Macrophage-Targeted Therapy: CD64-Based Immunotoxins for Treatment of Chronic Inflammatory Diseases

{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. The Role of Macrophages in Immunity
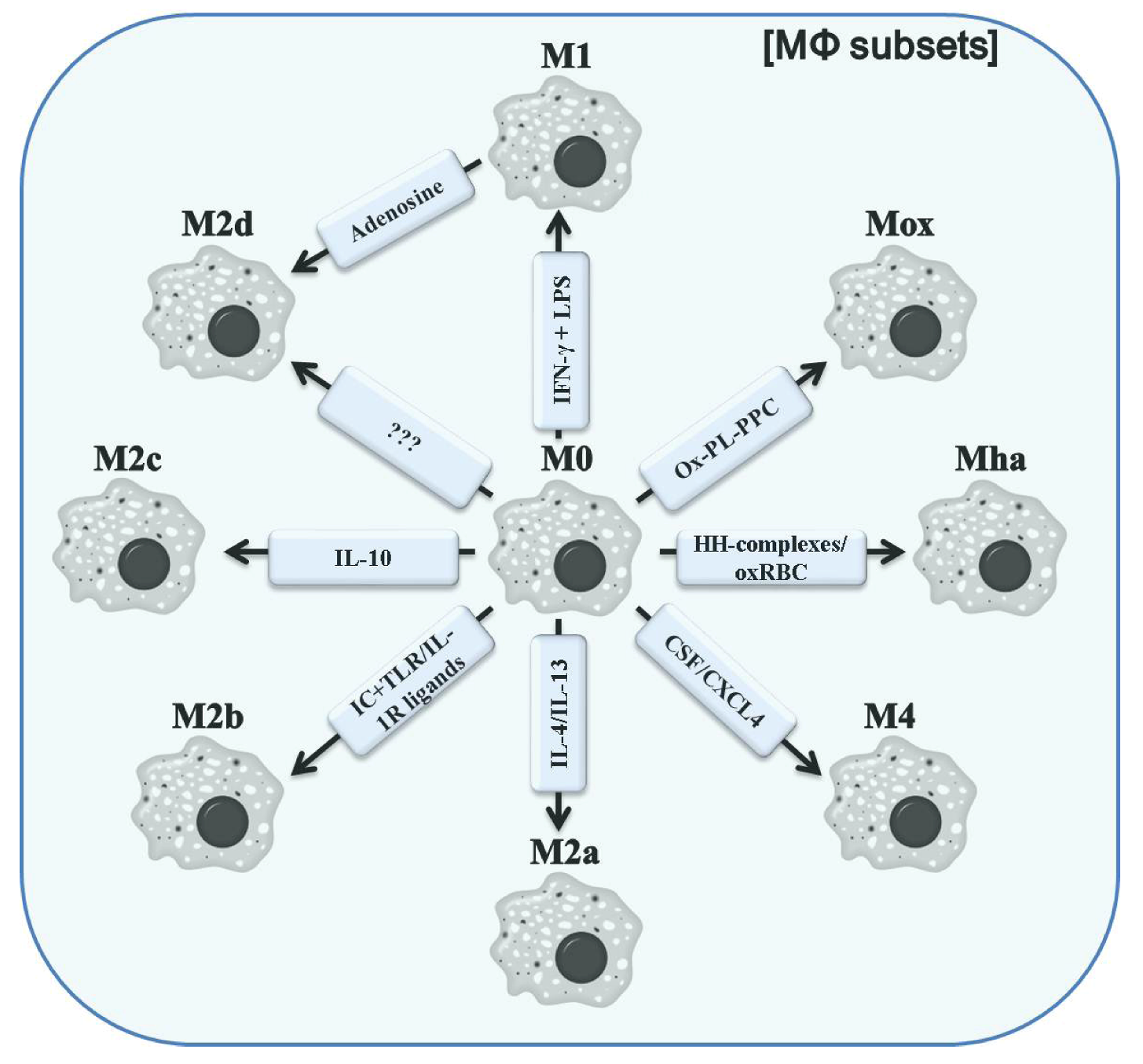
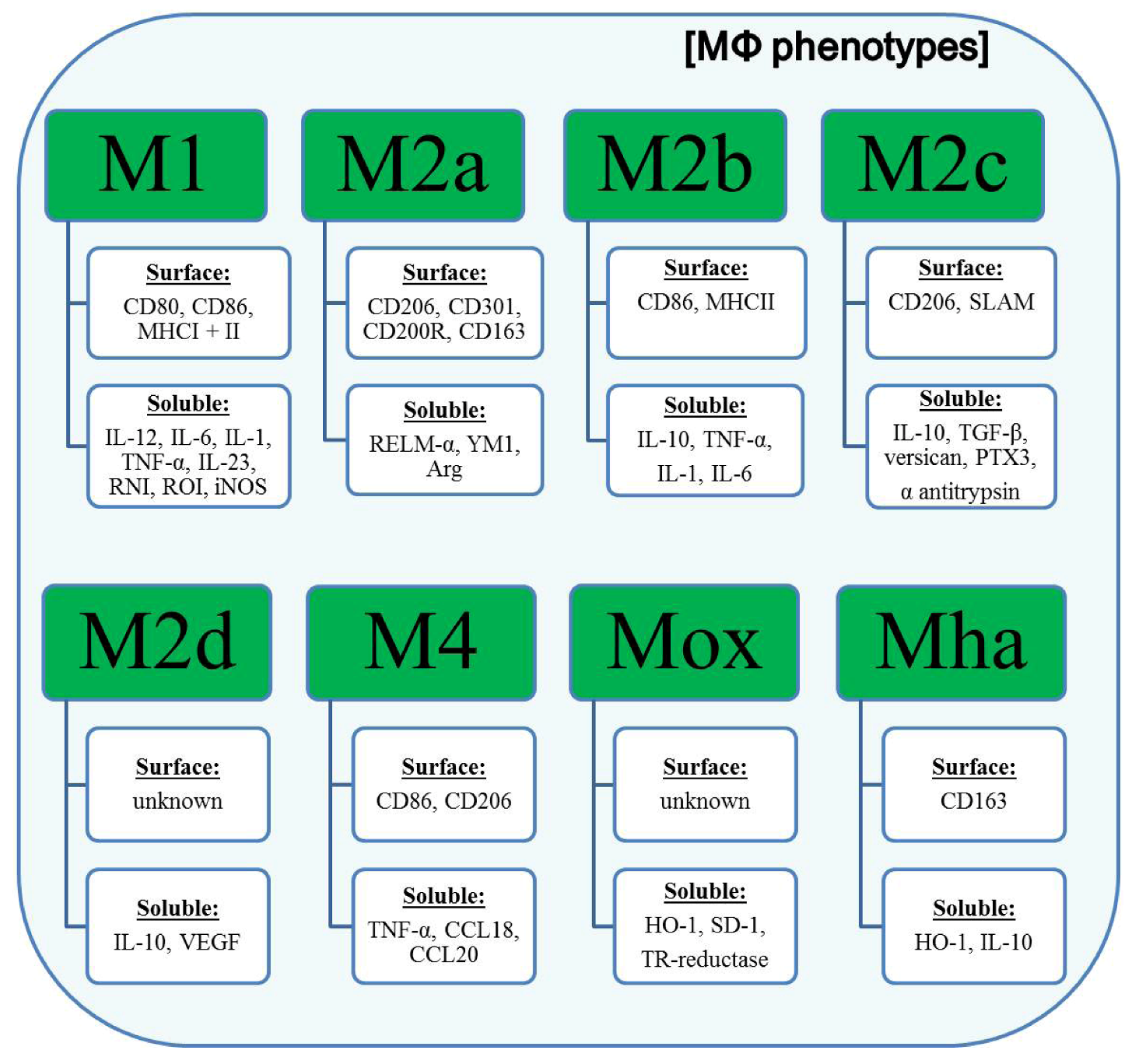
3. Classification of Polarized Macrophages and Their Contribution to Inflammation
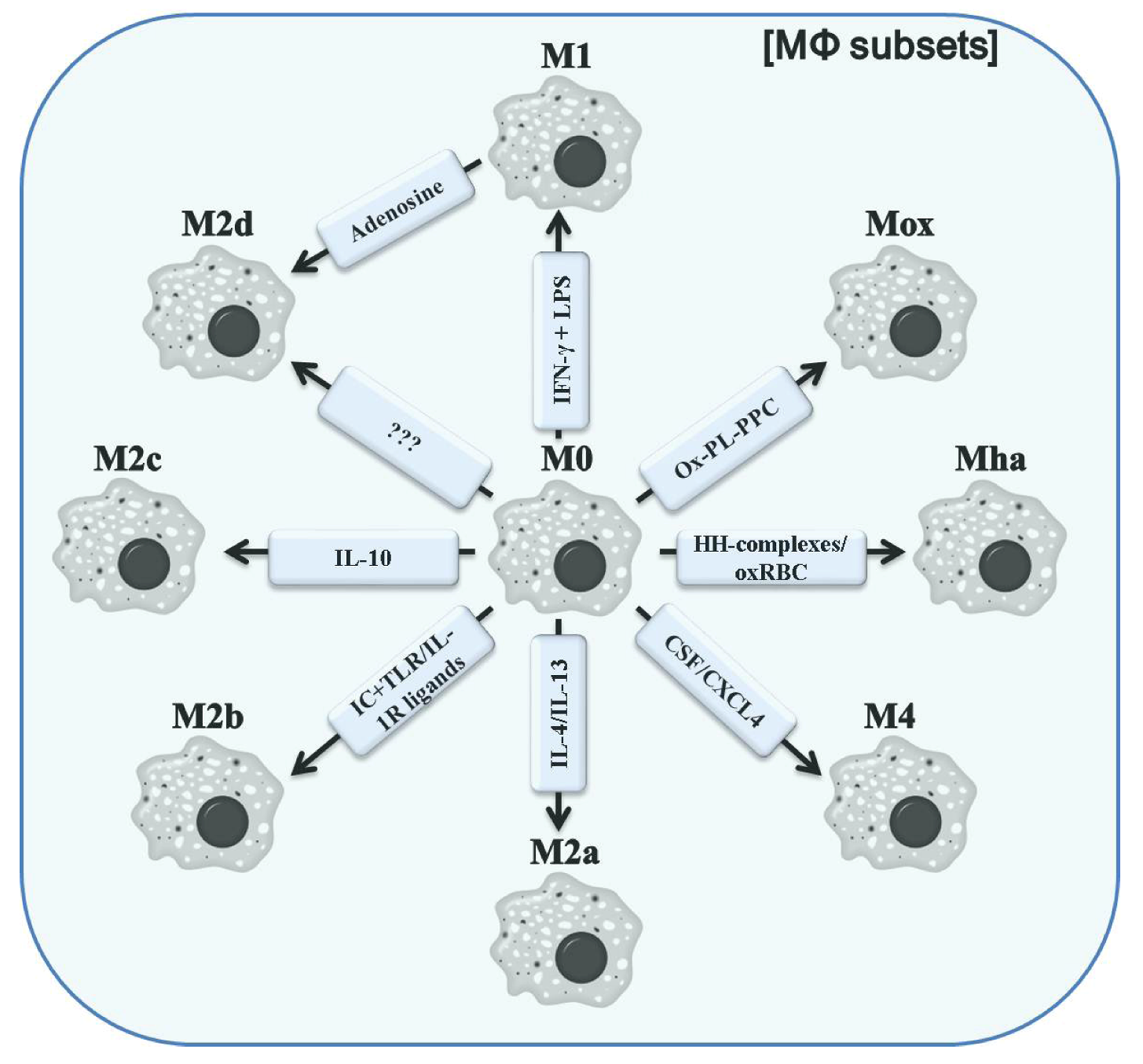
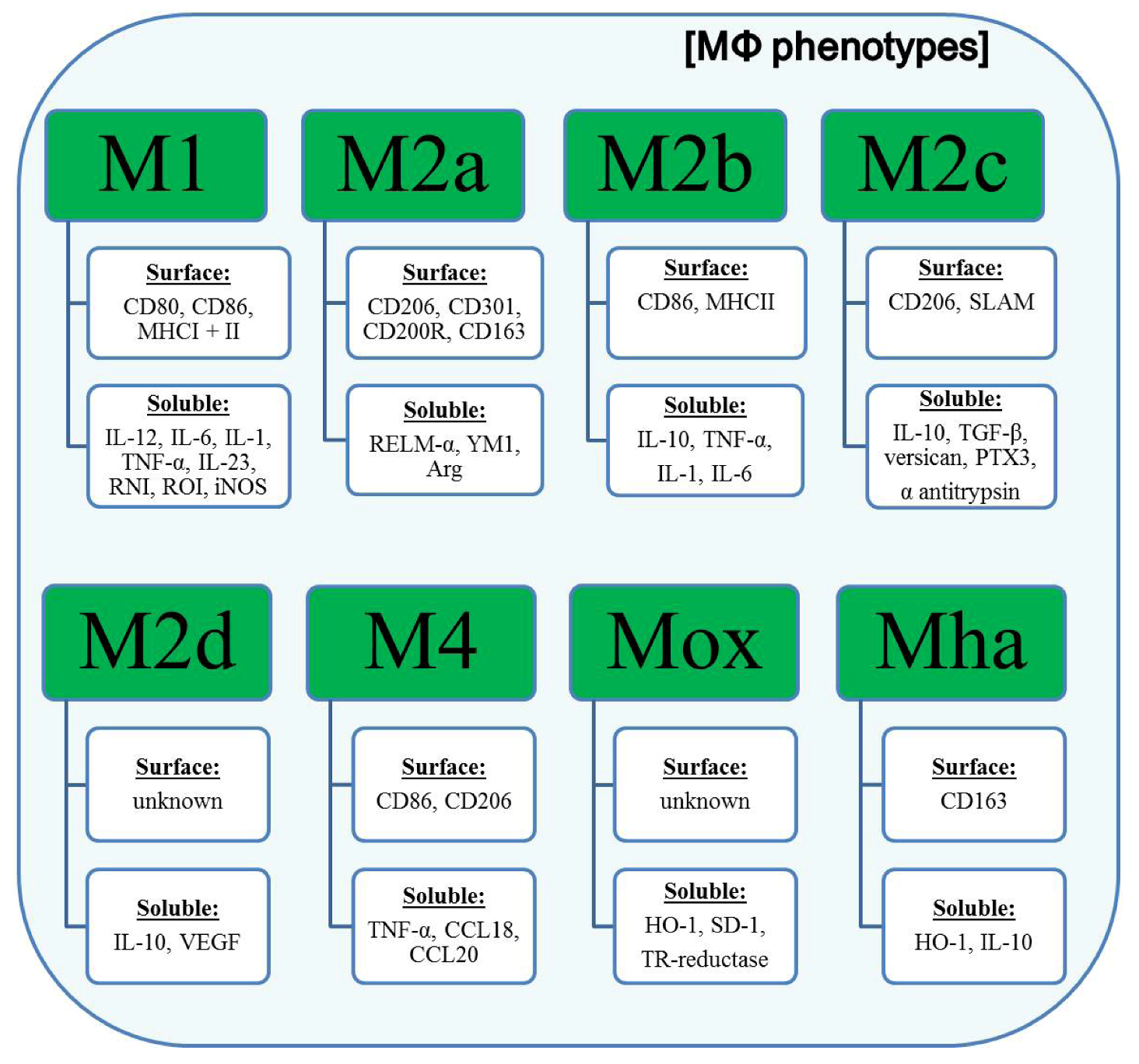
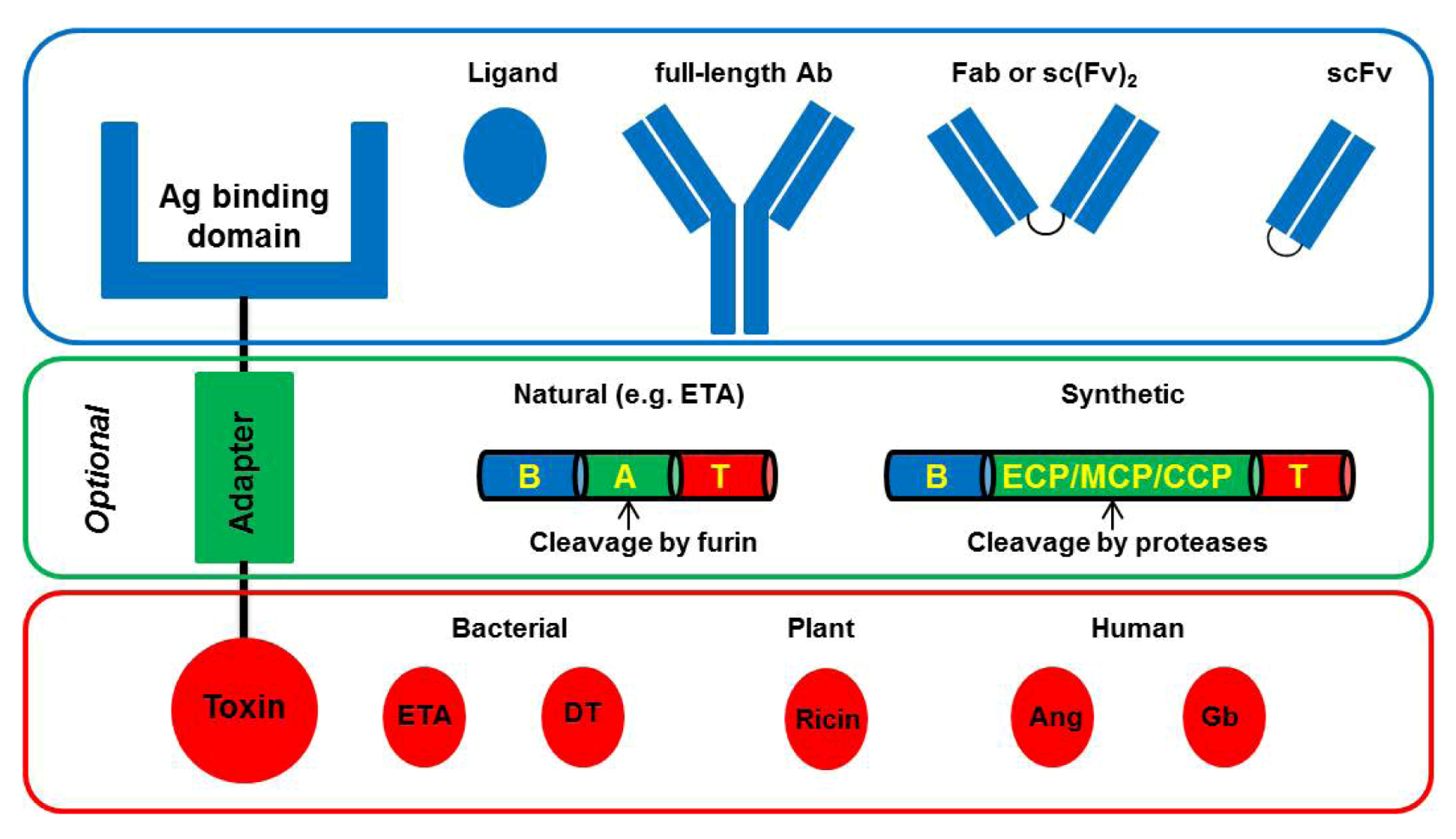
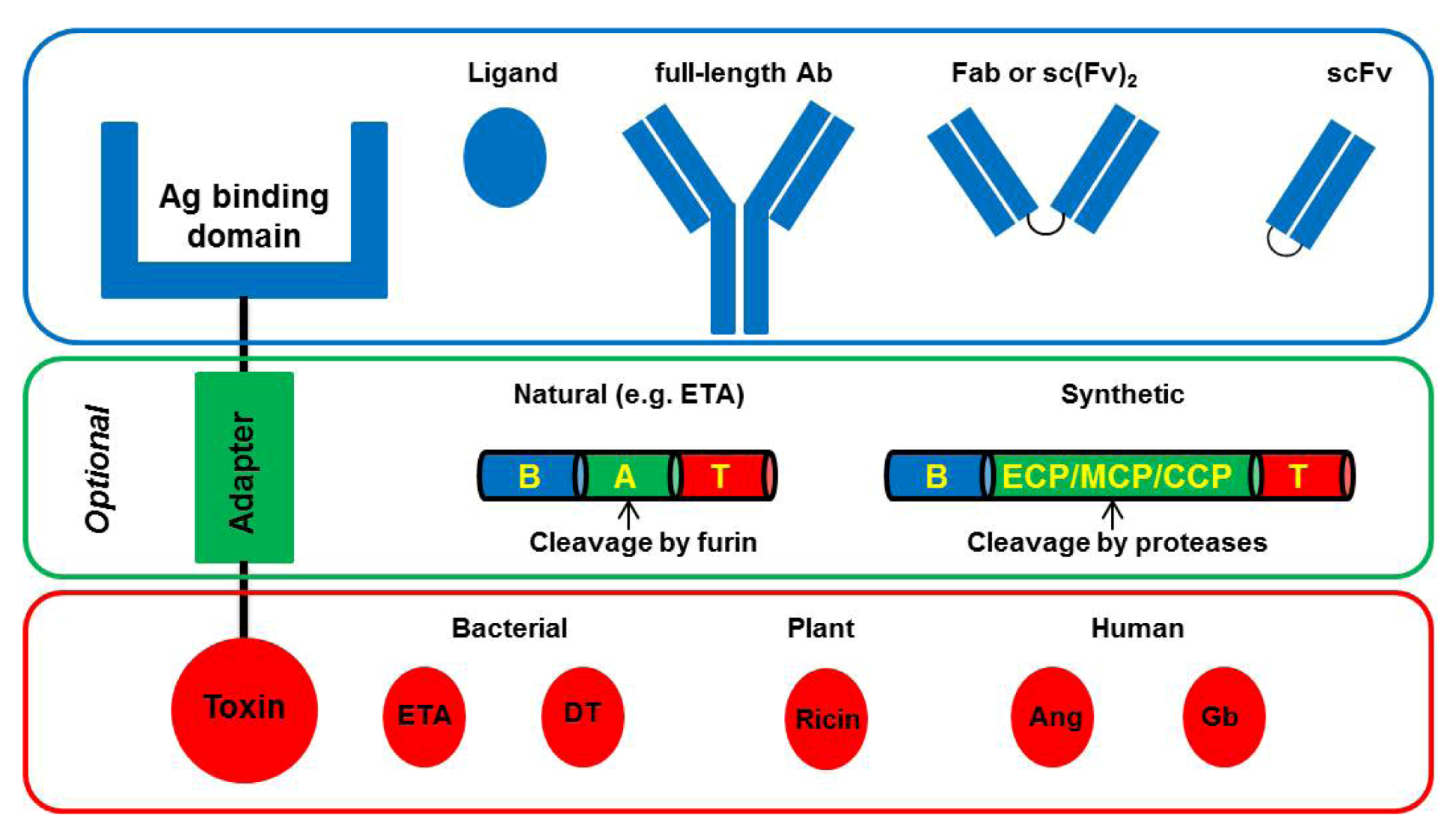
4. Improved CD64-Targeting Immunotoxins for Therapy of Chronic Inflammatory Diseases
5. Conclusions
Conflict of Interest
Acknowledgements
References
- Cawley, D.B.; Herschman, H.R.; Gilliland, D.G.; Collier, R.J. Epidermal growth factor-toxin A chain conjugates: EGF-ricin A is a potent toxin while EGF-diphtheria fragment A is nontoxic. Cell 1980, 22, 563–570. [Google Scholar] [CrossRef]
- Murphy, J.R.; Bishai, W.; Borowski, M.; Miyanohara, A.; Boyd, J.; Nagle, S. Genetic construction, expression, and melanoma-selective cytotoxicity of a diphtheria toxin-related alpha-melanocyte-stimulating hormone fusion protein. Proc. Natl. Acad. Sci. USA 1986, 83, 8258–8262. [Google Scholar] [CrossRef]
- FitzGerald, D.J.; Padmanabhan, R.; Pastan, I.; Willingham, M.C. Adenovirus-induced release of epidermal growth factor and pseudomonas toxin into the cytosol of KB cells during receptor-mediated endocytosis. Cell 1983, 32, 607–617. [Google Scholar] [CrossRef]
- Williams, D.P.; Parker, K.; Bacha, P.; Bishai, W.; Borowski, M.; Genbauffe, F.; Strom, T.B.; Murphy, J.R. Diphtheria toxin receptor binding domain substitution with interleukin-2: Genetic construction and properties of a diphtheria toxin-related interleukin-2 fusion protein. Protein. Eng. 1987, 1, 493–498. [Google Scholar] [CrossRef]
- Von Minckwitz, G.; Harder, S.; Hovelmann, S.; Jager, E.; Al-Batran, S.E.; Loibl, S.; Atmaca, A.; Cimpoiasu, C.; Neumann, A.; Abera, A.; et al. Phase I clinical study of the recombinant antibody toxin scFv(FRP5)-ETA specific for the ErbB2/HER2 receptor in patients with advanced solid malignomas. Breast Cancer Res. 2005, 7, R617–R626. [Google Scholar] [CrossRef] [Green Version]
- Zhou, X.X.; Ji, F.; Zhao, J.L.; Cheng, L.F.; Xu, C.F. Anti-cancer activity of anti-p185HER-2 ricin A chain immunotoxin on gastric cancer cells. J. Gastroenterol. Hepatol. 2010, 25, 1266–1275. [Google Scholar]
