Triggering of Suicidal Erythrocyte Death by Celecoxib

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
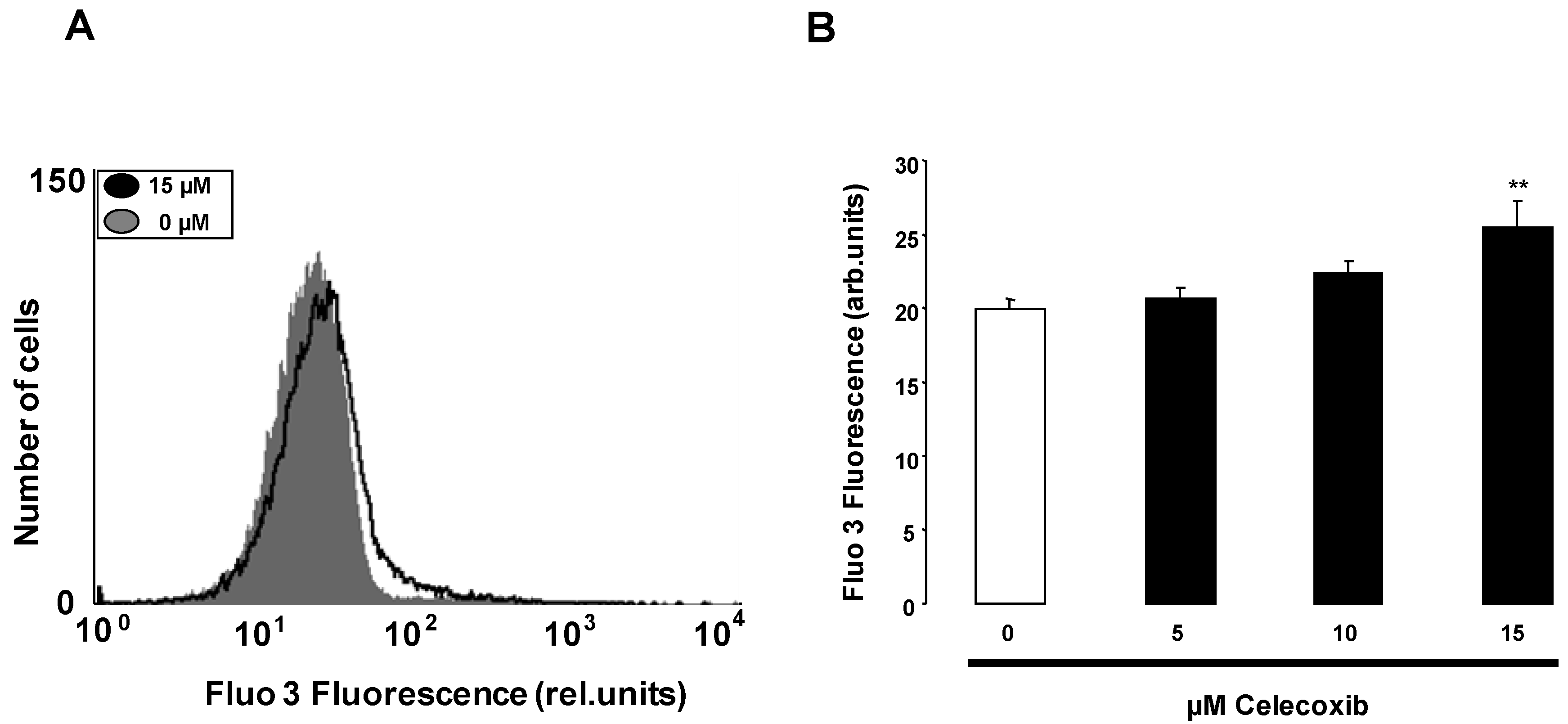
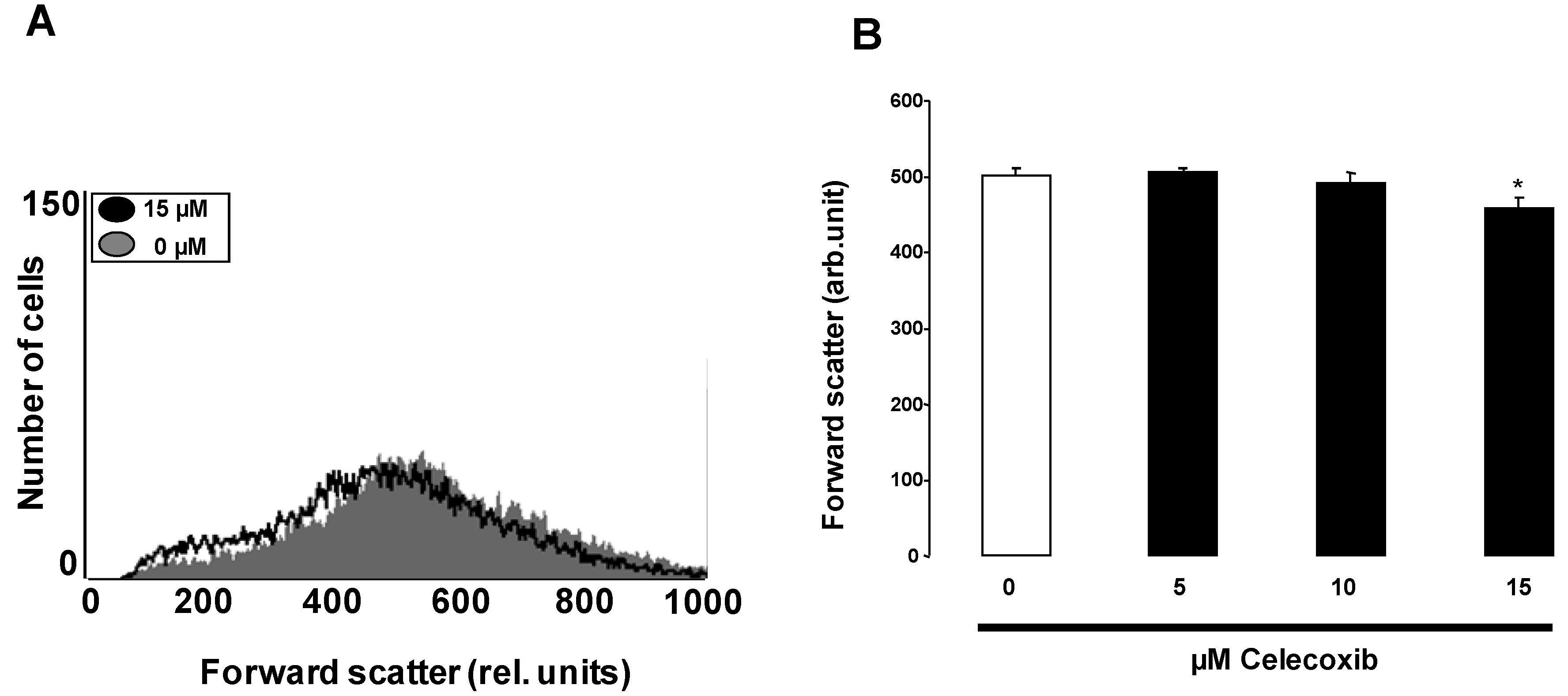
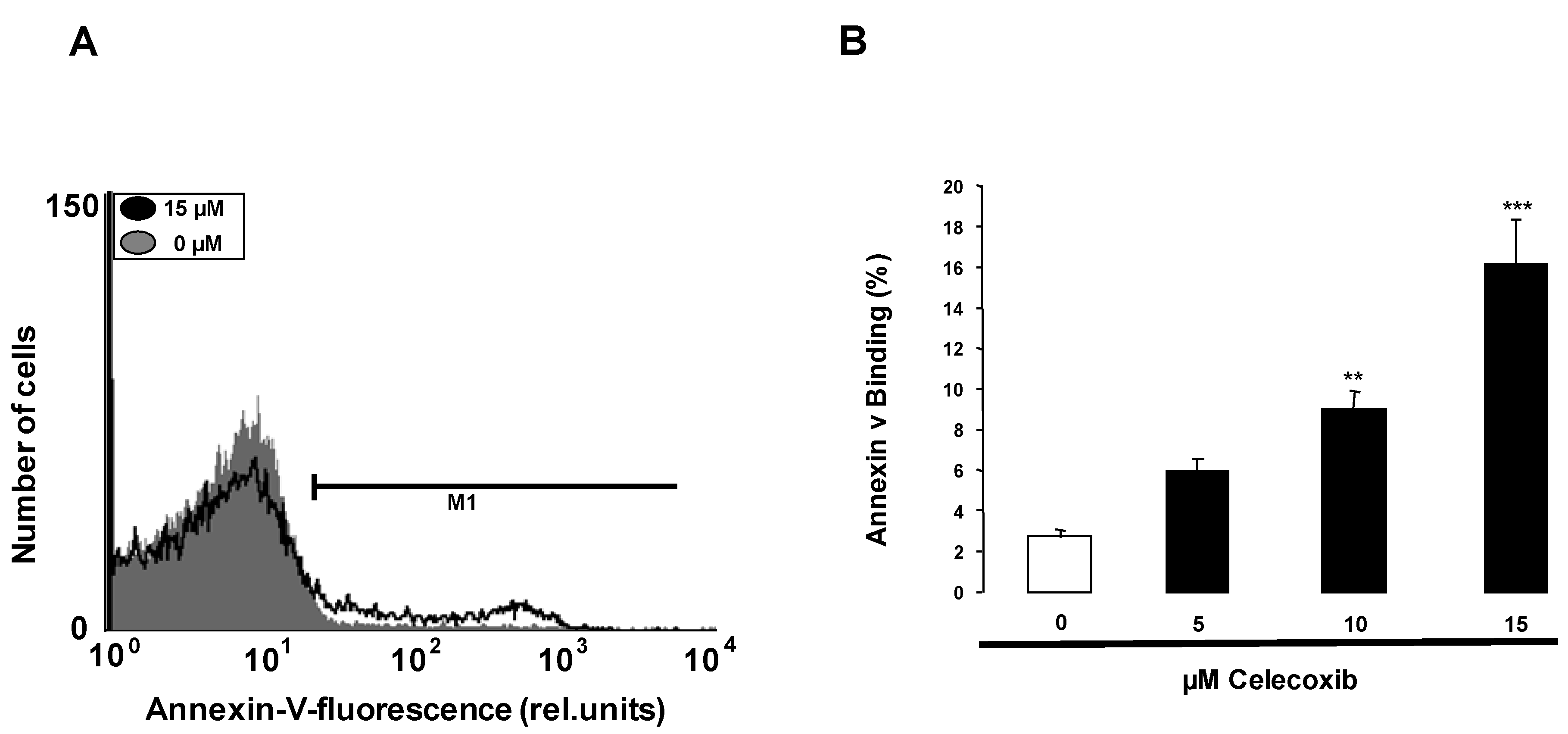
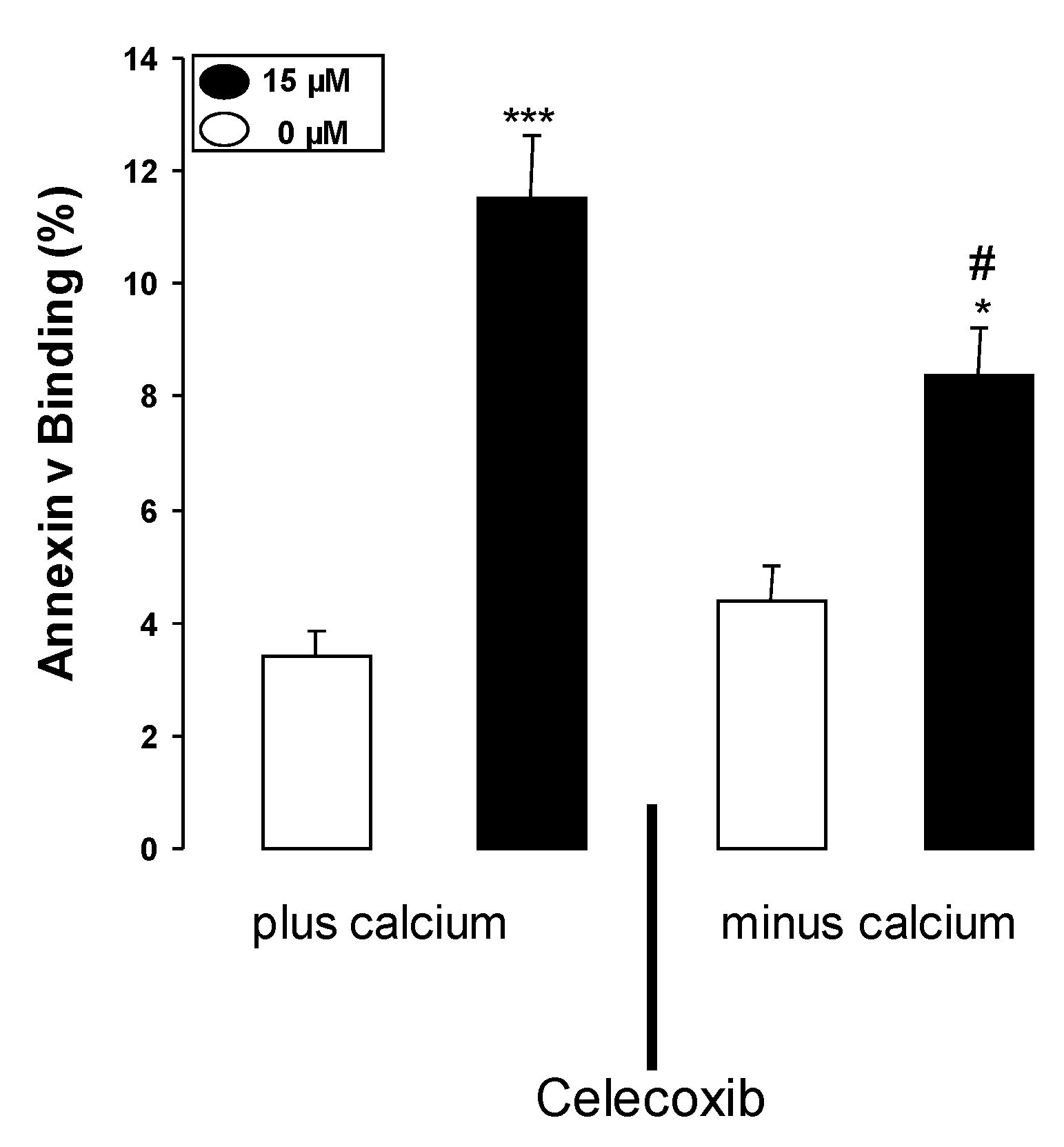
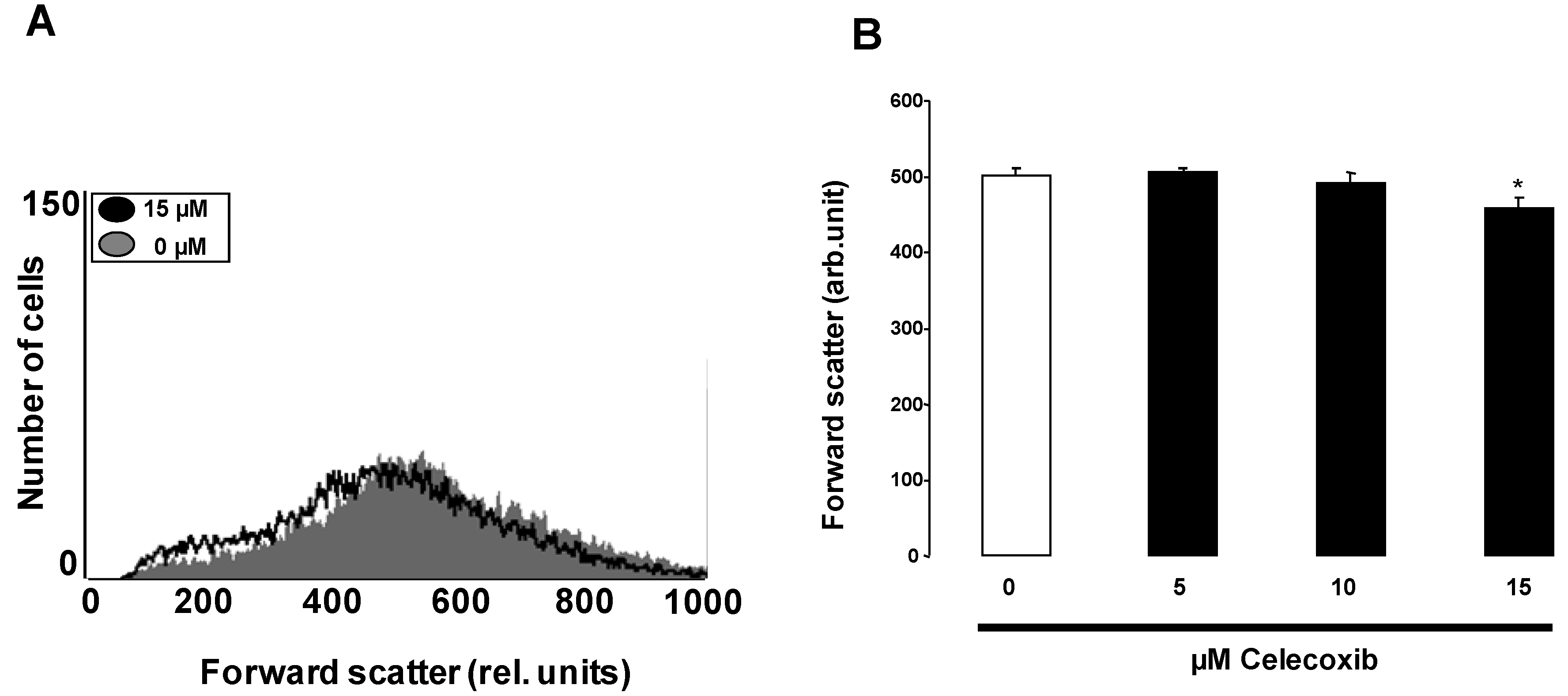
2. Results and Discussion



3. Methods
3.1. Erythrocytes, Solutions and Chemicals
3.2. FACS Analysis of Annexin-V-Binding and Forward Scatter
3.3. Measurement of Intracellular Ca2+
3.4. Measurement of Hemolysis
3.5. Statistics
4. Conclusions
Acknowledgements
Conflicts of Interest
References
- Jendrossek, V. Targeting apoptosis pathways by celecoxib in cancer. Cancer Lett. 2013, 332, 313–324. [Google Scholar] [CrossRef]
- Kismet, K.; Akay, M.T.; Abbasoglu, O.; Ercan, A. Celecoxib: A potent cyclooxygenase-2 inhibitor in cancer prevention. Cancer Detect. Prev. 2004, 28, 127–142. [Google Scholar] [CrossRef]
- Schonthal, A.H. Antitumor properties of dimethyl-celecoxib, a derivative of celecoxib that does not inhibit cyclooxygenase-2: Implications for glioma therapy. Neurosurg. Focus 2006, 20, E21. [Google Scholar] [CrossRef]
- Winfield, L.L.; Payton-Stewart, F. Celecoxib and bcl-2: Emerging possibilities for anticancer drug design. Future Med. Chem. 2012, 4, 361–383. [Google Scholar] [CrossRef]
- Blanke, C.D. Celecoxib with chemotherapy in colorectal cancer. Oncology 2002, 16, 17–21. [Google Scholar]
- Wang, J.L.; Lin, K.L.; Chou, C.T.; Kuo, C.C.; Cheng, J.S.; Hsu, S.S.; Chang, H.T.; Tsai, J.Y.; Liao, W.C.; Lu, Y.C.; et al. Effect of celecoxib on Ca(2+) handling and viability in human prostate cancer cells (pc3). Drug Chem. Toxicol. 2012, 35, 456–462. [Google Scholar] [CrossRef]
- Lang, F.; Gulbins, E.; Lerche, H.; Huber, S.M.; Kempe, D.S.; Foller, M. Eryptosis, a window to systemic disease. Cell. Physiol. Biochem. 2008, 22, 373–380. [Google Scholar] [CrossRef]
- Foller, M.; Kasinathan, R.S.; Koka, S.; Lang, C.; Shumilina, E.; Birnbaumer, L.; Lang, F.; Huber, S.M. Trpc6 contributes to the Ca(2+) leak of human erythrocytes. Cell. Physiol. Biochem. 2008, 21, 183–192. [Google Scholar] [CrossRef]
- Foller, M.; Sopjani, M.; Koka, S.; Gu, S.; Mahmud, H.; Wang, K.; Floride, E.; Schleicher, E.; Schulz, E.; Munzel, T.; et al. Regulation of erythrocyte survival by amp-activated protein kinase. FASEB J. 2009, 23, 1072–1080. [Google Scholar] [CrossRef]
