
From Mollusks to Medicine: A Venomics Approach for the Discovery and Characterization of Therapeutics from Terebridae Peptide Toxins

Abstract
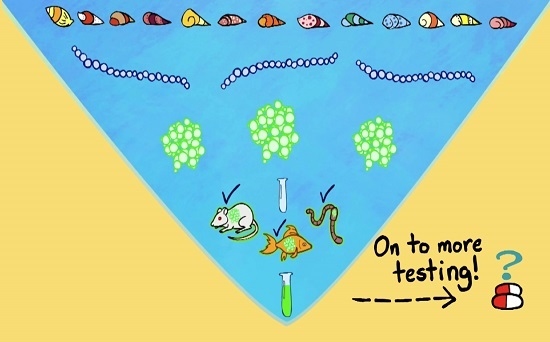
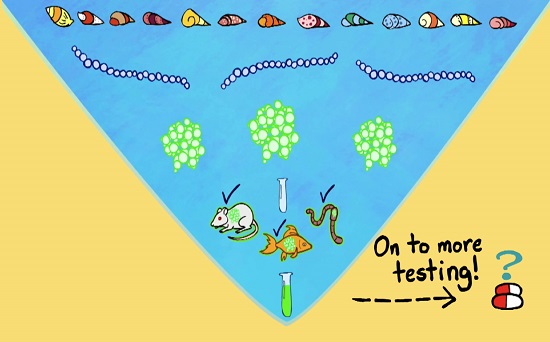
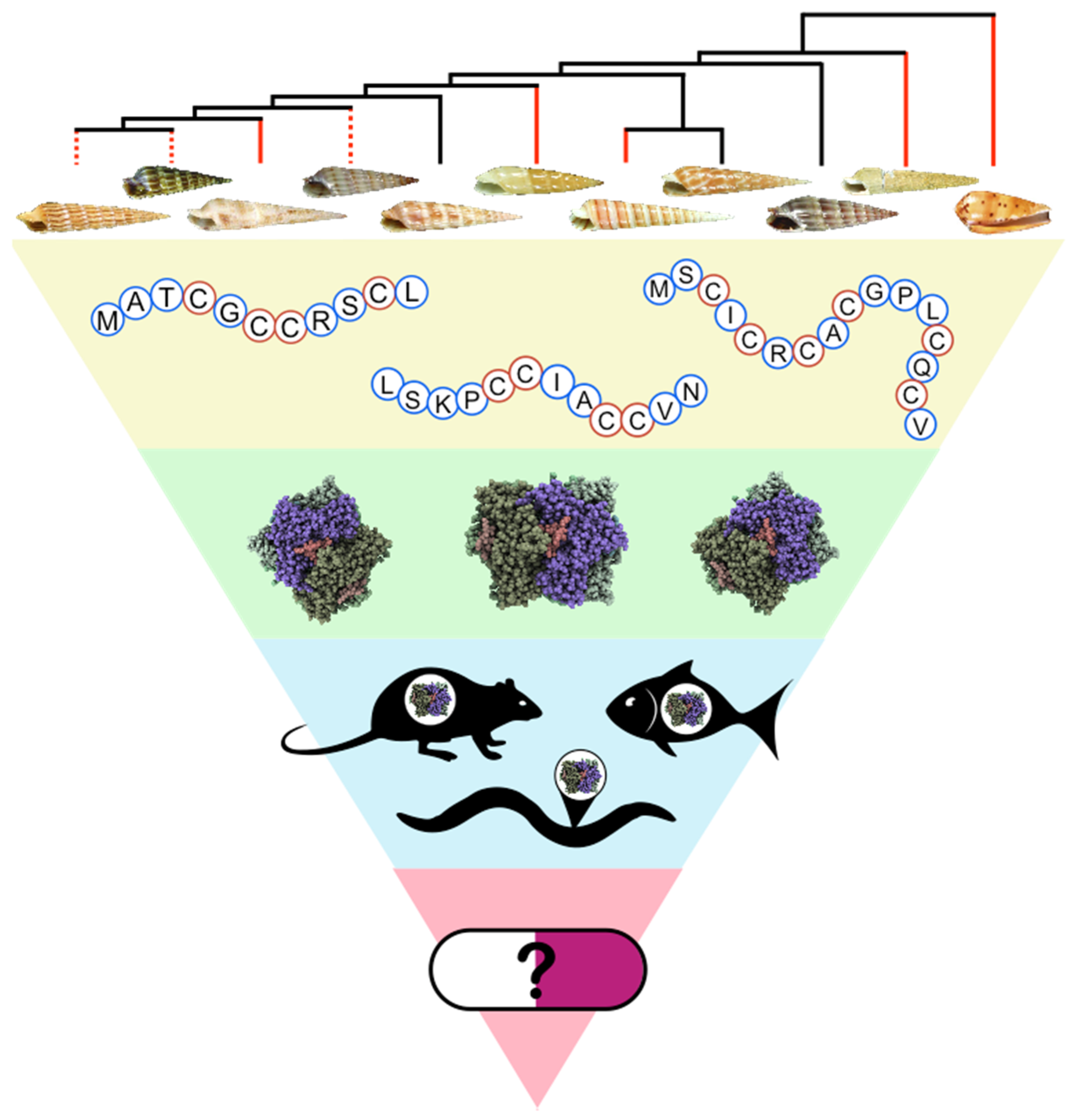
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
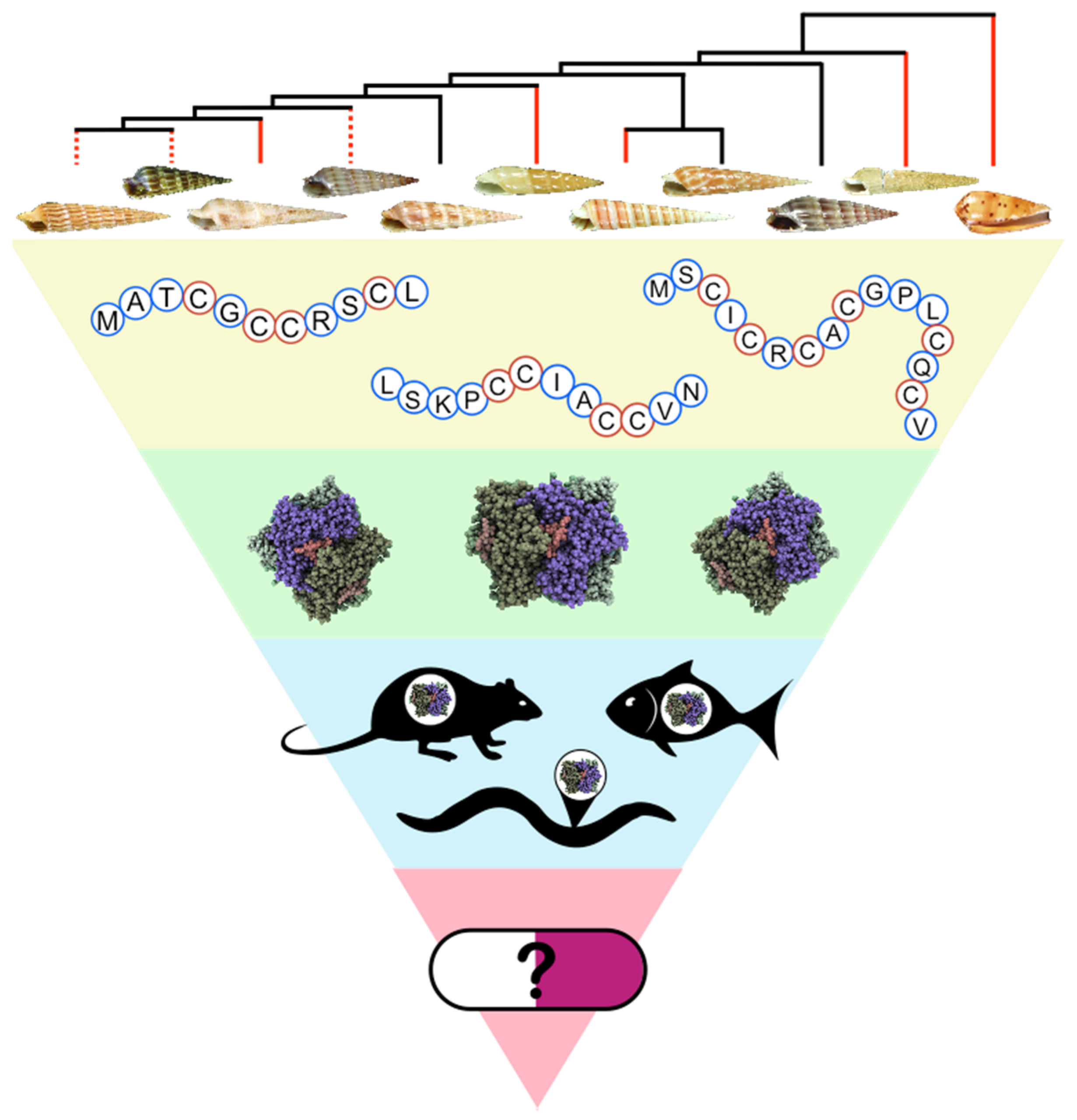
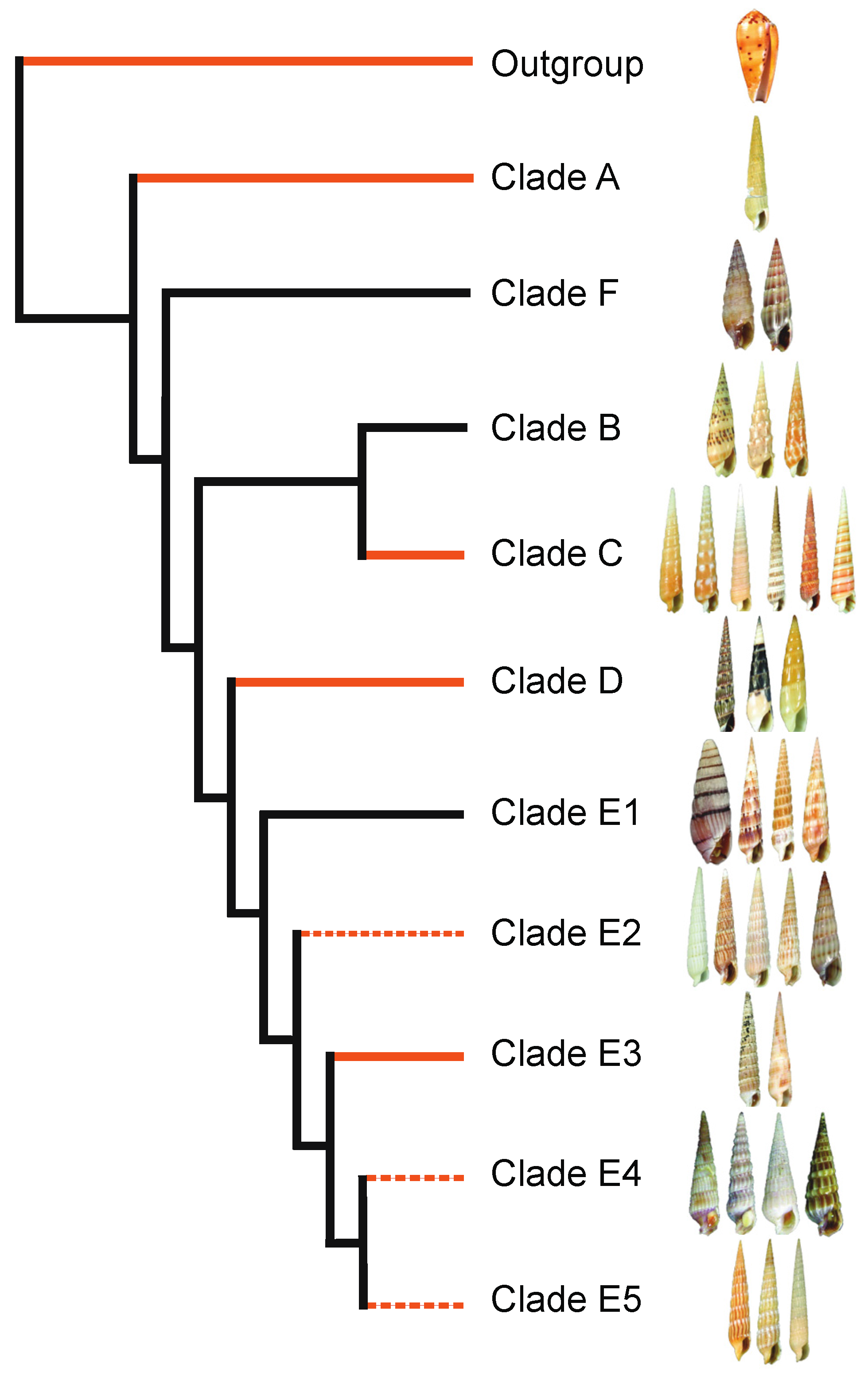
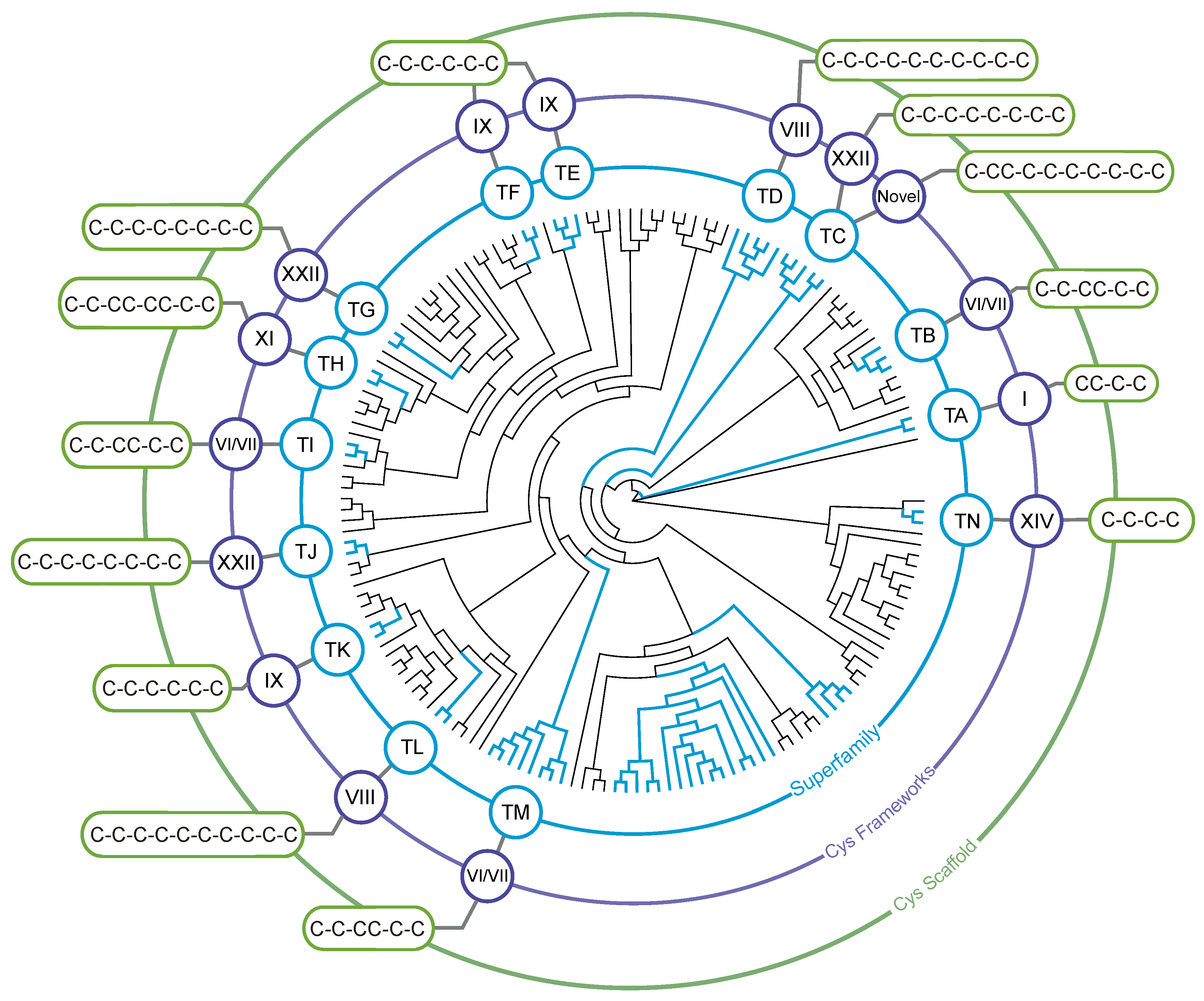
2. Phylogeny-Based Discovery of Teretoxins
2.1. Terebridae Phylogenetics
2.2. Venom Apparatus Evolution
3. Teretoxin Identification and Classification
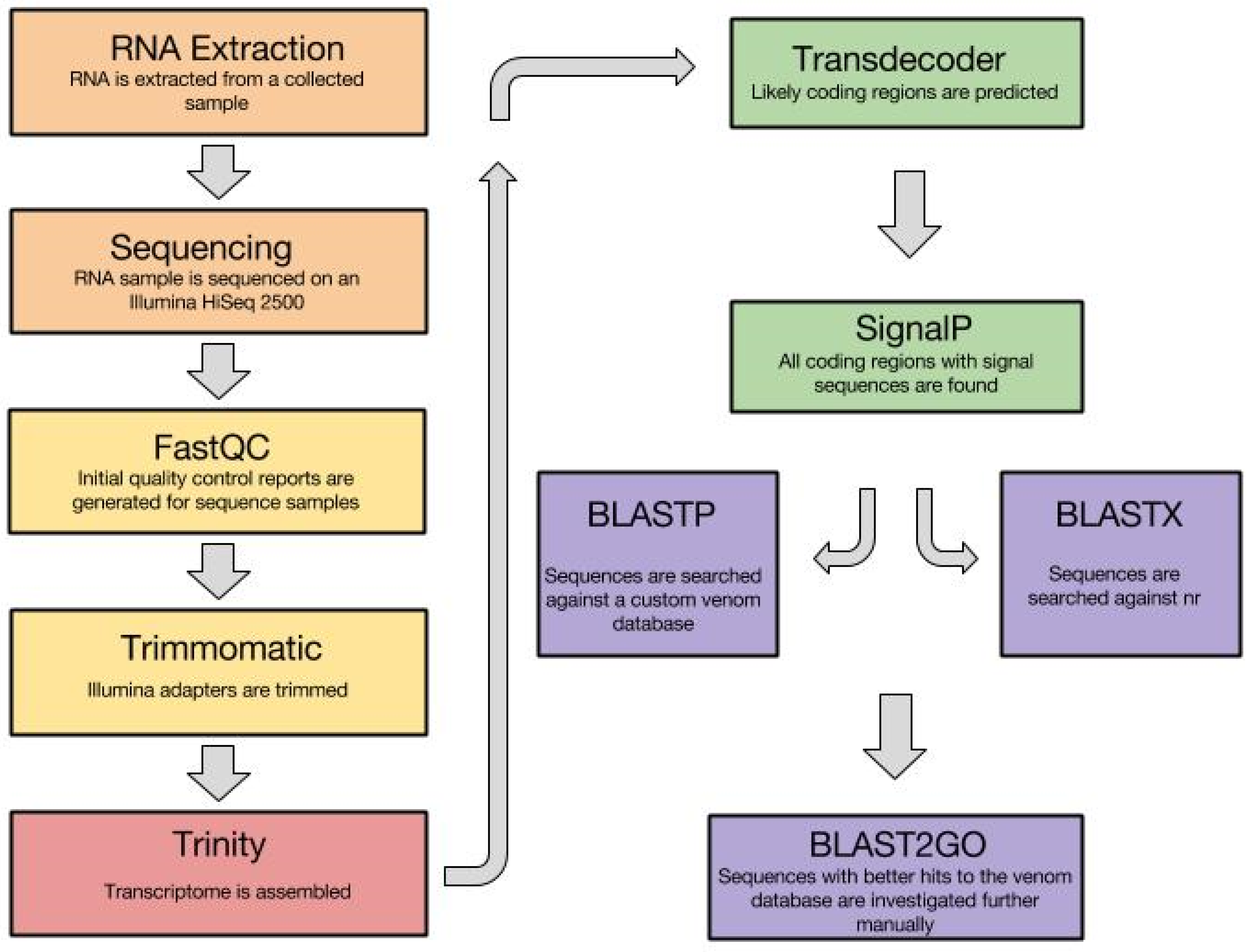
3.1. Venom Gland Transcriptomics
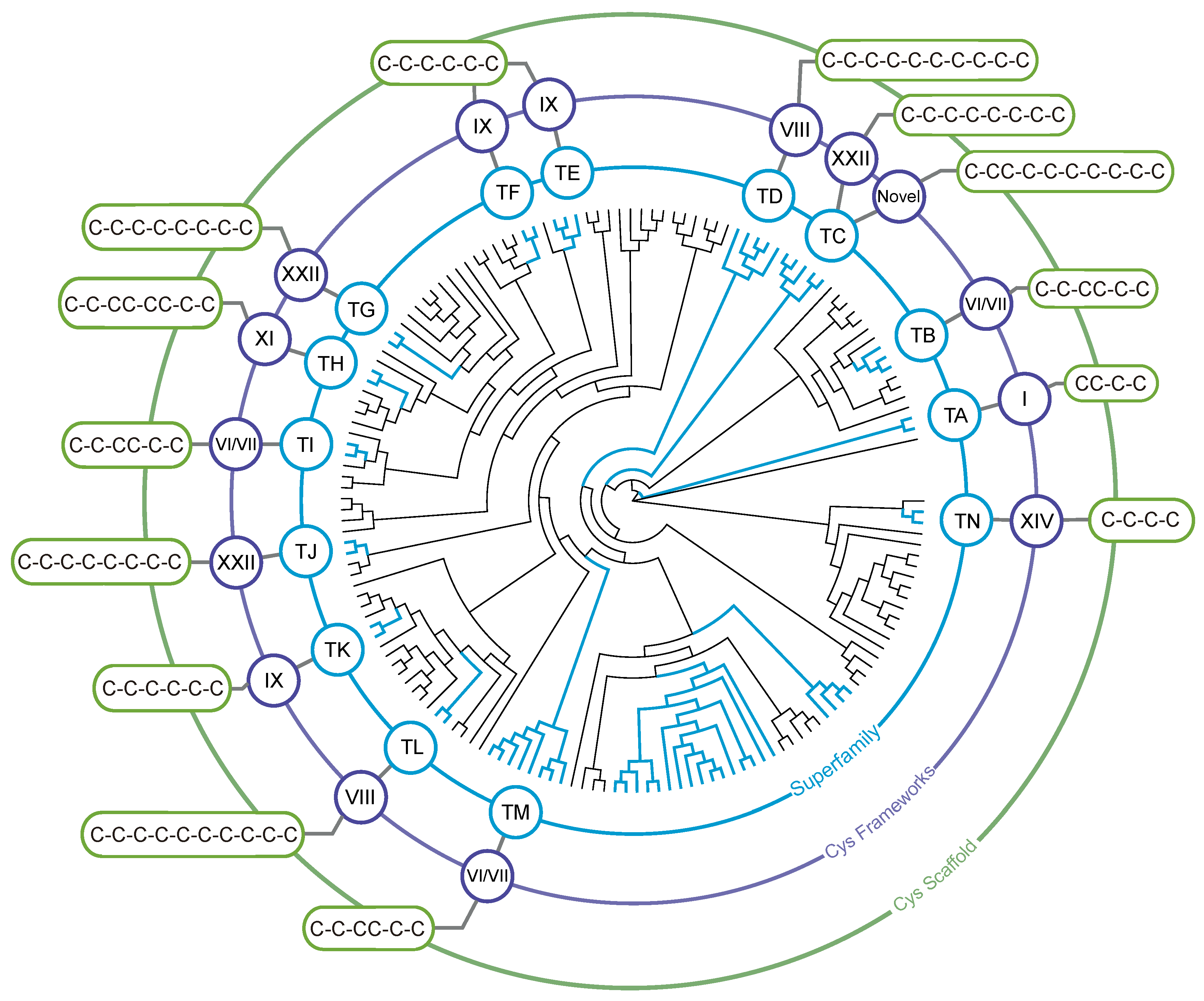
3.2. Identification of Teretoxin Superfamilies
3.3. Venom Proteomics and Proteogenomics Analyses
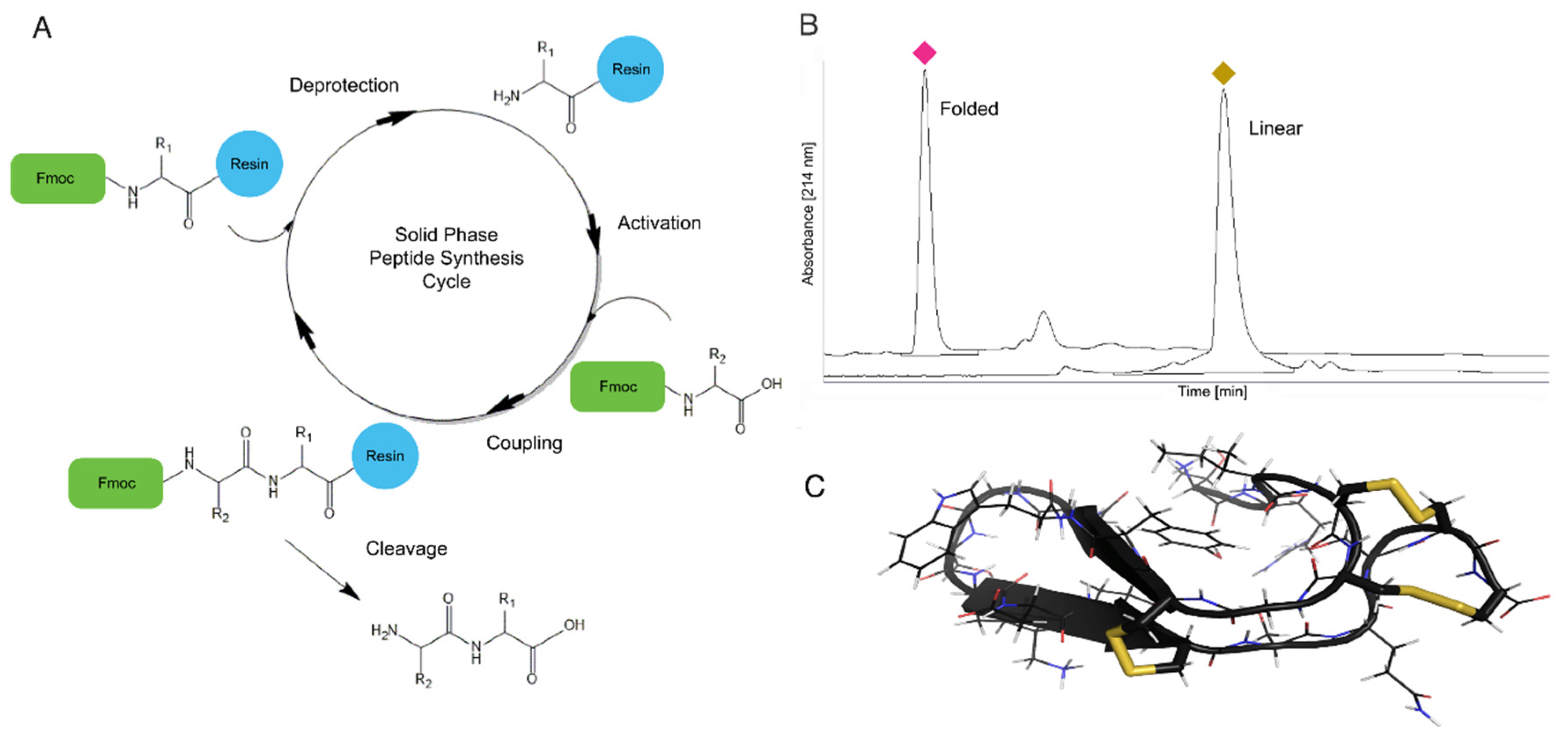
4. Chemical and Recombinant Peptide Synthesis of Teretoxins
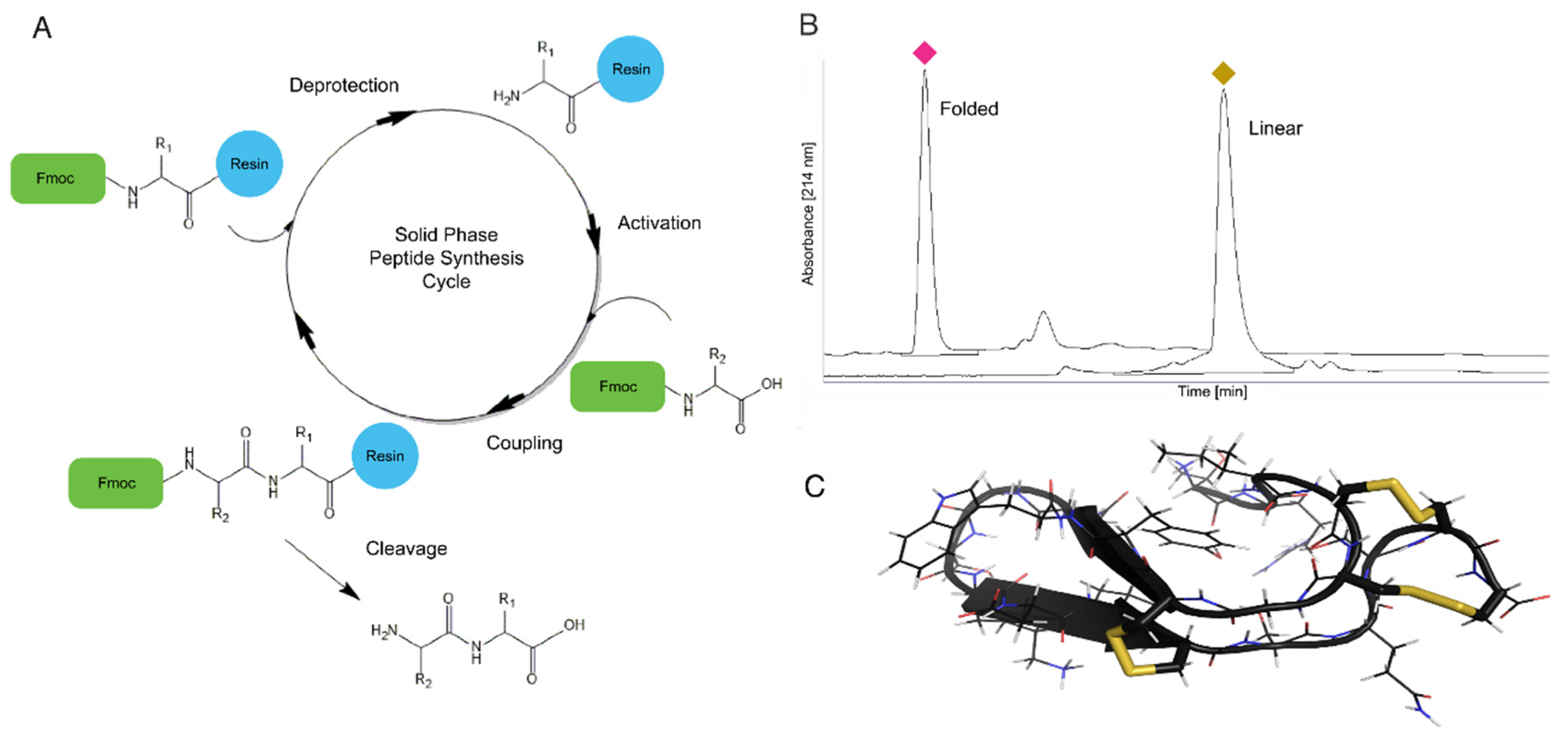
4.1. Solid Phase Peptide Synthesis
4.2. Recombinant Synthesis
5. Characterization of Teretoxin Structure
5.1. Characterization of Teretoxin Disulfide Motif
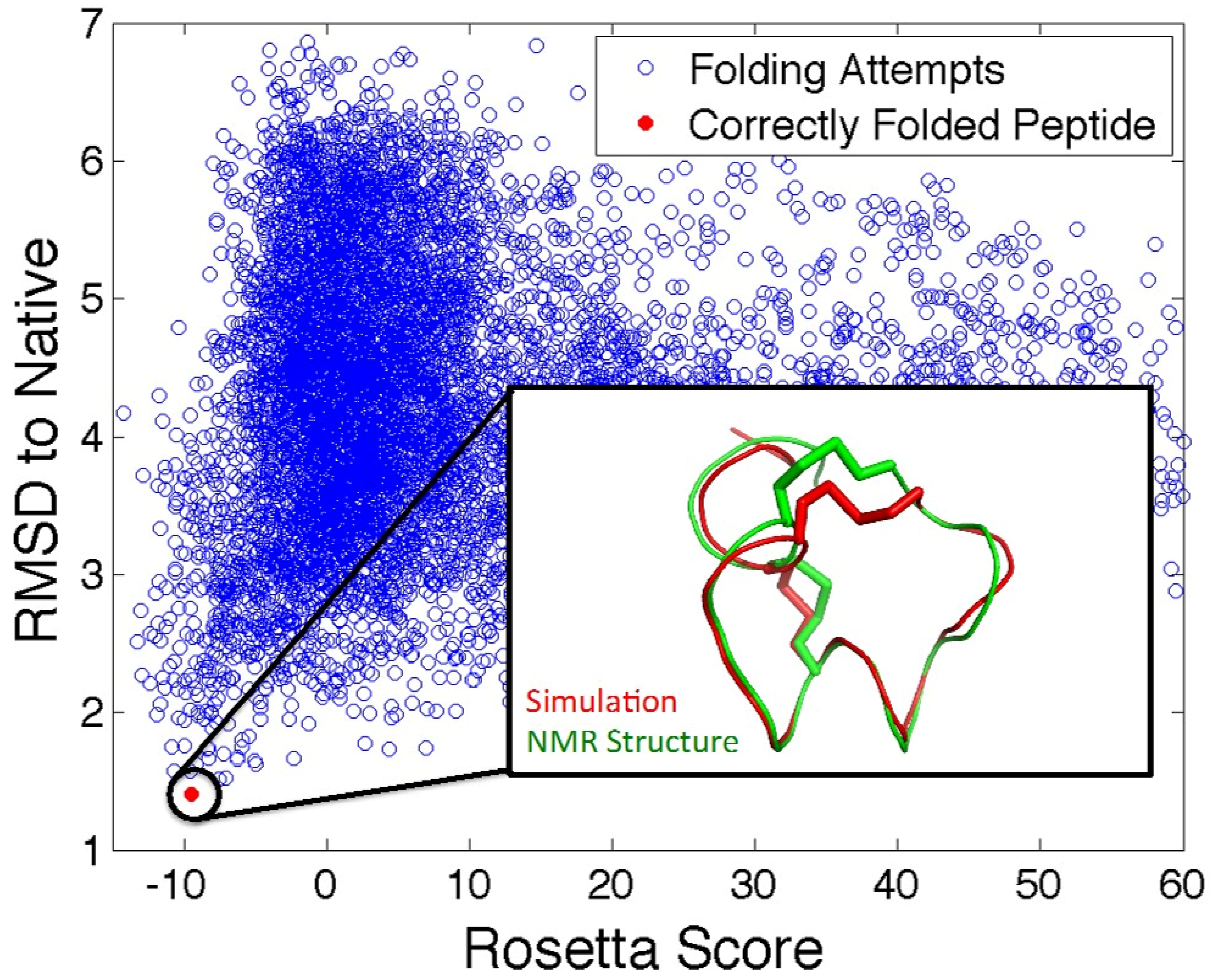
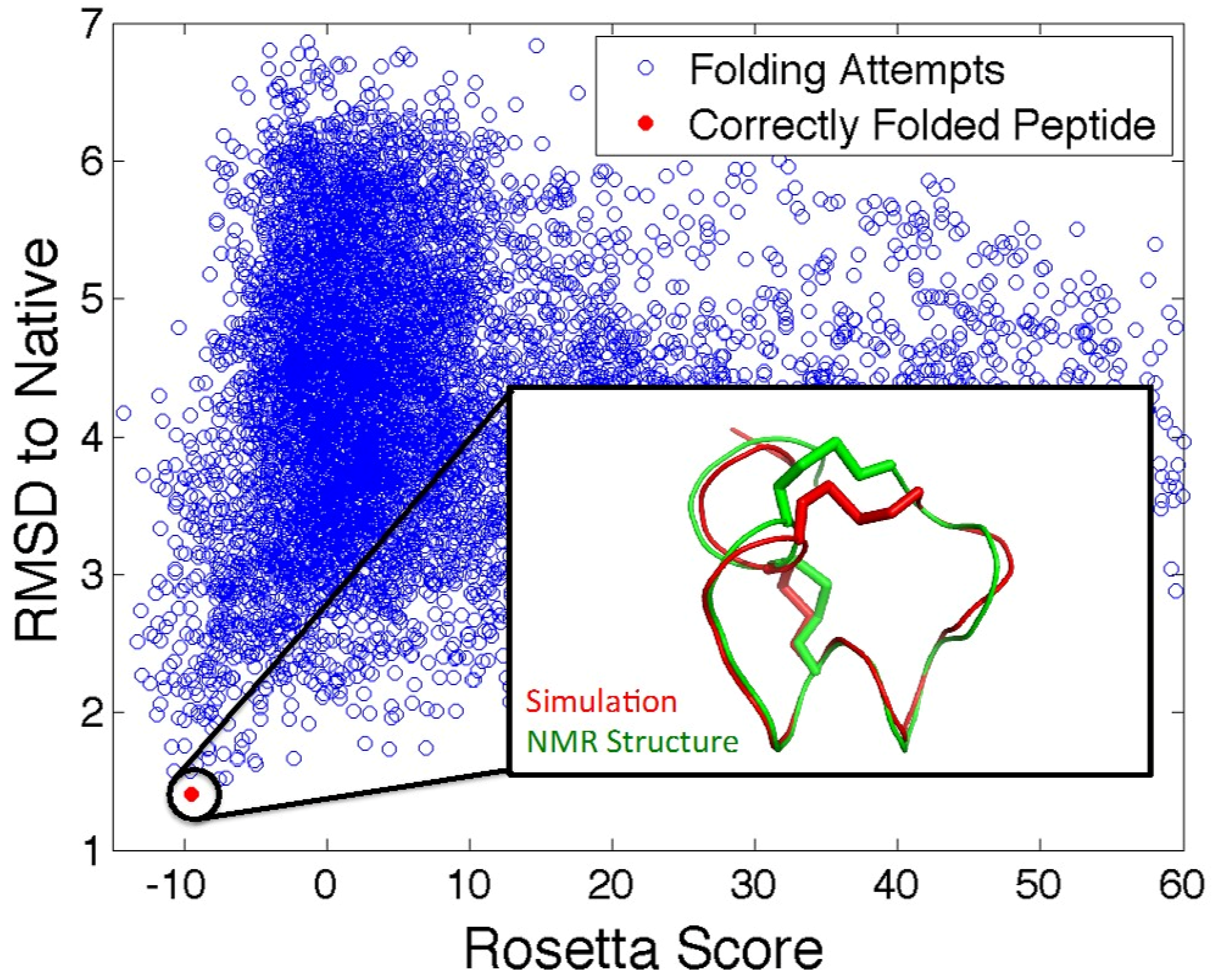
5.2. In Silico Peptide Structure Determination
6. Teretoxins Bioactivity Assays and Functionalization
6.1. Biological Assays
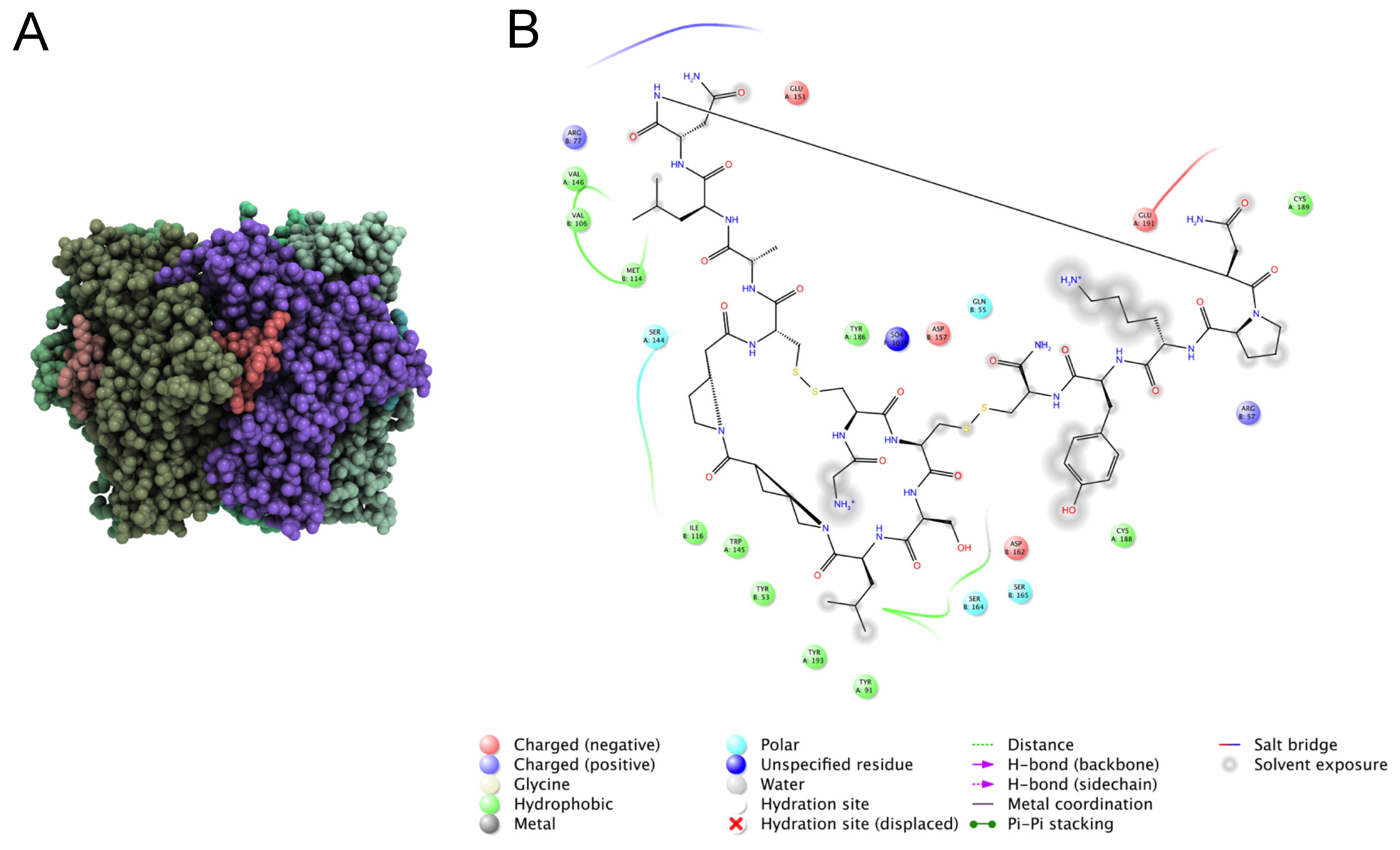
6.2. Characterizing Molecular Function
7. Optimization of Venom Peptides for Drug Development
7.1. Computational Design for Increased Affinity and Selectivity of Peptide Toxins
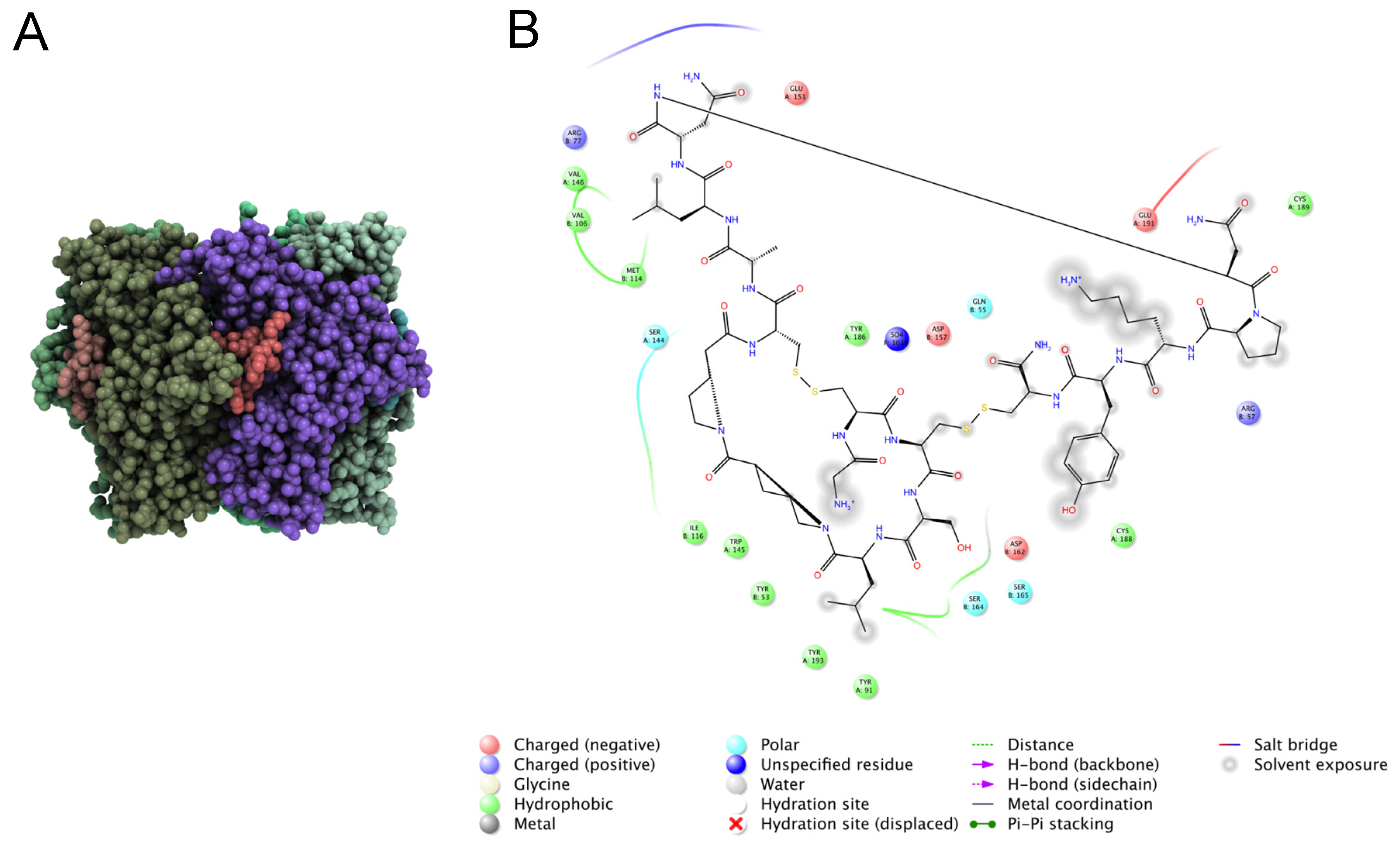
7.2. Identification of Key Residues in Venom Peptides
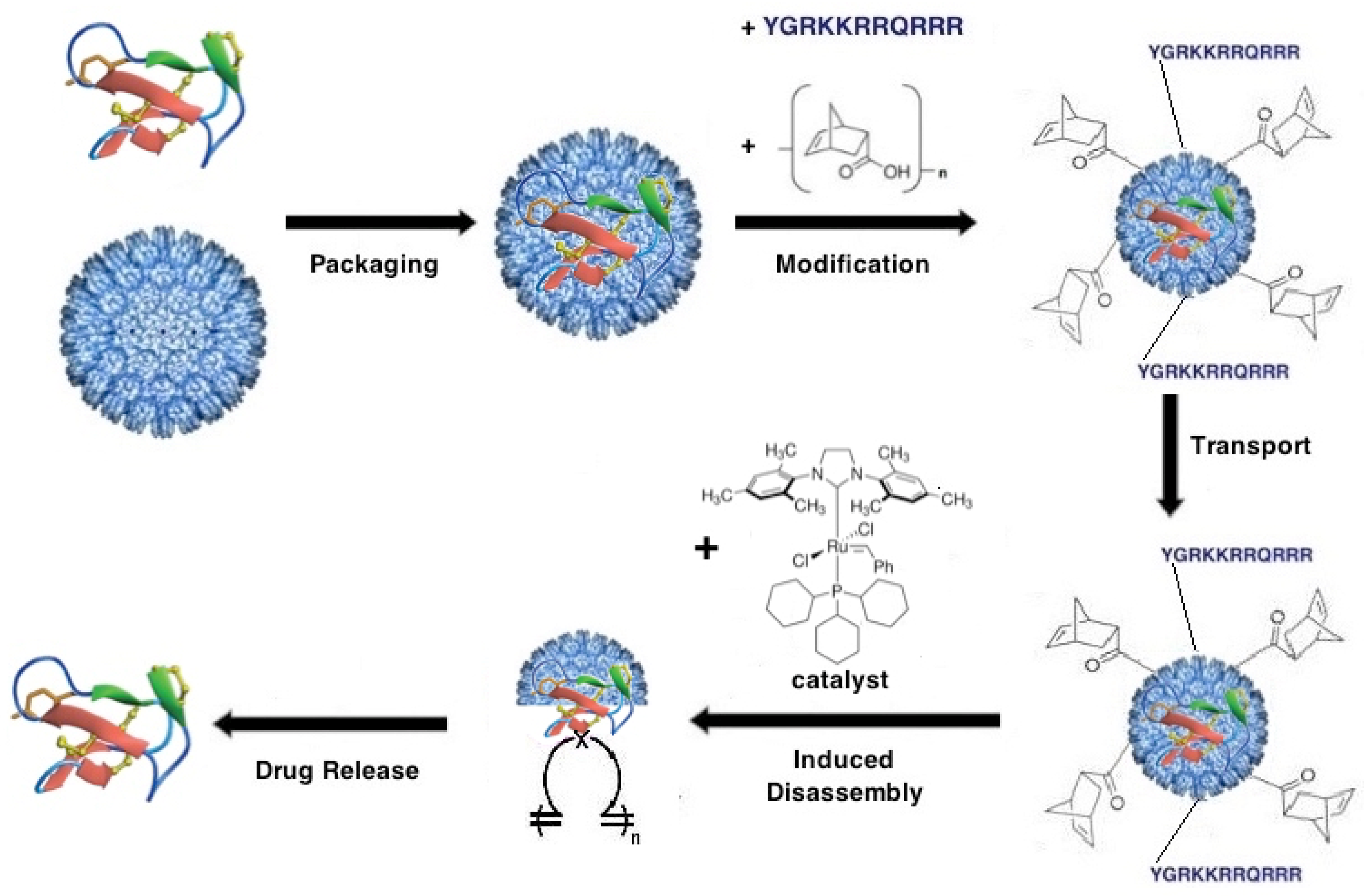
8. Venom Peptide Drug Delivery
8.1. P22 Nanocontainers for Venom Peptide Drug Delivery
8.2. Release of Venom Peptide at Molecular Target Site
9. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Mishra, B.B.; Tiwari, V.K. Natural products: An evolving role in future drug discovery. Eur. J. Med. Chem. 2011, 46, 4769–4807. [Google Scholar] [CrossRef] [PubMed]
- Harvey, A.L.; Edrada-Ebel, R.; Quinn, R.J. The re-emergence of natural products for drug discovery in the genomics era. Nat. Rev. Drug Discov. 2015, 14, 111–129. [Google Scholar] [CrossRef] [PubMed]
- Shen, B. A New Golden Age of Natural Products Drug Discovery. Cell 2016, 163, 1297–1300. [Google Scholar] [CrossRef] [PubMed]
- Fosgerau, K.; Hoffmann, T. Peptide therapeutics: Current status and future directions. Drug Discov. Today 2015, 20, 122–128. [Google Scholar] [CrossRef] [PubMed]
- Cushman, D.W.; Ondetti, M.A. History of the design of captopril and related inhibitors of angiotensin converting enzyme. Hypertension 1991, 17, 589–592. [Google Scholar] [CrossRef] [PubMed]
- Koh, C.Y.; Kini, R.M. From snake venom toxins to therapeutics—Cardiovascular examples. Toxicon 2012, 59, 497–506. [Google Scholar] [CrossRef] [PubMed]
- Furman, B.L. The development of Byetta (exenatide) from the venom of the Gila monster as an anti-diabetic agent. Toxicon 2012, 59, 464–471. [Google Scholar] [CrossRef] [PubMed]
- Miljanich, G.P. Ziconotide: Neuronal calcium channel blocker for treating severe chronic pain. Curr. Med. Chem. 2004, 11, 3029–3040. [Google Scholar] [CrossRef] [PubMed]
- Webster, L.R.; Fakata, K.L. Ziconotide for chronic severe pain. Pr. Pain Mgmt. 2005, 5, 47–51 and 55–56. [Google Scholar]
- Gorson, J.; Ramrattan, G.; Verdes, A.; Wright, M.; Kantor, Y.I.; Srinivasan, R.; Musunuri, R.; Packer, D.; Albano, G.; Qiu, W.; et al. Molecular Diversity and Gene Evolution of the Venom Arsenal of Terebridae Predatory Marine Snails. Genome Biol. Evol. 2015, 7, 1761–1778. [Google Scholar] [CrossRef] [PubMed]
