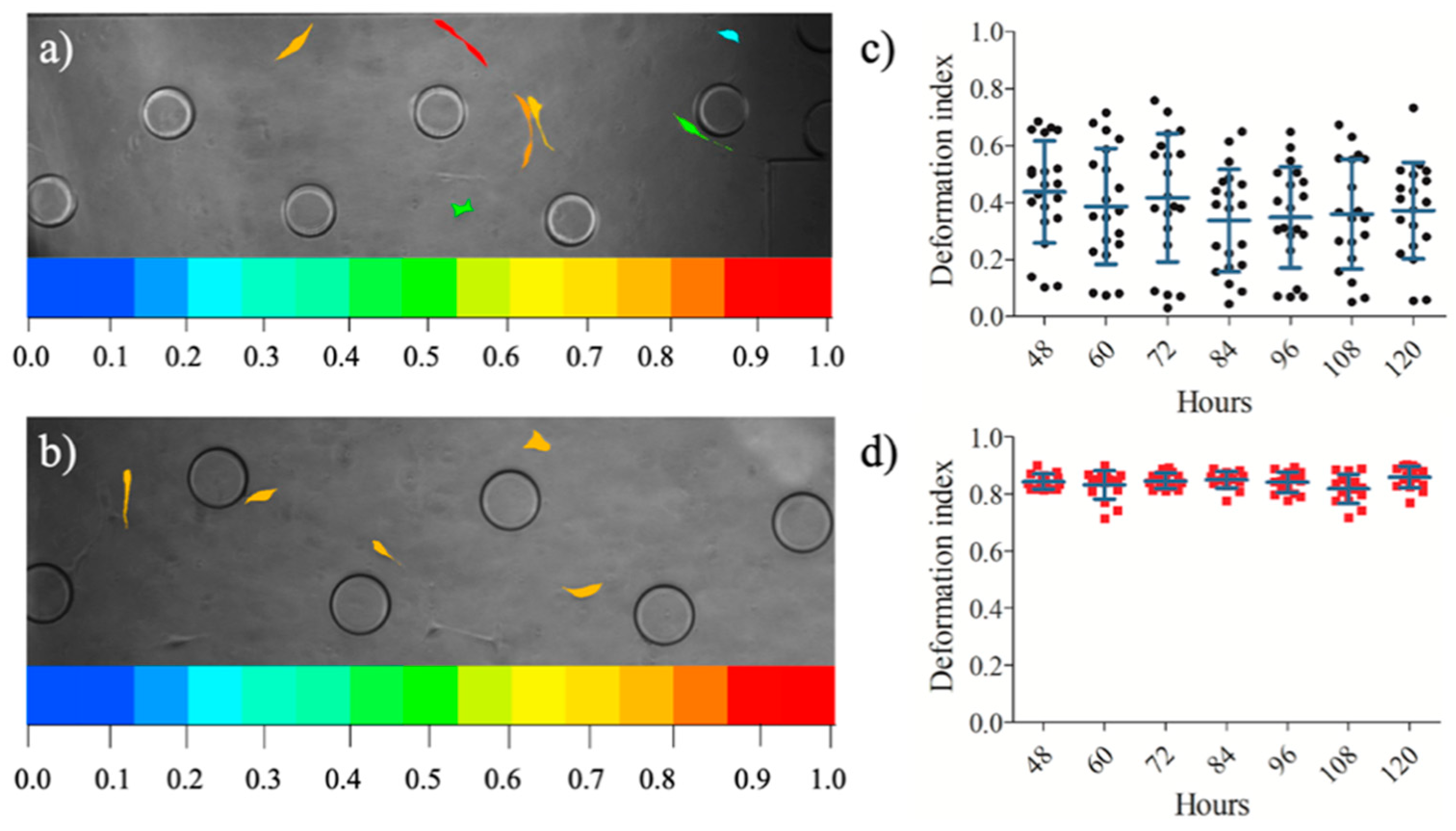
3.3. Influence of Conditional Medium on U87 Cell Migration Using a Microfluidic Device
3.4. Influence of Conditional Medium on U87 Cell Migration Using a Microfluidic Device
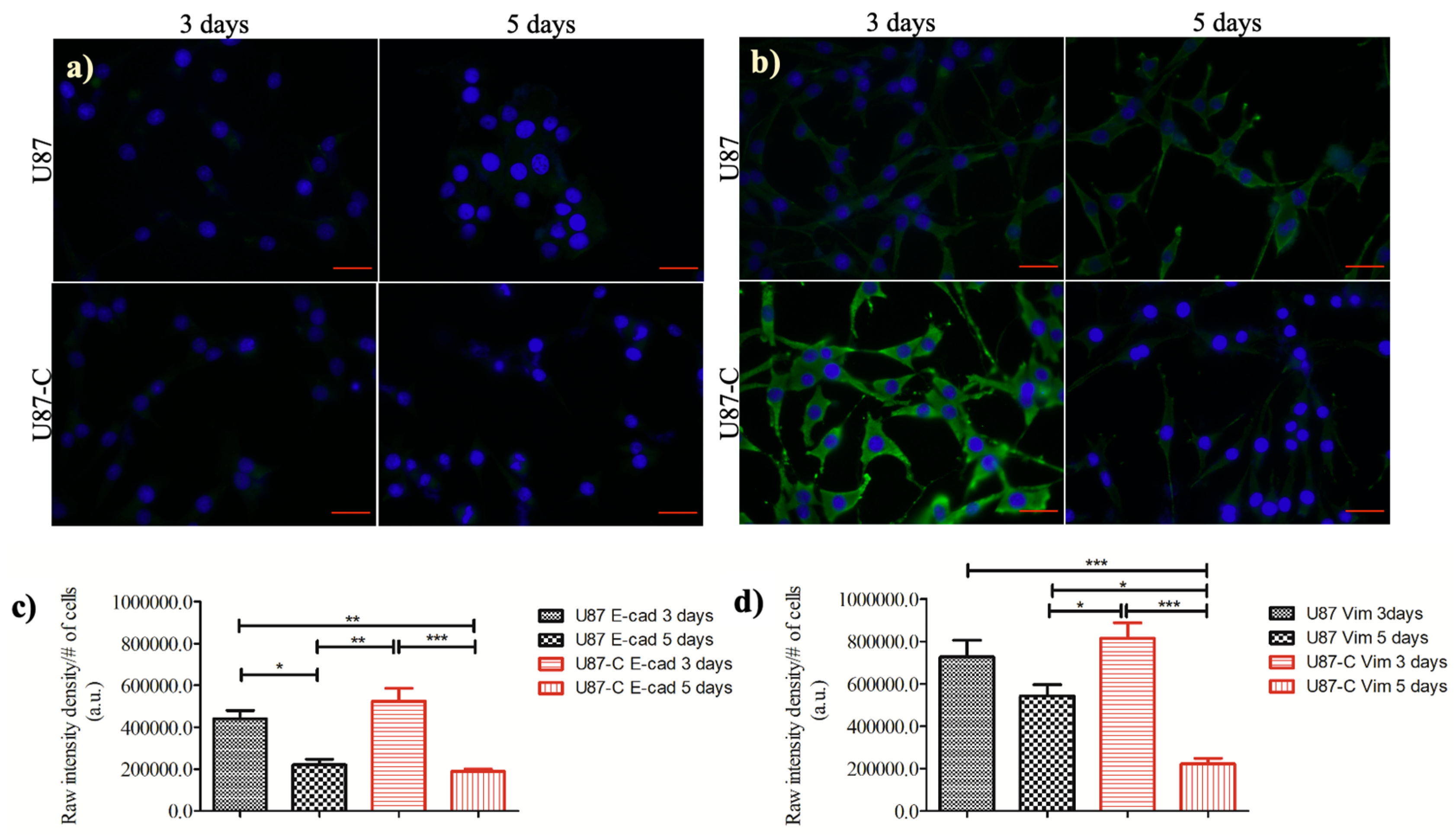
3.5. Influence of Conditional Medium on the Expression of E-cadherin and Vimentin
4. Discussion
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Ostrom, Q.T.; Gittleman, H.; Liao, P.; Rouse, C.; Chen, Y.; Dowling, J.; Wolinsky, Y.; Kruchko, C.; Barnholtz-Sloan, J. CBTRUS Statistical Report: Primary Brain and Central Nervous System Tumors Diagnosed in the United States in 2007-2011. Neuro-Oncol. 2014, 16 (Suppl. S4), iv1–iv63. [Google Scholar] [CrossRef] [PubMed]
- Komori, T. The 2016 WHO Classification of Tumours of the Central Nervous System: The Major Points of Revision. Neurol. Med.-Chir. 2017, 57, 301–311. [Google Scholar] [CrossRef]
- Holland, E.C. Glioblastoma Multiforme: The Terminator. Proc. Natl. Acad. Sci. USA 2000, 97, 6242–6244. [Google Scholar] [CrossRef] [PubMed]
- Gimple, R.C.; Kidwell, R.L.; Kim, L.J.; Sun, T.; Gromovsky, A.D.; Wu, Q.; Wolf, M.; Lv, D.; Bhargava, S.; Jiang, L.; et al. Glioma Stem Cell–Specific Superenhancer Promotes Polyunsaturated Fatty-Acid Synthesis to Support EGFR Signaling. Cancer Discov. 2019, 9, 1248–1267. [Google Scholar] [CrossRef] [PubMed]
- Schiffer, D.; Annovazzi, L.; Casalone, C.; Corona, C.; Mellai, M. Glioblastoma: Microenvironment and Niche Concept. Cancers 2018, 11, 5. [Google Scholar] [CrossRef] [PubMed]
- Takashima, Y.; Kawaguchi, A.; Yamanaka, R. Promising Prognosis Marker Candidates on the Status of Epithelial–Mesenchymal Transition and Glioma Stem Cells in Glioblastoma. Cells 2019, 8, 1312. [Google Scholar] [CrossRef]
- Phillips, H.S.; Kharbanda, S.; Chen, R.; Forrest, W.F.; Soriano, R.H.; Wu, T.D.; Misra, A.; Nigro, J.M.; Colman, H.; Soroceanu, L.; et al. Molecular Subclasses of High-Grade Glioma Predict Prognosis, Delineate a Pattern of Disease Progression, and Resemble Stages in Neurogenesis. Cancer Cell 2006, 9, 157–173. [Google Scholar] [CrossRef]
