1. Introduction
B. cereus sensu lato is a complex group of eight species genetically close but distinct in terms of pathogenicity. Indeed, this group comprises beneficial bacteria such as
Bacillus toyonensis, used as probiotic or
Bacillus thuringiensis, applied in agriculture as biopesticide, but also highly pathogenic species, like
Bacillus anthracis, responsible for the anthrax disease. Moreover, some strains of
B. cereus sensu stricto behave as opportunistic pathogens involved in non-gastrointestinal (e.g., endophtalmitis, periodontitis, meningitis or pneumonia) and gastrointestinal infections [
1] with two types of symptoms: emetic or diarrhoeal.
The emetic syndrome is characterized by nausea and vomiting that occur 0.5 to 6 h after the meal. The symptoms are generally mild and disappear after less than 24 h, even though several complication cases have been reported [
2,
3,
4,
5,
6]. The emetic syndrome is associated with a heat-stable toxin, the cereulide, which is generally preformed in food and resists to proteolysis and extreme pH [
7]. The emesis seems to be due to the stimulation, by cereulide, of the efferent vagus nerve receptors (5-HT3) [
8].
The diarrhoeal syndrome is characterized by abdominal pain, profuse diarrhoea with sometimes nausea. These symptoms occur 8 to 16 h after ingestion of
B. cereus contaminated meal (infection dose: 10
5–10
7 CFU) and are probably due to the production of one or several enterotoxins by the
B. cereus strains in the small intestine [
9]. However, full light has not yet been shed on the exact molecules responsible for these symptoms. The most regularly cited enterotoxin candidates are: haemolysin BL (Hbl) [
10], non-haemolytic enterotoxin (Nhe) [
11] or cytotoxin K (CytK) [
12]. Hbl is the only toxin for which a diarrhoeal activity was clearly demonstrated in vivo on rabbit intestines, using the purified toxin. Cytotoxic, haemolytic, vascular permeability and dermonecrotic activities of Hbl have also been demonstrated [
13]. Nhe was first isolated from a foodborne outbreak in Norway [
11]. A significant cytotoxicity effect of this toxin was observed on Vero and Caco-2 cells, although its haemolytic activity was limited [
14,
15]. Hbl and Nhe are tripartite enterotoxin complexes and belong to the α-helical pore-forming toxins family (α-PFT), like the Haemolysin E (HlyE) from
Escherichia coli [
15,
16,
17]. CytK is a single protein of 34 kDa belonging to the β-barrel pore-forming toxin family including other Gram-positive enterotoxins such as the α-haemolysin from
Staphylococcus aureus or the β-toxin from
Clostridium perfringens [
12]. Two CytK variants have been described, CytK-1 and CytK-2. They display different pathogenicity with CytK-1 much more cytotoxic than CytK-2 on Vero and Caco-2 cells. This difference has been explained by a better conductance of the CytK-1-dependent channels than those from CytK-2 [
18]. However, a strain (NVH 883/00) carrying the genetic determinants of CytK-1, but referenced as not toxic, has been found, which suggests that the virulence may also depend on the level of gene expression [
19].
In addition to these three putative enterotoxins, other
B. cereus haemolysins could participate to the gastrointestinal disease caused by
B. cereus including: cereolysin O (CerO) [
20], haemolysin II (HlyII) [
21] and haemolysin III (HlyIII) [
22]. Three phospholipases C have also been proposed to be involved in the
B. cereus gastrointestinal syndrome: the phosphatidylinositol-specific PLC (PI-PLC), phosphatidylcholine PLC (PC-PLC) and the sphingomyelinase (Smase) [
23,
24]. Phopholipases are known to contribute to the pathogenesis of bacteria, through degradation of the mucus layer, destruction of the tissues or deregulation of cellular signalling cascades [
25]. Therefore, it has been proposed that these molecules could act in concert with other enterotoxins to elicit diarrhoea [
26]. Similarly, several studies have suggested that metalloproteases (e.g., InhA1, InhA2, InhA3 or NprA) could also contribute to the bacterial infection [
27,
28,
29]. Finally, other compounds could also act as virulence factors contributing to the diarrhoeal pathotype of
B. cereus, like the enterotoxin S (EntS) or FM (EntFM), described as cell-wall peptidase [
30,
31].
