Intranasal Rapamycin Rescues Mice from Staphylococcal Enterotoxin B-Induced Shock

Abstract
:1. Introduction
2. Results and Discussion
2.1. Therapeutic Window of Rapamycin Treatment

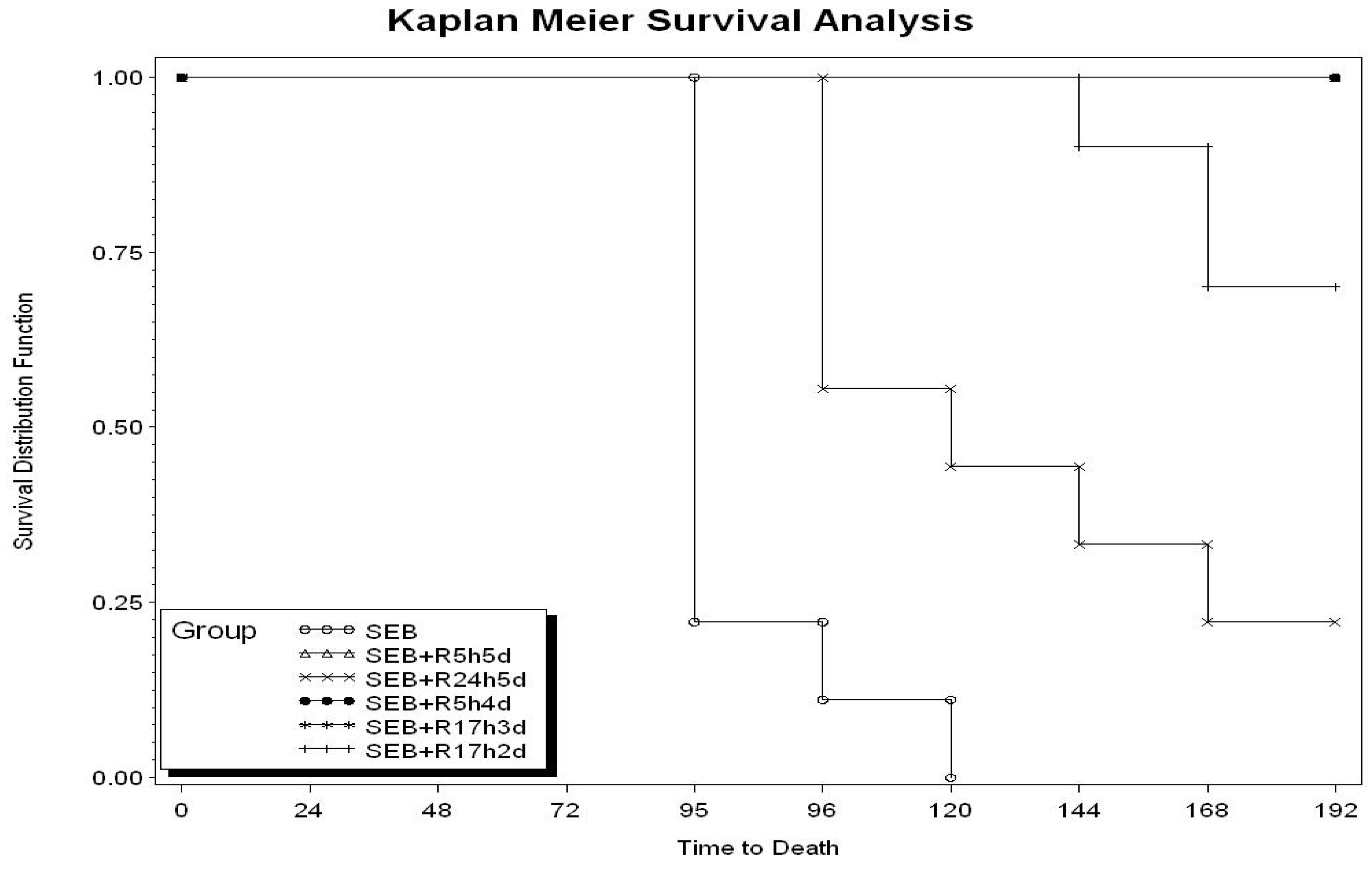{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Toxin + rapamycin (R) regiment a | Live/Total b |
|---|---|
| SEB | 0/9 |
| SEB + R at 5, 24, 48, 72, 96 h (R5h5d) | 9/9 |
| SEB + R at 24, 30, 48, 72, 96 h (R24h5d) | 2/9 |
| SEB | 0/10 |
| SEB + R at 5, 24, 48, 72 h (R5h4d) | 10/10 |
| SEB + R at 17, 23, 41 h (R17h3d) | 10/10 |
| SEB + R at 17, 23 h (R17h2d) | 7/10 |

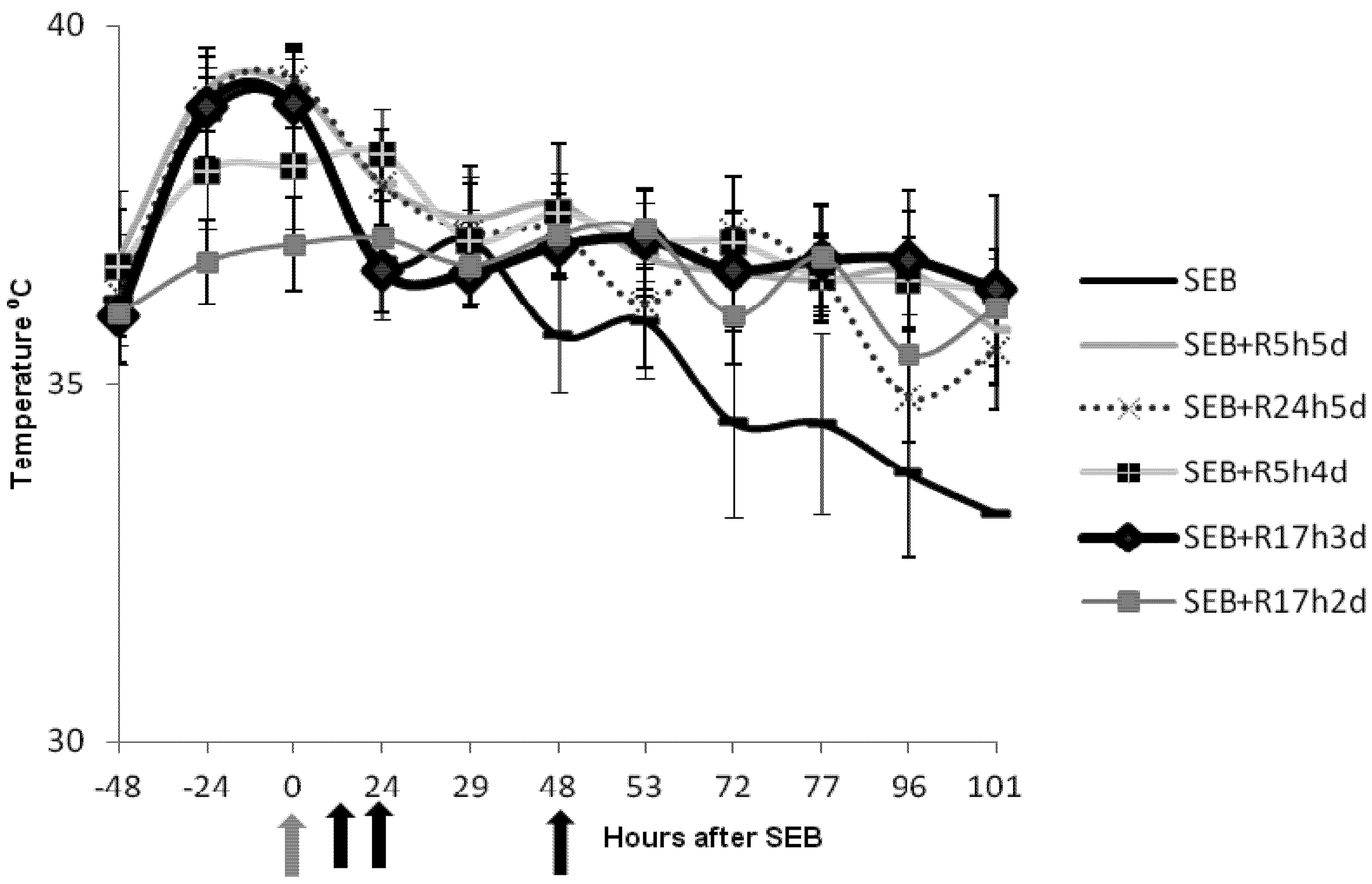
2.2. Rapamycin Prevents Hyperthermia in SEB-Induced Shock Model
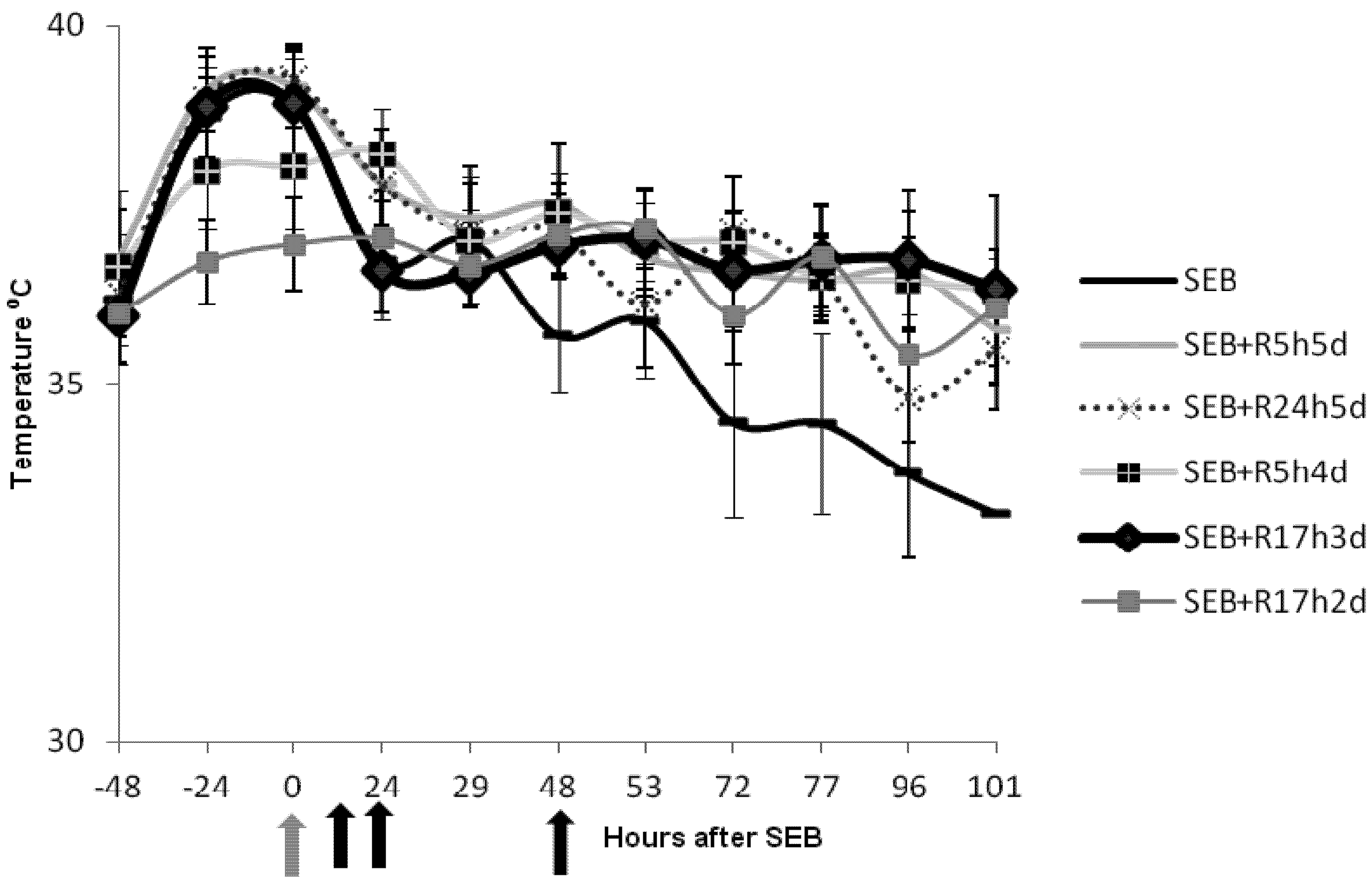
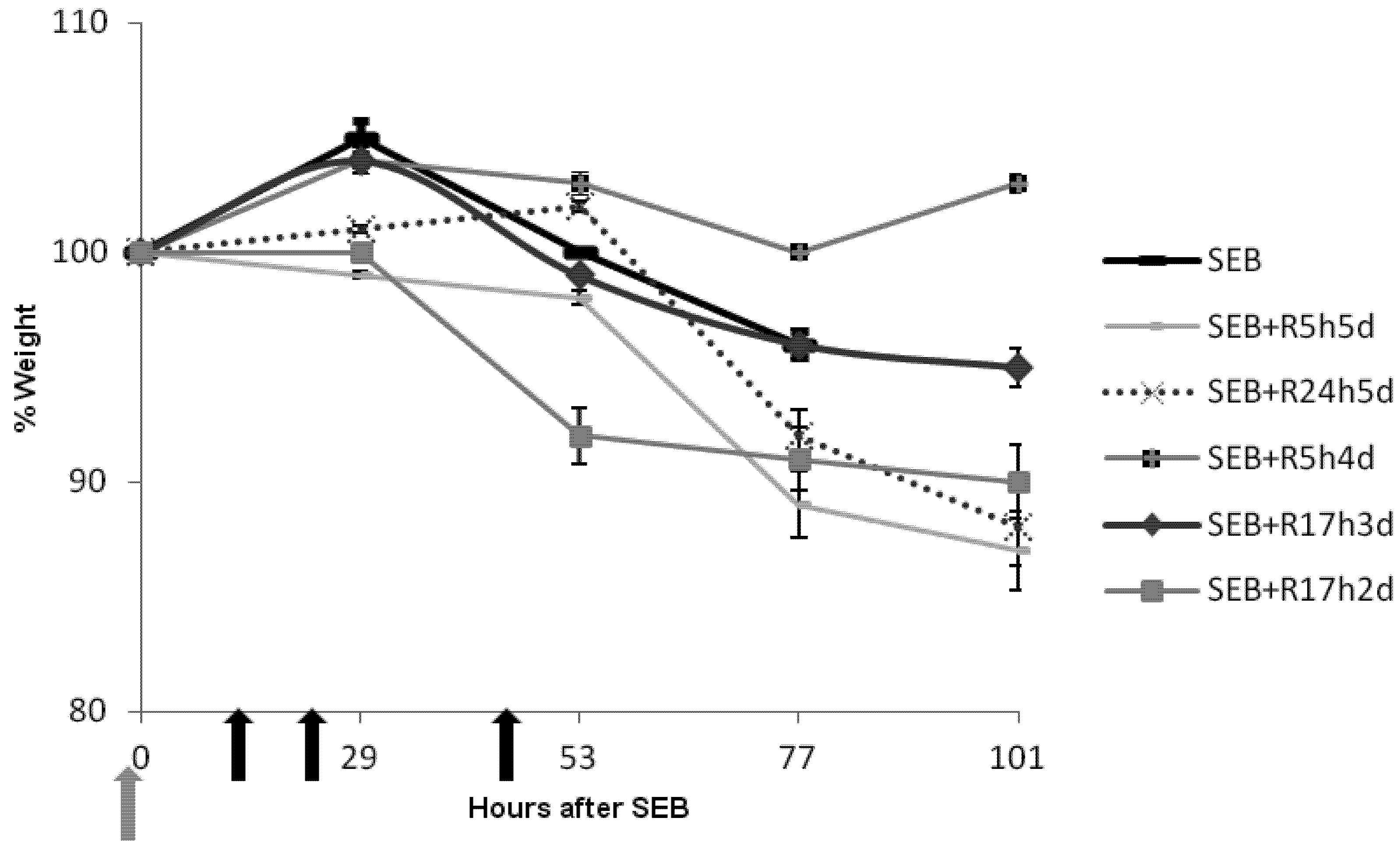
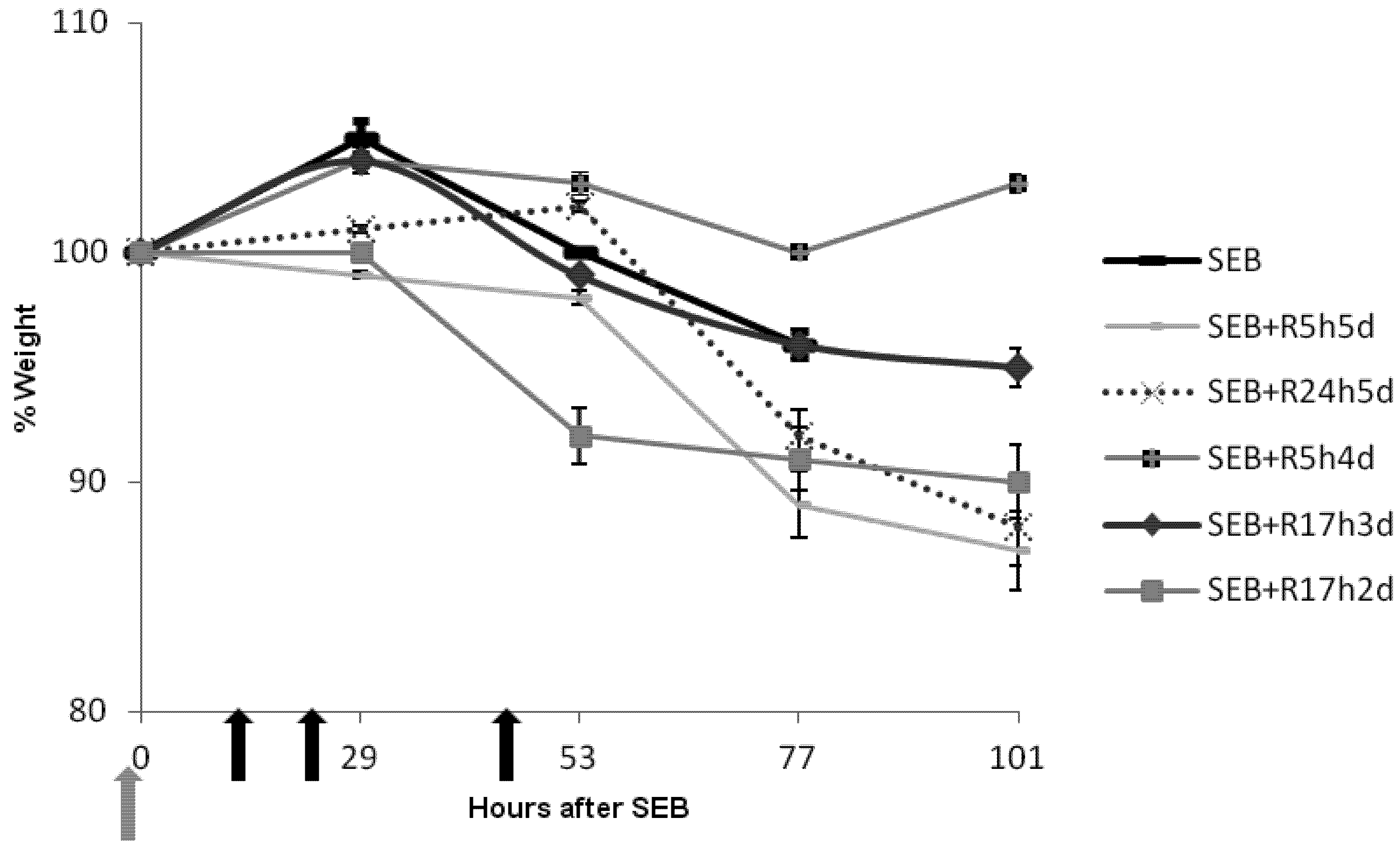
2.3. Rapamycin Prevents Weight Loss in SEB-Induced Shock Model
2.4. Rapamycin Attenuates Pulmonary Mediators

3. Experimental Section
3.1. Materials
3.2. Mouse Model of SEB-Induced Shock
3.3. Cytokine and Chemokine Assays
3.4. Statistical Analysis
4. Conclusions
Acknowledgments
Conflict of Interest
References
