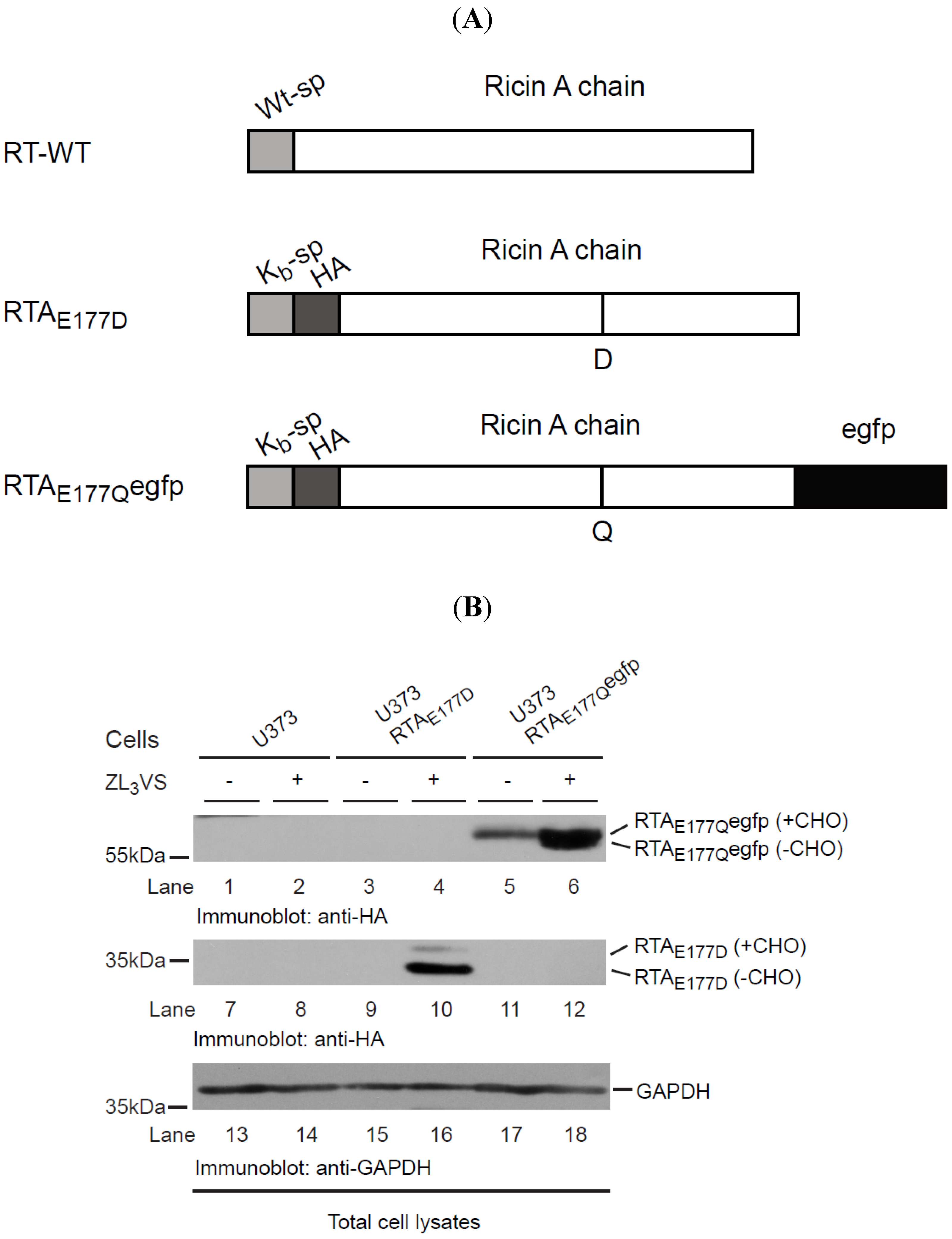
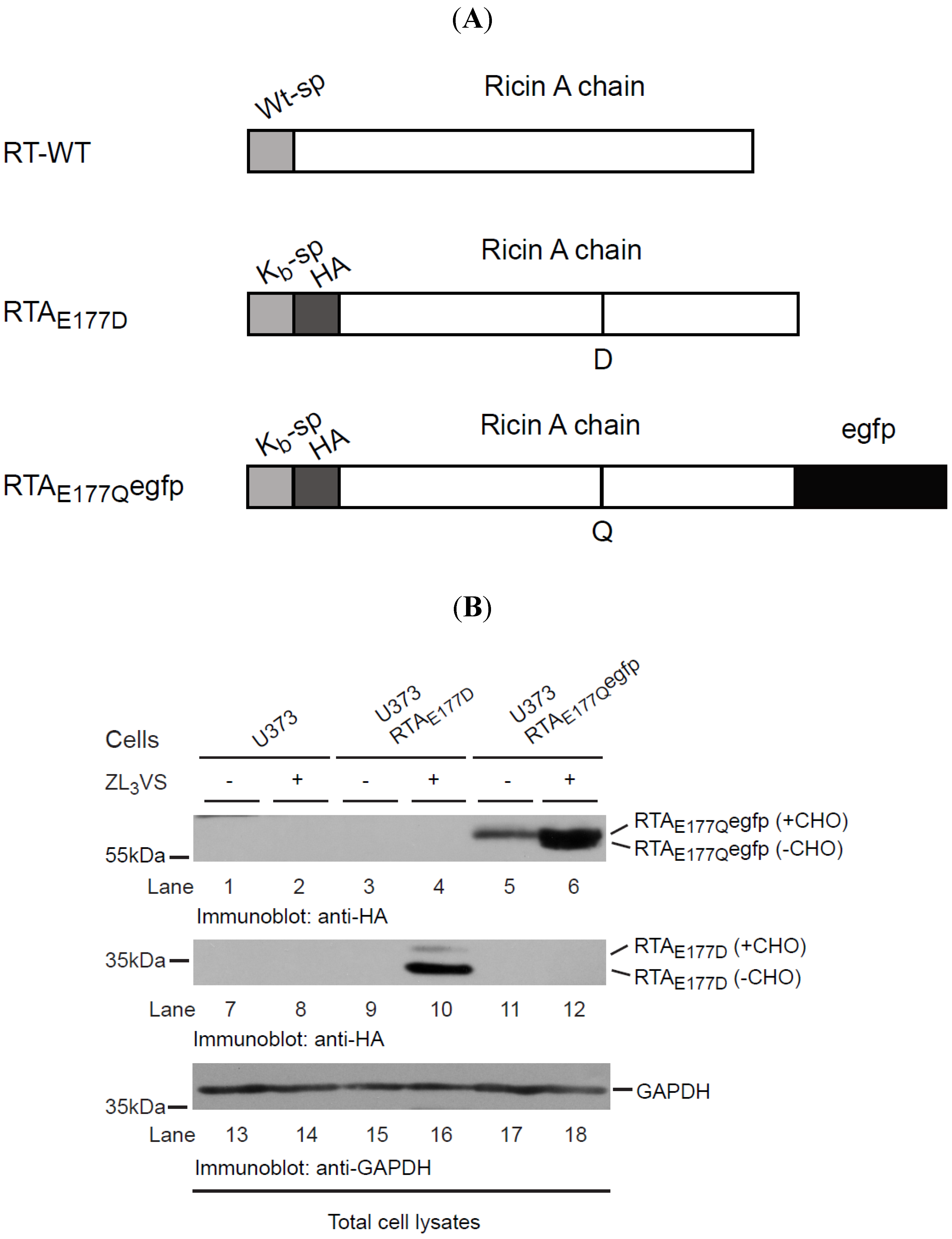
2.1. RTAE177Qegfp Molecules Are Stabilized by Inclusion of Proteasome Inhibitor
Ricin A chain is transported across the ER membrane to the cytoplasm where it inhibits protein translation through the inactivation of ribosomes [
19]. We previously established a human cell-based model to study the molecular requirements of RTA retrograde translocation using a catalytically inactive toxin because the wild type RTA inhibited protein synthesis [
18]. Utilizing this cell-based RTA assay, we planned to develop a high-throughput screen to identify cell-permeable compounds that stabilize RTA within intracellular compartments, preventing its access to the ribosome and thus limiting ricin intoxication. To that end, a chimeric RTA was generated comprised of an enzymatically-attenuated RTA molecule (RTA
E177Q) fused to an enhanced green fluorescent protein (egfp) (RTA
E177Qegfp) (
Figure 1A). Our initial experiments examined the stability of the chimeric RTA
E177Qegfp molecule expressed in human U373 cells (U373-RTA
E177Qegfp). U373 cells, U373-RTA
E177D cells, and U373-RTA
E177Qegfp cells were treated for 16 h with ZL
3VS (3 μM) and subjected to immunoblot analysis (
Figure 1B). Stabilization of RTA molecules in the ER upon inhibiting proteasome activity is a result of blocking its degradation and thus affecting the retrograde translocation event [
7,
18]. As expected, proteasome inhibitor treated U373-RTA
E177D cells induced the accumulation of two polypeptides consisting of glycosylated and deglycosylated RTA proteins (
Figure 1B, lane 10). Consistent with this result, proteasome inhibitor treatment caused an increase in RTA
E177Qegfp polypeptides consistent with glycosylated and deglycosylated polypeptides (
Figure 1B, lane 6 and
Supplemental Figure 1). The observation of diverse amounts of glycosylated and deglycosylated of RTA
E177Qegfp as compared to RTA
E177D suggests that RTA
E177Qegfp was probably dislocated with slower kinetics than RTA
E177D. Analysis of glyceraldehyde 3-phosphate dehydrogenase (GAPDH) levels confirmed equal protein loading (
Figure 1B, lanes 13–18). Collectively, the data demonstrate that RTA
E177Qegfp molecules, like RTA
E177D, gain a single N-linked glycan, and are targeted for retrograde translocation across the ER membrane where RTA species are eventually degraded in proteasome-dependent manner.