- Goswami, P.K.; Samant, M.; Srivastava, R.S. Snake venom, anti-snake venom & potential of snake venom. Int. J. Pharm. Pharm. Sci. 2014, 6, 4–7. [Google Scholar]
- Ortiz, E.; Gurrola, G.B.; Schwartz, E.F.; Possani, L.D. Scorpion venom components as potential candidates for drug development. Toxicon 2015, 93, 125–135. [Google Scholar] [CrossRef] [PubMed]
- Lewis, R.J.; Garcia, M.L. Therapeutic potential of venom peptides. Nat. Rev. Drug Discov. 2003, 2, 790–802. [Google Scholar] [CrossRef] [PubMed]
- King, G.F. Venoms as a platform for human drugs: Translating toxins into therapeutics. Expert Opin. Biol. Ther. 2011, 11, 1469–1484. [Google Scholar] [CrossRef] [PubMed]
- Olivera, B.M. Conus venom peptides, receptor and ion channel targets and drug design: 50 million years of neuropharmacology (E.E. Just Lecture, 1996). Mol. Biol. Cell 1997, 8, 2101–2109. [Google Scholar] [CrossRef] [PubMed]
- Vetter, I.; Davis, J.L.; Rash, L.D.; Anangi, R.; Mobli, M.; Alewood, P.F.; Lewis, R.J.; King, G.F. Venomics: A new paradigm for natural products-based drug discovery. Amino Acids 2011, 40, 15–28. [Google Scholar] [CrossRef] [PubMed]
- Von Reumont, B.; Campbell, L.; Jenner, R. Quo Vadis Venomics? A Roadmap to Neglected Venomous Invertebrates. Toxins 2014, 6, 3488–3551. [Google Scholar] [CrossRef] [PubMed]
- Modica, M.V.; Lombardo, F.; Franchini, P.; Oliverio, M. The venomous cocktail of the vampire snail Colubraria reticulata (Mollusca, Gastropoda). BMC Genom. 2015, 16, 441. [Google Scholar] [CrossRef] [PubMed]
- Puillandre, N.; Holford, M. The Terebridae and teretoxins: Combining phylogeny and anatomy for concerted discovery of bioactive compounds. BMC Chem. Biol. 2010, 10, 7. [Google Scholar] [CrossRef] [PubMed]
- Puillandre, N.; Kantor, Y.I.; Sysoev, A.; Couloux, A.; Meyer, C.; Rawlings, T.; Todd, J.A.; Bouchet, P. The Dragon Tamed? A Molecular Phylogeny of the Conoidea (Gastropoda). J. Molluscan Stud. 2011, 77, 259–272. [Google Scholar] [CrossRef]