- Brand, V.B.; Sandu, C.D.; Duranton, C.; Tanneur, V.; Lang, K.S.; Huber, S.M.; Lang, F. Dependence of plasmodium falciparum in vitro growth on the cation permeability of the human host erythrocyte. Cell. Physiol. Biochem. 2003, 13, 347–356. [Google Scholar] [CrossRef]
- Brugnara, C.; de Franceschi, L.; Alper, S.L. Inhibition of Ca(2+)-dependent K+ transport and cell dehydration in sickle erythrocytes by clotrimazole and other imidazole derivatives. J. Clin. Invest. 1993, 92, 520–526. [Google Scholar] [CrossRef]
- Lang, P.A.; Kaiser, S.; Myssina, S.; Wieder, T.; Lang, F.; Huber, S.M. Role of Ca2+-activated K+ channels in human erythrocyte apoptosis. Am. J. Physiol. Cell. Physiol. 2003, 285, C1553–C1560. [Google Scholar] [CrossRef]
- Berg, C.P.; Engels, I.H.; Rothbart, A.; Lauber, K.; Renz, A.; Schlosser, S.F.; Schulze-Osthoff, K.; Wesselborg, S. Human mature red blood cells express caspase-3 and caspase-8, but are devoid of mitochondrial regulators of apoptosis. Cell Death Differ. 2001, 8, 1197–1206. [Google Scholar] [CrossRef]
- Lang, F.; Gulbins, E.; Lang, P.A.; Zappulla, D.; Foller, M. Ceramide in suicidal death of erythrocytes. Cell. Physiol. Biochem. 2010, 26, 21–28. [Google Scholar] [CrossRef]
- Klarl, B.A.; Lang, P.A.; Kempe, D.S.; Niemoeller, O.M.; Akel, A.; Sobiesiak, M.; Eisele, K.; Podolski, M.; Huber, S.M.; Wieder, T.; et al. Protein kinase c mediates erythrocyte “programmed cell death” following glucose depletion. Am. J. Physiol. Cell. Physiol. 2006, 290, C244–C253. [Google Scholar]
- Bhavsar, S.K.; Bobbala, D.; Xuan, N.T.; Foller, M.; Lang, F. Stimulation of suicidal erythrocyte death by alpha-lipoic acid. Cell. Physiol. Biochem. 2010, 26, 859–868. [Google Scholar] [CrossRef]
- Foller, M.; Huber, S.M.; Lang, F. Erythrocyte programmed cell death. IUBMB Life 2008, 60, 661–668. [Google Scholar] [CrossRef]
- Foller, M.; Mahmud, H.; Gu, S.; Wang, K.; Floride, E.; Kucherenko, Y.; Luik, S.; Laufer, S.; Lang, F. Participation of leukotriene c(4) in the regulation of suicidal erythrocyte death. J. Physiol. Pharmacol. 2009, 60, 135–143. [Google Scholar]
- Lau, I.P.; Chen, H.; Wang, J.; Ong, H.C.; Leung, K.C.; Ho, H.P.; Kong, S.K. In vitro effect of ctab- and peg-coated gold nanorods on the induction of eryptosis/erythroptosis in human erythrocytes. Nanotoxicology 2011, in press. [Google Scholar] [CrossRef]