To date, no toxicological study has succeeded in demonstrating any correlation between one (or a combination) of these compounds and the diarrhoeal syndrome caused by
B. cereus. This might potentially be due to the fact that previous cytotoxicity studies of
B. cereus used CHO, Vero or Hep-2 cells. These cells lines are indeed very sensitive to toxins in general but do not necessarily reflect the specific interactions with the intestinal barrier and thereby with the diarrhoeal symptoms. Besides, several toxicological studies have been performed with human intestinal Caco-2 cells, but the cells were often not fully differentiated and therefore did not display the full characteristics of enterocytes [
32]. This is of major importance though since it was demonstrated that the toxicological response between undifferentiated and differentiated Caco-2 cell cultures differs substantially [
33]. Therefore, with the aim to better mimic the in vivo conditions of toxinogenesis, this study assessed the enterotoxicity of 70
B. cereus strains on fully differentiated Caco-2 cells. Additionally, this work also studied the interaction between the
B. cereus supernatants and the mucus layer, using a co-culture (Caco-2/HT29-MTX) cell model [
34].
4. Materials and Methods
4.1. B. cereus Collection and DNA Extraction
70
B. cereus strains selected for this study were isolated from diverse origins: foods (15 strains), food-poisonings (38), environmental (13), clinical (1) or with unknown origin (3) as summarized in
Table 1. These strains were collected within the framework of a Federal Public Service (FOD) Health, Food Chain Safety and Environment project (BACEREUS, RT09/2 project, Brussels, BE). The origin and the references related to these strains are described in
Table A3. For all the experiments, the bacteria were cultured in Luria–Bertani medium (Oxoid, Dardilly, France) at 30 °C, under agitation (120 rpm).
The strain genomic DNA was extracted and purified using the DNeasy® blood and tissue extraction kit (Qiagen, Hilden, Germany) with the recommended pre-treatment step for Gram-positive bacteria. In brief, 2 mL of an overnight culture of the bacteria in LB medium was centrifuged (10 min, 5000× g, 4 °C). The bacterial pellet was then resuspended in 180 μL of enzymatic lysis buffer (20 mM Tris-HCl, pH 8.0; 2 mM sodium EDTA; 1.2% Triton X-100; 20 mg/mL lysozyme) (Sigma-Aldrich, St. Louis, MO, USA) and incubated for 30 min at 37 °C. Then the extraction process was fully automated using the QIAcube System with the adequate protocol (Qiagen, Hilden, Germany). The purified DNA was resuspended in 150 μL of Buffer EA (10 mM Tris-HCl; 0.5 mM EDTA; pH 9.0) (Sigma-Aldrich, St. Louis, MO, USA) and stored at −20 °C for further use.
4.2. Detection of Virulence Factors and Enterotoxin Genes
The PCR screening of the genetic determinants of the main putative virulence factors involved in the
B. cereus gastrointestinal toxi-infections was performed using the GoTaq
® Green Master Mix (Promega, Fitchburg, MA, USA). The different primers used are shown in
Table 2. Thermal cycling parameters were 5 min at 94 °C, then 32 cycles of 1 min at 94 °C, 1 min at the annealing temperature (depending on the primer pair, see
Table 2) and 1 min at 68 °C, and finally 10 min at 68 °C. The success of amplification was confirmed by gel electrophoresis at 100 V for 28 min on 0.8% agarose gel (Santa Cruz Biotechnology, Dallas, TX, USA) in Tris-Acetate-EDTA buffer (Sigma-Aldrich, St. Louis, MO, USA).
For several B. cereus strains, the presence of Nhe and HBL toxins in the supernatant was also assessed using the immunodetection kit Duopath Cereus Enterotoxins (Merck, Darmstadt, Germany).
