1. Introduction
Malignant diseases are a major health concern worldwide [
1] being the leading cause of death in most US states [
2]. Although various treatment options including surgery, radiotherapy, chemotherapy, immunotherapy, and targeted therapeutics have been established [
3], results in terms of progression-free and overall survival in several cancer types are still unsatisfying [
4]. The most common limitations to currently-available approaches are severe adverse reactions and the development of multidrug resistance [
5,
6]. Therefore, there is a need for the development of new agents with novel mechanisms of action for cancer treatment [
4]. Compounds isolated from different natural sources are promising candidates for the development of novel anticancer drugs, as shown previously [
7]. Recently, naturally-occurring cyclic depsipeptides, which consist of hydroxyl- and amino acids linked by amide and ester bonds became of interest. They are secondary metabolites of bacteria, fungi and plants, or originate from algae, cyanobacteria, or sponges of the marine environment [
8]. These compounds are known to exert a broad spectrum of biological effects, such as immunosuppressive, antibiotic, antifungal, as well as anti-inflammatory activities. Importantly, they were also shown to exhibit anticancer effects in different tumor models [
9,
10,
11]. Recently, in vitro anticancer activity of the cyclodepsipeptides enniatins (ENNs) [
12,
13] and beauvericin (BEA) [
14,
15,
16,
17] were reported. In the case of ENNs, we showed high tumor cell specificity indicated by clearly-enhanced cytotoxicity against malignant, as compared to non-malignant, cells [
12]. In addition, in vivo synergism of enniatin B with sorafenib, a clinically-approved tyrosine kinase inhibitor was observed in a cervical cancer model [
18]. Similar anticancer effects were discussed for the structurally-related beauvericin. In the present pilot study we evaluated the cytotoxicity of beauvericin in normal versus malignant cell lines and assessed the in vivo anticancer activity of beauvericin to estimate its therapeutic potential.
3. Discussion
This study addressed in vivo anticancer efficacy and the therapeutic window of beauvericin. First, conforming to results from the structurally related enniatins [
12] we found that beauvericin exerts modestly stronger cytotoxic effects in some malignant versus non-malignant cell types in vitro. The molecular mechanisms underlying these different sensitivities of cancer cell models are not fully understood but might include ABC-transporter-mediated drug efflux mechanisms or altered activation of cell survival pathways [
23,