- Guardia, G.D.A.; Correa, B.R.; Araujo, P.R.; Qiao, M.; Burns, S.; Penalva, L.O.F.; Galante, P.A.F. Proneural and Mesenchymal Glioma Stem Cells Display Major Differences in Splicing and LncRNA Profiles. npj Genom. Med. 2020, 5. [Google Scholar] [CrossRef]
- Scherer, H.J. A Critical Review: The Pathology of Cerebral Gliomas. J. Neurol. Neurosur. Psychiatry 1940, 3, 147–177. [Google Scholar] [CrossRef]
- Kozminsky, M.; Sohn, L.L. The Promise of Single-Cell Mechanophenotyping for Clinical Applications. Biomicrofluidics 2020, 14, 031301. [Google Scholar] [CrossRef]
- Wu, P.-H.; Aroush, D.R.-B.; Asnacios, A.; Chen, W.-C.; Dokukin, M.E.; Doss, B.L.; Durand-Smet, P.; Ekpenyong, A.; Guck, J.; Guz, N.V.; et al. A Comparison of Methods to Assess Cell Mechanical Properties. Nat. Methods 2018, 15, 491–498. [Google Scholar] [CrossRef] [PubMed]
- Wirtz, D.; Konstantopoulos, K.; Searson, P.C. The Physics of Cancer: The Role of Physical Interactions and Mechanical Forces in Metastasis. Nat. Rev. Cancer 2011, 11, 512–522. [Google Scholar] [CrossRef] [PubMed]
- Guck, J.; Schinkinger, S.; Lincoln, B.; Wottawah, F.; Ebert, S.; Romeyke, M.; Lenz, D.; Erickson, H.M.; Ananthakrishnan, R.; Mitchell, D.; et al. Optical Deformability as an Inherent Cell Marker for Testing Malignant Transformation and Metastatic Competence. Biophys. J. 2005, 88, 3689–3698. [Google Scholar] [CrossRef] [PubMed]
- Tse, H.T.; Gosset, R.D.; Moon, Y.S.; Masaeli, M.; Sohsman, M.; Ying, Y.; Mislick, K.; Adams, P.; Rao, J.; Di Carlo, D. Quantitative Diagnosis of Malignant Pleural Effusions by Single-Cell Mechanophenotyping. Sci. Transit. Med. 2013, 5, 212ra163. [Google Scholar] [CrossRef] [PubMed]
- Urbanska, M.; Muñoz, H.E.; Bagnall, J.S.; Otto, O.; Manalis, S.R.; Carlo, D.D.; Guck, J. A Comparison of Microfluidic Methods for High-Throughput Cell Deformability Measurements. Nat. Methods 2020, 17, 587–593. [Google Scholar] [CrossRef]
- Eluru, G.; Srinivasan, R.; Gorthi, S.S. Deformability Measurement of Single-Cells at High-Throughput With Imaging Flow Cytometry. J. Lightwave. Technol. 2015, 33, 3475–3480. [Google Scholar] [CrossRef]
- Krutzik, P.O.; Nolan, G.P. Fluorescent Cell Barcoding in Flow Cytometry Allows High-Throughput Drug Screening and Signaling Profiling. Nat. Methods 2006, 3, 361–368. [Google Scholar] [CrossRef]