Figure 1.
RTAE177Qegfp molecules are stabilized by proteasome inhibition. (A) Schematic diagram of wild type RTA (RTA-WT) and enzymatically-defective constructs (RTAE177D and RTAE177Qegfp); (B) U373 cells and U373-RTAE177D- and U373-RTAE177Qegfp-expressing cells treated without or with ZL3VS (3 µM) were subjected to immunoblot analysis for RTAE177Qegfp (lanes 1–6), RTAE177D (lanes 7–12), and GAPDH (lanes 13–18). RTA polypeptides and molecular weight markers are indicated.
Figure 1.
RTAE177Qegfp molecules are stabilized by proteasome inhibition. (A) Schematic diagram of wild type RTA (RTA-WT) and enzymatically-defective constructs (RTAE177D and RTAE177Qegfp); (B) U373 cells and U373-RTAE177D- and U373-RTAE177Qegfp-expressing cells treated without or with ZL3VS (3 µM) were subjected to immunoblot analysis for RTAE177Qegfp (lanes 1–6), RTAE177D (lanes 7–12), and GAPDH (lanes 13–18). RTA polypeptides and molecular weight markers are indicated.
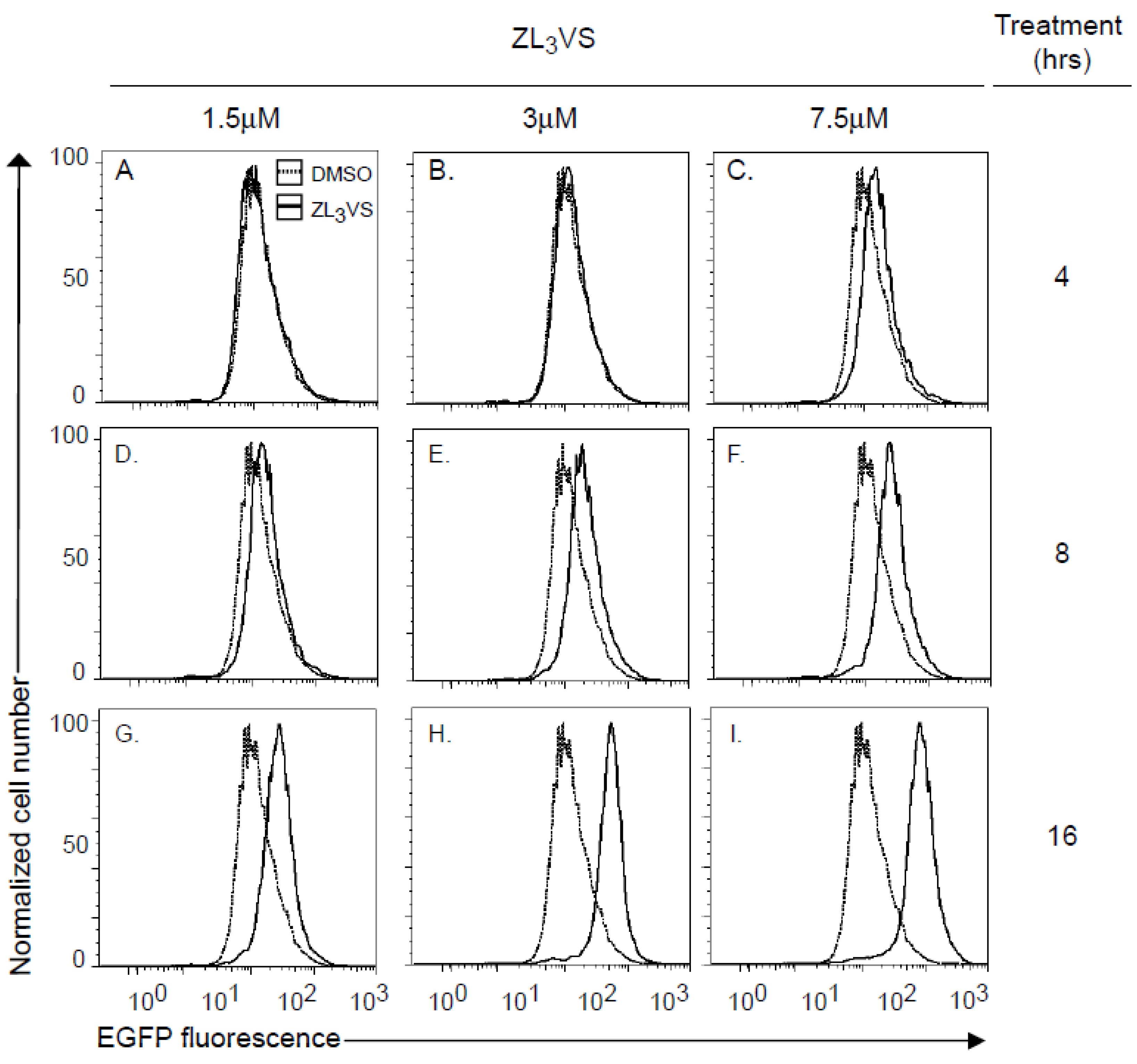
2.2. Development of a High-Content Assay to Identify Compounds that Stabilize Ricin A Chain
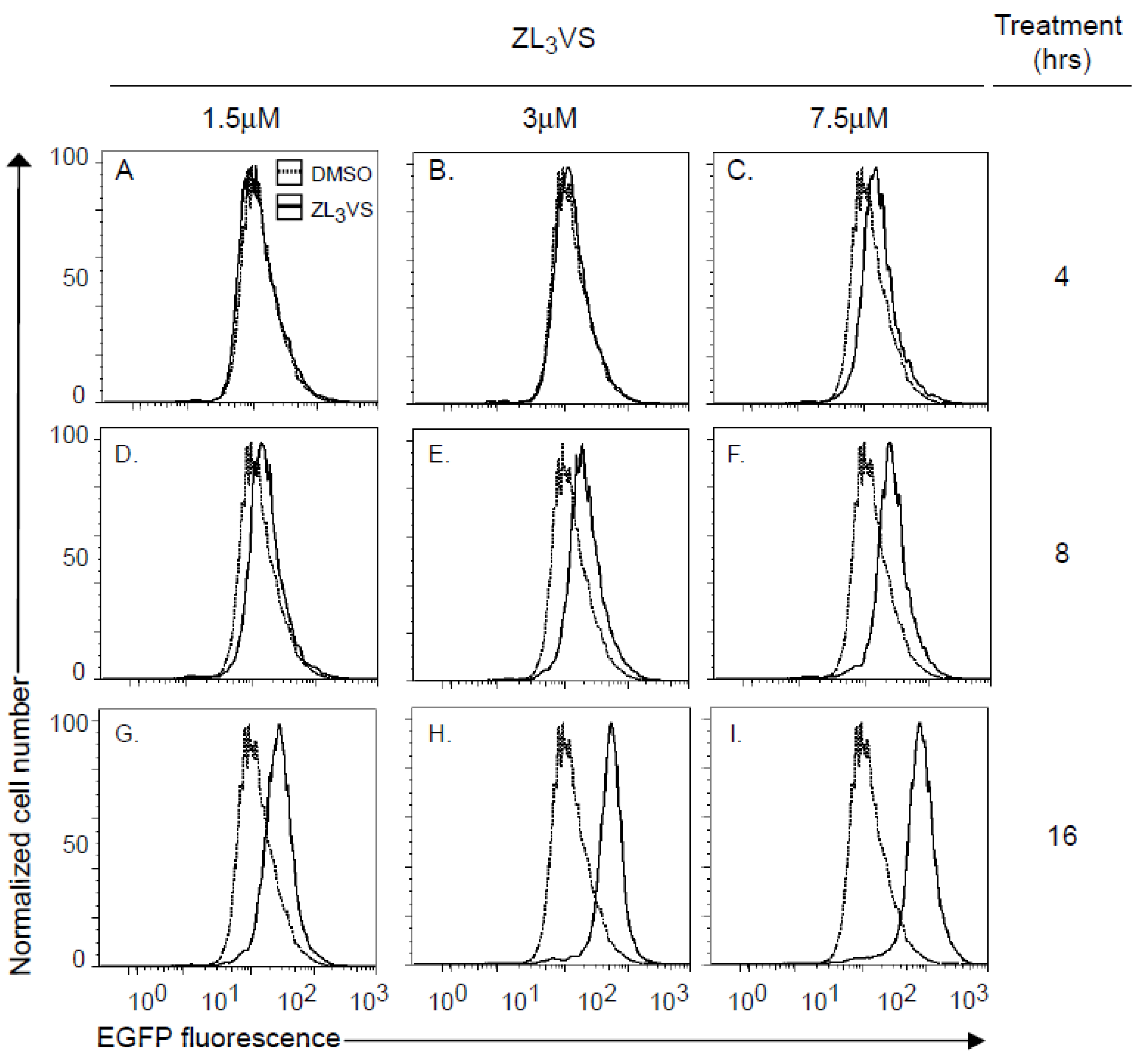
Using U373-RTA
E177Qegfp cells, we defined optimal conditions to measure the fluorescence signal from stabilized RTA
E177Qegfp molecules. U373-RTA
E177Qegfp cells untreated or treated for 4, 8, or 16 h with 1.5, 3 or 7.5 µM ZL
3VS were subjected to analysis by flow cytometry (
Figure 2). Cells treated with 1.5 or 3 µM ZL
3VS displayed a significant increase in fluorescence signal only after 16 h of treatment (
Figure 2G,H). Yet, the 7.5 µM ZL
3VS treatment caused a small change in signal after 8 h with a larger increase after 16 h of treatment (
Figure 2F,I). The increase in fluorescent signal was exclusively observed in RTA
E177Qegfp expressing cells and not from ZL
3VS-treated U373 cells (
Supplemental Figure 2). The data indicate that the largest increase in fluorescence signal was observed from cells treated for 16 h under all ZL
3VS concentrations (
Figure 2G–I). Considering cell morphology and toxicity of ZL
3VS-treated cells (data not shown), the optimal treatment time and concentration of proteasome inhibitor for subsequent assays was determined to be 3 µM ZL
3VS for 16 h (
Figure 2H).
Figure 2.