24]. In addition, when seeding non-malignant fibroblasts or keratinocytes, but also cancer cells at higher density to mimic the tissue situation, beauvericin cytotoxicity markedly dropped in non-malignant cell types and the differences to cancer cells became more distinct. This selectivity is important for potential in vivo applications where cell death should be triggered in cancer cells while leaving non-malignant cells and tissues unaffected. In addition, our data revealed higher cytotoxic potency of beauvericin in colon carcinoma SW-620 cells from a metastatic lesion compared to the moderately dedifferentiated primary tumor cell line SW-480 of the same patient [
20]. This is in agreement with migration inhibition by sublethal beauvericin concentrations in metastatic cancer cells (PC-3M, prostate cancer; MDA-MB-231, breast cancer) [
15]. Together this data indicates that beauvericin might target especially dedifferentiated and invasive cancer types.
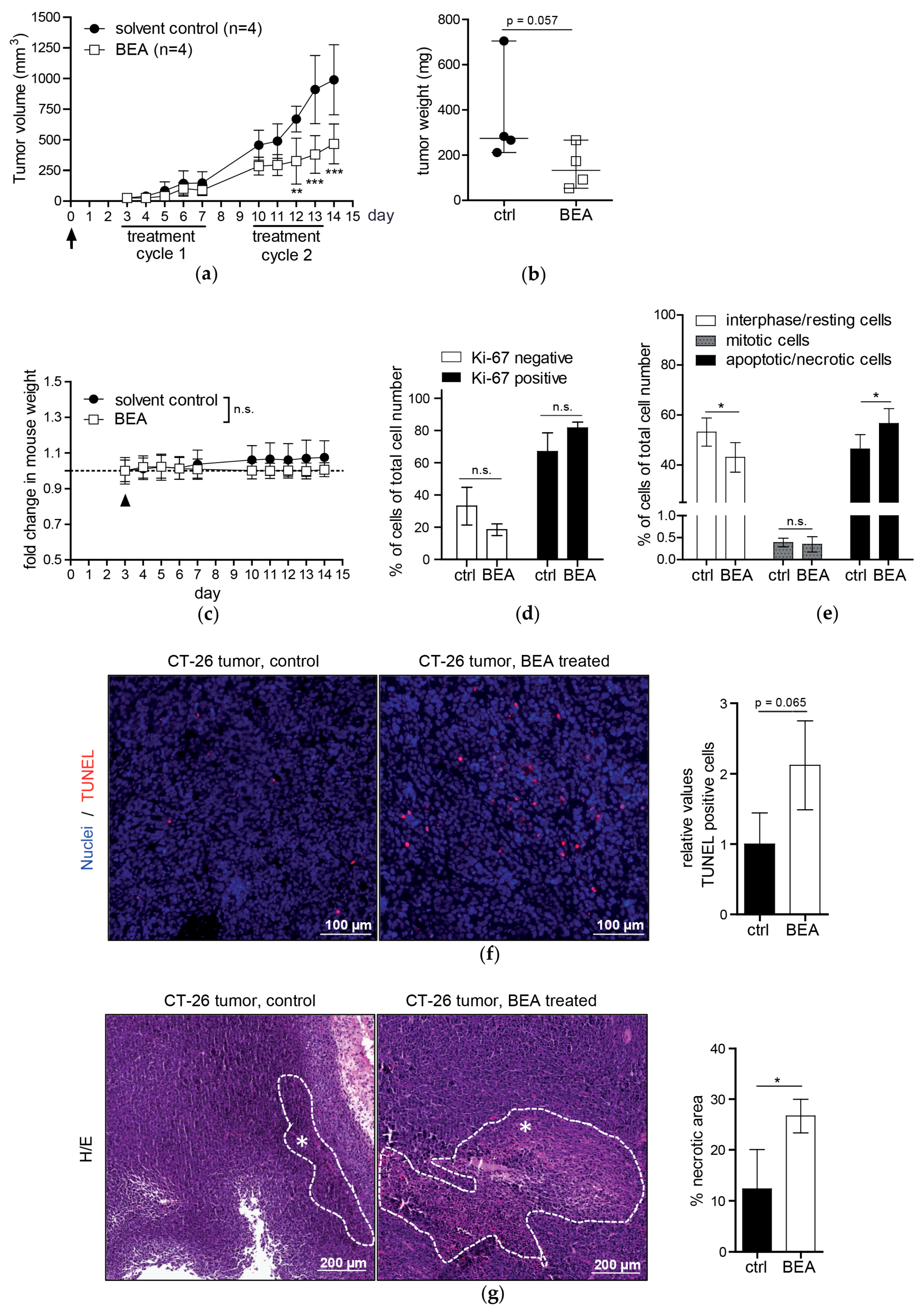
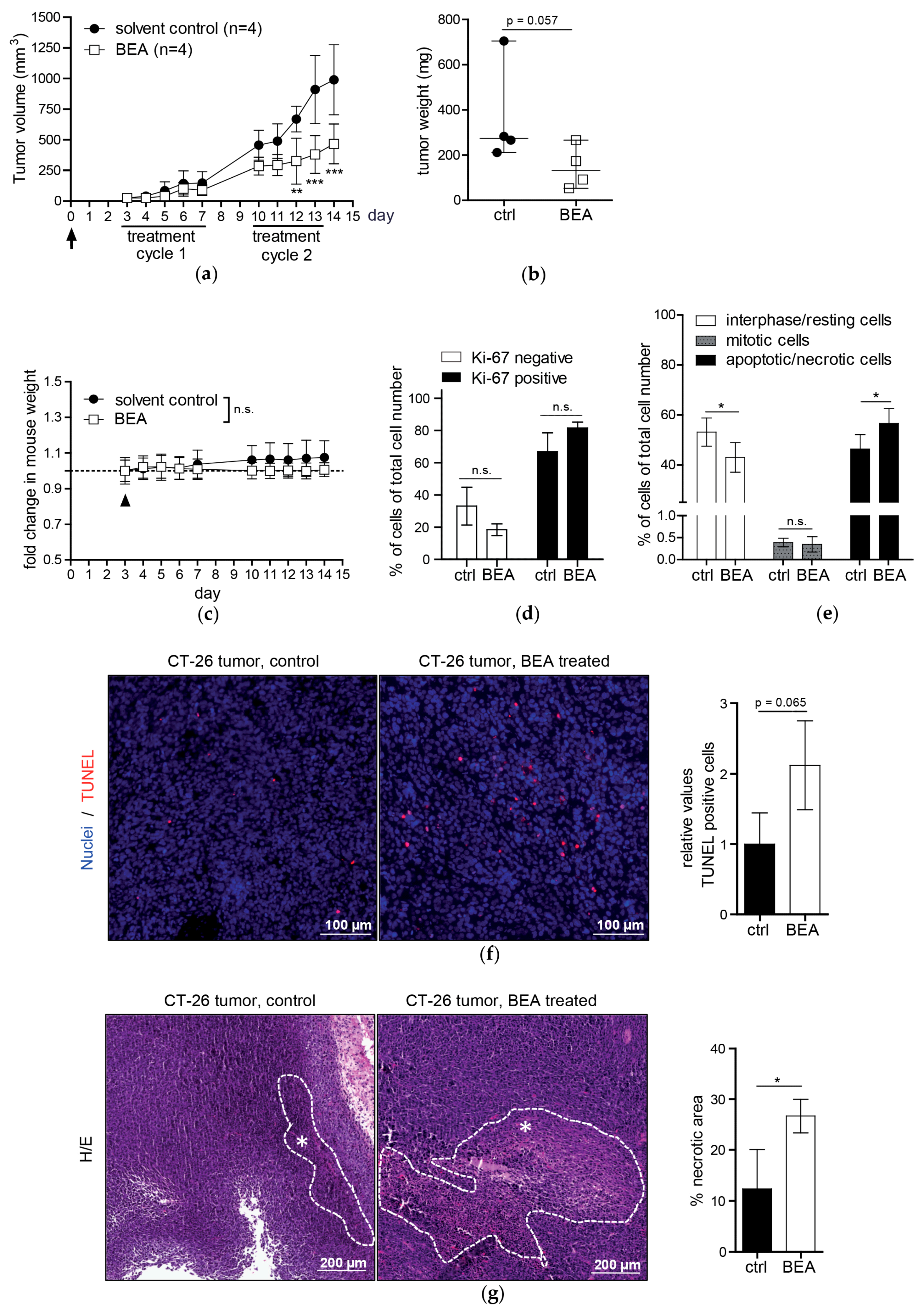
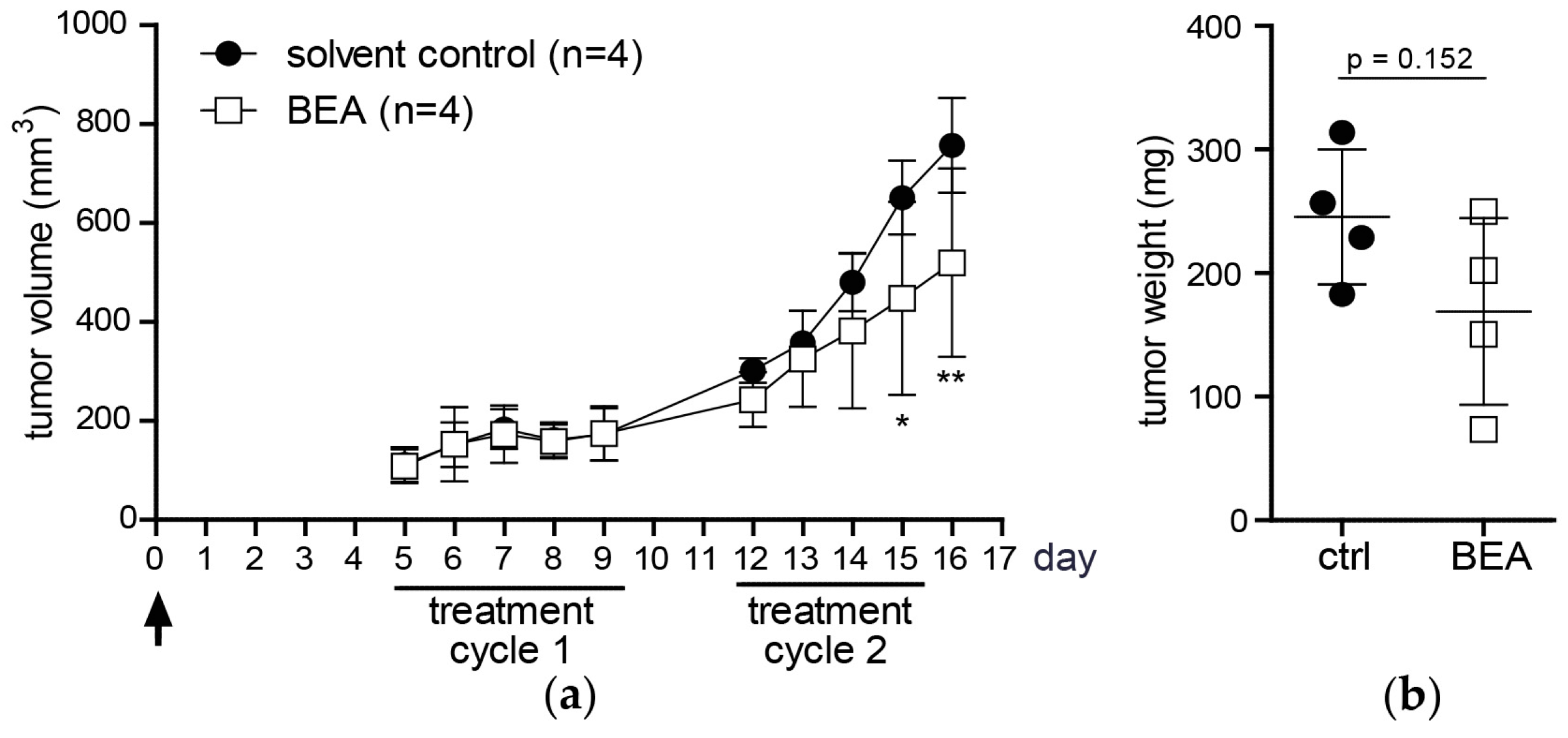
Investigating in vivo effects of beauvericin in mouse models of murine and human carcinomas, beauvericin treatment significantly reduced tumor volumes as compared to solvent-treated controls. Likewise, mean tumor weights were lower in the beauvericin-treated group of the xenograft and, even more pronounced, of the allograft model. Even though tumor growth was not completely inhibited, these effects suggest therapeutic potential of beauvericin, which may be further enhanced by optimization of the dose and the treatment schedule.