- Maellaro, E.; Leoncini, S.; Moretti, D.; del Bello, B.; Tanganelli, I.; de Felice, C.; Ciccoli, L. Erythrocyte caspase-3 activation and oxidative imbalance in erythrocytes and in plasma of type 2 diabetic patients. Acta Diabetol. 2011, in press. [Google Scholar] [CrossRef]
- Foller, M.; Feil, S.; Ghoreschi, K.; Koka, S.; Gerling, A.; Thunemann, M.; Hofmann, F.; Schuler, B.; Vogel, J.; Pichler, B.; et al. Anemia and splenomegaly in cgki-deficient mice. Proc. Natl. Acad. Sci. USA 2008, 105, 6771–6776. [Google Scholar]
- Bhavsar, S.K.; Gu, S.; Bobbala, D.; Lang, F. Janus kinase 3 is expressed in erythrocytes, phosphorylated upon energy depletion and involved in the regulation of suicidal erythrocyte death. Cell. Physiol. Biochem. 2011, 27, 547–556. [Google Scholar] [CrossRef]
- Kucherenko, Y.V.; Huber, S.M.; Nielsen, S.; Lang, F. Decreased redox-sensitive erythrocyte cation channel activity in aquaporin 9-deficient mice. J. Membr. Biol. 2012, 245, 797–805. [Google Scholar] [CrossRef]
- Zelenak, C.; Foller, M.; Velic, A.; Krug, K.; Qadri, S.M.; Viollet, B.; Lang, F.; Macek, B. Proteome analysis of erythrocytes lacking amp-activated protein kinase reveals a role of pak2 kinase in eryptosis. J. Proteome Res. 2011, 10, 1690–1697. [Google Scholar] [CrossRef]
- Gatidis, S.; Zelenak, C.; Fajol, A.; Lang, E.; Jilani, K.; Michael, D.; Qadri, S.M.; Lang, F. P38 mapk activation and function following osmotic shock of erythrocytes. Cell. Physiol. Biochem. 2011, 28, 1279–1286. [Google Scholar] [CrossRef]
- Lupescu, A.; Shaik, N.; Jilani, K.; Zelenak, C.; Lang, E.; Pasham, V.; Zbidah, M.; Plate, A.; Bitzer, M.; Foller, M.; et al. Enhanced erythrocyte membrane exposure of phosphatidylserine following sorafenib treatment: An in vivo and in vitro study. Cell. Physiol. Biochem. 2012, 30, 876–888. [Google Scholar] [CrossRef]
- Shaik, N.; Lupescu, A.; Lang, F. Sunitinib-sensitive suicidal erythrocyte death. Cell. Physiol. Biochem. 2012, 30, 512–522. [Google Scholar] [CrossRef]
- Jilani, K.; Enkel, S.; Bissinger, R.; Almilaji, A.; Abed, M.; Lang, F. Fluoxetine induced suicidal erythrocyte death. Toxins 2013, 5, 1230–1243. [Google Scholar] [CrossRef]
- Jilani, K.; Lang, F. Carmustine-induced phosphatidylserine translocation in the erythrocyte membrane. Toxins 2013, 5, 703–716. [Google Scholar] [CrossRef]
- Bottger, E.; Multhoff, G.; Kun, J.F.; Esen, M. Plasmodium falciparum-infected erythrocytes induce granzyme b by nk cells through expression of host-hsp70. PLoS One 2012, 7, e33774. [Google Scholar] [CrossRef]