- Hu, C.C.; Ji, H.M.; Chen, S.L.; Zhang, H.W.; Wang, B.Q.; Zhou, L.Y.; Zhang, Z.P.; Sun, X.L.; Chen, Z.Z.; Cai, Y.Q.; et al. Investigation of a plasmid containing a novel immunotoxin VEGF165-PE38 gene for antiangiogenic therapy in a malignant glioma model. Int. J. Cancer 2010, 12, 2222–2229. [Google Scholar]
- Wesche, J.; Rapak, A.; Olsnes, S. Dependence of ricin toxicity on translocation of the toxin A-chain from the endoplasmic reticulum to the cytosol. J. Biol. Chem. 1999, 27, 34443–34449. [Google Scholar]
- Hetzel, C.; Bachran, C.; Fischer, R.; Fuchs, H.; Barth, S.; Stocker, M. Small cleavable adapters enhance the specific cytotoxicity of a humanized immunotoxin directed against CD64-positive cells. J. Immunother. 2008, 31, 370–376. [Google Scholar] [CrossRef]
- Tagge, E.; Harris, B.; Burbage, C.; Hall, P.; Vesely, J.; Willingham, M.; Frankel, A. Synthesis of green fluorescent protein-ricin and monitoring of its intracellular trafficking. Bioconjug. Chem. 1997, 8, 743–750. [Google Scholar] [CrossRef]
- Mohanraj, D.; Ramakrishnan, S. Cytotoxic effects of ricin without an interchain disulfide bond: Genetic modification and chemical crosslinking studies. Biochim. Biophys. Acta 1995, 1243, 399–406. [Google Scholar] [CrossRef]
- Chiron, M.F.; Fryling, C.M.; FitzGerald, D.J. Cleavage of pseudomonas exotoxin and diphtheria toxin by a furin-like enzyme prepared from beef liver. J. Biol. Chem. 1994, 269, 18167–18176. [Google Scholar]
- Kaul, P.; Silverman, J.; Shen, W.H.; Blanke, S.R.; Huynh, P.D.; Finkelstein, A.; Collier, R.J. Roles of Glu 349 and Asp 352 in membrane insertion and translocation by diphtheria toxin. Protein Sci. 1996, 5, 687–692. [Google Scholar]
- Lemichez, E.; Bomsel, M.; Devilliers, G.; vanderSpek, J.; Murphy, J.R.; Lukianov, E.V.; Olsnes, S.; Boquet, P. Membrane translocation of diphtheria toxin fragment A exploits early to late endosome trafficking machinery. Mol. Microbiol. 1997, 23, 445–457. [Google Scholar] [CrossRef]
- Kreitman, R.J. Recombinant immunotoxins for the treatment of chemoresistant hematologic malignancies. Curr. Pharm. Des. 2009, 15, 2652–2664. [Google Scholar] [CrossRef]