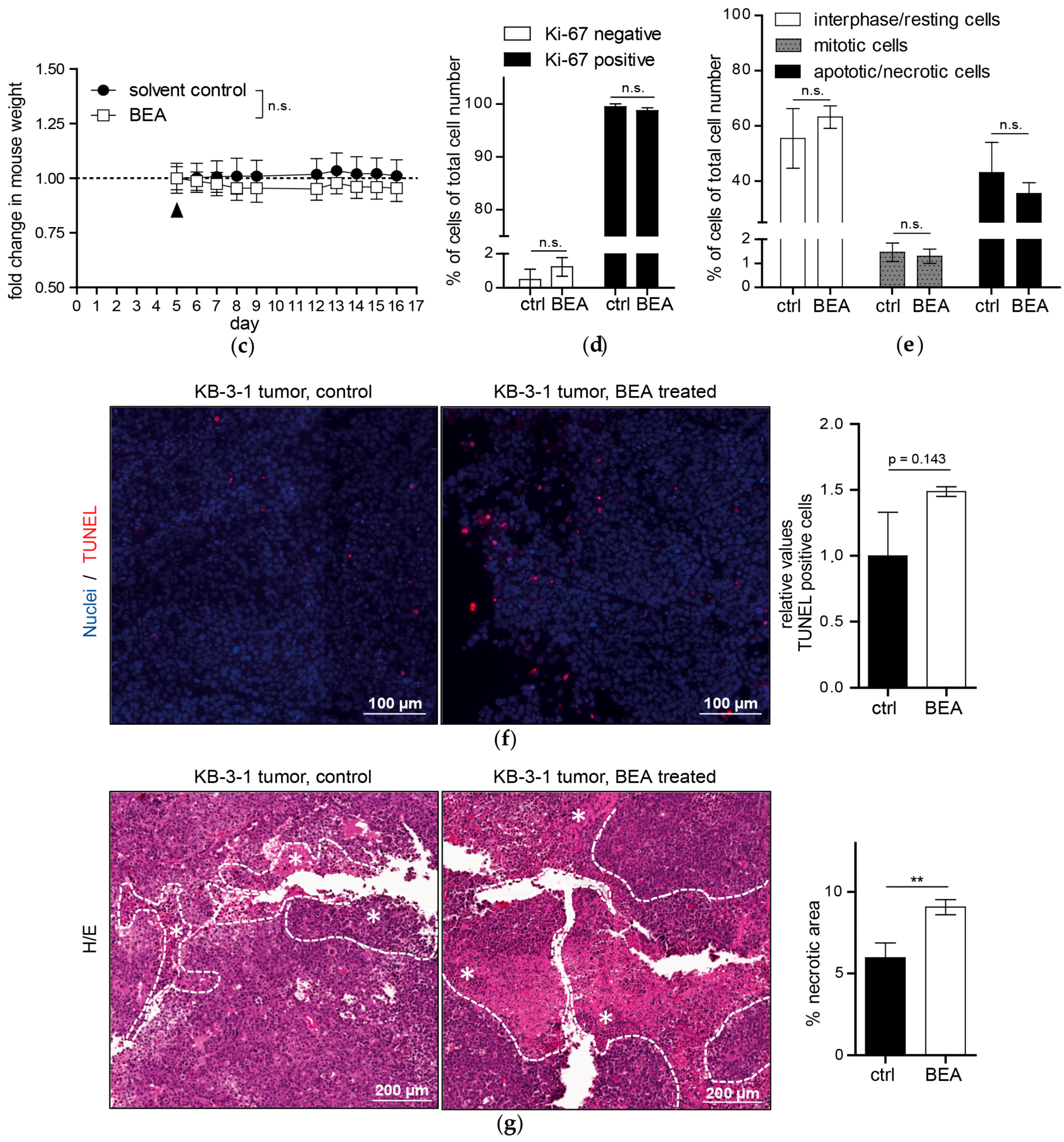
To examine mechanisms underlying the therapeutic effect of beauvericin histological stainings of tumor specimens were performed. In Ki-67-stained tissues the fractions of proliferating (Ki-67-positive) and resting (G0) cells (Ki-67-negative) did not show significant differences between treated and untreated groups in either model corroborated by similar rates of mitotic cells. Therefore, tumor growth reduction by beauvericin treatment was not caused by altered proliferation rates of malignant cells. Counting apoptotic/necrotic cells in H/E-stained tumor section, we observed a higher percentage of cells exhibiting apoptotic and necrotic features in beauvericin-treated mice, especially in the allograft model. Correspondingly, a distinct increase in TUNEL-positive cells, characteristic of apoptosis, was detected. Albeit, due to high variability between different regions of viable tumor parts, this alteration was not statistically significant. Whether uneven distribution of beauvericin in tumor nodules caused this variability needs to be determined. However, our results confirmed previous studies where beauvericin-induced apoptosis in diverse cancer cell types was shown in vitro [
16,
17,
23,
24].
The exact mechanisms of cell death induction by beauvericin is not yet established. However, in cervix carcinoma cells, we detected a G0/G1 phase arrest at subtoxic concentrations of beauvericin, followed by apoptosis induction at higher concentrations which was accompanied by the activation of the intrinsic mitochondrial cell death pathway. Additionally, the cytotoxic effects of beauvericin were discussed to be based on its ionophoric characteristics increasing cytoplasmic calcium concentrations and stimulating calcium-dependent endonucleases finally resulting in DNA fragmentation and apoptosis [
25]. Furthermore, others have also shown an influence on mitochondrial membrane potential [
26,
27], increased cytochrome C release followed by caspase 3 activation [
17,
27], a boost of reactive oxygen species (ROS) production [
26], interaction with NF-KB [
28] and/or MAPK pathways [
16,
28], as well as necrotic cell death [
28] were suggested to underlie the multifaceted actions of beauvericin. Several of these suggested modes of action, like the boost of ROS production and the interaction with oncogenic NF-KB and/or MAPK pathways might have distinctly stronger impacts on malignant as compared to non-malignant cells and, hence, might contribute to the observed cancer selectivity. Cancer cells, for example, suffer from enhanced oxidative stress and are vulnerable to ROS-generating compounds [
29]. For the MAP kinase pathway, inhibitory compounds are already in clinical use against cancer [
30]. However, further investigations are necessary to estimate which cancer types might be primary targets for beauvericin treatment and which biomarkers might help to stratify respective patient subgroups.