4.3. Cell Line Cultures
Human colon carcinoma Caco-2 cells used to model the intestinal mucosa were provided by Dr. M. Rescigno (clone C2E; passage 10–30; University of Milano, Milan, Italy) and were seeded in 48-well plates (Corning-Costar, Corning, NY, USA) (6 × 104 cells/well) pre-coated with type I collagen (Sigma-Aldrich, Saint-Louis, MO, USA) and incubated during 21 days after confluence. Cells were grown in Dulbecco’s modified Eagle’s minimal essential (DMEM) medium with 4.5 g/L glucose (Lonza, Basel, Switzerland). Media were supplemented with 10% (v/v) fetal bovine serum (FBS) (GE, Healthcare, Cramlington, UK), 1% (v/v) non-essential amino acids (Lonza, Basel, Switzerland), 1% (v/v) L-glutamine (Lonza, Basel, Switzerland) and 1% (v/v) penicillin-streptomycin (Lonza, Basel, Switzerland). Caco-2 cells were cultured at 37 °C under 10% CO2 and water saturated atmosphere.
Co-cultures of Caco-2 and HT29-MTX cells were used to assess the protective effect of mucin secretion. HT29 cells adapted to methotrexate (MTX, 10
−5 M) were obtained from Dr. T. Lesuffleur (INSERM U505, Villejuif, France) were used between passages 30–50 with a seeding ratio Caco-2/HT29-MTX of 3:1, as previously described by Nollevaux et al. (2006) [
34]. Together with Caco-2 cells, they form a co-culture that reproduces the two main cellular types encountered in the human intestinal epithelium.
4.4. Cytotoxicity Assays
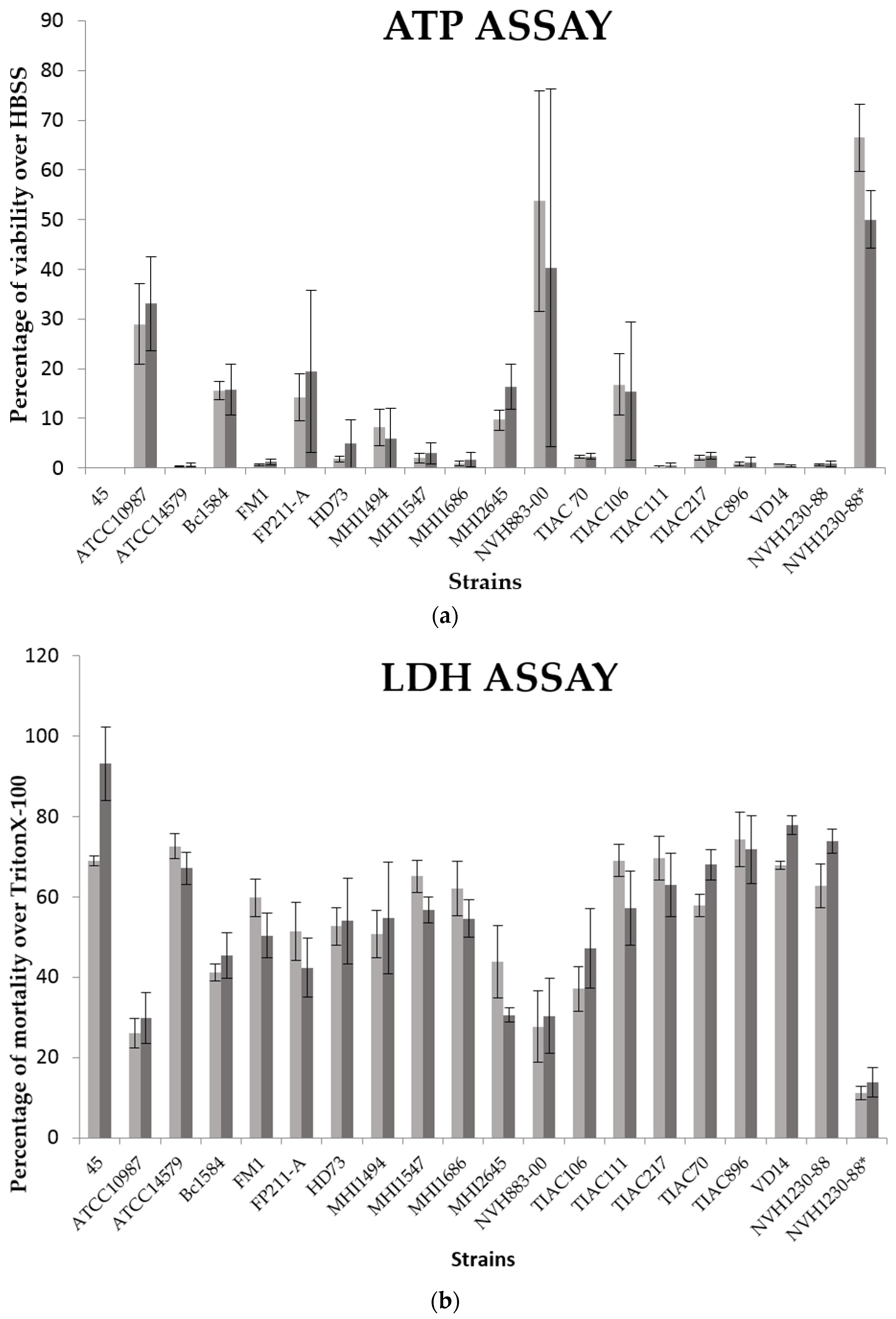