- Felder, K.M.; Hoelzle, K.; Ritzmann, M.; Kilchling, T.; Schiele, D.; Heinritzi, K.; Groebel, K.; Hoelzle, L.E. Hemotrophic mycoplasmas induce programmed cell death in red blood cells. Cell. Physiol. Biochem. 2011, 27, 557–564. [Google Scholar] [CrossRef] [Green Version]
- Firat, U.; Kaya, S.; Cim, A.; Buyukbayram, H.; Gokalp, O.; Dal, M.S.; Tamer, M.N. Increased caspase-3 immunoreactivity of erythrocytes in stz diabetic rats. Exp. Diabetes Res. 2012, 2012, 316384. [Google Scholar]
- Ganesan, S.; Chaurasiya, N.D.; Sahu, R.; Walker, L.A.; Tekwani, B.L. Understanding the mechanisms for metabolism-linked hemolytic toxicity of primaquine against glucose 6-phosphate dehydrogenase deficient human erythrocytes: Evaluation of eryptotic pathway. Toxicology 2012, 294, 54–60. [Google Scholar] [CrossRef]
- Gao, M.; Cheung, K.L.; Lau, I.P.; Yu, W.S.; Fung, K.P.; Yu, B.; Loo, J.F.; Kong, S.K. Polyphyllin d induces apoptosis in human erythrocytes through Ca(2)(+) rise and membrane permeabilization. Arch. Toxicol. 2012, 86, 741–752. [Google Scholar] [CrossRef]
- Ghashghaeinia, M.; Toulany, M.; Saki, M.; Bobbala, D.; Fehrenbacher, B.; Rupec, R.; Rodemann, H.P.; Ghoreschi, K.; Rocken, M.; Schaller, M.; et al. The nfkb pathway inhibitors bay 11-7082 and parthenolide induce programmed cell death in anucleated erythrocytes. Cell. Physiol. Biochem. 2011, 27, 45–54. [Google Scholar] [CrossRef]
- Jilani, K.; Lupescu, A.; Zbidah, M.; Abed, M.; Shaik, N.; Lang, F. Enhanced apoptotic death of erythrocytes induced by the mycotoxin ochratoxin a. Kidney Blood Press. Res. 2012, 36, 107–118. [Google Scholar]
- Kucherenko, Y.V.; Lang, F. Inhibitory effect of furosemide on non-selective voltage-independent cation channels in human erythrocytes. Cell. Physiol. Biochem. 2012, 30, 863–875. [Google Scholar] [CrossRef]
- Lang, E.; Jilani, K.; Zelenak, C.; Pasham, V.; Bobbala, D.; Qadri, S.M.; Lang, F. Stimulation of suicidal erythrocyte death by benzethonium. Cell. Physiol. Biochem. 2011, 28, 347–354. [Google Scholar] [CrossRef]
- Lang, E.; Qadri, S.M.; Jilani, K.; Zelenak, C.; Lupescu, A.; Schleicher, E.; Lang, F. Carbon monoxide-sensitive apoptotic death of erythrocytes. Basic Clin. Pharmacol. Toxicol. 2012, 111, 348–355. [Google Scholar]
- Lang, F.; Qadri, S.M. Mechanisms and significance of eryptosis, the suicidal death of erythrocytes. Blood Purif. 2012, 33, 125–130. [Google Scholar] [CrossRef]