Analysis of RTAE177Qegfp stabilization by flow cytometry. U373-RTAE177Qegfp cells treated with ZL3VS (1.5, 3 or 7.5 µM) for 4 (A–C), 8 (D–F) or 16 (G–I) h were analyzed for EGFP fluorescence intensity using flow cytometry. The data are plotted as normalized cell number versus EGFP fluorescence signal.
Figure 2.
Analysis of RTAE177Qegfp stabilization by flow cytometry. U373-RTAE177Qegfp cells treated with ZL3VS (1.5, 3 or 7.5 µM) for 4 (A–C), 8 (D–F) or 16 (G–I) h were analyzed for EGFP fluorescence intensity using flow cytometry. The data are plotted as normalized cell number versus EGFP fluorescence signal.
An initial experiment to analyze ZL
3VS-treated U373-RTA
E177Qegfp cells using a fluorescent plate reader revealed no significant difference in EGFP fluorescence signal upon ZL
3VS treatment of U373-RTA
E177Qegfp cells (
Supplemental Figure 3). Subsequently, U373 and U373-RTA
E177Qegfp cells treated with ZL
3VS were analyzed using plate-scanning confocal fluorescence microscope (
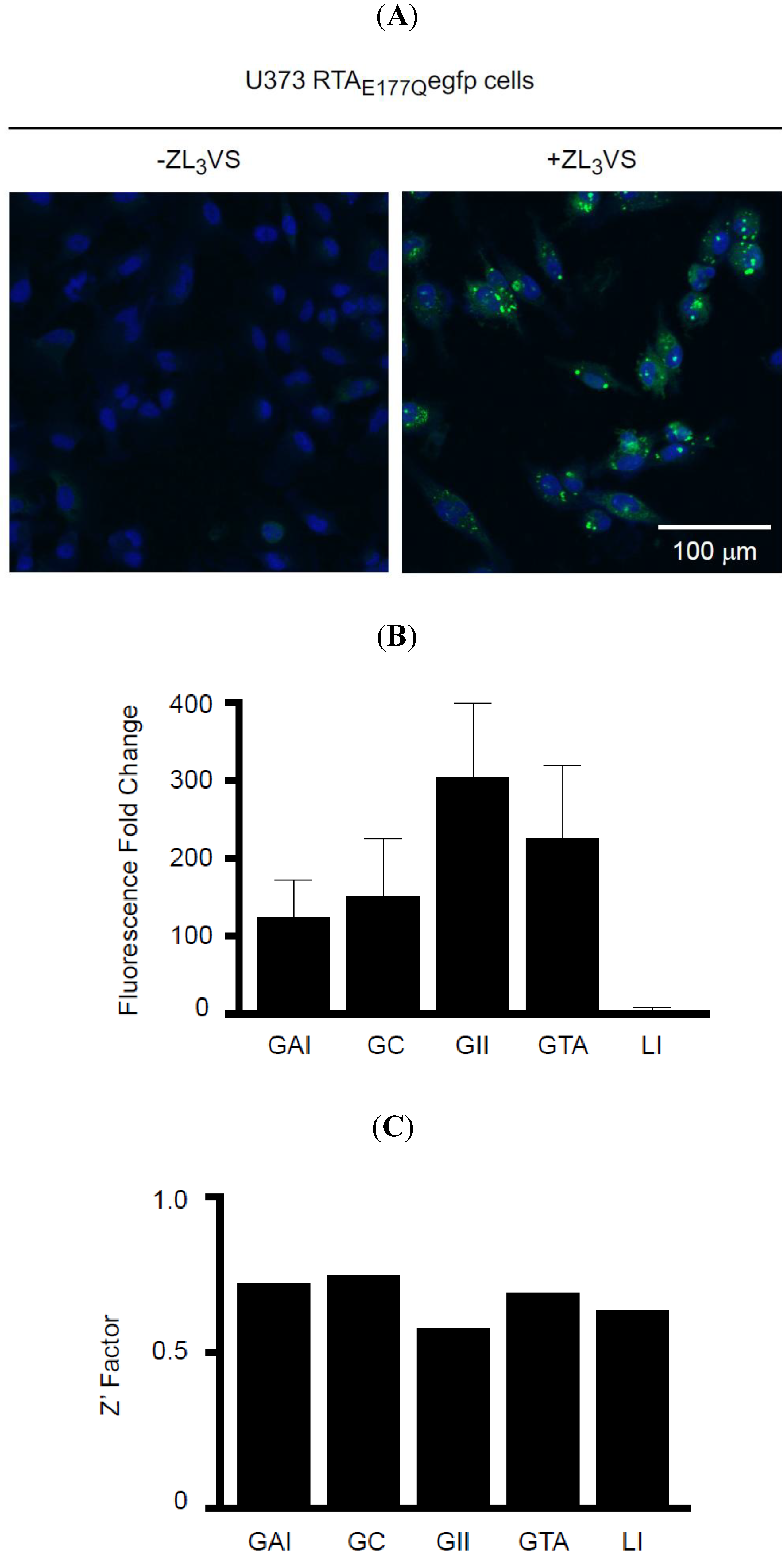
Figure 3A). Following proteasome inhibitor treatment, cells were fixed and stained with Hoechst reagent to visualize the nucleus of the cell. Strikingly, U373-RTA
E177Qegfp cells treated with ZL
3VS induced distinct peri-nuclear granules (
Figure 3A). The fluorescent intensity of these granules were quantified as granule average intensity (GAI), granule count (GC), granule integrated intensity (GII), granule total area (GTA), and Laplacian index (LI) (
Figure 3B,C) to determine the most appropriate analysis parameter to determine RTA
E177Qegfp stability. The comparison of these granularity parameters using non-treated cells as a control demonstrated relative fold change ranging from 3 (LI) to 302 (GII) (
Figure 3B). All of the parameters yielded good Z’ factor values > 0.5 with GAI, GC, and GTA generating Z’ factor values > 0.7 (
Figure 3C). The largest fold change of GII between untreated or ZL
3VS treated cells did not produce the highest Z’ Factor (
Figure 3B,C). We subsequently selected granule average intensity (GAI) as our analysis parameter which induced a ~124 fold increase over background, a 0.72 Z’ Factor value (
Figure 3B,C), and was more consistent among different plates (data not shown). The observation of distinct fluorescent granules upon stabilization of RTA
E177Qegfp in ZL
3VS-treated cells provided the basis to perform a high-content screen to identify compounds that stabilize RTA.