- Chesney, P.J.; Davis, J.P.; Purday, W.K.; Wand, P.J.; Chesney, R.W. Clinical manifestations of toxic shock syndrome. J. Am. Med. Assoc. 1981, 246, 741–748. [Google Scholar] [CrossRef]
- Holmberg, S.D.; Blake, P.A. Staphylococcal food poisoning in the United States. New facts and old misconceptions. JAMA 1984, 251, 487–489. [Google Scholar] [CrossRef]
- Wieneke, A.; Roberts, D.; Gilbert, R.J. Staphyloccal food poisoning in the United Kingdom. Epidemiol. Infect. 1990, 110, 519–531. [Google Scholar]
- Kotzin, B.L.; Leung, D.Y.M.; Kappler, J.; Marrack, P.A. Superantigens and their potential role in human disease. Adv. Immunol. 1993, 54, 99–166. [Google Scholar] [CrossRef]
- McCormick, J.K.; Yarwood, J.M.; Schlievert, P.M. Toxic shock syndrome and bacterial superantigens: an update. Ann. Rev. Microbiol. 2001, 55, 77–104. [Google Scholar] [CrossRef]
- Stevens, D.L. The toxic shock syndromes. Infect. Dis. Clin. North Am. 1996, 10, 727–746. [Google Scholar] [CrossRef]
- Proft, T.; Fraser, J.D. Bacterial superantigens. Clin. Exp. Immunol. 2003, 133, 299–306. [Google Scholar] [CrossRef]
- Choi, Y.; Kotzin, B.; Hernon, L.; Callahan, J.; Marrack, P.; Kappler, J. Interaction of Staphyloccocus aureus toxin “superantigens” with human T cells. Proc. Natl. Acad. Sci. USA 1989, 86, 8941–8945. [Google Scholar]