- Terlau, H.; Olivera, B.M. Conus venoms: A rich source of novel ion channel-targeted peptides. Physiol. Rev. 2004, 84, 41–68. [Google Scholar] [CrossRef] [PubMed]
- Olivera, B.M. Conus Peptides: Biodiversity-based Discovery and Exogenomics. J. Biol. Chem. 2006, 281, 31173–31177. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Wang, L.; Zhou, M.; You, Y.; Zhu, X.; Qiang, Y.; Qin, M.; Luo, S.; Ren, Z.; Xu, A. Molecular evolution and diversity of Conus peptide toxins, as revealed by gene structure and intron sequence analyses. PLoS ONE 2013, 8, e82495. [Google Scholar]
- Robinson, S.D.; Safavi-Hemami, H.; McIntosh, L.D.; Purcell, A.W.; Norton, R.S.; Papenfuss, A.T. Diversity of conotoxin gene superfamilies in the venomous snail, Conus victoriae. PLoS ONE 2014, 9, e87648. [Google Scholar] [CrossRef] [PubMed]
- Robinson, S.D.; Norton, R.S. Conotoxin Gene Superfamilies. Mar. Drugs 2014, 12, 6058–6101. [Google Scholar] [CrossRef] [PubMed]
- Dutertre, S.; Jin, A.-H.; Kaas, Q.; Jones, A.; Alewood, P.F.; Lewis, R.J. Deep venomics reveals the mechanism for expanded peptide diversity in cone snail venom. Mol. Cell. Proteom. 2013, 12, 312–329. [Google Scholar] [CrossRef] [PubMed]
- Lavergne, V.; Harliwong, I.; Jones, A.; Miller, D.; Taft, R.J.; Alewood, P.F. Optimized deep-targeted proteotranscriptomic profiling reveals unexplored Conus toxin diversity and novel cysteine frameworks. Proc. Natl. Acad. Sci. USA 2015, 112, 3782–3791. [Google Scholar] [CrossRef] [PubMed]
- Boxshall, G.A.; Mees, J.; Costello, M.J.; Hernandez, F.; Bailly, N.; Boury-Esnault, N.; Gofas, S.; Horton, T.; Klautau, M.; Kroh, A.; et al. World Register of Marine Species (WoRMS). Available online: http://www.marinespecies.org (accessed on 12 April 2016).