Figure 3.
Stabilization of RTAE177Qegfp in cells. (A) U373 RTAE177Qegfp cells treated without or with ZL3VS (3 µM, 16h) were fixed, stained with Hoechst reagent, and subjected to confocal fluorescent microscopy. The merged images of the nucleus (blue) and EGFP fluorescent signal from stabilized RTAE177Qegfp molecules are shown; (B) Fluorescence signal from stabilized RTAE177Qegfp molecules was quantified into granule average intensity (GAI), granule count (GC), granule integrated intensity (GII), granule total area (GTA), and Laplacian index (LI). These fluorescence intensity-based values were plotted as fluorescence fold change using DMSO treated cells as background value. The error bars represent calculated fold change from eight independent samples; (C) The Z’ factor was determined using the various fluorescent intensity parameters.
Figure 3.
Stabilization of RTAE177Qegfp in cells. (A) U373 RTAE177Qegfp cells treated without or with ZL3VS (3 µM, 16h) were fixed, stained with Hoechst reagent, and subjected to confocal fluorescent microscopy. The merged images of the nucleus (blue) and EGFP fluorescent signal from stabilized RTAE177Qegfp molecules are shown; (B) Fluorescence signal from stabilized RTAE177Qegfp molecules was quantified into granule average intensity (GAI), granule count (GC), granule integrated intensity (GII), granule total area (GTA), and Laplacian index (LI). These fluorescence intensity-based values were plotted as fluorescence fold change using DMSO treated cells as background value. The error bars represent calculated fold change from eight independent samples; (C) The Z’ factor was determined using the various fluorescent intensity parameters.
2.3. Identification of Hit Compounds from a High-Content Screen that Stabilize RTAE177Qegfp Molecules
We performed a high-content screen using U373-RTA
E177Q-egfp cells with a bioactive chemical library (2080 compounds, Microsource Discovery Systems, Inc.) using the optimized assay conditions (Material and Methods). In general, U373-RTA
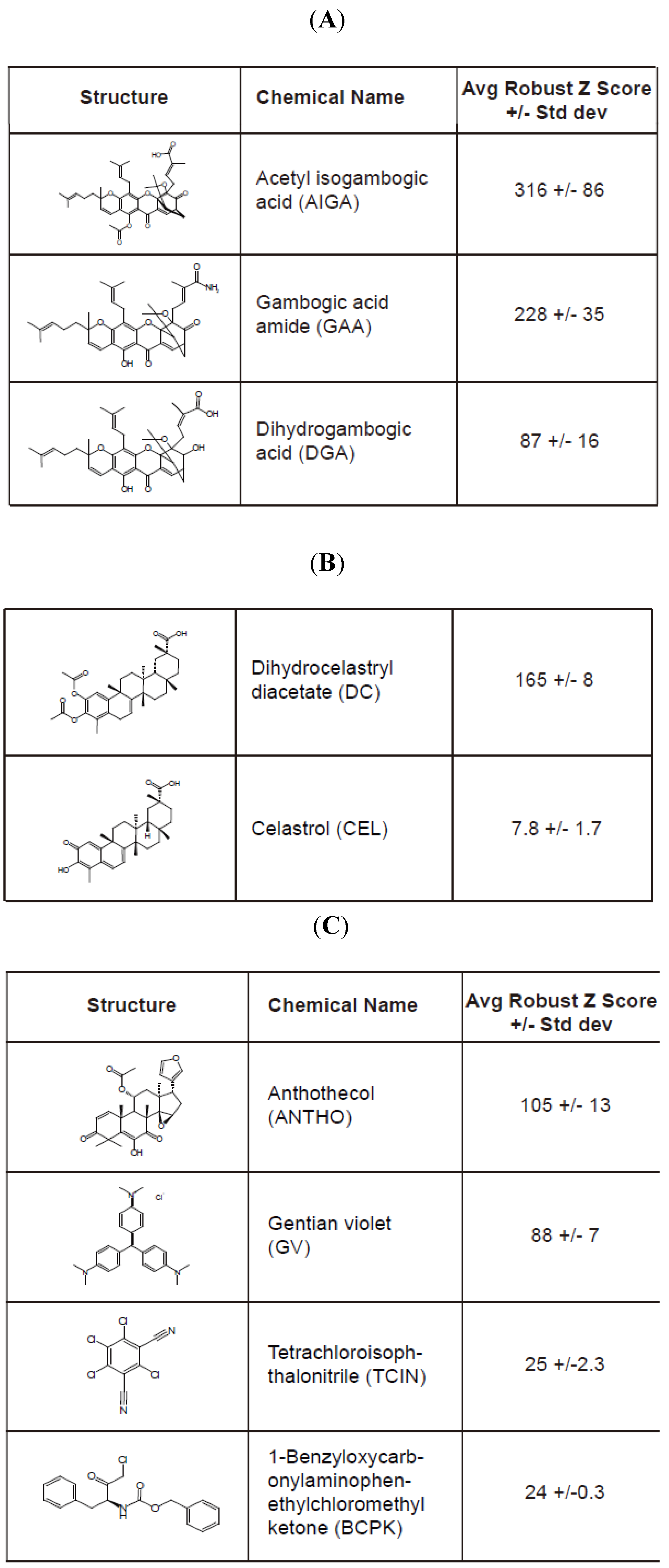
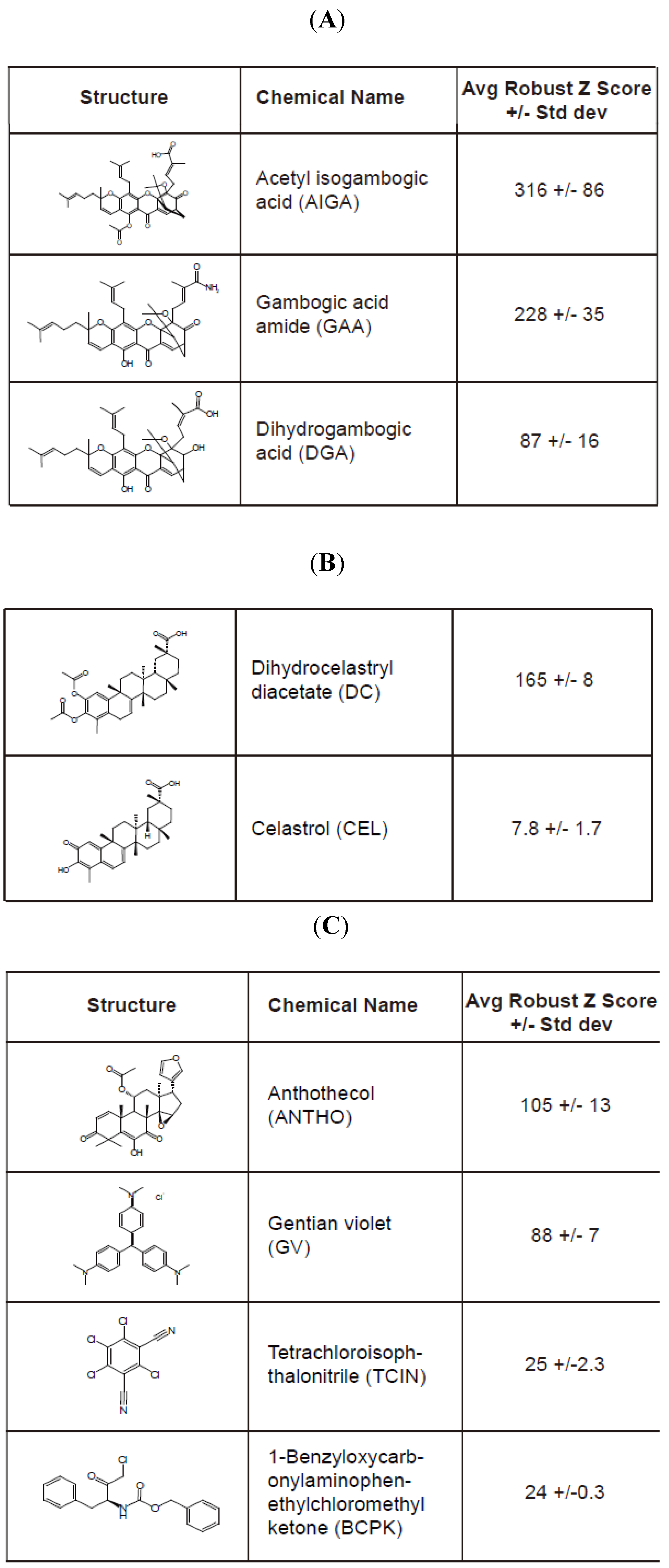
E177Qegfp cells plated in a 384-well Aurora black clear-bottom microplate were pinned with the compound library for 16 h. Cells were subsequently fixed and analyzed by confocal microscopy to determine the granule average intensity (GAI)/well. The GAI values (in triplicate) were utilized to calculate the Robust Z score for the compound library and eight compounds that produced an average Robust Z score > 20 were considered hit compounds (
Figure 4). A large range of values was observed among the hit compounds in which acetyl isogambogic acid (AIGA) yielded a Robust Z score of 316, while 1-benzyloxycarbonylaminophenethylchloromethyl ketone (BCPK) yielded a Robust Z score of 24 (
Figure 4). Interestingly, some of the hit compounds clustered into two groups that were structurally related (