- Fleischer, B.; Schrezenmeier, H.; Conradt, P. T-lymphocyte activation by staphylococcal enterotoxins: Role of class II molecules and T cell surface structures. Cell. Immunol. 1989, 120, 92–101. [Google Scholar] [CrossRef]
- Marrack, P.; Kappler, J. The staphylococcal enterotoxins and their relatives. Science 1990, 248, 705–709. [Google Scholar]
- Mollick, J.A.; Chintagumpala, M.; Cook, R.G.; Rich, R.R. Staphylococcal exotoxin activation of T cells. Role of exotoxin-MHC class II binding affinity and class II isotype. J. Immunol. 1991, 146, 463–468. [Google Scholar]
- Scholl, P.; Diez, A.; Mourad, W.; Parsonnet, J.; Geha, R.S.; Chatila, T. Toxic shock syndrome toxin-1 binds to major histocompatibility complex class II molecules. Proc. Natl. Acad. Sci. USA 1989, 86, 4210–4214. [Google Scholar] [CrossRef]
- Fraser, J.D.; Proft, T. The bacterial superantigens and superantigen-like proteins. Immunol. Rev. 2008, 225, 226–243. [Google Scholar] [CrossRef]
- Krakauer, T. Therapeutic down-modulators of staphylococcal superantigen-induced inflammation and toxic shock. Toxins 2010, 2, 1963–1983. [Google Scholar] [CrossRef]