- Krysko, D.V.; D’Herde, K.; Vandenabeele, P. Clearance of apoptotic and necrotic cells and its immunological consequences. Apoptosis 2006, 11, 1709–1726. [Google Scholar] [CrossRef]
- Krysko, D.V.; Denecker, G.; Festjens, N.; Gabriels, S.; Parthoens, E.; D’Herde, K.; Vandenabeele, P. Macrophages use different internalization mechanisms to clear apoptotic and necrotic cells. Cell Death Differ. 2006, 13, 2011–2022. [Google Scholar] [CrossRef]
- Blythman, H.E.; Casellas, P.; Gros, O.; Gros, P.; Jansen, F.K.; Paolucci, F.; Pau, B.; Vidal, H. Immunotoxins: Hybrid molecules of monoclonal antibodies and a toxin subunit specifically kill tumour cells. Nature 1981, 290, 145–146. [Google Scholar]
- Surendranath, K.; Karande, A.A. A neutralizing antibody to the a chain of abrin inhibits abrin toxicity both in vitro and in vivo. Clin. Vaccine Immunol. 2008, 15, 737–743. [Google Scholar] [CrossRef]
- Bolognesi, A.; Polito, L.; Tazzari, P.L.; Lemoli, R.M.; Lubelli, C.; Fogli, M.; Boon, L.; de Boer, M.; Stirpe, F. In vitro anti-tumour activity of anti-CD80 and anti-CD86 immunotoxins containing type 1 ribosome-inactivating proteins. Br. J. Haematol. 2000, 110, 351–361. [Google Scholar] [CrossRef]
- Endo, Y.; Mitsui, K.; Motizuki, M.; Tsurugi, K. The mechanism of action of ricin and related toxic lectins on eukaryotic ribosomes. The site and the characteristics of the modification in 28 S ribosomal RNA caused by the toxins. J. Biol. Chem. 1987, 262, 5908–5912. [Google Scholar]
- Zamboni, M.; Brigotti, M.; Rambelli, F.; Montanaro, L.; Sperti, S. High-pressure-liquid-chromatographic and fluorimetric methods for the determination of adenine released from ribosomes by ricin and gelonin. Biochem. J. 1989, 259, 639–643. [Google Scholar]
- Iglewski, B.H.; Liu, P.V.; Kabat, D. Mechanism of action of Pseudomonas aeruginosa exotoxin Aiadenosine diphosphate-ribosylation of mammalian elongation factor 2 in vitro and in vivo. Infect. Immun. 1977, 15, 138–144. [Google Scholar]
- Van Ness, B.G.; Howard, J.B.; Bodley, J.W. ADP-ribosylation of elongation factor 2 by diphtheria toxin. Isolation and properties of the novel ribosyl-amino acid and its hydrolysis products. J. Biol. Chem. 1980, 255, 10717–10720. [Google Scholar]
- Kondo, T.; FitzGerald, D.; Chaudhary, V.K.; Adhya, S.; Pastan, I. Activity of immunotoxins constructed with modified Pseudomonas exotoxin A lacking the cell recognition domain. J. Biol. Chem. 1988, 263, 9470–9475. [Google Scholar]