Figure 4A,B). Note, celastrol (CEL) was selected due to its structurally similarity to dihydrocelastryl diacetate (DC) despite its low Robust Z score. Other structurally similar compounds consisted of acetyl isogambogic acid (AIGA), gambogic acid amide (GAA) and dihydrogambogic acid (DGA). The identification of chemical families that stabilize RTA molecules supports their effectiveness to stabilize RTA molecules. Overall, the RTA
E177Qegfp-based high-content screen can identify compounds that effectively stabilize RTA polypeptides.
Figure 4.
Hit compounds identified from the high-content screen of small chemical library. The hit compounds (A–C) that induced a reproducible and significant increase of the granule average intensity (GAI) in U373-RTAE177Qegfp cells are indicated by their structure, name, and average Robust Z score. The standard deviation value is from three replicates. Two chemically distinct groups of compounds comprised of acetyl isogambogic acid (AIGA), gambogic acid amide (GAA), and dihydrogambogic acid (DGA) (A) and dihydrocelastryl diacetate (DC) and celastrol (CEL) (B) were identified as hit compounds. Other hit compounds (C) include gentian violet (GV), anthothecol (ANTHO), tetrachloroisophthalonitrile (TCIN) and 1-benzyloxycarbonylaminophenethylchloromethyl ketone (BCPK).
Figure 4.
Hit compounds identified from the high-content screen of small chemical library. The hit compounds (A–C) that induced a reproducible and significant increase of the granule average intensity (GAI) in U373-RTAE177Qegfp cells are indicated by their structure, name, and average Robust Z score. The standard deviation value is from three replicates. Two chemically distinct groups of compounds comprised of acetyl isogambogic acid (AIGA), gambogic acid amide (GAA), and dihydrogambogic acid (DGA) (A) and dihydrocelastryl diacetate (DC) and celastrol (CEL) (B) were identified as hit compounds. Other hit compounds (C) include gentian violet (GV), anthothecol (ANTHO), tetrachloroisophthalonitrile (TCIN) and 1-benzyloxycarbonylaminophenethylchloromethyl ketone (BCPK).
2.4. Characterization of Hit Compounds that Stabilize RTAE177Qegfp Molecules
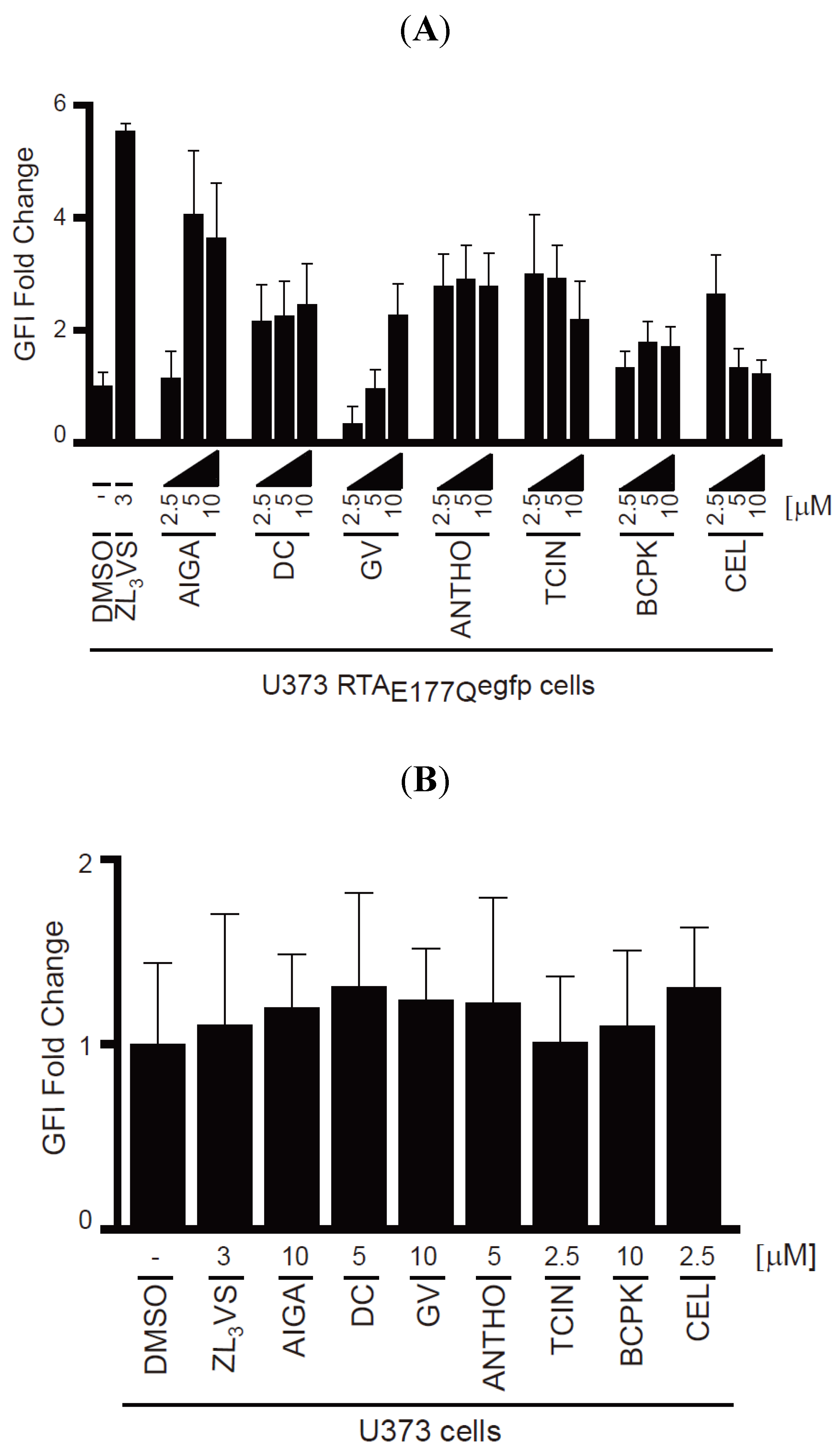
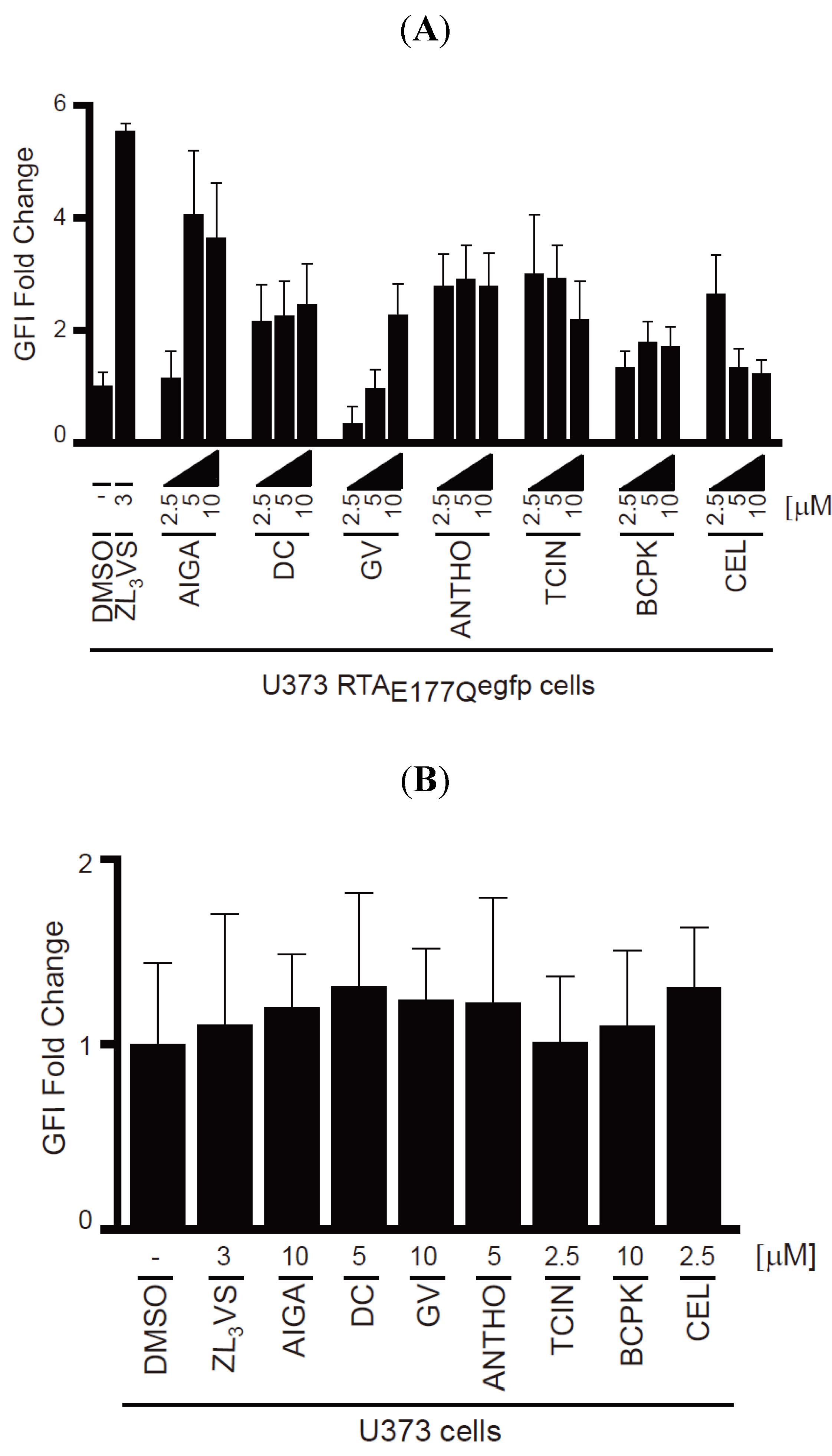
To validate the hit compounds, U373-RTA
E177Qegfp cells treated with DMSO, 3 µM ZL
3VS or 2.5, 5 or 10 µM of the hit compounds (
Figure 4) for 16 h were assessed by flow cytometry using EGFP fluorescent intensity (
Figure 5). The EGFP fold change of U373-RTA
E177Qegfp cells treated with hit compounds was determined from the peak fluorescent signal of treated cells compared to DMSO-treated cells (
Figure 5A). Note, acetyl isogambogic acid was examined as a representative of the gambogic acid family. As expected, ZL
3VS treatment caused the largest increase in fluorescence signal, quantified as EGFP fluorescent intensity (GFI) fold change, when compared to DMSO treated cells (
Figure 5A). Interestingly, the increase in fluorescence signal was concentration dependent for acetyl isogambogic acid (AIGA)- and gentian violet (GV)-treated cells. In contrast, dihydrocelastryl diacetate (DC), anthothecol (ANTHO), tetrachloroisophthalonitrile (TCIN), 1-benzyloxycarbonylaminophenethylchloromethyl ketone (BCPK), and celastrol (CEL) were effective at all concentrations (