- Krakauer, T.; Vilcek, J.; Oppenheim, J.J. TNF family cytokines, chemokines and other pro- or anti-inflammatory cytokines. In Fundamental Immunology, 4th; Paul, W.E., Ed.; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 1999; pp. 783–811. [Google Scholar]
- Miethke, T.; Wahl, C.; Heeg, K.; Echtenacher, B.; Krammer, P.H.; Wagner, H. T cell-mediated lethal shock triggered in mice by the superantigen SEB: Critical role of TNF. J. Exp. Med. 1992, 175, 91–98. [Google Scholar] [CrossRef]
- Stiles, B.G.; Bavari, S.; Krakauer, T.; Ulrich, R.G. Toxicity of staphylococcal enterotoxins potentiated by lipopolysaccharide: Major histocompatibility complex class II molecule dependency and cytokine release. Infect. Immun. 1993, 61, 5333–5338. [Google Scholar]
- Blank, C.; Luz, A.; Bendigs, S.; Erdmann, A.; Wagner, H.; Heeg, K. Superantigen and endotoxin synergize in the induction of lethal shock. Eur. J. Immunol. 1997, 27, 825–833. [Google Scholar] [CrossRef]
- Arad, G.; Levy, R.; Hillman, D.; Kaempfer, R. Superantigen antagonist protects against lethal shock and defines a new domain for T-cell activation. Nat. Med. 2000, 6, 414–421. [Google Scholar] [CrossRef]
- Visvanathan, K.; Charles, A.; Bannan, J.; Pugach, P.; Kashfi, K.; Zabriskie, J.B. Inhibition of bacterial superantigens by peptides and antibodies. Infect. Immun. 2001, 69, 875–884. [Google Scholar] [CrossRef]