B. cereus cell-free supernatants were prepared from a 15 h liquid culture in Luria–Bertani broth medium (Oxoid, Dardilly, France) at 30 °C, under agitation (120 rpm). This culture was centrifuged (4 °C, 15 min, 9000× g) and then filtrated with a 0.22 µm porosity filter. Strain NVH1230-88, known to produce at least the Nhe, Hbl and CytK toxins, was used as positive control. The negative control consisted on the same strain (NVH1230-88) for which the toxins were degraded by heat treatment (80 °C, 20 min).
After 21 days in 48-well plates, Caco-2 cells (or Caco-2 and HT-29 cells) were washed with phosphate buffered saline (PBS) (137 mM NaCl; 2.68 mM KCl; 1.14 mM KH2PO4; 8 mM Na2HPO4.2H2O; pH 7.2) (Sigma-Aldrich, St. Louis, MO, USA), incubated during 3 h with the B. cereus supernatants, diluted two times in HBSS buffer (Lonza, Basel, Switzerland) and then washed with PBS before assays.
To assess the cytotoxic effect of these supernatants several cytotoxicity assays were performed including ATP, NR and LDH assays. The first assesses the cell viability by the cytoplasmic ATP dosage. This quantification is based on the measurement of the luminescent signal generated by the conversion of luciferin by luciferase and the cytoplasmic ATP. The second assay determines the cell viability by the spectrophotometric determination of NR (3-amino-2-methyl-phenazine hydrochloride) taken up by viable cells and stored in their lysosomes [
61]. The third test evaluates the lactate dehydrogenase (LDH) released by cells in necrosis [
62]. For each experiment, several wells of each plate were dedicated to the positive (incubation with 1% of Triton X-100) (Sigma-Aldrich, St. Louis, MO, USA) and negative (incubation with Hank’s Balanced Salt Solution (HBSS), 100%) controls.