Additionally to dispersed apoptotic/necrotic cells in the viable tumor, necrotic tissue areas—especially in the centers of the tumors—were significantly enhanced by beauvericin treatment in both tumor models. This is not necessarily a consequence of necrotic cell death but might be induced by focal, but massive, apoptosis of cancer cells or tissue breakdown due to starvation and lack of oxygen. This would suggest an interference of beauvericin with nutrient and oxygen delivery into malignant tissues probably based on inhibition of angiogenic processes. In accordance with this finding, anti-angiogenic activity of subtoxic beauvericin concentrations on human umbilical vein endothelial cells (HUVEC-2) has been described [
15]. Likewise, in our hands both vessel forming and migratory ability of HUVEC cells were distinctly inhibited by subtoxic concentrations of beauvericin (unpublished data). Hence, reduction of blood supply in combination with the cytotoxic activity of beauvericin against tumor cells are likely to underlie the extensive necrosis detected in tumors of beauvericin-treated mice. Therefore, experiments investigating the impact of beauvericin on blood supply of tumors are currently initiated.
In our in vivo studies we observed a more pronounced anticancer activity of beauvericin on tumors in the allograft than in the xenograft model. This might result from the different cell-types used, i.e., cells derived from a colon carcinoma (CT-26) in the allograft and from a cervix carcinoma (KB-3-1) in the xenograft model. Accordingly, the former proved to be more susceptible to beauvericin also in vitro (IC
50 for CT-26: 1.8 µM, for KB-3-1: 3.1 µM). Alternatively, the stronger activity in the allograft model might indicate a contribution of immune-related factors to the anticancer activity of beauvericin. Several chemotherapeutic agents support activation of tumor-targeting T-cell subclones partly based on enhanced tumor antigen presentation [
31]. This immune-stimulating effect—synergizing with direct cytotoxic activity against malignant cells—is definitely lacking in SCID mice without functional B- and T-cells. However, immune-inhibitory effects of beauvericin have also been described in a Crohn’s disease model [
32]. Consequently, beauvericin might have an effect on invasion of immune cells e.g., T-cells, into the malignant tissue which is currently addressed in our allograft model. Generally, we did not see any signs of adverse effects or immune-related reactions in beauvericin-treated mice indicated by stable body weight, unaltered general physiological conditions (e.g., activity and coat appearance) or lack of abnormal behavior (e.g., grooming, fatigue). In addition, no signs of tissue alteration or inflammatory responses in H/E stained tissue sections of diverse organs were detected. Thus, we conclude that major adverse effects of beauvericin are unlikely.
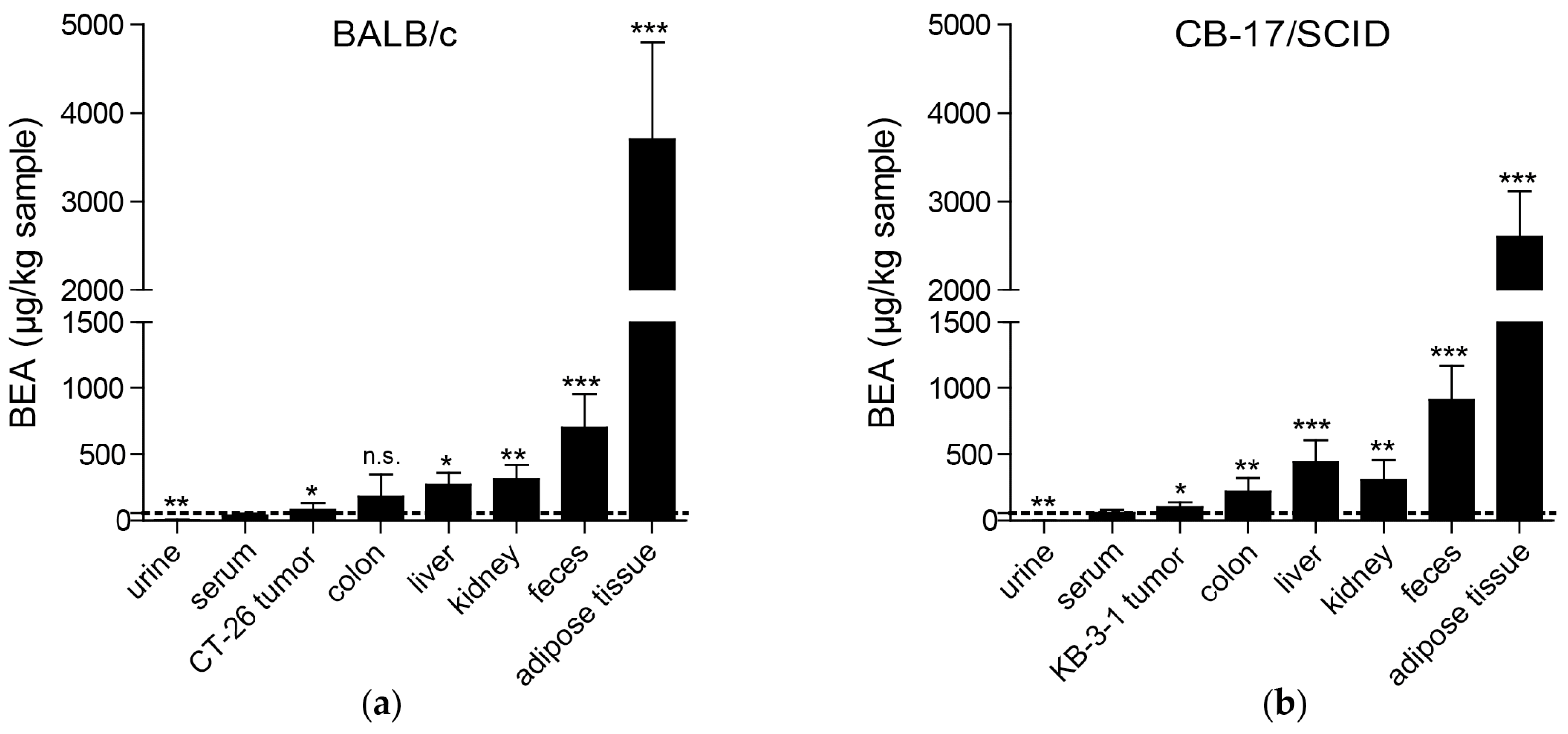
As already suggested based on the lipophilicity of beauvericin [
25], we found the highest beauvericin concentrations in adipose tissue followed by feces, kidney, and liver tissue. However, we also detected moderate beauvericin accumulation in tumor tissues in both mouse models in comparison to serum levels. In urine samples, we only detected minor beauvericin concentrations implying negligible renal clearance of the compound. Fecal enrichment, however, was also reported [
25] suggesting elimination of beauvericin mainly through feces.