- Madsen, J.M. Toxins as weapons of mass destruction. A comparison and contrast with biological-warfare and chemical-warfare agents. Clin. Lab. Med 2001, 21, 593–605. [Google Scholar]
- Darenberg, J.; Soderquist, B.; Normark, B.H.; Norrby-Tegland, A. Differences in potency of intravenous polyspecific immunoglobin G against streptoccocal and staphyloccocal superantigen: implications for therapy of toxic shock syndrome. Clin. Infect. Dis. 2004, 38, 836–842. [Google Scholar] [CrossRef]
- Nagaki, M.; Muto, Y.; Ohnishi, H.; Yasuda, S.; Sano, K.; Naito, T.; Maeda, T.; Yamada, T.; Moriwaki, H. Hepatic injury and lethal shock in galactosamine-sensitized mice induced by the superantigen staphylococcal enterotoxin B. Gastroenterology 1994, 106, 450–458. [Google Scholar]
- Chen, J.Y.; Qiao, Y.; Komisar, J.L.; Bazem, W.B.; Hsu, I.C.; Tseng, J. Increased susceptibility to staphylococcal enterotoxin B intoxication in mice primed with actinomycin D. Infect. Immun. 1994, 62, 4626–4631. [Google Scholar]
- Sarwar, S.R.; Blackman, M.A.; Doherty, P.C. Superantigen shock in mice with an inapparent viral infection. J. Infect. Dis. 1994, 170, 1189–1194. [Google Scholar] [CrossRef]