- Pelling, A.E.; Veraitch, F.S.; Chu, C.P.-K.; Mason, C.; Horton, M.A. Mechanical Dynamics of Single Cells during Early Apoptosis. Cell Motil. Cytoskeleton. 2009, 66, 409–422. [Google Scholar] [CrossRef]
- Radmacher, M. Studying the Mechanics of Cellular Processes by Atomic Force Microscopy. Methods Cell Biol. 2007, 347–372. [Google Scholar] [CrossRef]
- Puig-De-Morales, M.; Grabulosa, M.; Alcaraz, J.; Mullol, J.; Maksym, G.N.; Fredberg, J.J.; Navajas, D. Measurement of Cell Microrheology by Magnetic Twisting Cytometry with Frequency Domain Demodulation. J. Appl. Physiol. 2001, 91, 1152–1159. [Google Scholar] [CrossRef]
- Thoumine, O.; Ott, A.; Cardoso, O.; Meister, J.-J. Microplates: A New Tool for Manipulation and Mechanical Perturbation of Individual Cells. J. Biochem. Bioph. Meth. 1999, 39, 47–62. [Google Scholar] [CrossRef]
- Guck, J.; Ananthakrishnan, R.; Mahmood, H.; Moon, T.J.; Cunningham, C.C.; Käs, J. The Optical Stretcher: A Novel Laser Tool to Micromanipulate Cells. Biophys. J. 2001, 81, 767–784. [Google Scholar] [CrossRef]
- Huang, N.-T.; Zhang, H.-L.; Chung, M.-T.; Seo, J.H.; Kurabayashi, K. Recent Advancements in Optofluidics-Based Single-Cell Analysis: Optical on-Chip Cellular Manipulation, Treatment, and Property Detection. Lab. Chip. 2014, 14, 1230–1245. [Google Scholar] [CrossRef] [PubMed]
- Musielak, M. Red Blood Cell-Deformability Measurement: Review of Techniques. Clin. Hemorheol. Microcirc. 2009, 42, 47–64. [Google Scholar] [CrossRef] [PubMed]
- Artmann, G. Microscopic Photometric Quantification of Stiffness and Relaxation Time of Red Blood Cells in a Flow Chamber. Biorheology 1995, 32, 553–570. [Google Scholar] [CrossRef] [PubMed]
- Scott, M.D.; Matthews, K.; Ma, H. Assessing the Vascular Deformability of Erythrocytes and Leukocytes: From Micropipettes to Microfluidics. In Current and Future Aspects of Nanomedicine; IntechOpen: London, UK, 2019. [Google Scholar] [CrossRef]
- Zhao, R.; Sider, K.L.; Simmons, C.A. Measurement of Layer-Specific Mechanical Properties in Multilayered Biomaterials by Micropipette Aspiration. Acta Biomater. 2011, 7, 1220–1227. [Google Scholar] [CrossRef]