- Bouchet, P. Evolution of larval development in Eastern Atlantic Terebridae (Gastropoda), Neogene to Recent. Malacologia 1981, 21, 363–369. [Google Scholar]
- Miller, B.A. The Biology of Terebra gouldi Deshayes, 1859, and a Discussion of Life History Similarities among Other Terebrids of Similar Proboscis Type. Pac. Sci. 1975, 29, 227–241. [Google Scholar]
- Oyama, K. On some new facts of the taxonomy of Terebridae. Venus Jap. J. Malacol. 1961, 21, 176–189. [Google Scholar]
- Taylor, J.D. The anatomy of the foregut and relationships in the Terebridae. Malacologia 1990, 32, 19–34. [Google Scholar]
- Terryn, Y.; Holford, M. The Terebridae of the Vanuatu Archipelago with a Revision of the Genus Granuliterebra Oyama 1961, the Description of a New Genus and a Three New Species. Visaya 2008, 3, 1–96. [Google Scholar]
- Miller, B.A. The Biology of Hastula inconstans (Hinds, 1844) and a Discussion of Life History Similarities among other Hastulas of Similar Proboscis Type. Pac. Sci. 1979, 33, 289–306. [Google Scholar]
- Terryn, Y. A Collectors Guide to Recent Terebridae (Mollusca: Neogastropoda); Conchbooks & NaturalArt: Hackenheim, Germany, 2007. [Google Scholar]
- Powell, A.W.B. The molluscan families Speightiidae and Turridae. Bull. Auckl. Inst. Museum 1966, 5, 1–184. [Google Scholar]
- Kendel, Y.; Melaun, C.; Kurz, A.; Nicke, A.; Peigneur, S.; Tytgat, J.; Wunder, C.; Mebs, D.; Kauferstein, S. Venomous secretions from marine snails of the Terebridae family target acetylcholine receptors. Toxins 2013, 5, 1043–1050. [Google Scholar] [CrossRef] [PubMed]
- Imperial, J.S.; Kantor, Y.; Watkins, M.; Heralde, F.M., 3rd; Stevenson, B.; Chen, P.; Hansson, K.; Stenflo, J.; Ownby, J.P.; Bouchet, P.; et al. Venomous auger snail Hastula (Impages) hectica (Linnaeus, 1758): Molecular phylogeny, foregut anatomy and comparative toxinology. J. Exp. Zool. B Mol. Dev. Evol. 2007, 308, 744–756. [Google Scholar] [CrossRef] [PubMed]
- Imperial, J.S.; Watkins, M.; Chen, P.; Hillyard, D.R.; Cruz, L.J.; Olivera, B.M. The augertoxins: Biochemical characterization of venom components from the toxoglossate gastropod Terebra subulata. Toxicon 2003, 42, 391–398. [Google Scholar] [CrossRef]
- Modica, M.V.; Holford, M. The Neogastropoda: Evolutionary Innovations of Predatory Marine Snails with Remarkable Pharmacological Potential. In Evolutionary Biology–Concepts, Molecular and Morphological Evolution; Springer Berlin Heidelberg: Heidelberg, Alemania, 2010; pp. 249–270. [Google Scholar]
- Fry, B.G.; Koludarov, I.; Jackson, T.N.W.; Holford, M.; Terrat, Y.; Casewell, N.R.; Undheim, E.A.B.; Vetter, I.; Ali, S.A.; Low, D.H.W.; et al. “Seeing the Woods for the Trees: Understanding Venom Evolution as a Guide for Biodiscovery” Venoms to Drugs: Venom as a Source for the Development of Human Therapeutics; King, G.F., Ed.; Royal Society of Chemistry: London, UK, 2013. [Google Scholar]
- Escoubas, P.; King, G.F. Venomics as a drug discovery platform. Expert Rev. Proteom. 2009, 6, 221–224. [Google Scholar] [CrossRef] [PubMed]
- Sunagar, K.; Morgenstern, D.; Reitzel, A.M.; Moran, Y. Ecological venomics: How genomics, transcriptomics and proteomics can shed new light on the ecology and evolution of venom. J. Proteom. 2015, 135, 62–72. [Google Scholar] [CrossRef] [PubMed]
- Escoubas, P.; Quinton, L.; Nicholson, G.M. Venomics: Unravelling the complexity of animal venoms with mass spectrometry. J. Mass Spectrom. 2008, 43, 279–295. [Google Scholar] [CrossRef] [PubMed]
- Holford, M.; Zhang, M.M.; Gowd, K.H.; Azam, L.; Green, B.R.; Watkins, M.; Ownby, J.P.; Yoshikami, D.; Bulaj, G.; Olivera, B.M. Pruning Nature: Biodiversity-Derived Discovery of Novel Sodium Channel Blocking Conotoxins from Conus bullatus. Toxicon 2009, 53, 90–98. [Google Scholar] [CrossRef] [PubMed]
- Olivera, B.M.; Teichert, R.W. Diversity of the neurotoxic Conus peptides: A model for concerted pharmacological discovery. Mol. Interv. 2007, 7, 251–260. [Google Scholar] [CrossRef] [PubMed]
- King, G.F. Venoms to Drugs: Venom as a Source for the Development of Human Therapeutics, RSC Drug Discovery; The Royal Society of Chemistry: London, UK, 2015. [Google Scholar]
- Castelin, M.; Puillandre, N.; Kantor, Y.I.; Modica, M.V.; Terryn, Y.; Cruaud, C.; Bouchet, P.; Holford, M. Macroevolution of venom apparatus innovations in auger snails (Gastropoda; Conoidea; Terebridae). Mol. Phylogenet. Evol. 2012, 64, 21–44. [Google Scholar] [CrossRef] [PubMed]
- Harasewych, M.G.; Adamkewicz, S.L.; Blake, J.A.; Saudek, D.; Spriggs, T.; Bult, C.J. Neogastropod phylogeny: A molecular perspective. J. Molluscan Stud. 1997, 63, 327–351. [Google Scholar] [CrossRef]
- Zou, S.; Li, Q.; Kong, L. Additional gene data and increased sampling give new insights into the phylogenetic relationships of Neogastropoda, within the caenogastropod phylogenetic framework. Mol. Phylogenet. Evol. 2011, 61, 425–435. [Google Scholar] [CrossRef] [PubMed]
- Cunha, R.L.; Grande, C.; Zardoya, R. Neogastropod phylogenetic relationships based on entire mitochondrial genomes. BMC Evol. Biol. 2009, 9, 1. [Google Scholar] [CrossRef] [PubMed]
- Holford, M.; Puillandre, N.; Terryn, Y.; Cruaud, C.; Olivera, B.; Bouchet, P. Evolution of the toxoglossa venom apparatus as inferred by molecular phylogeny of the Terebridae. Mol. Biol. Evol. 2009, 26, 15–25. [Google Scholar] [CrossRef] [PubMed]
- Holford, M.; Puillandre, N.; Modica, M.V.; Watkins, M.; Collin, R.; Bermingham, E.; Olivera, B.M. Correlating Molecular Phylogeny with Venom Apparatus Occurrence in Panamic Auger Snails (Terebridae). PLoS ONE 2009, 4, e7667. [Google Scholar] [CrossRef] [PubMed]
- Heath, T.A.; Hedtke, S.M.; Hillis, D.M. Taxon sampling and the accuracy of phylogenetic analyses. J. Syst. Evol. 2008, 46, 239–257. [Google Scholar]
- Hedtke, S.M.; Townsend, T.M.; Hillis, D.M. Resolution of phylogenetic conflict in large data sets by increased taxon sampling. Syst. Biol. 2006, 55, 522–529. [Google Scholar] [CrossRef] [PubMed]
- Huelsenbeck, J.P.; Bollback, J.P.; Levine, A.M. Inferring the root of a phylogenetic tree. Syst. Biol. 2002, 51, 32–43. [Google Scholar] [CrossRef] [PubMed]
- Graham, S.W.; Olmstead, R.G.; Barrett, S.C.H. Rooting phylogenetic trees with distant outgroups: A case study from the commelinoid monocots. Mol. Biol. Evol. 2002, 19, 1769–1781. [Google Scholar] [CrossRef] [PubMed]
- Puillandre, N.; Bouchet, P.; Duda, T.F.; Kauferstein, S.; Kohn, A.J.; Olivera, B.M.; Watkins, M.; Meyer, C. Molecular phylogeny and evolution of the cone snails (Gastropoda, Conoidea). Mol. Phylogenet. Evol. 2014, 78, 290–303. [Google Scholar] [CrossRef] [PubMed]
- Miller, B.A. Feeding mechanisms of the family Terebridae. Ann. Rep. Am. Mal. Union 1971, 1970, 72–74. [Google Scholar]
- Miller, B.A. Studies on the Biology of Indo-Pacific Terebra. Ph.D. Dissertation, University of New Hampshire, Durham, NH, USA, 1970. [Google Scholar]
- Fedosov, A.E.; Tiunov, A.V.; Kiyashko, S.I.; Kantor, Y.I. Trophic diversification in the evolution of the predatory marine gastropods of the family Terebridae as inferred from stable isotope data. Mar. Ecol. Prog. Ser. 2014, 497, 143–156. [Google Scholar] [CrossRef]
- Kantor, Y.I.; Puillandre, N. Evolution of the Radular Apparatus in Conoidea (Gastropoda: Neogastropoda) as Inferred from a Molecular Phylogeny. Malacologia 2012, 55, 55–90. [Google Scholar] [CrossRef]
- Kantor, Y.I.; Taylor, J.D. Formation of marginal radular teeth in Conoidea (Neogastropoda) and the evolution of the hypodermic envenomation mechanism. J. Zool. 2000, 252, 251–262. [Google Scholar] [CrossRef]
- Smith, W.L.; Wheeler, W.C. Venom evolution widespread in fishes: A phylogenetic road map for the bioprospecting of piscine venoms. J. Hered. 2006, 97, 206–217. [Google Scholar] [CrossRef] [PubMed]
- Fry, B.G.; Vidal, N.; Norman, J.A.; Vonk, F.J.; Scheib, H.; Ramjan, S.F.R.; Kuruppu, S.; Fung, K.; Hedges, S.B.; Richardson, M.K.; et al. Early evolution of the venom system in lizards and snakes. Nature 2006, 439, 584–588. [Google Scholar] [CrossRef] [PubMed]
- Hargreaves, A.D.; Swain, M.T.; Logan, D.W.; Mulley, J.F. Testing the Toxicofera: Comparative transcriptomics casts doubt on the single, early evolution of the reptile venom system. Toxicon 2014, 92, 140–156. [Google Scholar] [CrossRef] [PubMed]
- Marcus, E.; Marcus, E. On Hastula cinerea. Boletins da Faculdade de Filosofia, Ciencias e Letras. Universidade de Sao Paulo, Zoologica 1960, 23, 25–66. [Google Scholar]
- Taylor, J.; Miller, J. A new type of gastropod proboscis: The foregut of Hastula bacillus (Gastropoda: Terebridae). J. Zool. 1990, 220, 603–617. [Google Scholar] [CrossRef]
- Barghi, N.; Concepcion, G.P.; Olivera, B.M.; Lluisma, A.O. Comparison of the Venom Peptides and Their Expression in Closely Related Conus Species: Insights into Adaptive Post-speciation Evolution of Conus Exogenomes. Genome Biol. Evol. 2015, 7, 1797–1814. [Google Scholar] [CrossRef] [PubMed]
- Duda, T.F., Jr.; Kohn, A.J.; Palumbi, S.R. Origins of diverse feeding ecologies within Conus, a genus of venomous marine gastropods. Biol. J. Linn. Soc. 2001, 73, 391–409. [Google Scholar] [CrossRef]
- Dutertre, S.; Jin, A.H.; Vetter, I.; Hamilton, B.; Sunagar, K.; Lavergne, V.; Dutertre, V.; Fry, B.G.; Antunes, A.; Venter, D.J.; et al. Evolution of separate predation- and defence-evoked venoms in carnivorous cone snails. Nat. Commun. 2014, 5, 3521. [Google Scholar] [CrossRef] [PubMed]
- Anand, P.; Grigoryan, A.; Bhuiyan, M.H.; Ueberheide, B.; Russell, V.; Quinoñez, J.; Moy, P.; Chait, B.T.; Poget, S.F.; Holford, M. Sample Limited Characterization of a Novel Disulfide-Rich Venom Peptide Toxin from Terebrid Marine Snail Terebra variegata. PLoS ONE 2014, 9, e94122. [Google Scholar] [CrossRef] [PubMed]
- Lluisma, A.O.; Milash, B.A.; Moore, B.; Olivera, B.M.; Bandyopadhyay, P.K. Novel venom peptides from the cone snail Conus pulicaricus discovered throguh next-generation sequencing of its venom duct transcriptome. Mar. Genom. 2012, 5, 43–51. [Google Scholar] [CrossRef] [PubMed]