- Roy, C.J.; Warfield, K.L.; Welcher, B.C.; Gonzales, R.F.; Larsen, T.; Hanson, J.; David, C.S.; Krakauer, T.; Bavari, S. Human leukocyte antigen-DQ8 transgenic mice: a model to examine the toxicity of aerosolized staphylococcal enterotoxin B. Infect. Immun. 2005, 73, 2452–2460. [Google Scholar]
- Yeung, R.S.; Penninger, J.M.; Kundig, T.; Khoo, W.; Ohashi, P.S.; Kroemer, G.; Mak, T.W. Human CD4 and human major histocompatibility complex class II (DQ6) transgenic mice: Supersensitivity to superantigen-induced septic shock. Eur. J. Immunol. 1996, 26, 1074–1082. [Google Scholar] [CrossRef]
- Huzella, L.M.; Buckley, M.J.; Alves, D.A.; Stiles, B.G.; Krakauer, T. Central roles for IL-2 and MCP-1 following intranasal exposure to SEB: A new mouse model. Res. Vet. Sci. 2009, 86, 241–247. [Google Scholar] [CrossRef]
- Flechner, S.M. Sirolimus in kidney transplantation indications and practical guidelines: Denovo sirolimus-based therapy without calcineurin inhibitors. Transplantation 2009, 87, S1–S6. [Google Scholar] [CrossRef]
- Thomson, A.W.; Turnquist, H.R.; Raimondi, G. Immunoregulatory functions of mTOR inhibition. Nat. Rev. Immunol. 2009, 9, 324–337. [Google Scholar] [CrossRef]