- Lee, L.M.; Lee, J.W.; Chase, D.; Gebrezgiabhier, D.; Liu, A.P. Development of an Advanced Microfluidic Micropipette Aspiration Device for Single Cell Mechanics Studies. Biomicrofluidics 2016, 10, 054105. [Google Scholar] [CrossRef]
- Tee, S.-Y.; Bausch, A.R.; Janmey, P.A. The Mechanical Cell. Curr. Biol. 2009, 19. [Google Scholar] [CrossRef]
- Leggett, S.E.; Patel, M.; Valentin, T.M.; Gamboa, L.; Khoo, A.S.; Williams, E.K.; Franck, C.; Wong, I.Y. Mechanophenotyping of 3D Multicellular Clusters Using Displacement Arrays of Rendered Tractions. Proc. Natl. Acad. Sci. USA 2020, 117, 5655–5663. [Google Scholar] [CrossRef]
- Shah, M.K.; Garcia-Pak, I.H.; Darling, E.M. Influence of Inherent Mechanophenotype on Competitive Cellular Adherence. Ann. Biomed. Eng. 2017, 45, 2036–2047. [Google Scholar] [CrossRef]
- Diao, W.; Tong, X.; Yang, C.; Zhang, F.; Bao, C.; Chen, H.; Liu, L.; Li, M.; Ye, F.; Fan, Q.; et al. Behaviors of Glioblastoma Cells in in Vitro Microenvironments. Sci. Rep. 2019, 9. [Google Scholar] [CrossRef] [PubMed]
- Andolfi, L.; Bourkoula, E.; Migliorini, E.; Palma, A.; Pucer, A.; Skrap, M.; Scoles, G.; Beltrami, A.P.; Cesselli, D.; Lazzarino, M. Investigation of Adhesion and Mechanical Properties of Human Glioma Cells by Single Cell Force Spectroscopy and Atomic Force Microscopy. PLoS ONE 2014, 9, e112582. [Google Scholar] [CrossRef] [PubMed]
- Kaufman, L.; Brangwynne, C.; Kasza, K.; Filippidi, E.; Gordon, V.; Deisboeck, T.; Weitz, D. Glioma Expansion in Collagen I Matrices: Analyzing Collagen Concentration-Dependent Growth and Motility Patterns. Biophys. J. 2005, 89, 635–650. [Google Scholar] [CrossRef] [PubMed]
- Ulrich, T.A.; Pardo, E.M.D.J.; Kumar, S. The Mechanical Rigidity of the Extracellular Matrix Regulates the Structure, Motility, and Proliferation of Glioma Cells. Cancer Res. 2009, 69, 4167–4174. [Google Scholar] [CrossRef] [PubMed]
- Memmel, S.; Sukhorukov, V.L.; Höring, M.; Westerling, K.; Fiedler, V.; Katzer, A.; Krohne, G.; Flentje, M.; Djuzenova, C.S. Cell Surface Area and Membrane Folding in Glioblastoma Cell Lines Differing in PTEN and p53 Status. PLoS ONE 2014, 9, e87052. [Google Scholar] [CrossRef]