- Von Reumont, B.M.; Campbell, L.I.; Richter, S.; Hering, L.; Sykes, D.; Hetmank, J.; Jenner, R.A.; Bleidorn, C. A Polychaete’s Powerful Punch: Venom Gland Transcriptomics of Glycera Reveals a Complex Cocktail of Toxin Homologs. Genome Biol. Evol. 2014, 6, 2406–2423. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Gerstein, M.; Snyder, M. RNA-Seq: A revolutionary tool for transcriptomics. Nat. Rev. Genet. 2009, 10, 57–63. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Yu, H.; Xue, W.; Yue, Y.; Liu, S.; Xing, R.; Li, P. Jellyfish venomics and venom gland transcriptomics analysis of Stomolophus meleagris to reveal the toxins associated with sting. J. Proteom. 2014, 106C, 17–29. [Google Scholar] [CrossRef] [PubMed]
- Violette, A.; Biass, D.; Dutertre, S.; Koua, D.; Piquemal, D.; Pierrat, F.; Stöcklin, R.; Favreau, P. Large-scale discovery of conopeptides and conoproteins in the injectable venom of a fish-hunting cone snail using a combined proteomic and transcriptomic approach. J. Proteom. 2012, 75, 5215–5225. [Google Scholar] [CrossRef] [PubMed]
- Tayo, L.L.; Lu, B.; Cruz, L.J.; Yates, J.R. Proteomic Analysis Provides Insights on Venom Processing in Conus textile. J. Proteome Res. 2010, 9, 2292–2301. [Google Scholar] [CrossRef] [PubMed]
- Petras, D.; Heiss, P.; Süssmuth, R.D.; Calvete, J.J. Venom Proteomics of Indonesian King Cobra, Ophiophagus hannah: Integrating Top-Down and Bottom-Up Approaches. J. Proteome Res. 2015, 14, 2539–2556. [Google Scholar] [CrossRef] [PubMed]
- Wong, E.S.W.; Morgenstern, D.; Mofiz, E.; Gombert, S.; Morris, K.M.; Temple-Smith, P.; Renfree, M.B.; Whittington, C.M.; King, G.F.; Warren, W.C.; et al. Proteomics and deep sequencing comparison of seasonally active venom glands in the platypus reveals novel venom peptides and distinct expression profiles. Mol. Cell. Proteom. 2012, 11, 1354–1364. [Google Scholar] [CrossRef] [PubMed]
- Margres, M.J.; McGivern, J.J.; Wray, K.P.; Seavy, M.; Calvin, K.; Rokyta, D.R. Linking the transcriptome and proteome to characterize the venom of the eastern diamondback rattlesnake (Crotalus adamanteus). J. Proteom. 2014, 96, 145–158. [Google Scholar] [CrossRef] [PubMed]
- Sanz, L.; Lisle Gibbs, H.; Mackessy, S.P.; Calvete, J.J. Venom proteomes of closely related Sistrurus rattlesnakes with divergent diets. J. Proteome Res. 2006, 5, 2098–2112. [Google Scholar] [CrossRef] [PubMed]
- Calvete, J.J.; Sanz, L.; Pérez, A.; Borges, A.; Vargas, A.M.; Lomonte, B.; Angulo, Y.; Gutiérrez, J.M.; Chalkidis, H.M.; Mourão, R.H.V.; et al. Snake population venomics and antivenomics of Bothrops atrox: Paedomorphism along its transamazonian dispersal and implications of geographic venom variability on snakebite management. J. Proteom. 2011, 74, 510–527. [Google Scholar] [CrossRef] [PubMed]
- Nesvizhskii, A.I. Proteogenomics: Concepts, applications and computational strategies. Nat. Methods 2014, 11, 1114–1125. [Google Scholar] [CrossRef] [PubMed]
- Kaas, Q.; Westermann, J.C.; Halai, R.; Wang, C.K.L.; Craik, D.J. ConoServer, a database for conopeptide sequences and structures. Bioinformatics 2008, 24, 445–446. [Google Scholar] [CrossRef] [PubMed]
- Kaas, Q.; Yu, R.; Jin, A.-H.; Dutertre, S.; Craik, D.J. ConoServer: Updated content, knowledge, and discovery tools in the conopeptide database. Nucleic Acids Res. 2012, 40, D325–D330. [Google Scholar] [CrossRef] [PubMed]
- Jungo, F.; Bougueleret, L.; Xenarios, I.; Poux, S. The UniProtKB/Swiss-Prot Tox-Prot program: A central hub of integrated venom protein data. Toxicon 2012, 60, 551–557. [Google Scholar] [CrossRef] [PubMed]
- Andrews, S. FastQC: A Quality Control Tool for High Throughput Sequence Data. Available online: http://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 12 April 2016).
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Haas, B.J.; Papanicolaou, A.; Yassour, M.; Grabherr, M.; Blood, P.D.; Bowden, J.; Couger, M.B.; Eccles, D.; Li, B.; Lieber, M.; et al. De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nat. Protoc. 2013, 8, 1494–1512. [Google Scholar] [CrossRef] [PubMed]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.; et al. Trinity: Recontructing a full-length transcriptome assembly without a genome from RNA-Seq data. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef] [PubMed]
- Petersen, T.N.; Brunak, S.; von Heijne, G.; Nielsen, H. SignalP 4.0: Discriminating signal peptides from transmembrane regions. Nat. Methods 2011, 8, 785–786. [Google Scholar] [CrossRef] [PubMed]
- Johnson, M.; Zaretskaya, I.; Raytselis, Y.; Merezhuk, Y.; McGinnis, S.; Madden, T.L. NCBI BLAST: A better web interface. Nucleic Acids Res. 2008, 36, W5–W9. [Google Scholar] [CrossRef] [PubMed]
- Conesa, A.; Götz, S.; García-Gómez, J.M.; Terol, J.; Talón, M.; Robles, M. Blast2GO: A universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 2005, 21, 3674–3676. [Google Scholar] [CrossRef] [PubMed]
- Götz, S.; García-Gómez, J.M.; Terol, J.; Williams, T.D.; Nagaraj, S.H.; Nueda, M.J.; Robles, M.; Talón, M.; Dopazo, J.; Conesa, A. High-throughput functional annotation and data mining with the Blast2GO suite. Nucleic Acids Res. 2008, 36, 3420–3435. [Google Scholar] [CrossRef] [PubMed]
- Mulder, N.J.; Apweiler, R. The InterPro database and tools for protein domain analysis. Curr. Protoc. Bioinform. 2008. [Google Scholar] [CrossRef]
- Zdobnov, E.M.; Apweiler, R. InterProScan–an integration platform for the signature-recognition methods in InterPro. Bioinformatics 2001, 17, 847–848. [Google Scholar] [CrossRef] [PubMed]
- Kaas, Q.; Westermann, J.C.; Craik, D.J. Conopeptide characterization and classifications: An analysis using ConoServer. Toxicon 2010, 55, 1491–1509. [Google Scholar] [CrossRef] [PubMed]
- Puillandre, N.; Koua, D.; Favreau, P.; Olivera, B.M.; Stocklin, R. Molecular phylogeny, classification and evolution of conopeptides. J. Mol. Evol. 2012, 74, 297–309. [Google Scholar] [CrossRef] [PubMed]
- Quinton, L.; Demeure, K.; Dobson, R.; Gilles, N.; Gabelica, V.; De Pauw, E. New Method for Characterizing Highly Disulfide-Bridged Peptides in Complex Mixtures: Application to Toxin Identification from Crude research articles. J. Proteome Res. 2007, 6, 3216–3223. [Google Scholar] [CrossRef] [PubMed]
- Weinstock-Zlotnick, G.; Hinojosa, J. Bottom-up or top-down evaluation: Is one better than the other? Am. J. Occup. Ther. 2004, 58, 594–599. [Google Scholar] [CrossRef] [PubMed]
- Ueberheide, B.M.; Fenyö, D.; Alewood, P.F.; Chait, B.T. Rapid sensitive analysis of cysteine rich peptide venom components. Proc. Natl. Acad. Sci. USA 2009, 106, 6910–6915. [Google Scholar] [CrossRef] [PubMed]
- Safavi-Hemami, H.; Gajewiak, J.; Karanth, S.; Robinson, S.D.; Ueberheide, B.; Douglass, A.D.; Schlegel, A.; Imperial, J.S.; Watkins, M.; Bandyopadhyay, P.K.; et al. Specialized insulin is used for chemical warfare by fish-hunting cone snails. Proc. Natl. Acad. Sci. USA 2014. [Google Scholar] [CrossRef] [PubMed]
- Craig, A.G.; Bandyopadhyay, P.; Olivera, B.M. Post-translationally modified neuropeptides from Conus venoms. Eur. J. Biochem. 1999, 264, 271–275. [Google Scholar] [CrossRef] [PubMed]
- Buczek, O.; Bulaj, G.; Olivera, B.M. Conotoxins and the posttranslational modification of secreted gene products. Cell. Mol. Life Sci. 2005, 62, 3067–3079. [Google Scholar] [CrossRef] [PubMed]
- Safavi-Hemami, H.; Bulaj, G.; Olivera, B.M.; Williamson, N.A.; Purcell, A.W. Identification of Conus peptidylprolyl cis-trans isomerases (PPIases) and assessment of their role in the oxidative folding of conotoxins. J. Biol. Chem. 2010, 285, 12735–12746. [Google Scholar] [CrossRef] [PubMed]
- Barghi, N.; Concepcion, G.P.; Olivera, B.M.; Lluisma, A.O. High Conopeptide Diversity in Conus tribblei Revealed Through Analysis of Venom Duct Transcriptome Using Two High-Throughput Sequencing Platforms. Mar. Biotechnol. 2014, 17, 81–98. [Google Scholar] [CrossRef] [PubMed]
- Klint, J.K.; Senff, S.; Saez, N.J.; Seshadri, R.; Lau, H.Y.; Bende, N.S.; Undheim, E.A.B.; Rash, L.D.; Mobli, M.; King, G.F. Production of Recombinant Disulfide-Rich Venom Peptides for Structural and Functional Analysis via Expression in the Periplasm of E. coli. PLoS ONE 2013, 8, e63865. [Google Scholar] [CrossRef] [PubMed]
- Craik, D.J. Microwave-assisted Boc-solid phase peptide synthesis of cyclic cysteine-rich peptides. J. Pept. Sci. 2008, 14, 683–689. [Google Scholar]
- Han, T.S.; Zhang, M.-M.; Gowd, K.H.; Walewska, A.; Yoshikami, D.; Olivera, B.M.; Bulaj, G. Disulfide-Depleted Selenoconopeptides: Simplified Oxidative Folding of Cysteine-Rich Peptides. ACS Med. Chem. Lett. 2010, 1, 140–144. [Google Scholar] [CrossRef] [PubMed]
- Moon, J.; Gorson, J.; Wright, M.E.; Yee, L.; Khawaja, S.; Shin, H.; Karma, Y.; Lochan, R.; Michelle Yun, M.; Holford, M. Characterization and recombinant expression of terebrid venom peptide from Terebra guttata. Toxins 2016, 8, 63–75. [Google Scholar] [CrossRef] [PubMed]
- Merrifield, R.B. Solid Phase Peptide Synthesis. I. The Synthesis of Tetrapeptide. J. Am. Chem. Soc. 1963, 85, 2149. [Google Scholar] [CrossRef]
- Cheneval, O.; Schroeder, C.I.; Durek, T.; Walsh, P.; Huang, Y.H.; Liras, S.; Price, D.A.; Craik, D.J. Fmoc-based synthesis of disulfide-rich cyclic peptides. J. Org. Chem. 2014, 79, 5538–5544. [Google Scholar] [CrossRef] [PubMed]
- Chan, W.C.; White, P.D. Fmoc Solid Phase Peptide Synthesis; Oxford University Press: Oxford, UK, 2000. [Google Scholar]
- Krause, A.; Neitz, S.; Mägert, H.-J.; Schulz, A.; Forssmann, W.-G.; Schulz-Knappe, P.; Adermann, K. LEAP-1, a novel highly disulfide-bonded human peptide, exhibits antimicrobial activity. FEBS Lett. 2000, 480, 147–150. [Google Scholar] [CrossRef]
- Carstens, B.B.; Rosengren, K.J.; Gunasekera, S.; Schempp, S.; Bohlin, L.; Dahlström, M.; Clark, R.J.; Göransson, U. Isolation, Characterization, and Synthesis of the Barrettides: Disulfide-Containing Peptides from the Marine Sponge Geodia barretti. J. Nat. Prod. 2015, 78, 1886–1893. [Google Scholar] [CrossRef] [PubMed]
- Luo, S.; Zhangsun, D.; Harvey, P.J.; Kaas, Q.; Wu, Y.; Zhu, X.; Hu, Y.; Li, X.; Tsetlin, V.I.; Christensen, S.; et al. Cloning, synthesis, and characterization of αO-conotoxin GeXIVA, a potent α9α10 nicotinic acetylcholine receptor antagonist. Proc. Natl. Acad. Sci. USA 2015, 112, E4026–E4035. [Google Scholar] [CrossRef] [PubMed]
- Buczek, O.; Yoshikami, D.; Bulaj, G.; Jimenez, E.C.; Olivera, B.M. Posttranslational amino acid isomerization: A functionally important D-amino acid in an excitatory peptide. J. Biol. Chem. 2005, 280, 4247–4253. [Google Scholar] [CrossRef] [PubMed]