- Polak-Jonkisz, D.; Purzyc, L. Ca(2+) influx versus efflux during eryptosis in uremic erythrocytes. Blood Purif. 2012, 34, 209–210. [Google Scholar] [CrossRef]
- Qadri, S.M.; Kucherenko, Y.; Zelenak, C.; Jilani, K.; Lang, E.; Lang, F. Dicoumarol activates Ca2+-permeable cation channels triggering erythrocyte cell membrane scrambling. Cell. Physiol. Biochem. 2011, 28, 857–864. [Google Scholar] [CrossRef]
- Qadri, S.M.; Bauer, J.; Zelenak, C.; Mahmud, H.; Kucherenko, Y.; Lee, S.H.; Ferlinz, K.; Lang, F. Sphingosine but not sphingosine-1-phosphate stimulates suicidal erythrocyte death. Cell. Physiol. Biochem. 2011, 28, 339–346. [Google Scholar] [CrossRef]
- Qian, E.W.; Ge, D.T.; Kong, S.K. Salidroside protects human erythrocytes against hydrogen peroxide-induced apoptosis. J. Nat. Prod. 2012, 75, 531–537. [Google Scholar] [CrossRef]
- Shaik, N.; Zbidah, M.; Lang, F. Inhibition of Ca(2+) entry and suicidal erythrocyte death by naringin. Cell. Physiol. Biochem. 2012, 30, 678–686. [Google Scholar] [CrossRef]
- Weiss, E.; Cytlak, U.M.; Rees, D.C.; Osei, A.; Gibson, J.S. Deoxygenation-induced and Ca(2+) dependent phosphatidylserine externalisation in red blood cells from normal individuals and sickle cell patients. Cell Calcium 2012, 51, 51–56. [Google Scholar] [CrossRef]
- Zelenak, C.; Pasham, V.; Jilani, K.; Tripodi, P.M.; Rosaclerio, L.; Pathare, G.; Lupescu, A.; Faggio, C.; Qadri, S.M.; Lang, F. Tanshinone iia stimulates erythrocyte phosphatidylserine exposure. Cell. Physiol. Biochem. 2012, 30, 282–294. [Google Scholar] [CrossRef]
- Lang, E.; Qadri, S.M.; Lang, F. Killing me softly—Suicidal erythrocyte death. Int. J. Biochem. Cell. Biol. 2012, 44, 1236–1243. [Google Scholar] [CrossRef]
- Vota, D.M.; Maltaneri, R.E.; Wenker, S.D.; Nesse, A.B.; Vittori, D.C. Differential erythropoietin action upon cells induced to eryptosis by different agents. Cell. Biochem. Biophys. 2013, 65, 145–157. [Google Scholar] [CrossRef]
- Vota, D.M.; Crisp, R.L.; Nesse, A.B.; Vittori, D.C. Oxidative stress due to aluminum exposure induces eryptosis which is prevented by erythropoietin. J. Cell. Biochem. 2012, 113, 1581–1589. [Google Scholar]
- Zappulla, D. Environmental stress, erythrocyte dysfunctions, inflammation, and the metabolic syndrome: Adaptations to co2 increases? Cardiometab. Syndr. 2008, 3, 30–34. [Google Scholar] [CrossRef]