- Kreitman, R.J.; Batra, J.K.; Seetharam, S.; Chaudhary, V.K.; FitzGerald, D.J.; Pastan, I. Single-chain immunotoxin fusions between anti-Tac and Pseudomonas exotoxin: Relative importance of the two toxin disulfide bonds. Bioconjug. Chem. 1993, 4, 112–120. [Google Scholar] [CrossRef]
- Siegall, C.B.; Chaudhary, V.K.; FitzGerald, D.J.; Pastan, I. Functional analysis of domains II, Ib, and III of Pseudomonas exotoxin. J. Biol. Chem. 1989, 264, 14256–14261. [Google Scholar]
- Williams, D.P.; Snider, C.E.; Strom, T.B.; Murphy, J.R. Structure/function analysis of interleukin-2-toxin (DAB486-IL-2). Fragment B sequences required for the delivery of fragment A to the cytosol of target cells. J. Biol. Chem. 1990, 265, 11885–11889. [Google Scholar]
- Chaudhary, V.K.; FitzGerald, D.J.; Pastan, I. A proper amino terminus of diphtheria toxin is important for cytotoxicity. Biochem. Biophys. Res. Commun. 1991, 180, 545–551. [Google Scholar] [CrossRef]
- Madhumathi, J.; Verma, R.S. Therapeutic targets and recent advances in protein immunotoxins. Curr. Opin. Microbiol. 2012, 1, 300–309. [Google Scholar]
- Kreitman, R.J. Immunotoxins for targeted cancer therapy. AAPS J. 2006, 8, E532–E551. [Google Scholar] [CrossRef]
- Roscoe, D.M.; Jung, S.H.; Benhar, I.; Pai, L.; Lee, B.K.; Pastan, I. Primate antibody response to immunotoxin: Serological and computer-aided analysis of epitopes on a truncated form of Pseudomonas exotoxin. Infect. Immun. 1994, 62, 5055–5065. [Google Scholar]
- Roscoe, D.M.; Pai, L.H.; Pastan, I. Identification of epitopes on a mutant form of Pseudomonas exotoxin using serum from humans treated with Pseudomonas exotoxin containing immunotoxins. Eur. J. Immunol. 1997, 27, 1459–1468. [Google Scholar] [CrossRef]
- Liu, W.; Onda, M.; Lee, B.; Kreitman, R.J.; Hassan, R.; Xiang, L.; Pastan, I. Recombinant immunotoxin engineered for low immunogenicity and antigenicity by identifying and silencing human B-cell epitopes. Proc. Natl. Acad. Sci. USA 2012, 109, 11782–11787. [Google Scholar]
- Reddy, K.R. Development and pharmacokinetics and pharmacodynamics of pegylated interferon alfa-2a (40 kD). Semin. Liver Dis. 2004, 2, 33–38. [Google Scholar] [CrossRef]