Figure 5A) suggesting that lower concentrations of these compounds may stabilize RTA molecules. In general, the hit compounds induced a one to four fold increase in fluorescence signal (
Figure 5A). These data support the screening assay results that the identified hit compounds induce an increase in fluorescence signal in U373-RTA
E177Qegfp cells.
To exclude the possibility that the increase in fluorescence signal in U373-RTA
E177Qegfp cells treated with hit compounds was due to fluorescent properties of the compound, U373 cells treated with the hit compounds were assessed by flow cytometry (
Figure 5B). None of the compounds caused a significant increase in fluorescence intensity upon treatment. Thus, the increase in fluorescent intensity observed in treated-U373-RTA
E177Qegfp cells can be attributed to stabilization of RTA
E177Qegfp molecules and not the possible fluorescence property of a compound.
Figure 5.
Validation of hit compounds to induce RTAE177Qegfp fluorescence intensity. (A) U373-RTAE177Qegfp cells treated with DMSO, ZL3VS (3 µM) or hit compounds (2.5, 5, or 10 µM) for 16 h were subjected to flow cytometry for the analysis of EGFP fluorescence intensity (GFI fold change); (B) U373 cells treated with DMSO, ZL3VS (3 µM) or hit compounds (indicated concentration) for 16 h were subjected to flow cytometry for the analysis of GFI. The fluorescence signal from the respective treated cells was plotted as GFI fold change utilizing the peak fluorescent signal from cells treated with the various chemicals compared to DMSO treated. Error bars represent the standard deviation of the fluorescent signal from 50% of the peak GFI.
Figure 5.
Validation of hit compounds to induce RTAE177Qegfp fluorescence intensity. (A) U373-RTAE177Qegfp cells treated with DMSO, ZL3VS (3 µM) or hit compounds (2.5, 5, or 10 µM) for 16 h were subjected to flow cytometry for the analysis of EGFP fluorescence intensity (GFI fold change); (B) U373 cells treated with DMSO, ZL3VS (3 µM) or hit compounds (indicated concentration) for 16 h were subjected to flow cytometry for the analysis of GFI. The fluorescence signal from the respective treated cells was plotted as GFI fold change utilizing the peak fluorescent signal from cells treated with the various chemicals compared to DMSO treated. Error bars represent the standard deviation of the fluorescent signal from 50% of the peak GFI.
To further define the effectiveness of the hit compounds, we determined the half maximal effective concentration (EC
50) of the hit compounds using U373-RTA
E177Qegfp cells (
Supplemental Figure 4). In general, the EC
50 values of most compounds ranged from ~1–3 μM with the exception of BCPK and GV whose EC
50 values were >10 mM. These data provide additional support for the ability of the hit compounds to stabilize RTA polypeptides.