- Calderon-Salinas, J.V.; Munoz-Reyes, E.G.; Guerrero-Romero, J.F.; Rodriguez-Moran, M.; Bracho-Riquelme, R.L.; Carrera-Gracia, M.A.; Quintanar-Escorza, M.A. Eryptosis and oxidative damage in type 2 diabetic mellitus patients with chronic kidney disease. Mol. Cell. Biochem. 2011, 357, 171–179. [Google Scholar] [CrossRef]
- Nicolay, J.P.; Schneider, J.; Niemoeller, O.M.; Artunc, F.; Portero-Otin, M.; Haik, G., Jr.; Thornalley, P.J.; Schleicher, E.; Wieder, T.; Lang, F. Stimulation of suicidal erythrocyte death by methylglyoxal. Cell. Physiol. Biochem. 2006, 18, 223–232. [Google Scholar] [CrossRef]
- Myssina, S.; Huber, S.M.; Birka, C.; Lang, P.A.; Lang, K.S.; Friedrich, B.; Risler, T.; Wieder, T.; Lang, F. Inhibition of erythrocyte cation channels by erythropoietin. J. Am. Soc. Nephrol. 2003, 14, 2750–2757. [Google Scholar] [CrossRef]
- Lang, P.A.; Beringer, O.; Nicolay, J.P.; Amon, O.; Kempe, D.S.; Hermle, T.; Attanasio, P.; Akel, A.; Schafer, R.; Friedrich, B.; et al. Suicidal death of erythrocytes in recurrent hemolytic uremic syndrome. J. Mol. Med. 2006, 84, 378–388. [Google Scholar] [CrossRef]
- Kempe, D.S.; Akel, A.; Lang, P.A.; Hermle, T.; Biswas, R.; Muresanu, J.; Friedrich, B.; Dreischer, P.; Wolz, C.; Schumacher, U.; et al. Suicidal erythrocyte death in sepsis. J. Mol. Med. 2007, 85, 273–281. [Google Scholar] [CrossRef]
- Lang, P.A.; Kasinathan, R.S.; Brand, V.B.; Duranton, C.; Lang, C.; Koka, S.; Shumilina, E.; Kempe, D.S.; Tanneur, V.; Akel, A.; et al. Accelerated clearance of plasmodium-infected erythrocytes in sickle cell trait and annexin-a7 deficiency. Cell. Physiol. Biochem. 2009, 24, 415–428. [Google Scholar] [CrossRef]
- Siraskar, B.; Ballal, A.; Bobbala, D.; Foller, M.; Lang, F. Effect of amphotericin b on parasitemia and survival of plasmodium berghei-infected mice. Cell. Physiol. Biochem. 2010, 26, 347–354. [Google Scholar] [CrossRef]
- Bobbala, D.; Alesutan, I.; Foller, M.; Huber, S.M.; Lang, F. Effect of anandamide in plasmodium berghei-infected mice. Cell. Physiol. Biochem. 2010, 26, 355–362. [Google Scholar] [CrossRef]
- Foller, M.; Bobbala, D.; Koka, S.; Huber, S.M.; Gulbins, E.; Lang, F. Suicide for survival—Death of infected erythrocytes as a host mechanism to survive malaria. Cell. Physiol. Biochem. 2009, 24, 133–140. [Google Scholar] [CrossRef]
- Koka, S.; Bobbala, D.; Lang, C.; Boini, K.M.; Huber, S.M.; Lang, F. Influence of paclitaxel on parasitemia and survival of plasmodium berghei infected mice. Cell. Physiol. Biochem. 2009, 23, 191–198. [Google Scholar] [CrossRef]