- Rosano, G.L.; Ceccarelli, E.A. Recombinant protein expression in Escherichia coli: Advances and challenges. Front. Microbiol. 2014, 5, 172. [Google Scholar] [CrossRef] [PubMed]
- Zhan, J.; Chen, X.; Wang, C.; Qiu, J.; Ma, F.; Wang, K.; Zheng, S. A fusion protein of conotoxin MVIIA and thioredoxin expressed in Escherichia coli has significant analgesic activity. Biochem. Biophys. Res. Commun. 2003, 311, 495–500. [Google Scholar] [CrossRef] [PubMed]
- Braud, S.; Belin, P.; Dassa, J.; Pardo, L.; Mourier, G.; Caruana, A.; Priest, B.T.; Dulski, P.; Garcia, M.L.; Ménez, A.; et al. BgK, a disulfide-containing sea anemone toxin blocking K+ channels, can be produced in Escherichia coli cytoplasm as a functional tagged protein. Protein Expr. Purif. 2004, 38, 69–78. [Google Scholar] [CrossRef] [PubMed]
- Kumar, G.S.; Ramasamy, P.; Sikdar, S.K.; Sarma, S.P. Overexpression, purification, and pharmacological activity of a biosynthetically derived conopeptide. Biochem. Biophys. Res. Commun. 2005, 335, 965–972. [Google Scholar] [CrossRef] [PubMed]
- Bayrhuber, M.; Graf, R.; Ferber, M.; Zweckstetter, M.; Imperial, J.; Garrett, J.E.; Olivera, B.M.; Terlau, H.; Becker, S. Production of recombinant Conkunitzin-S1 in Escherichia coli. Protein Expr. Purif. 2006, 47, 640–644. [Google Scholar] [CrossRef] [PubMed]
- Peng, C.; Tang, S.; Pi, C.; Liu, J.; Wang, F.; Wang, L.; Zhou, W.; Xu, A. Discovery of a novel class of conotoxin from Conus litteratus, lt14a, with a unique cysteine pattern. Peptides 2006, 27, 2174–2181. [Google Scholar] [CrossRef] [PubMed]
- Maher, C.A.; Kumar-Sinha, C.; Cao, X.; Kalyana-Sundaram, S.; Han, B.; Jing, X.; Sam, L.; Barrette, T.; Palanisamy, N.; Chinnaiyan, A.M. Transcriptome sequencing to detect gene fusions in cancer. Nature 2009, 458, 97–101. [Google Scholar] [CrossRef] [PubMed]
- Spiezia, M.C.; Chiarabelli, C.; Polticelli, F. Recombinant expression and insecticidal properties of a Conus ventricosus conotoxin-GST fusion protein. Toxicon 2012, 60, 744–751. [Google Scholar] [CrossRef] [PubMed]
- Hernandez-Cuebas, L.M.; White, M.M. Expression of a biologically-active conotoxin PrIIIE in Escherichia coli. Protein Expr. Purif. 2012, 82, 6–10. [Google Scholar] [CrossRef] [PubMed]
- Gao, B.; Zhangsun, D.; Hu, Y.; Wu, Y.; Sheng, L.; Fang, L.; Wu, X.; Yu, J.; Luo, S. Expression and secretion of functional recombinant μO-conotoxin MrVIB-His-tag in Escherichia coli. Toxicon 2013, 72, 81–89. [Google Scholar] [CrossRef] [PubMed]
- Sermadiras, I.; Revell, J.; Linley, J.E.; Sandercock, A.; Ravn, P. Recombinant expression and in vitro characterisation of active Huwentoxin-IV. PLoS ONE 2013, 8, e83202. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Wang, L.; Zhou, M.; Jiang, X.; Zhu, X.; Chen, Y.; Luo, S.; You, Y.; Ren, Z.; Xu, A. Soluble expression, purification and functional identification of the framework XV conotoxins derived from different Conus species. Peptides 2014, 56, 77–83. [Google Scholar] [CrossRef] [PubMed]
- Olivera, B.M. Conus Venom Peptides: Reflections from the Biology of Clades and Species. Annu. Rev. Ecol. Syst. 2002, 33, 25–47. [Google Scholar] [CrossRef]
- Shu, Q.; Lu, S.Y.; Gu, X.C.; Liang, S.P. The structure of spider toxin huwentoxin-II with unique disulfide linkage: Evidence for structural evolution. Protein Sci. 2002, 11, 245–252. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharyya, M.; Gupta, K.; Gowd, K.H.; Balaram, P. Rapid mass spectrometric determination of disulfide connectivity in peptides and proteins. Mol. Biosyst. 2013, 9, 1340–1350. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.R.; Hartley, B.S. Location of disulphide bridges by diagonal paper electrophoresis. The disulphide bridges of bovine chymotrypsinogen A. Biochem. J. 1966, 101, 214–228. [Google Scholar] [CrossRef] [PubMed]
- Mobli, M.; King, G.F. NMR methods for determining disulfide-bond connectivities. Toxicon 2010, 56, 849–854. [Google Scholar] [CrossRef] [PubMed]
- Gray, W.R. Disulfide structures of highly bridged peptides: A new strategy for analysis. Protein Sci. 1993, 2, 1732–1748. [Google Scholar] [CrossRef] [PubMed]
- Gorman, J.J.; Wallis, T.P.; Pitt, J.J. Protein disulfide bond determination by mass spectrometry. Mass Spectrom. Rev. 2002, 21, 183–216. [Google Scholar] [CrossRef] [PubMed]
- Kuhlman, B.; Dantas, G.; Ireton, G.C.; Varani, G.; Stoddard, B.L.; Baker, D. Design of a Novel Globular Protein Fold with Atomic-Level Accuracy. Science 2003, 302, 1364–1368. [Google Scholar] [CrossRef] [PubMed]
- Das, R.; Baker, D. Macromolecular Modeling with Rosetta. Annu. Rev. Biochem. 2008, 77, 363–382. [Google Scholar] [CrossRef] [PubMed]
- Das, R. Four Small Puzzles That Rosetta Doesn’t Solve. PLoS ONE 2011, 6, e20044. [Google Scholar] [CrossRef] [PubMed]
- Lluisma, A.O.; López-Vera, E.; Bulaj, G.; Watkins, M.; Olivera, B.M. Characterization of a novel ψ-conotoxin from Conus parius Reeve. Toxicon 2008, 51, 174–180. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.F.; Ma, R.L.; Wang, S.L.; Duan, Z.Y.; Zhang, J.H.; Wu, L.J.; Wu, C.F. Expression of an antitumor-analgesic peptide from the venom of Chinese scorpion Buthus martensii karsch in Escherichia coli. Protein Expr. Purif. 2003, 27, 253–258. [Google Scholar] [CrossRef]
- Martin, M.F.; Rochat, H. Purification of thirteen toxins active on mice from the venom of the North African scorpion Buthus occitanus tunetanus. Toxicon 1984, 22, 279–291. [Google Scholar] [CrossRef]
- Malmberg, A.B.; Gilbert, H.; McCabe, R.T.; Basbaum, A.I. Powerful antinociceptive effects of the cone snail venom-derived subtype-selective NMDA receptor antagonists conantokins G and T. Pain 2003, 101, 109–116. [Google Scholar] [CrossRef]
- Xiong, Y.M.; Lan, Z.D.; Wang, M.; Liu, B.; Liu, X.Q.; Fei, H.; Xu, L.G.; Xia, Q.C.; Wang, C.G.; Wang, D.C.; Chi, C.W. Molecular characterization of a new excitatory insect neurotoxin with an analgesic effect on mice from the scorpion Buthus martensi Karsch. Toxicon 1999, 37, 1165–1180. [Google Scholar] [CrossRef]
- Kayano, A.M.; Simões-Silva, R.; Medeiros, P.S.M.; Maltarollo, V.G.; Honorio, K.M.; Oliveira, E.; Albericio, F.; Da Silva, S.L.; Aguiar, A.C.C.; Krettli, A.U.; et al. BbMP-1, a new metalloproteinase isolated from Bothrops brazili snake venom with in vitro antiplasmodial properties. Toxicon 2015, 106, 30–41. [Google Scholar] [CrossRef] [PubMed]
- Memar, B.; Jamili, S.; Shahbazzadeh, D.; Bagheri, K.P. The first report on coagulation and phospholipase A2 activities of Persian Gulf lionfish, Pterois russelli, an Iranian venomous fish. Toxicon 2016, 113, 25–31. [Google Scholar] [CrossRef] [PubMed]
- Otvos, R.A.; Heus, F.; Vonk, F.J.; Halff, J.; Bruyneel, B.; Paliukhovich, I.; Smit, A.B.; Niessen, W.M.A.; Kool, J. Analytical workflow for rapid screening and purification of bioactives from venom proteomes. Toxicon 2013, 76, 270–281. [Google Scholar] [CrossRef] [PubMed]
- Heus, F.; Vonk, F.; Otvos, R.A.; Bruyneel, B.; Smit, A.B.; Lingeman, H.; Richardson, M.; Niessen, W.M.A.; Kool, J. An efficient analytical platform for on-line microfluidic profiling of neuroactive snake venoms towards nicotinic receptor affinity. Toxicon 2013, 61, 112–124. [Google Scholar] [CrossRef] [PubMed]
- Strong, P.N. Potassium channel toxins. Pharmacol. Ther. 1990, 46, 137–162. [Google Scholar] [CrossRef]
- Garcia, M.L.; Galvez, A.; Garcia-Calvo, M.; King, V.F.; Vazquez, J.; Kaczorowski, G.J. Use of toxins to study potassium channels. J. Bioenerg. Biomembr. 1991, 23, 615–646. [Google Scholar] [CrossRef] [PubMed]
- Dror, R.O.; Pan, A.C.; Arlow, D.H.; Borhani, D.W.; Maragakis, P.; Shan, Y.; Xu, H.; Shaw, D.E.; Lefkowitz, R.J. Pathway and mechanism of drug binding to G-protein-coupled receptors. Proc. Natl. Acad. Sci. USA 2011, 108, 13118–13123. [Google Scholar] [CrossRef] [PubMed]
- Marx, V. Watching peptide drugs grow up. Chem. Eng. News 2005, 83, 17–24. [Google Scholar]
- Craik, D.J.; Fairlie, D.P.; Liras, S.; Price, D. The Future of Peptide-based Drugs. Chem. Biol. Drug Des. 2013, 81, 136–147. [Google Scholar] [CrossRef] [PubMed]
- Kasheverov, I.E.; Zhmak, M.N.; Vulfius, C.A.; Gorbacheva, E.V.; Mordvintsev, D.Y.; Utkin, Y.N.; Van Elk, R.; Smit, A.B.; Tsetlin, V.I. α-Conotoxin analogs with additional positive charge show increased selectivity towards Torpedo californica and some neuronal subtypes of nicotinic acetylcholine receptors. FEBS J. 2006, 273, 4470–4481. [Google Scholar] [CrossRef] [PubMed]
- Pucci, L.; Grazioso, G.; Dallanoce, C.; Rizzi, L.; De Micheli, C.; Clementi, F.; Bertrand, S.; Bertrand, D.; Longhi, R.; De Amici, M.; et al. Engineering of α-conotoxin MII-derived peptides with increased selectivity for native α6β2* nicotinic acetylcholine receptors. FASEB J. 2011, 25, 3775–3789. [Google Scholar] [CrossRef] [PubMed]
- Clark, R.J.; Fischer, H.; Dempster, L.; Daly, N.L.; Rosengren, K.J.; Nevin, S.T.; Meunier, F.A.; Adams, D.J.; Craik, D.J. Engineering stable peptide toxins by means of backbone cyclization: Stabilization of the α-conotoxin MII. Proc. Natl. Acad. Sci. USA 2005, 102, 13767–13772. [Google Scholar] [CrossRef] [PubMed]
- Lovelace, E.S.; Armishaw, C.J.; Colgrave, M.L.; Wahlstrom, M.E.; Alewood, P.F.; Daly, N.L.; Craik, D.J. Cyclic MrIA: A stable and potent cyclic conotoxin with a novel topological fold that targets the norepinephrine transporter. J. Med. Chem. 2006, 49, 6561–6568. [Google Scholar] [CrossRef] [PubMed]
- Blanchfield, J.T.; Gallagher, O.P.; Cros, C.; Lewis, R.J.; Alewood, P.F.; Toth, I. Oral absorption and in vivo biodistribution of α-conotoxin MII and a lipidic analogue. Biochem. Biophys. Res. Commun. 2007, 361, 97–102. [Google Scholar] [CrossRef] [PubMed]
- Clark, R.J.; Jensen, J.; Nevin, S.T.; Callaghan, B.P.; Adams, D.J.; Craik, D.J. The engineering of an orally active conotoxin for the treatment of neuropathic pain. Angew. Chem. Int. Ed. 2010, 49, 6545–6548. [Google Scholar] [CrossRef] [PubMed]
- Murray, J.K.; Ligutti, J.; Liu, D.; Zou, A.; Poppe, L.; Li, H.; Andrews, K.L.; Moyer, B.D.; McDonough, S.I.; Favreau, P.; et al. Engineering potent and selective analogues of GpTx-1, a tarantula venom peptide antagonist of the Na(V)1.7 sodium channel. J. Med. Chem. 2015, 58, 2299–2314. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, J.; Yongye, A.B.; Chang, Y.P.; Gyanda, R.; Medina-Franco, J.L.; Armishaw, C.J. Design and synthesis of α-conotoxin GID analogues as selective α4β2 nicotinic acetylcholine receptor antagonists. Biopolymers 2014, 102, 78–87. [Google Scholar] [CrossRef] [PubMed]