2.5. Stabilization of RTAE177D in Cells Treated with Hit Compounds
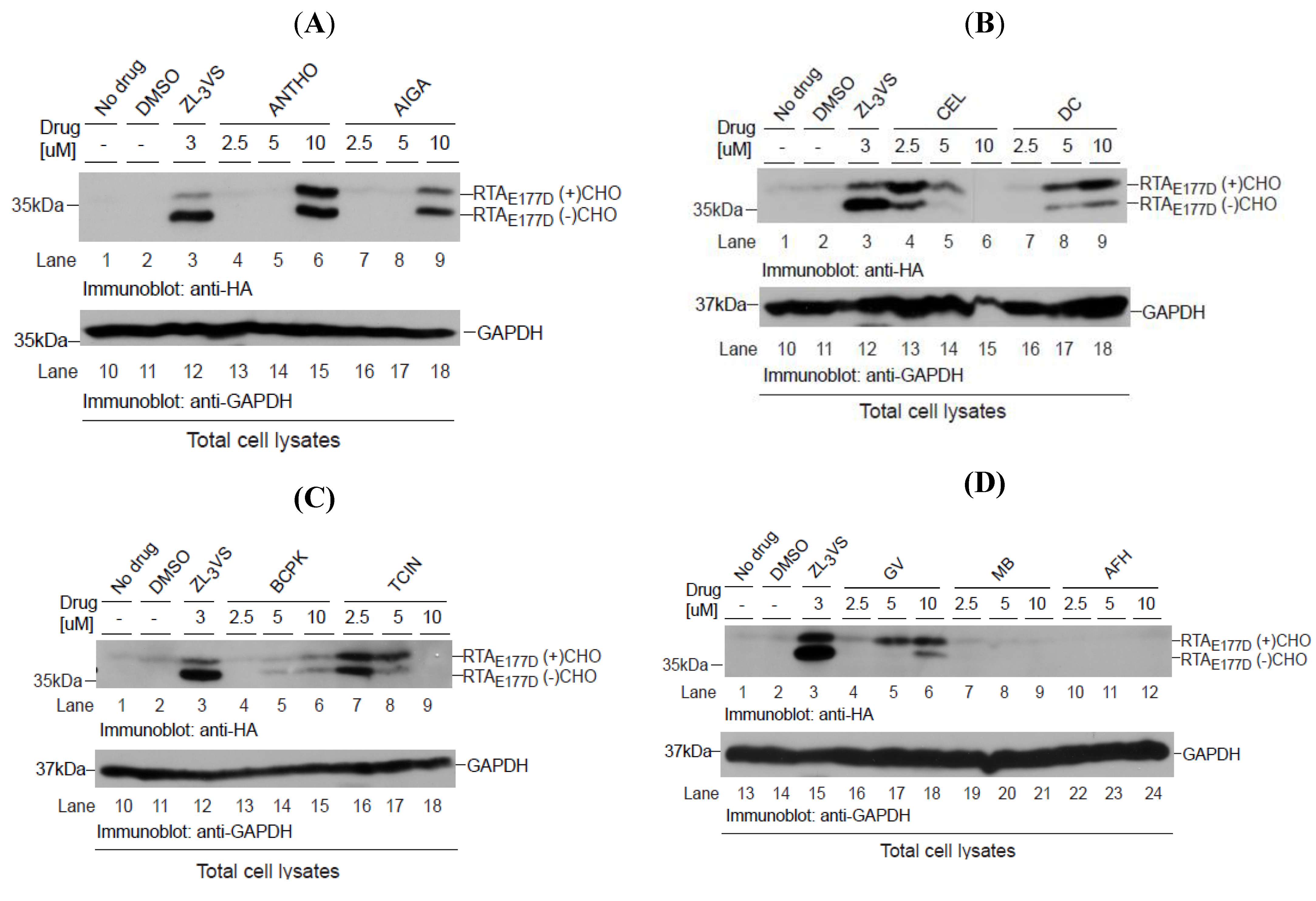
Can the hit compounds stabilize RTA
E177D polypeptides? To address this question, U373-RTA
E177D cells treated with DMSO, 3 µM ZL
3VS or 2.5, 5 or 10 µM of hit compounds were subjected to immunoblot analysis (
Figure 6). As expected, ZL
3VS-treated U373-RTA
E177D cells accumulated both glycosylated and deglycosylated forms of RTA
E177D (
Figure 6A–D, lane 3). Strikingly, all of the hit compounds at varying degrees stabilized glycosylated and deglycosylated RTA
E177D polypeptides (
Figure 6A–C, lanes 4–9 and 6D, lanes 4–6). As controls, U373-RTA
E177D cells treated with merbromin (MB) and acriflavinium hydrochloride (AFH), compounds that increased the general nuclear fluorescent signal during the primary high-content screen (data not shown), did not stabilize RTA
E177D polypeptides (
Figure 6D, lanes 7–12). As a loading control, an immunoblot for GAPDH examined protein levels (
Figure 6A–C, lanes 10–18 and 6D, lanes 13–24). Celastrol (CEL) and tetrachloroisophthalonitrile (TCIN) were effective at lower concentrations, yet the higher concentrations may be toxic to cells due to the loss of GAPDH protein (
Figure 6A, lanes 4–6, 13–15 and 6C, lanes 7–9, 16–18). Interestingly, celastrol (CV), dihydrocelastryl diacetate (DC) and gentian violet (GV) preferentially stabilized the glycosylated species of RTA
E177D (
Figure 6B, lanes 4–9 and 6D, lanes 4–6), implying that these compounds interfere with the retrograde translocation step of RTA. Anthothecol (ANTHO), acetyl isogambogic acid (AIGA), 1-Benzyloxycarbonylaminophenethylchloromethyl ketone (BCPK), and tetrachloroisophthalonitrile (TCIN) treatment induced the accumulation of equivalent levels of glycosylated and deglycosylated forms of RTA
E177D proteins (
Figure 6A, lanes 6 and 9 and 6C, lanes 4–9). The observation of similar amounts of glycosylated and deglycosylated RTA species suggests that these compounds likely target a post-retrograde translocation step. Interestingly, the concentrations required to stabilize RTA
E177D were slightly higher than GFP tagged RTA mutant probably due to the faster kinetics of RTA
E177D retrograde translocation. In conclusion, the hit compounds were effective reagents to stabilize non-egfp tagged RTA polypeptides.
2.6. Inhibition of Ricin Induced Translation Shutdown by Selected Hit Compounds
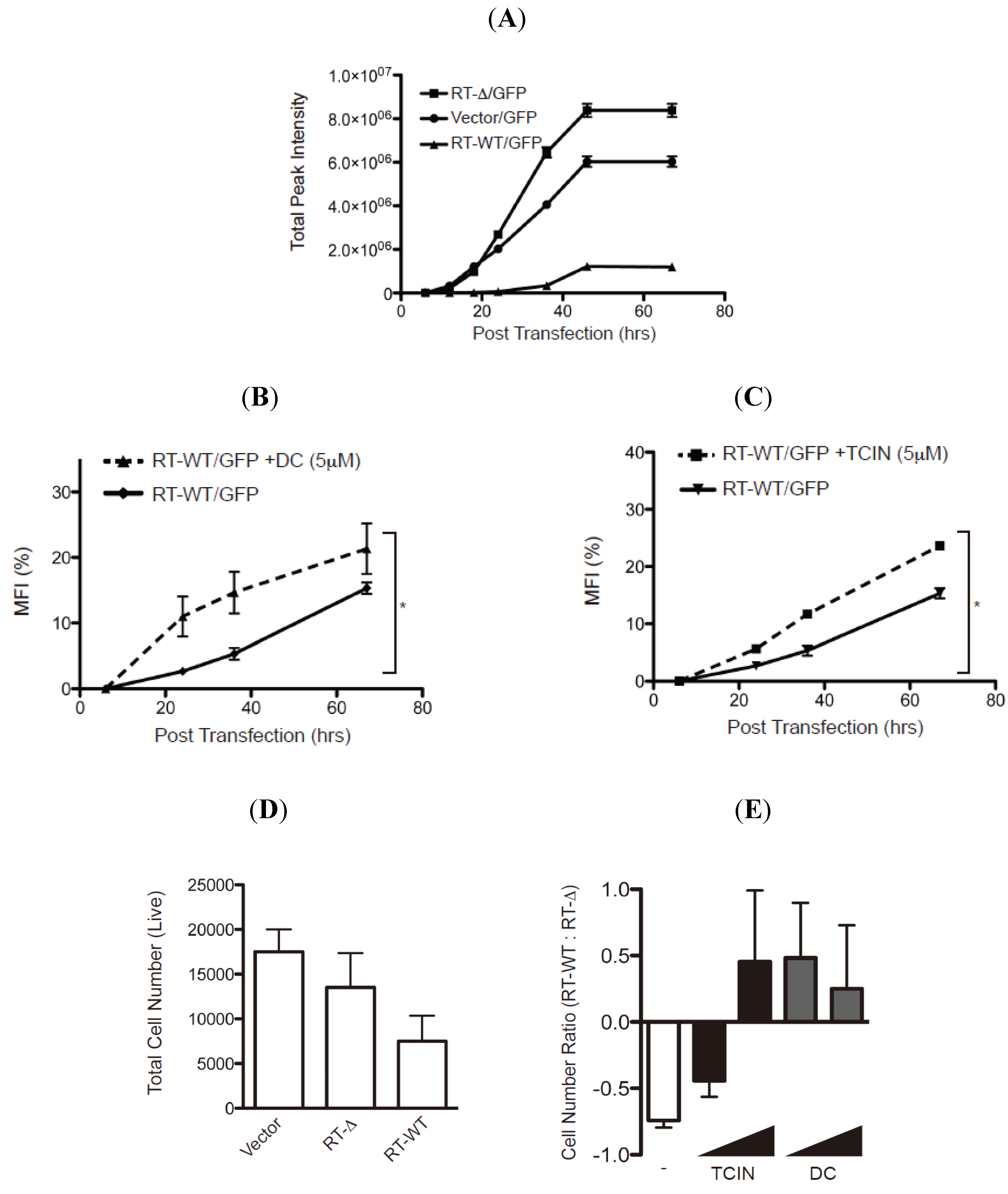
The stabilization of RTA mutants pre- or post-retrograde translocation by the hit compounds suggests that these reagents may indeed limit the enzymatic activity of wild type ricin to inhibit protein translation. To evaluate the ability of the hit compounds to attenuate enzymatic activity, we established a RTA-activity assay by measuring the GFP fluorescent signal from HEK-293 cells transfected with plasmids encoding GFP with either empty vector, wild type RTA (RT-WT) or an enzymatically inactive RTA molecule lacking residues 177–181 (RT-Δ (
Figure 7 and [
18]). The GFP fluorescent signal was evaluated for up to 68 h post-transfection (hpt) and as expected a robust increase in total fluorescent signal was observed in the cells transfected with vector alone or RT-Δ (
Figure 7A). In contrast, there was only a modest increase in fluorescent intensity in cells transfected with RT-WT validating that enzymatically active RTA inhibits protein synthesis (
Figure 7A).