- Lang, P.A.; Schenck, M.; Nicolay, J.P.; Becker, J.U.; Kempe, D.S.; Lupescu, A.; Koka, S.; Eisele, K.; Klarl, B.A.; Rubben, H.; et al. Liver cell death and anemia in wilson disease involve acid sphingomyelinase and ceramide. Nat. Med. 2007, 13, 164–170. [Google Scholar] [CrossRef]
- Kempe, D.S.; Lang, P.A.; Duranton, C.; Akel, A.; Lang, K.S.; Huber, S.M.; Wieder, T.; Lang, F. Enhanced programmed cell death of iron-deficient erythrocytes. FASEB J. 2006, 20, 368–370. [Google Scholar]
- Birka, C.; Lang, P.A.; Kempe, D.S.; Hoefling, L.; Tanneur, V.; Duranton, C.; Nammi, S.; Henke, G.; Myssina, S.; Krikov, M.; et al. Enhanced susceptibility to erythrocyte “apoptosis” following phosphate depletion. Pflugers Arch. 2004, 448, 471–477. [Google Scholar]
- Niederberger, E.; Tegeder, I.; Vetter, G.; Schmidtko, A.; Schmidt, H.; Euchenhofer, C.; Brautigam, L.; Grosch, S.; Geisslinger, G. Celecoxib loses its anti-inflammatory efficacy at high doses through activation of nf-kappab. FASEB J. 2001, 15, 1622–1624. [Google Scholar]
- Kim, C.H.; Chung, C.W.; Lee, H.M.; Kim, D.H.; Kwak, T.W.; Jeong, Y.I.; Kang, D.H. Synergistic effects of 5-aminolevulinic acid based photodynamic therapy and celecoxib via oxidative stress in human cholangiocarcinoma cells. Int. J. Nanomed. 2013, 8, 2173–2185. [Google Scholar]
- Lampiasi, N.; Azzolina, A.; Umezawa, K.; Montalto, G.; McCubrey, J.A.; Cervello, M. The novel nf-kappab inhibitor dhmeq synergizes with celecoxib to exert antitumor effects on human liver cancer cells by a ros-dependent mechanism. Cancer Lett. 2012, 322, 35–44. [Google Scholar] [CrossRef]
- Bookchin, R.M.; Ortiz, O.E.; Lew, V.L. Activation of calcium-dependent potassium channels in deoxygenated sickled red cells. Prog. Clin. Biol. Res. 1987, 240, 193–200. [Google Scholar]
- Lang, P.A.; Kempe, D.S.; Myssina, S.; Tanneur, V.; Birka, C.; Laufer, S.; Lang, F.; Wieder, T.; Huber, S.M. Pge(2) in the regulation of programmed erythrocyte death. Cell Death Differ. 2005, 12, 415–428. [Google Scholar] [CrossRef]
- Borst, O.; Abed, M.; Alesutan, I.; Towhid, S.T.; Qadri, S.M.; Foller, M.; Gawaz, M.; Lang, F. Dynamic adhesion of eryptotic erythrocytes to endothelial cells via cxcl16/sr-psox. Am. J. Physiol. Cell. Physiol. 2012, 302, C644–C651. [Google Scholar] [CrossRef]
- Andrews, D.A.; Low, P.S. Role of red blood cells in thrombosis. Curr. Opin. Hematol. 1999, 6, 76–82. [Google Scholar] [CrossRef]