4.4.1. Neutral Red Accumulation Assays
The treated cells (after washings) were incubated for 3 h with NR (750 µL at 0.33 mg/mL). Cells were then washed with PBS and NR was extracted using 300 µL of extracting solution (50% ethanol: 1% acetic acid) (Merck, Darmstadt, Germany). After 5–10 min of plate agitation the absorbance was measured by spectrophotometry at 540 nm [
63]. The results were then expressed as percentages of Caco-2 cells viability over cells incubated with HBSS (toxicity negative control). The mean of the replicates and standard deviation (SD) were calculated for each
B. cereus supernatant (
Table A1 and
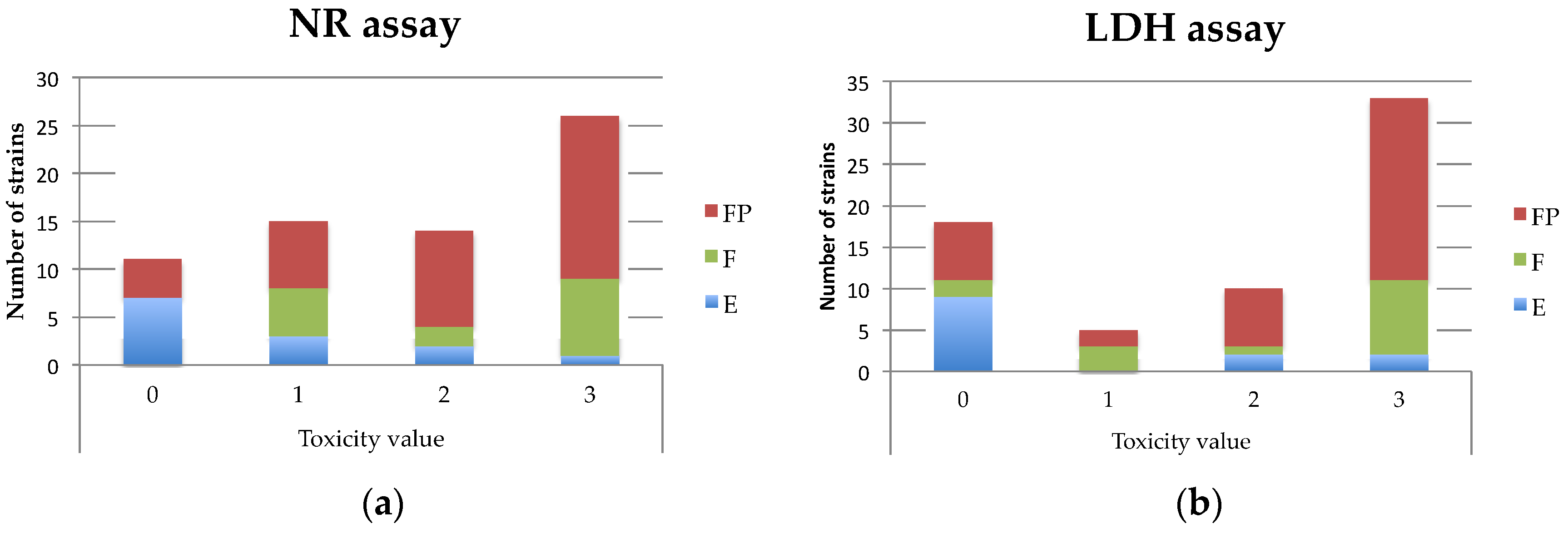
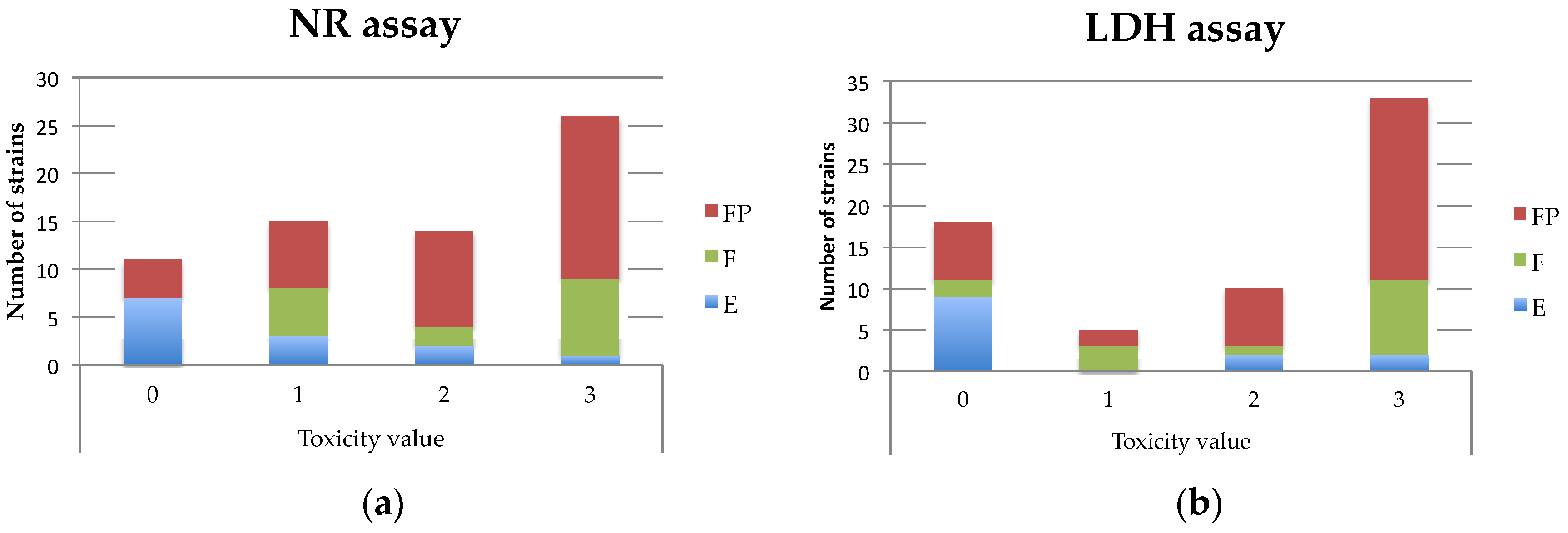
Table A3). For the monoculture assays, with the aim to facilitate the comparison with the LDH assay, a toxicity value (from 0 to 3) was attributed to each supernatant as follows: 0 (non-cytotoxic), % of viability higher than 70%; 1, % of viability between 50% and 70%; 2, % of viability between 20% and 50%; 3, % of viability less than 20% (
Table 1).
4.4.2. LDH Release Assays
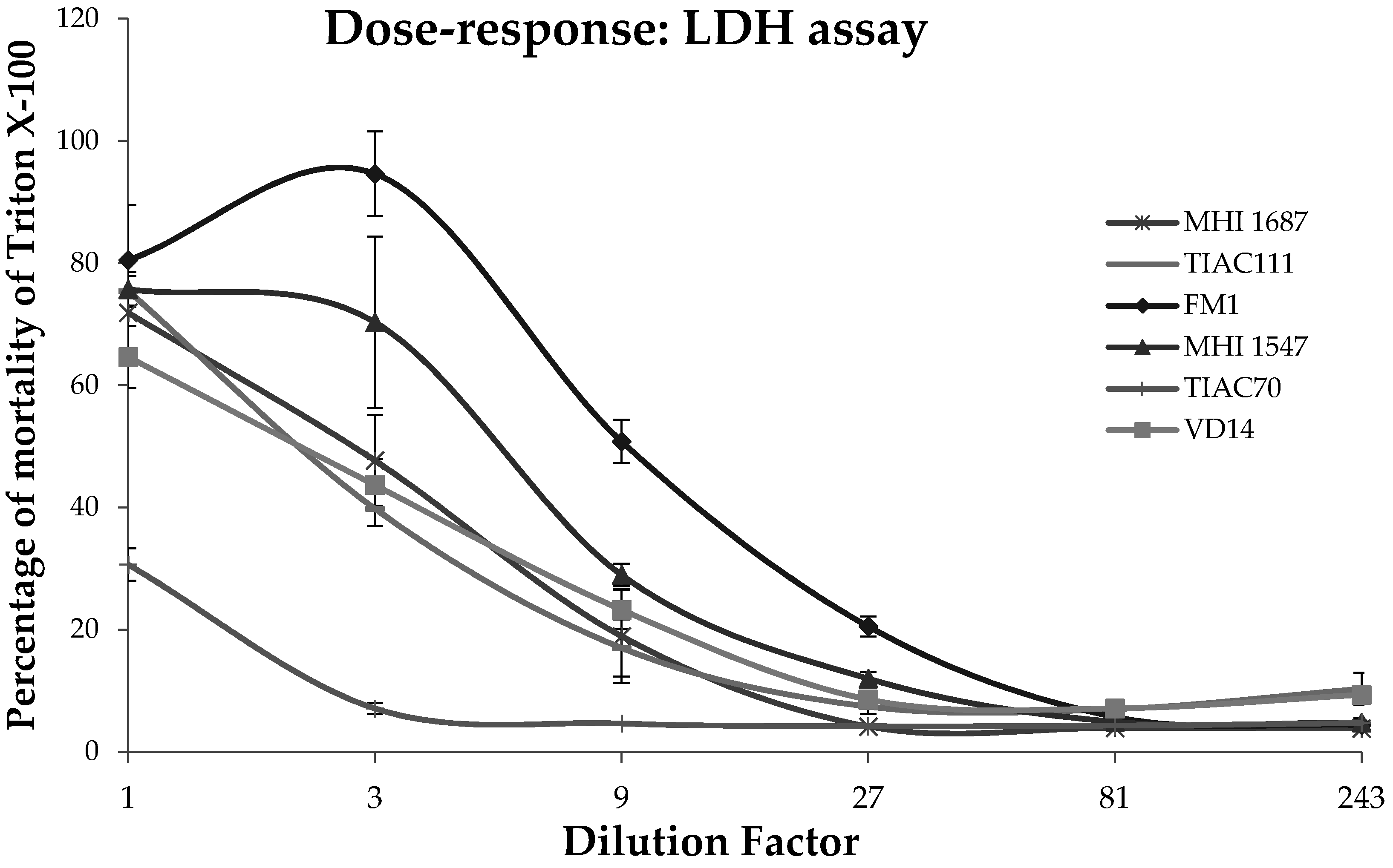
After the 3 h of exposure of the intestinal cells to the