- Texier, B.D.; Laurent, P.; Stoukatch, S.; Dorbolo, S. Wicking through a confined micropillary array. Microfluid. Nanofluid. 2016, 20, 53. [Google Scholar] [CrossRef]
- Elitas, M.; Sadeghi, S.; Karamahmutoglu, H.; Gozuacik, D.; Turhal, N.S. Microfabricated platforms to quantitatively investigate cellular behavior under the influence of chemical gradients. Biomed. Phys. Eng. Express 2017, 3, 03023. [Google Scholar] [CrossRef]
- Nawas, A.A.; Ubanska, M.; Herbig, M.; Nötzel, M.; Kräter, M.; Rosendahk, P.; Herold, C.; Toepfner, N.; Kubánková, M.; Goswami, R.; et al. Intelligent Image-based Deformation Assisted Cell Sorting with Molecular Specificity. Nat. Methods 2020, 17, 595–599. [Google Scholar] [CrossRef]
- Elitas, M.; Sengul, E. Quantifying Heterogeneity According to Deformation of the U937 Monocytes and U937-Differentiated Macrophages Using 3D Carbon Dielectrophoresis in Microfluidics. Micromachines 2020, 11, 576. [Google Scholar] [CrossRef]
- Shankar, J.; Nabi, I.R. Actin Cytoskeleton Regulation of Epithelial Mesenchymal Transition in Metastatic Cancer Cells. PLoS ONE 2015, 10, e0119954. [Google Scholar] [CrossRef]
- Paguirigan, A.L.; Beebe, D.J. Microfluidics Meet Cell Biology: Bridging the Gap by Validation and Application of Microscale Techniques for Cell Biological Assays. BioEssays 2008, 30, 811–821. [Google Scholar] [CrossRef] [PubMed]
- Duncombe, T.A.; Tentori, A.M.; Herr, A.E. Microfluidics: Reframing Biological Enquiry. Nat. Rev. Mol. Cell Biol. 2015, 16, 5540567. [Google Scholar] [CrossRef] [PubMed]
- Chiu, D.T.; deMello, A.J.; Di Carlo, D.; Doyle, P.S.; Hansen, C.; Maceiczyk, R.M.; Wooton, R.C.R. Small but Perfectly Formed? Successes, Challenges, and Opportunities for Microfluidics in the Chemical and Biological Sciences. Chem 2017, 2, 201–223. [Google Scholar] [CrossRef]
- Bangasser, B.L.; Shamsan, G.A.; Chan, C.E.; Opoku, J.N.; Tüzel, E.; Schlichtmann, B.W.; Kasim, J.A.; Fuller, B.J.; McCullough, B.R.; Rosenfeld, S.S.; et al. Shifting the Optimal Stiffness for Cell Migration. Nat. Commun. 2017, 8, 15313. [Google Scholar] [CrossRef] [PubMed]