- Closse, C.; Dachary-Prigent, J.; Boisseau, M.R. Phosphatidylserine-related adhesion of human erythrocytes to vascular endothelium. Br. J. Haematol. 1999, 107, 300–302. [Google Scholar] [CrossRef]
- Gallagher, P.G.; Chang, S.H.; Rettig, M.P.; Neely, J.E.; Hillery, C.A.; Smith, B.D.; Low, P.S. Altered erythrocyte endothelial adherence and membrane phospholipid asymmetry in hereditary hydrocytosis. Blood 2003, 101, 4625–4627. [Google Scholar] [CrossRef]
- Pandolfi, A.; di Pietro, N.; Sirolli, V.; Giardinelli, A.; di Silvestre, S.; Amoroso, L.; di Tomo, P.; Capani, F.; Consoli, A.; Bonomini, M. Mechanisms of uremic erythrocyte-induced adhesion of human monocytes to cultured endothelial cells. J. Cell. Physiol. 2007, 213, 699–709. [Google Scholar] [CrossRef]
- Wood, B.L.; Gibson, D.F.; Tait, J.F. Increased erythrocyte phosphatidylserine exposure in sickle cell disease: Flow-cytometric measurement and clinical associations. Blood 1996, 88, 1873–1880. [Google Scholar]
- Chung, S.M.; Bae, O.N.; Lim, K.M.; Noh, J.Y.; Lee, M.Y.; Jung, Y.S.; Chung, J.H. Lysophosphatidic acid induces thrombogenic activity through phosphatidylserine exposure and procoagulant microvesicle generation in human erythrocytes. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 414–421. [Google Scholar]
- Zwaal, R.F.; Comfurius, P.; Bevers, E.M. Surface exposure of phosphatidylserine in pathological cells. Cell. Mol. Life Sci. 2005, 62, 971–988. [Google Scholar] [CrossRef]
- Chan, A.L. Celecoxib-induced deep-vein thrombosis. Ann. Pharmacother. 2005, 39, 1138. [Google Scholar] [CrossRef]
- Sands, G.; Shell, B.; Zhang, R. Adverse events in patients with blood loss: A pooled analysis of 51 clinical studies from the celecoxib clinical trial database. Open Rheumatol. J. 2012, 6, 44–49. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Lupescu, A.; Bissinger, R.; Jilani, K.; Lang, F. Triggering of Suicidal Erythrocyte Death by Celecoxib. Toxins 2013, 5, 1543-1554. https://doi.org/10.3390/toxins5091543
Lupescu A, Bissinger R, Jilani K, Lang F. Triggering of Suicidal Erythrocyte Death by Celecoxib. Toxins. 2013; 5(9):1543-1554. https://doi.org/10.3390/toxins5091543
Chicago/Turabian StyleLupescu, Adrian, Rosi Bissinger, Kashif Jilani, and Florian Lang. 2013. "Triggering of Suicidal Erythrocyte Death by Celecoxib" Toxins 5, no. 9: 1543-1554. https://doi.org/10.3390/toxins5091543