B. cereus supernatant, the lactate dehydrogenase activity released in the culture medium was assessed using the Cytotoxicity Detection Kit (LDH) (Roche, Basel, Switzerland). The liquid culture was centrifuged to remove the cell debris. Then, 1 µL of cell-free liquid culture was mixed with 99 µL of PBS in a 96 wells plate. 250 µL of Catalyst solution (containing diaphorase and NAD
+) was mixed with 11, 25 mL of Dye solution (containing iodonitrotetrazolium and sodium lactate) and 100 µL of this reagent were added to each well. The reduction by the LDH enzyme of NAD
+ to NADH was measured by spectrophotometry at 500 nm. The results of this test were expressed in percentage of Caco-2 cell mortality over cells treated with Triton X-100 (positive control). The mean over replicates as well as their SD were calculated for each supernatant (
Table A1 and
Table A3). Similarly to NR and for the Caco-2 monoculture assays, a toxicity value (from 0 to 3) was attributed to each
B. cereus supernatant as follows: 0 (non-cytotoxic), % of mortality less than 20%; 1, % of mortality between 20% and 50%; 2, % of mortality between 50 and 70%; 3, % of mortality higher than 70% (
Table 1).
4.4.3. ATP Content Assays
The intracellular ATP content was monitored to assess the cytotoxicity of B. cereus supernatant for the co-culture assays instead of NR. Indeed, the mucin produced by the HT-29 cells could interfere with the NR uptake. The ATP content was measured using the luminescent CellTiter-Glo® assay kit (Promega, Fitchburg, WI, USA). After the B. cereus supernatant exposition, the eukaryotic cells were rinsed twice with PBS. Then, 150 μL of HBSS were added to each well during 15 min at room temperature. The CellTiter-Glo® buffer was mixed with the CellTiter-Glo® substrate and 150 μL of this reagent mix was added to the wells. The plate was agitated during 2 min to homogenize the solution and accelerate cell lysis. In order to measure the highest signal possible, 200 μL of the cell solutions were transferred into a 96 wells white polystyrene plate, specific for luminescence measurements (Nunc™, Thermo Fisher Scientific, Waltham, MA, USA). The plate was shaken for 30 s and the luminescence recorded with a fluorimeter-luminometer (Fluoroskan Ascent™ FL, Thermo Fisher Scientific, Waltham, MA, USA) every 2 min during 20 min (0.5 s of integration time). This kinetics allowed identifying the time lapse where the maximum of luminescence was observed. The results of this test were expressed in percentage of Caco-2 cell viability over cells incubated with HBSS (negative control).