- Graham, M.L. Pegaspargase: A review of clinical studies. Adv. Drug Deliv. Rev. 2003, 55, 1293–1302. [Google Scholar] [CrossRef]
- Tsutsumi, Y.; Onda, M.; Nagata, S.; Lee, B.; Kreitman, R.J.; Pastan, I. Site-specific chemical modification with polyethylene glycol of recombinant immunotoxin anti-Tac(Fv)-PE38 (LMB-2) improves antitumor activity and reduces animal toxicity and immunogenicity. Proc. Natl. Acad. Sci. USA 2000, 97, 8548–8553. [Google Scholar]
- Li, Z.; Yu, T.; Zhao, P.; Ma, J. Immunotoxins and cancer therapy. Cell Mol. Immunol. 2005, 2, 106–112. [Google Scholar]
- Keating, S.E.; Baran, M.; Bowie, A.G. Cytosolic DNA sensors regulating type I interferon induction. Trends Immunol. 2011, 32, 574–581. [Google Scholar] [CrossRef]
- Lavelle, E.C.; Jarnicki, A.; McNeela, E.; Armstrong, M.E.; Higgins, S.C.; Leavy, O.; Mills, K.H. Effects of cholera toxin on innate and adaptive immunity and its application as an immunomodulatory agent. J. Leukoc Biol. 2004, 75, 756–763. [Google Scholar]
- Peterson, M.L.; Ault, K.; Kremer, M.J.; Klingelhutz, A.J.; Davis, C.C.; Squier, C.A.; Schlievert, P.M. The innate immune system is activated by stimulation of vaginal epithelial cells with Staphylococcus aureus and toxic shock syndrome toxin 1. Infect. Immun. 2005, 73, 2164–2174. [Google Scholar]
- Mathew, M.; Verma, R.S. Humanized immunotoxins: A new generation of immunotoxins for targeted cancer therapy. Cancer Sci. 2009, 100, 1359–1365. [Google Scholar] [CrossRef]
- Huhn, M.; Sasse, S.; Tur, M.K.; Matthey, B.; Schinkothe, T.; Rybak, S.M.; Barth, S.; Engert, A. Human angiogenin fused to human CD30 ligand (Ang-CD30L) exhibits specific cytotoxicity against CD30-positive lymphoma. Cancer Res. 2001, 61, 8737–8742. [Google Scholar]
- Tur, M.K.; Neef, I.; Jager, G.; Teubner, A.; Stocker, M.; Melmer, G.; Barth, S. Immunokinases, a novel class of immunotherapeutics for targeted cancer therapy. Curr. Pharm. Des. 2009, 15, 2693–2699. [Google Scholar] [CrossRef]
- Tur, M.K.; Neef, I.; Jost, E.; Galm, O.; Jager, G.; Stocker, M.; Ribbert, M.; Osieka, R.; Klinge, U.; Barth, S. Targeted restoration of down-regulated DAPK2 tumor suppressor activity induces apoptosis in Hodgkin lymphoma cells. J. Immunother. 2009, 32, 431–441. [Google Scholar] [CrossRef]