- Millard, E.L.; Nevin, S.T.; Loughnan, M.L.; Nicke, A.; Clark, R.J.; Alewood, P.F.; Lewis, R.J.; Adams, D.J.; Craik, D.J.; Daly, N.L. Inhibition of neuronal nicotinic acetylcholine receptor subtypes by α-Conotoxin GID and analogues. J. Biol. Chem. 2009, 284, 4944–4951. [Google Scholar] [CrossRef] [PubMed]
- Kortemme, T.; Kim, D.E.; Baker, D. Computational alanine scanning of protein-protein interfaces. Sci. STKE 2004, 2004, pl2. [Google Scholar] [CrossRef] [PubMed]
- Bastianelli, G.; Bouillon, A.; Nguyen, C.; Crublet, E.; Pêtres, S.; Gorgette, O.; Le-Nguyen, D.; Barale, J.C.; Nilges, M. Computational Reverse-Engineering of a spider-venom derived peptide active against plasmodium falciparum SUB1. PLoS ONE 2011, 6, e21812. [Google Scholar] [CrossRef] [PubMed]
- Tyka, M.D.; Keedy, D.A.; André, I.; Dimaio, F.; Song, Y.; Richardson, D.C.; Richardson, J.S.; Baker, D. Alternate States of Proteins Revealed by Detailed Energy Landscape Mapping. J. Mol. Biol. 2011, 405, 607–618. [Google Scholar] [CrossRef] [PubMed]
- Raveh, B.; London, N.; Zimmerman, L.; Schueler-Furman, O. Rosetta FlexPepDock ab-initio: Simultaneous Folding, Docking and Refinement of Peptides onto Their Receptors. PLoS ONE 2011, 6, e18934. [Google Scholar] [CrossRef] [PubMed]
- Smith, C.A.; Kortemme, T. Backrub-Like Backbone Simulation Recapitulates Natural Protein Conformational Variability and Improves Mutant Side-Chain Prediction. J. Mol. Biol. 2008, 380, 742–756. [Google Scholar] [CrossRef] [PubMed]
- Renfrew, P.D.; Choi, E.J.; Bonneau, R.; Kuhlman, B. Incorporation of noncanonical amino acids into rosetta and use in computational protein-peptide interface design. PLoS ONE 2012, 7, e32637. [Google Scholar] [CrossRef] [PubMed]
- Lyskov, S.; Chou, F.-C.; Der, B.S.; Drew, K.; Kuroda, D.; Xu, J.; Weitzner, B.D.; Renfrew, P.D.; Sripakdeevong, P.; Borgo, B.; et al. Serverification of Molecular Modeling Applications: The Rosetta Online Server That Includes Everyone (ROSIE). PLoS ONE 2013, 8, e63906. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z. PAML 4: Phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 2007, 24, 1586–1591. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z. PAML: A program package for phylogenetic analysis by maximum likelihood. Comput. Appl. Biosci. 1997, 13, 555–556. [Google Scholar] [CrossRef] [PubMed]
- Pond, S.L.K.; Frost, S.D.W.; Muse, S. V HyPhy: Hypothesis testing using phylogenies. Bioinformatics 2005, 21, 676–679. [Google Scholar] [CrossRef] [PubMed]
- Zhu, S.; Bosmans, F.; Tytgat, J. Adaptive Evolution of Scorpion Sodium Channel Toxins. J. Mol. Evol. 2004, 58, 145–153. [Google Scholar] [CrossRef] [PubMed]
- Nakashima, K.; Ogawa, T.; Oda, N.; Hattori, M.; Sakaki, Y.; Kihara, H.; Ohno, M. Accelerated evolution of Trimeresurus flavoviridis venom gland phospholipase A2 isozymes. Proc. Natl. Acad. Sci. USA 1993, 90, 5964–5968. [Google Scholar] [CrossRef] [PubMed]
- Duda, T.F.; Palumbi, S.R. Molecular genetics of ecological diversification: Duplication and rapid evolution of toxin genes of the venomous gastropod Conus. Proc. Natl. Acad. Sci. USA 1999, 96, 6820–6823. [Google Scholar] [CrossRef] [PubMed]
- Starrett, J.; Waters, E.R. Positive natural selection has driven the evolution of the Hsp70s in Diguetia spiders. Biol. Lett. 2007, 3, 439–444. [Google Scholar] [CrossRef] [PubMed]
- Zhu, S. Positive selection targeting the cathelin-like domain of the antimicrobial cathelicidin family. Cell. Mol. Life Sci. 2008, 65, 1285–1294. [Google Scholar] [CrossRef] [PubMed]
- Sunagar, K.; Moran, Y. The Rise and Fall of an Evolutionary Innovation: Contrasting Strategies of Venom Evolution in Ancient and Young Animals. PLoS Genet. 2015, 11, e1005596. [Google Scholar] [CrossRef] [PubMed]
- Margres, M.J.; Aronow, K.; Loyacano, J.; Rokyta, D.R. The venom-gland transcriptome of the eastern coral snake (Micrurus fulvius) reveals high venom complexity in the intragenomic evolution of venoms. BMC Genom. 2013, 14, 531. [Google Scholar] [CrossRef] [PubMed]
- Guest, L.; Herbert, G.S.; Gastaldo, R.A.; Harries, P.J.; Oches, E.A.; Portell, R.; Dietl, G. Can predator-prey arms races intensify during a mass extinction event: Strombid gastropods from the Late Neogene of Florida. Geol. Soc. Am. Abstr. Progr. 2008, 40, 142. [Google Scholar]
- Weinberger, H.; Moran, Y.; Gordon, D.; Turkov, M.; Kahn, R.; Gurevitz, M. Positions under positive selection-key for selectivity and potency of scorpion α-toxins. Mol. Biol. Evol. 2010, 27, 1025–1034. [Google Scholar] [CrossRef] [PubMed]
- Gurevitz, M.; Zilberberg, N. Advances in molecular genetics of scorpion neurotoxins. J. Toxicol. 1994, 13, 65–100. [Google Scholar] [CrossRef]
- Gordon, D.; Karbat, I.; Ilan, N.; Cohen, L.; Kahn, R.; Gilles, N.; Dong, K.; Stühmer, W.; Tytgat, J.; Gurevitz, M. The differential preference of scorpion α-toxins for insect or mammalian sodium channels: Implications for improved insect control. Toxicon 2007, 49, 452–472. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Gao, B.; Zhu, S. Target-Driven Evolution of Scorpion Toxins. Sci. Rep. 2015, 5, 14973. [Google Scholar] [CrossRef] [PubMed]
- Anand, P.; O’Neil, A.; Lin, E.; Douglas, T.; Holford, M. Tailored delivery of analgesic ziconotide across a blood brain barrier model using viral nanocontainers. Sci. Rep. 2015, 5, 12497. [Google Scholar] [CrossRef] [PubMed]
- Schmidtko, A.; Lötsch, J.; Freynhagen, R.; Geisslinger, G. Ziconotide for treatment of severe chronic pain. Lancet 2010, 375, 1569–1577. [Google Scholar] [CrossRef]
- Tsomaia, N. Peptide therapeutics: Targeting the undruggable space. Eur. J. Med. Chem. 2015, 94, 459–470. [Google Scholar] [CrossRef] [PubMed]
- Lian, T.; Ho, R.J. Trends and developments in liposome drug delivery systems. J. Pharm. Sci. 2001, 90, 667–680. [Google Scholar] [CrossRef] [PubMed]
- Fleige, E.; Quadir, M.A.; Haag, R. Stimuli-responsive polymeric nanocarriers for the controlled transport of active compounds: Concepts and applications. Adv. Drug Deliv. Rev. 2012, 64, 866–884. [Google Scholar] [CrossRef] [PubMed]
- Tamanoi, F.; Zink, J.I. Multifunctional Inorganic Nanoparticles for Imaging, Targeting, and Drug Delivery. ACS Nano 2008, 2, 889–896. [Google Scholar]
- Wei, Z.; Matsui, H. Synthesis in reverse micelle reactors. Nat. Commun. 2014, 5, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Douglas, S.M.; Bachelet, I.; Church, G.M. A Logic-Gated Nanorobot for Targeted Transport of Molecular Payloads. Science 2012, 335, 831–834. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Nolte, R.J.M.; Cornelissen, J.J.L.M. Virus-based nanocarriers for drug delivery. Adv. Drug Deliv. Rev. 2012, 64, 811–825. [Google Scholar] [CrossRef] [PubMed]
- Schoonen, L.; van Hest, J.C.M. Functionalization of protein-based nanocages for drug-delivery applications. Nanoscale 2014, 6, 7124–7141. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.; Uchida, M.; Oneil, A.; Li, R.; Prevelige, P.E.; Douglas, T. Implementation of P22 viral capsids as nanoplatforms. Biomacromolecules 2010, 11, 2804–2809. [Google Scholar] [CrossRef] [PubMed]
- O’Neil, A.; Reichhardt, C.; Johnson, B.; Prevelige, P.E.; Douglas, T. Genetically programmed in vivo packaging of protein cargo and its controlled release from bacteriophage P22. Angew. Chem. Int. Ed. Engl. 2011, 50, 7425–7428. [Google Scholar] [CrossRef] [PubMed]
- O’Neil, A.L. Engineering Bacteriophage P22 as Nanomaterial; Montana State University: Bozeman, MT, USA, 2013. [Google Scholar]
- Kelly, P.; Anand, P.; Uvaydov, A.; Chakravartula, S.; Sherpa, C.; Pires, E.; O’Neil, A.; Douglas, T.; Holford, M. Developing a Dissociative Nanocontainer for Peptide Drug Delivery. Int. J. Environ. Res. Public Health 2015, 12, 12543–12555. [Google Scholar] [CrossRef] [PubMed]
- Wörsdörfer, B.; Woycechowsky, K.J.; Hilvert, D. Directed evolution of a protein container. Science 2011, 331, 589–592. [Google Scholar] [CrossRef] [PubMed]
- Sutthasupa, S.; Shiotsuki, M.; Sanda, F. Recent advances in ring-opening metathesis polymerization, and application to synthesis of functional materials. Polym. J. 2010, 42, 905–915. [Google Scholar] [CrossRef]
- Leitgeb, A.; Wappel, J.; Slugovc, C. The ROMP toolbox upgraded. Polym. J. 2010, 51, 2927–2946. [Google Scholar] [CrossRef]
- France, M.B.; Uffelman, E.S. Ring-Opening Metatheiss with a Well-Defined Ruthenium Carbene Complex an Experiment for the Undergraduate Inorganic or Polymer Laboratory. J. Chem. Educ. 1999, 76, 661–665. [Google Scholar] [CrossRef]
- Hong, S.H.; Grubbs, R.H. Highly Active Water-Soluble Olefin Metathesis Catalyst. J. Am. Chem. Soc. 2006, 10, 3508–3509. [Google Scholar] [CrossRef] [PubMed]
- Schrock, R.R. Olefin Metathesis by Well-Defined Complexes of Molybdenum and Tungsten. In Alkene Metathesis in Organic Synthesis; Springer Berlin Heidelberg: Heidelberg, Germany, 2001; pp. 1–36. [Google Scholar]
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Verdes, A.; Anand, P.; Gorson, J.; Jannetti, S.; Kelly, P.; Leffler, A.; Simpson, D.; Ramrattan, G.; Holford, M. From Mollusks to Medicine: A Venomics Approach for the Discovery and Characterization of Therapeutics from Terebridae Peptide Toxins. Toxins 2016, 8, 117. https://doi.org/10.3390/toxins8040117
Verdes A, Anand P, Gorson J, Jannetti S, Kelly P, Leffler A, Simpson D, Ramrattan G, Holford M. From Mollusks to Medicine: A Venomics Approach for the Discovery and Characterization of Therapeutics from Terebridae Peptide Toxins. Toxins. 2016; 8(4):117. https://doi.org/10.3390/toxins8040117
Chicago/Turabian StyleVerdes, Aida, Prachi Anand, Juliette Gorson, Stephen Jannetti, Patrick Kelly, Abba Leffler, Danny Simpson, Girish Ramrattan, and Mandë Holford. 2016. "From Mollusks to Medicine: A Venomics Approach for the Discovery and Characterization of Therapeutics from Terebridae Peptide Toxins" Toxins 8, no. 4: 117. https://doi.org/10.3390/toxins8040117
APA StyleVerdes, A., Anand, P., Gorson, J., Jannetti, S., Kelly, P., Leffler, A., Simpson, D., Ramrattan, G., & Holford, M. (2016). From Mollusks to Medicine: A Venomics Approach for the Discovery and Characterization of Therapeutics from Terebridae Peptide Toxins. Toxins, 8(4), 117. https://doi.org/10.3390/toxins8040117