4.5. Statistical Analysis
For the Caco-2 cells toxicity assays, all measures were made in triplicates and each experiment was independently repeated three times (
n = 9). Concerning the cytotoxicity comparison between co- and monocultures, all measures were made in triplicates and each experiment was repeated twice (
n = 6). The normal distribution of the values was verified. Finally, the results were expressed as mean ± SD (see
Table A1 and
Table A2) or ±SEM (
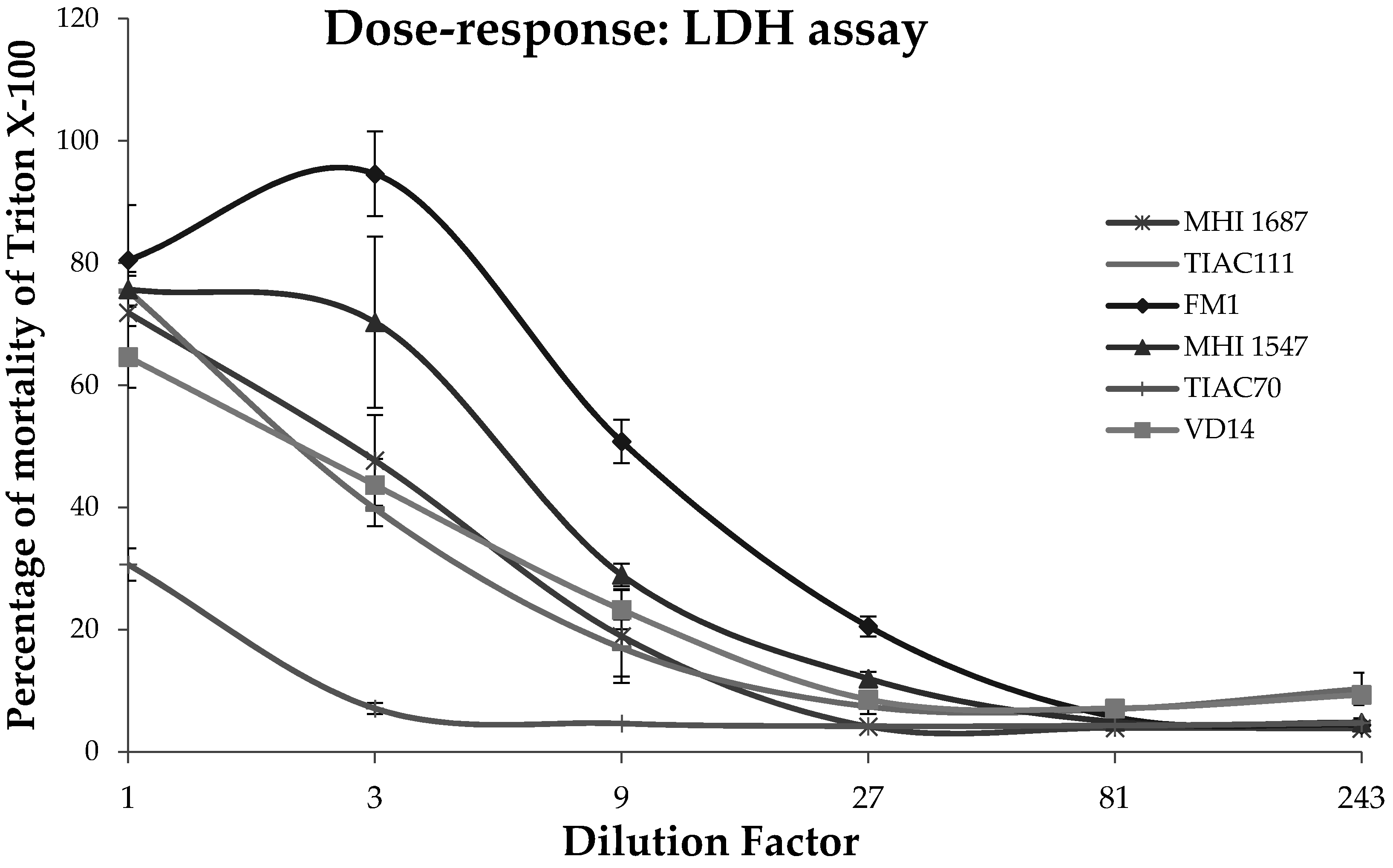
Figure 2). All the statistical correlation analyses including the contingency tables, the chi-square values and the MCA (Multi correspondence analyses) were performed with JMP 12 software (SAS Institute, Cary, NC, USA) and R statistics.



{kind=link}
{kind=link}
{kind=link}