- Ten Cate, B.; de Bruyn, M.; Wei, Y.; Bremer, E.; Helfrich, W. Targeted elimination of leukemia stem cells; a new therapeutic approach in hemato-oncology. Curr. Drug Targets 2010, 11, 95–110. [Google Scholar] [CrossRef]
- Wan, L.; Zeng, L.; Chen, L.; Huang, Q.; Li, S.; Lu, Y.; Li, Y.; Cheng, J.; Lu, X. Expression, purification, and refolding of a novel immunotoxin containing humanized single-chain fragment variable antibody against CTLA4 and the N-terminal fragment of human perforin. Protein Exp. Purif. 2006, 48, 307–313. [Google Scholar] [CrossRef]
- Gordon, S.; Taylor, P.R. Monocyte and macrophage heterogeneity. Nat. Rev. Immunol. 2005, 5, 953–964. [Google Scholar] [CrossRef]
- Mosser, D.M.; Edwards, J.P. Exploring the full spectrum of macrophage activation. Nat. Rev. Immunol. 2008, 8, 958–969. [Google Scholar] [CrossRef]
- Metchnikoff, E. Leçons sur la Pathologie Comparée de L’inflammation; Masson: Paris, France, 1892. [Google Scholar]
- Biswas, S.K.; Mantovani, A. Macrophage plasticity and interaction with lymphocyte subsets: Cancer as a paradigm. Nat. Immunol. 2010, 11, 889–896. [Google Scholar] [CrossRef]
- Gordon, S.; Martinez, F.O. Alternative activation of macrophages: Mechanism and functions. Immunity 2010, 32, 593–604. [Google Scholar] [CrossRef]
- Sica, A.; Bronte, V. Altered macrophage differentiation and immune dysfunction in tumor development. J. Clin. Invest. 2007, 11, 1155–1166. [Google Scholar] [CrossRef]
- Ross, R.; Odland, G. Human wound repair. II. Inflammatory cells, epithelial-mesenchymal interrelations, and fibrogenesis. J. Cell. Biol. 1968, 39, 152–168. [Google Scholar] [CrossRef]
- Banno, T.; Gazel, A.; Blumenberg, M. Effects of tumor necrosis factor-alpha (TNF alpha) in epidermal keratinocytes revealed using global transcriptional profiling. J. Biol. Chem. 2004, 279, 32633–32642. [Google Scholar]
- McKay, I.A.; Leigh, I.M. Epidermal cytokines and their roles in cutaneous wound healing. Br. J. Dermatol. 1991, 12, 513–518. [Google Scholar]
- Hancock, G.E.; Kaplan, G.; Cohn, Z.A. Keratinocyte growth regulation by the products of immune cells. J. Exp. Med. 1988, 16, 1395–1402. [Google Scholar]
- Brauchle, M.; Angermeyer, K.; Hubner, G.; Werner, S. Large induction of keratinocyte growth factor expression by serum growth factors and pro-inflammatory cytokines in cultured fibroblasts. Oncogene 1994, 9, 3199–3204. [Google Scholar]