- Coenen, J.J.; Koenen, H.J.; van Rijssen, E.; Hilbrands, L.B.; Joosten, I. Rapamycin, and not cyclosporin A, preserves the highly suppressive CD27+ subset of human CD4+CD25+ regulatory T cells. Blood 2006, 107, 1018–1023. [Google Scholar]
- Krakauer, T.; Buckley, M.; Issaq, H.J.; Fox, S.D. Rapamycin protects mice from staphylococcal enterotoxin B-induced toxic shock and blocks cytokine release in vitro and in vivo. Antimicrob. Agents Chemother. 2010, 54, 1125–1131. [Google Scholar] [CrossRef]
- Stiles, B.G.; Campbell, Y.G.; Castle, R.M.; Grove, S.A. Correlation of temperature and toxicity in murine studies of staphylococcal enterotoxins and toxic shock syndrome toxin 1. Infect. Immun. 1999, 67, 1521–1525. [Google Scholar]
- Vlach, K.D.; Boles, J.W.; Stiles, B.G. Telemetric evaluation of body temperature and physical activity as predictors of mortality in a murine model of staphylococcal enterotoxic shock. Comp. Med. 2000, 50, 160–166. [Google Scholar]
- Mattthys, P.; Mitera, T.; Heremans, H.; Van Damme, J.; Billiau, A. Anti-gamma interferon and anti-interleukin-6 antibodies affect staphylococcal enterotoxin B-induced weight loss, hypoglycemia, and cytokine release in D-galctosamine-sensitized and unsensitized mice. Infect. Immun. 1995, 63, 1158–1164. [Google Scholar]