- Koh, I.; Cha, J.; Park, J.; Choi, J.; Kang, S.-G.; Kim, P. The Mode and Dynamics of Glioblastoma Cell Invasion into a Decellularized Tissue-Derived Extracellular Matrix-Based Three-Dimensional Tumor Model. Sci. Rep. 2018, 8, 4608. [Google Scholar] [CrossRef] [PubMed]
- Pogoda, K.; Bucki, R.; Byfield, F.J.; Cruz, K.; Lee, T.; Marcinkiewicz, C.; Janmey, P.A. Soft Substrates Containing Hyaluronan Mimic the Effects of Increased Stiffness on Morphology, Motility, and Proliferation of Glioma Cells. Biomacromolecules 2017, 18, 3040–3051. [Google Scholar] [CrossRef] [PubMed]
- Manini, L.; Caponnetto, F.; Bartolini, A.; Ius, T.; Mariuzzi, L.; Di Loreto, C.; Beltrami, A.P.; Cesselli, D. Role of Microenvironment in Glioma Invasivness: What We Learned from In Vitro Models. Int. J. Mol. Sci. 2018, 19, 147. [Google Scholar] [CrossRef]
- Fayzullin, A.; Sandberg, C.J.; Spreadbury, M.; Saberniak, B.M.; Grieg, Z.; Skaga, E.; Langmoen, I.A.; Vik-Mo, E.O. Phenotypic and Expressional Heterogeneity in the Invasive Glioma Cells. Transl. Oncol. 2019, 12, 122–133. [Google Scholar] [CrossRef]
- Parker, J.J.; Canoll, P.; Niswander, L.; Kleinschmidt-DeMasters, B.K.; Foshay, K.; Waziri, A. Intratumoral Heterogeneity of Endogenous Tumor Cell Invasive Behavior in Human Glioblastoma. Sci. Rep. 2018, 8, 18002. [Google Scholar] [CrossRef]








© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sengul, E.; Elitas, M. Single-Cell Mechanophenotyping in Microfluidics to Evaluate Behavior of U87 Glioma Cells. Micromachines 2020, 11, 845. https://doi.org/10.3390/mi11090845
Sengul E, Elitas M. Single-Cell Mechanophenotyping in Microfluidics to Evaluate Behavior of U87 Glioma Cells. Micromachines. 2020; 11(9):845. https://doi.org/10.3390/mi11090845
Chicago/Turabian StyleSengul, Esra, and Meltem Elitas. 2020. "Single-Cell Mechanophenotyping in Microfluidics to Evaluate Behavior of U87 Glioma Cells" Micromachines 11, no. 9: 845. https://doi.org/10.3390/mi11090845
APA StyleSengul, E., & Elitas, M. (2020). Single-Cell Mechanophenotyping in Microfluidics to Evaluate Behavior of U87 Glioma Cells. Micromachines, 11(9), 845. https://doi.org/10.3390/mi11090845