- Hubner, G.; Brauchle, M.; Smola, H.; Madlener, M.; Fassler, R.; Werner, S. Differential regulation of pro-inflammatory cytokines during wound healing in normal and glucocorticoid-treated mice. Cytokine 1996, 8, 548–556. [Google Scholar] [CrossRef]
- Mori, R.; Shaw, T.J.; Martin, P. Molecular mechanisms linking wound inflammation and fibrosis: Knockdown of osteopontin leads to rapid repair and reduced scarring. J. Exp. Med. 2008, 20, 43–51. [Google Scholar]
- Takehara, K. Growth regulation of skin fibroblasts. J. Dermatol. Sci. 2000, 2, S70–S77. [Google Scholar] [CrossRef]
- Stramer, B.M.; Mori, R.; Martin, P. The inflammation-fibrosis link? A Jekyll and Hyde role for blood cells during wound repair. J. Invest. Dermatol. 2007, 12, 1009–1017. [Google Scholar]
- Brown, L.F.; Yeo, K.T.; Berse, B.; Yeo, T.K.; Senger, D.R.; Dvorak, H.F.; van de Water, L. Expression of vascular permeability factor (vascular endothelial growth factor) by epidermal keratinocytes during wound healing. J. Exp. Med. 1992, 17, 1375–1379. [Google Scholar]
- Frank, S.; Hubner, G.; Breier, G.; Longaker, M.T.; Greenhalgh, D.G.; Werner, S. Regulation of vascular endothelial growth factor expression in cultured keratinocytes. Implications for normal and impaired wound healing. J. Biol. Chem. 1995, 270, 12607–12613. [Google Scholar]
- Maruyama, K.; Asai, J.; Ii, M.; Thorne, T.; Losordo, D.W.; D’Amore, P.A. Decreased macrophage number and activation lead to reduced lymphatic vessel formation and contribute to impaired diabetic wound healing. Am. J. Pathol. 2007, 170, 1178–1191. [Google Scholar]
- Schledzewski, K.; Falkowski, M.; Moldenhauer, G.; Metharom, P.; Kzhyshkowska, J.; Ganss, R.; Demory, A.; Falkowska-Hansen, B.; Kurzen, H.; Ugurel, S.; et al. Lymphatic endothelium-specific hyaluronan receptor LYVE-1 is expressed by stabilin-1+, F4/80+, CD11b+ macrophages in malignant tumours and wound healing tissue in vivo and in bone marrow cultures in vitro: Implications for the assessment of lymphangiogenesis. J. Pathol. 2006, 20, 67–77. [Google Scholar]
- Sica, A.; Mantovani, A. Macrophage plasticity and polarization: In vivo veritas. J. Clin. Invest. 2012, 12, 787–795. [Google Scholar]
- Hamilton, T.A.; Ohmori, Y.; Tebo, J. Regulation of chemokine expression by antiinflammatory cytokines. Immunol. Res. 2002, 25, 229–245. [Google Scholar] [CrossRef]