- Nilsson, A.; Sköld, K.; Sjögren, B.; Svensson, M.; Pierson, J.; Zhang, X.; Caprioli, R.M.; Buijs, J.; Persson, B.; Svenningsson, P.; et al. Increased striatal mRNA and protein levels of the immunophilin FKBP-12 in experimental Parkinson’s disease and identification of FKBP-12-binding proteins. J. Proteome Res. 2007, 6, 3952–3961. [Google Scholar]
- Sternberg, E.M. Neural regulation of innate immunity: A coordinated nonspecific host response to pathogens. Nat. Rev. Immunol. 2006, 6, 318–328. [Google Scholar] [CrossRef]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Krakauer, T.; Buckley, M. Intranasal Rapamycin Rescues Mice from Staphylococcal Enterotoxin B-Induced Shock. Toxins 2012, 4, 718-728. https://doi.org/10.3390/toxins4090718
Krakauer T, Buckley M. Intranasal Rapamycin Rescues Mice from Staphylococcal Enterotoxin B-Induced Shock. Toxins. 2012; 4(9):718-728. https://doi.org/10.3390/toxins4090718
Chicago/Turabian StyleKrakauer, Teresa, and Marilyn Buckley. 2012. "Intranasal Rapamycin Rescues Mice from Staphylococcal Enterotoxin B-Induced Shock" Toxins 4, no. 9: 718-728. https://doi.org/10.3390/toxins4090718