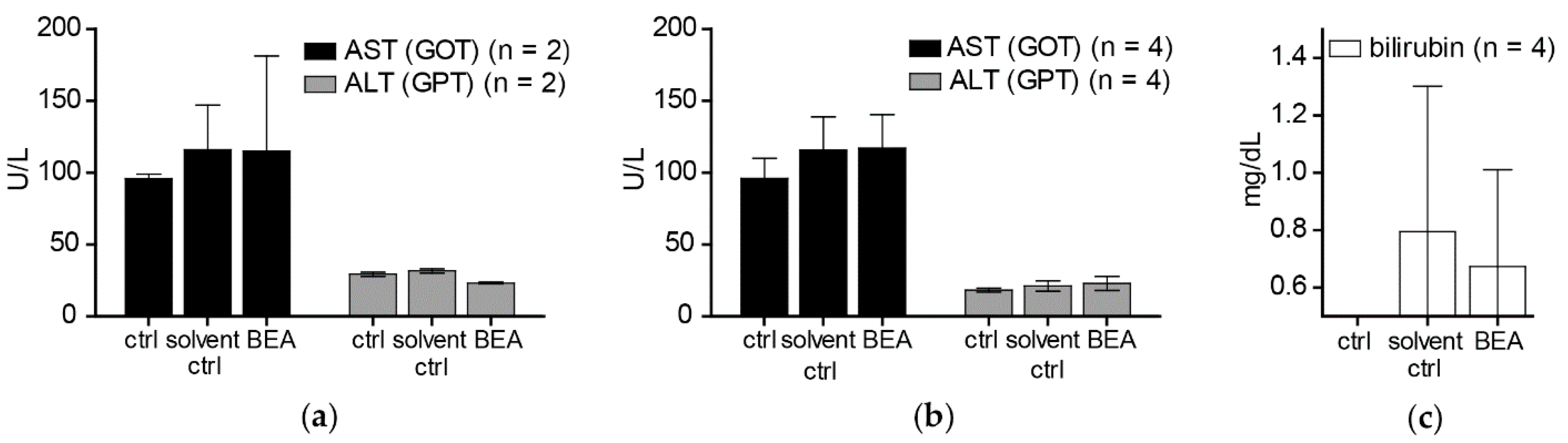
In contrast to the cytotoxic activity of beauvericin on non-malignant fibroblasts and keratinocytes shown in our in vitro experiments, no macro- and microstructural tissue changes were observed in liver and kidney [
19], despite accumulation of beauvericin in these organs. In line with this observation, no alterations of serum markers indicating tissue damage (AST, ALT) or impairment of kidney or liver function (bilirubin, creatinine) were detected both in treated and in control mice immediately and two weeks after the treatment period. This discrepancy between cytotoxic effects of beauvericin in vitro and lack of obvious adverse reactions in vivo might be explained at least in part by the loss of beauvericin cytotoxicity against non-malignant but not against malignant cell types at higher cell density. Additionally, pharmacokinetic parameters and blood vessel integrity effects might lead to tissue-specific drug exposure alterations in the in vivo situation.
Although in this study we observed a significant enrichment of beauvericin in tumors compared to serum levels, the accumulation was much more pronounced in adipose tissue, liver, kidney, or colon. In general, several approaches are possible to improve such sub-optimal tumor accumulation. Hence, drugs might be chemically modified to obtain derivatives with enhanced pharmacological characteristics. Alternatively, nanoformulations of several anticancer drugs were proven to be superior compared to the native substances also in clinical applications, such as the approved liposomal doxorubicin (Doxil
®) [
33]. Therefore, we plan to develop beauvericin derivatives and/or nanoformulations with improved therapeutic windows.
Beauvericin exerted a more pronounced anticancer activity in single drug regimens as compared to the closely related compound ENN B [
18], maybe due to differences in metabolization. While no metabolites of beauvericin could be detected in mice after three days of treatment, ENN B was processed to three phase I metabolites [
19]. Similar results were gained in vitro [
34]. Due to this obviously higher metabolic stability sustained concentrations of beauvericin may be achieved in vivo. Additionally, rapid acquisition of beauvericin resistance seems unlikely as we did not induce beauvericin-unresponsiveness of KB-3-1 cells during a two-year in vitro selection process [
24]. These favorable pharmacokinetic characteristics endorse the therapeutic potential of beauvericin.