Figure 6.
Stabilization of RTAE177D by hit compounds. U373RTAE177D cells treated with DMSO, ZL3VS (3 µM) or hit compounds (2.5, 5, and 10 µM) were subjected to immunoblot analysis for RTAE177D (A–C, lanes 1–9; D, lanes 1–12) and GAPDH (A–C, lanes 10–18; D, lanes 13–24). We analyzed the compounds anthothecol (ANTHO), acetyl isogambogic acid (AIGA) (A); celastrol (CEL), dihydrocelastryl diacetate (DC) (B); 1-benzyloxycarbonylaminophenethylchloromethyl ketone (BCPK), tetrachloroisophthalonitrile (TCIN) (C); gentian violet, merbromin (MB), and acriflavinium hydrochloride (AFH) (D). RTA polypeptides, GAPDH and molecular weight markers are indicated.
Figure 6.
Stabilization of RTAE177D by hit compounds. U373RTAE177D cells treated with DMSO, ZL3VS (3 µM) or hit compounds (2.5, 5, and 10 µM) were subjected to immunoblot analysis for RTAE177D (A–C, lanes 1–9; D, lanes 1–12) and GAPDH (A–C, lanes 10–18; D, lanes 13–24). We analyzed the compounds anthothecol (ANTHO), acetyl isogambogic acid (AIGA) (A); celastrol (CEL), dihydrocelastryl diacetate (DC) (B); 1-benzyloxycarbonylaminophenethylchloromethyl ketone (BCPK), tetrachloroisophthalonitrile (TCIN) (C); gentian violet, merbromin (MB), and acriflavinium hydrochloride (AFH) (D). RTA polypeptides, GAPDH and molecular weight markers are indicated.
Can the hit compounds limit the enzymatic activity and cytotoxicity of RTA? To address RTA enzymatic activity, HEK-293 cells co-transfected with plasmids encoding GFP and RT-WT or RT-Δ were treated with either dihydrocelastryl diacetate (DC) or tetrachloroisophthalonitrile (TCIN) at either 6 or 18 hpt and evaluated for GFP fluorescence intensity (
Figure 7B,C). These experiments examined DC and TCIN because they represent diverse chemical compounds that effectively stabilize RTA (
Figure 5 and
Figure 6). The percentage mean fluorescent intensity (MFI) from wild type RTA was determined using the fluorescence intensity from the respective RT-Δ expressing cells as 100%. As previously observed (
Figure 7A), the MFI from RT-WT expressing cells was >10% MFI at 68 hpt (
Figure 7B,C, solid lines). Remarkably, inclusion of dihydrocelastryl diacetate (DC) at 6 hpt or tetrachloroisophthalonitrile (TCIN) at 18 hpt caused a statistical significant increase in MFI in RT-WT expressing cells at all time points post-addition (
Figure 7B,C, dashed lines). The results imply that these compounds can attenuate the enzymatic activity of RTA by stabilizing the RTA molecule within the cell.
Figure 7.
Hit compounds attenuate RTA enzymatic activity. (A) HEK-293 cells co-transfected with an GFP-expressing plasmid with vector alone or vectors expressing wild type RTA (RT-WT) or enzymatically inactive mutant RTA-Δ177-181 (RT-Δ) were analyzed for GFP fluorescence over 68 h post-transfection (hpt) using a fluorescence cytometer. The total fluorescence intensity was plotted over time post transfection (h) and the error bars represent signal from six samples. (B and C) HEK-293 transfected-cells (see above) were treated with dihydrocelastryl diacetate (DC) 6 hpt (B) or tetrachloroisophthalonitrile (TCIN) 18 hpt (C) followed by analysis of GFP fluorescent signal from six independent samples. The percentage of the mean fluorescent intensity (MFI) from RT-WT transfected cells was calculated using the intensity from RT-Δ transfected cells as 100%. Human fibroblasts transfected with control plasmid, RT-WT, or RT-Δ were untreated (D) or treated with TCIN and DC (1 and 5μM) at 18 or 6 hpt (E). The total number of viable cells was measured (in quadruple) 48 hpt (D). The ratio of viable cells from RT-WT transfected cells to RT-Δ transfected cells from untreated and treated cells was plotted to compare the effect of the respective compound (E). The error bars represent the standard deviation between six samples. The data were subjected to a one-tail Student T-test and statistical significance was indicated by an * with p values < 0.05.
Figure 7.
Hit compounds attenuate RTA enzymatic activity. (A) HEK-293 cells co-transfected with an GFP-expressing plasmid with vector alone or vectors expressing wild type RTA (RT-WT) or enzymatically inactive mutant RTA-Δ177-181 (RT-Δ) were analyzed for GFP fluorescence over 68 h post-transfection (hpt) using a fluorescence cytometer. The total fluorescence intensity was plotted over time post transfection (h) and the error bars represent signal from six samples. (B and C) HEK-293 transfected-cells (see above) were treated with dihydrocelastryl diacetate (DC) 6 hpt (B) or tetrachloroisophthalonitrile (TCIN) 18 hpt (C) followed by analysis of GFP fluorescent signal from six independent samples. The percentage of the mean fluorescent intensity (MFI) from RT-WT transfected cells was calculated using the intensity from RT-Δ transfected cells as 100%. Human fibroblasts transfected with control plasmid, RT-WT, or RT-Δ were untreated (D) or treated with TCIN and DC (1 and 5μM) at 18 or 6 hpt (E). The total number of viable cells was measured (in quadruple) 48 hpt (D). The ratio of viable cells from RT-WT transfected cells to RT-Δ transfected cells from untreated and treated cells was plotted to compare the effect of the respective compound (E). The error bars represent the standard deviation between six samples. The data were subjected to a one-tail Student T-test and statistical significance was indicated by an * with p values < 0.05.
![Toxins 06 00033 g007]()
To examine the effectiveness of TCIN and DC to attenuate RTA-induced cytotoxicity, we performed an initial experiment by determining the number of viable cells followed by the transfection of empty, RT-WT, or RT-Δ expressing plasmids into human fibroblasts (
Figure 7D,E). As expected, the most significant decrease in viable cells was from RT-WT-transfected cells. The number of viable cells in drug treated samples was determined as a ratio of viable cells from RT-WT transfected cells to RT-Δ transfected cells (
Figure 7E). Hence, untreated cells yielded a negative value (
Figure 7E, white bar). Strikingly, the addition of TCIN and DC at increasing concentrations (1 and 5 μM) caused an increase in the RT-WT/ RT-Δ ratio (
Figure 7E, black and gray bars). Note, TCIN was more effective at a higher concentration. Collectively, the data supports the paradigm that targeting ER-localized RTA would limit ricin-intoxication.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}