5. Materials and Methods
5.1. Chemicals
Beauvericin was purchased from BioAustralis (Smithfield, Australia) and, for animal experiments, purified from
Beauveria bassiana (ATCC 7159). The culture conditions were adopted from Xu et al. [
14] and the biomass harvested by suction filtration. The mycelium was lyophilized and extracted with ethyl acetate. The solvent was evaporated and the brownish residue resolved in methanol. Insoluble residues were removed by filtration and the solvent evaporated. The residues were dissolved in acetonitrile/water (80:20
v/v) and the solution was centrifuged to remove insoluble particles. The supernatant was then subjected to reversed phase chromatography using a GROM-Sil 120 ODS-5 HE, 10 µm, 250 × 20 mm column (Grace GmbH and Co KG, Worms, Germany) on an Agilent 1100 series preparative HPLC system (Agilent Technologies, Waldbronn, Germany) running isocratically on acetonitrile (+0.1% formic acid)/water (+0.1% formic acid) (70:30
v/v) with a flow rate of 15 mL/min. Beauvericin containing fractions were pooled, acetonitrile was evaporated and water was removed by lyophilization. Purity of the compound was verified by LC-MS on an Agilent ESI-Triple-Quadrupol-MS, 6460 Series (Agilent Technologies, Waldbronn, Germany) and by nuclear magnetic resonance (NMR) on a Bruker Avance III 700 MHz-NMR spectrometer (Bruker, Karlsruhe, Germany). Stock solutions of beauvericin were prepared in DMSO and stored at −20 °C.
5.2. Cell Culture
All cancer cell lines used for this study are described in
Table S1. Cultures were regularly screened for
Mycoplasma contamination.
5.3. Cell Viability Assay
For the cell lines KB-3-1 and SW480 2 × 103 cells, for NIH/3T3, CT-26, HaCaT, and GH354 cells 3 × 103 cells and for ME-180 and SW620 cells 4 × 103 cells were seeded into 96-well pates and incubated at 37 °C (5% CO2) overnight. All cell numbers corresponded to a cell monolayer confluency of 5–10% 24 h after seeding and immediately before beauvericin treatment. For higher cell confluency, NIH/3T3, CT-26, HaCaT, KB-3-1, and SW480 cells were grown to a cell density of approximately 50–60% before treatment. Cells were exposed to increasing concentrations of beauvericin for 72 h. The percentage of viable cells was detected after incubation with 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) at 37 °C for 1–4 h, depending on the cell line, according to the user manual (EZ4U, Biomedica, Vienna, Austria). Cell viability after 72 h was determined and concentrations of beauvericin leading to a reduction of cell number by 25%, 50%, and 75% (IC25, IC50, IC75), respectively, were calculated from whole dose-response curves. All experiments were conducted using full-growth media with 10% FBS in triplicate and repeated three times. The cell confluency was analyzed by Image J 1.50i (NIH, New York, NY, USA).
5.4. In Vivo Allo- and Xenograft Experiments
For the allograft experiment, 4 × 10
5 CT-26 cells were resuspended in 50 µL RPMI medium and injected subcutaneously into the right flank of eight, 6–8 weeks old, male BALB/c mice that were obtained from Harlan Laboratories (San Pietro al Natisone, Italy). Likewise, in the xenograft experiment, 1 × 10
6 KB-3-1 cells in 50 µL RPMI medium were injected subcutaneously into the right flank of eight, 6–8 weeks old, male CB-17/IcrHanHsd-Prkdc severe combined immunodeficiency (SCID) mice. Of each group, four animals were randomly assigned to the control or to the treatment group. After the tumor was palpable and reached an approximate size of 25 mm
3 the respective mouse was either treated intraperitoneally with 5 mg/kg bw/day beauvericin (dissolved in 10% DMSO) as described previously [
19] or, for the control group, with solvent alone (10% DMSO). All animals were kept under pathogen-free conditions and all procedures were performed in a laminar flow hood. Effects of the treatment were assessed by daily recording of tumor size with a microcaliper and parameters indicating the animals’ overall health condition (e.g., body weight, fatigue, grooming, ragged coat, food and fluid consumption). Tumor volumes (mm
3) were calculated using the formula: (length × width
2)/2. After two therapy cycles of four to five days, and 24 h after the last beauvericin injection, animals were sacrificed by cervical dislocation after anesthesia (Ketavet
®/Rompun
® mix) to collect blood by heart puncture. The tumor mass was weighed and organs and tissues for immunohistochemical experiments were fixed in 4% formalin/PBS (Roti
®-Histofix 4%, Roth, Karlsruhe, Germany) or shock-frozen in liquid nitrogen and stored at −80 °C until the samples were prepared for LC-MS/MS analysis. The experiments were approved by the ethics committee of the Austrian Federal Ministry of Science, Research, and Economy (BMWF-66.009/0084-II/3b/2013, date of approval: 5 September 2013) and performed in line with the Arrive guidelines for animal care and protection and with guidelines from the Austrian Animal Science Association and from the Federation of European Laboratory Animal Science Associations (FELASA).
5.5. Immunohistochemistry
From each mouse from both the control (n = 4) and the treatment group (n = 4), tissue samples were formalin-fixed, paraffin-embedded and used to prepare serial 3 µm sections. Then, slices were deparaffinized and rehydrated. To evaluate the percentages of interphase/resting, mitotic and dead (apoptotic/necrotic) cell fractions in tumor specimens, sections were stained with hematoxylin and eosin (H/E) by means of standard protocols. Numbers of the three cell fractions were analyzed in a blinded setup in at least four images of H/E-stained tumor sections, taken under a 40 × objective microscope lens. In addition, for the quantification of areas of dead tumor tissue in whole tumor sections, the H/E-stained slides were scanned with a Pannoramic MIDI automated slide scanner (3DHISTECH, Budapest, Hungary) and evaluated using Definiens’ TissueStudio® 4.0 (Definiens®, Munich, Germany) software and Pannoramic Viewer (3DHISTECH) software. To evaluate the fraction of apoptotic cells in tumor sections a terminal deoxynucleotidyl transferase (TdT) deoxyuridine triphosphate (dUTP) nick end labeling (TUNEL) assay was performed applying the In Situ Cell Death Kit TMR (red) (Roche Diagnostics, Mannheim, Germany) according to the manufacturer’s protocol. Afterwards, slides were covered with Vectashield anti-fade mounting medium with DAPI (H-1200, Vector Laboratories, Burlingame, CA, USA). Slides were protected from light until they were scanned with a Pannoramic MIDI automated slide scanner (3DHISTECH) and analyzed via digital image analysis software (Definiens’ TissueStudio® 4.0, Definiens®). In order to visualize the proliferative cell fraction (cells in the interphase or M-phase) of tumors, KB-3-1 tumor sections were stained with the Ki-67 (clone MiB-1) antibody from DAKO (1:100; Glostrup, Denmark) and for CT-26 tumor sections with Ki-67 (1:100; D3B5, rabbit, Cell Signaling, Danvers, USA). The primary antibody was incubated for 30 min at room temperature in a humid chamber and after rinsing the slides for 3 min in PBS + 0.1% Tween, the UltraVision LP detection system was applied according to the manufacturer’s instructions (Thermo Fisher Scientific, Massachusetts, MA, USA). Sections were counterstained with hematoxylin. For analyzing the percentages of Ki-67-negative and -positive cells in the tumor sections, slides were scanned and analyzed via digital image analysis software (Definiens’ TissueStudio® 4.0, Definiens®).
5.6. LC-MS/MS Analysis
Concentrations of beauvericin in tissue samples were analyzed according to Rodríguez-Carrasco et al. [
19]. Briefly, tissues were collected from each mouse of both the control (
n = 4) and the treatment group (
n = 4). Samples of which were thawed on ice, divided and weighted. Afterwards, each slice was placed into a Precellys
® hard tissue homogenizing CK28 tube (VWR, Radnor, PA, USA). After adding 2 mL of acetonitrile (LC-MS LiChrosolv
®, Merck Millipore, Darmstadt, Germany) to each tube, samples were centrifuged for four cycles with 6000 rpm for 30 s with a 30 s break. Beauvericin from blood and urine samples was extracted by mixing 50 µL of sample with 1.5 mL acetonitrile and vortexing for 15 s. The supernatant was transferred into a fresh tube and stored at −80 °C until the LC-MS/MS analysis was performed. Therefore, a QTrap 5500MS/MS system (Applied Biosystems, Foster City, CA, USA) coupled to a TurboV electrospray ionization (ESI) source and a 1290 series UHPLC system (Agilent Technologies, Waldbronn, Germany) were used. Chromatographic separation was achieved at 25 °C on a Gemini
® C18 column (150 × 4.6 mm i.d., 5 µm particle size) connected to a C18 security guard cartridge (4 × 3 mm i.d.; all from Phenomenex, Torrance, CA, USA) with a flow rate of 1 mL/min. Elution was performed in binary gradient mode and both mobile phases contained 5 mM ammonium acetate and were composed of methanol/water/acetic acid 10:89:1 (
v/
v/
v; eluent A) and 97:2:1 (
v/
v/
v; eluent B), respectively. In the first 2 min of elution 100% eluent A was used, afterwards the proportion of eluent B was increased linearly to 50% within 3 min followed by a linear increase of B to 100% within 9 min. Finally, after a hold-time of 4 min with 100% of eluent B, the column was re-equilibrated with 100% eluent A for 2.5 min. ESI-MS/MS was performed in the scheduled selected reaction monitoring (sSRM) mode in positive mode and the target scan time was set to 1 s. The ESI source was set as follows: source temperature 550 °C, curtain gas 30 psi (206.8 kPa of max. 99.5% nitrogen), ion source gas 1 (sheath gas) 80 psi (551.6 kPa of nitrogen), ion source gas 2 (drying gas) 80 psi (551.6 kPa of nitrogen), ion-spray voltage + 5500, collision gas (nitrogen) medium.
5.7. Quantitative Determination of Aspartate Aminotransferase (AST), Alanine Aminotransferase (ALT), Bilirubin, and Serum Creatinine
For the measurement of AST, ALT, bilirubin, and creatinine, ten male CB-17/IcrHanHsd-Prkdcscid mice (Harlan Laboratories, San Pietro al Natisone, Italy) that were 6–8 weeks old, were randomly assigned to three groups: two to the control (i.e., no treatment), four to the solvent control (10% DMSO), and four to the beauvericin treatment group (5 mg/kg bw/day beauvericin). Mice were treated in two cycles of five and four days, respectively. One day and two weeks after the last treatment, blood was collected from mice. Samples were allowed to coagulate at room temperature and serum was obtained by two centrifugation steps (1000 rpm, 10 min, 4 °C). Concentrations of AST, ALT, bilirubin, and serum creatinine were determined in serum samples by Reflotron® Plus System (Roche, Basel, Switzerland) according to the manufacturer’s instructions.
5.8. Statistics
Data were analyzed using GraphPadPrism 5 software (GraphPad Software Inc., La Jolla, CA, USA). Results are given as mean ± standard deviation (SD), if not indicated otherwise. Tumor volumes of the two groups were compared for each day by two-way ANOVA followed by Bonferroni post-test. For all other statistical analyses an unpaired two-tailed Student’s t-test or, for non-parametric data-distribution, a Mann-Whitney test was performed.


 ,
, 






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}