- Mantovani, A.; Sica, A.; Sozzani, S.; Allavena, P.; Vecchi, A.; Locati, M. The chemokine system in diverse forms of macrophage activation and polarization. Trends Immunol. 2004, 25, 677–686. [Google Scholar] [CrossRef]
- Verreck, F.A.; de Boer, T.; Langenberg, D.M.; Hoeve, M.A.; Kramer, M.; Vaisberg, E.; Kastelein, R.; Kolk, A.; de Waal-Malefyt, R.; Ottenhoff, T.H. Human IL-23-producing type 1 macrophages promote but IL-10-producing type 2 macrophages subvert immunity to (myco)bacteria. Proc. Natl. Acad. Sci. USA 2004, 101, 4560–4565. [Google Scholar]
- Dale, D.C.; Boxer, L.; Liles, W.C. The phagocytes: Neutrophils and monocytes. Blood 2008, 112, 935–945. [Google Scholar] [CrossRef]
- Mackaness, G.B. Cellular immunity and the parasite. Adv. Exp. Med. Biol. 1977, 9, 65–73. [Google Scholar]
- O’Shea, J.J.; Murray, P.J. Cytokine signaling modules in inflammatory responses. Immunity 2008, 28, 477–487. [Google Scholar] [CrossRef]
- Martinez, F.O.; Sica, A.; Mantovani, A.; Locati, M. Macrophage activation and polarization. Front. Biosci. 2008, 1, 453–461. [Google Scholar]
- Edwards, J.P.; Zhang, X.; Frauwirth, K.A.; Mosser, D.M. Biochemical and functional characterization of three activated macrophage populations. J. Leukoc. Biol. 2006, 80, 1298–1307. [Google Scholar] [CrossRef]
- Martinez, F.O.; Helming, L.; Gordon, S. Alternative activation of macrophages: An immunologic functional perspective. Annu. Rev. Immunol. 2009, 2, 451–483. [Google Scholar]
- Barner, M.; Mohrs, M.; Brombacher, F.; Kopf, M. Differences between IL-4R alpha-deficient and IL-4-deficient mice reveal a role for IL-13 in the regulation of Th2 responses. Curr. Biol. 1998, 8, 669–672. [Google Scholar] [CrossRef]
- Noben-Trauth, N.; Shultz, L.D.; Brombacher, F.; Urban, J.F., Jr.; Gu, H.; Paul, W.E. An interleukin 4 (IL-4)-independent pathway for CD4+ T cell IL-4 production is revealed in IL-4 receptor-deficient mice. Proc. Natl. Acad. Sci. USA 1997, 94, 10838–10843. [Google Scholar]
- Gordon, S. Alternative activation of macrophages. Nat. Rev. Immunol. 2003, 3, 23–35. [Google Scholar] [CrossRef]
- Stein, M.; Keshav, S.; Harris, N.; Gordon, S. Interleukin 4 potently enhances murine macrophage mannose receptor activity: A marker of alternative immunologic macrophage activation. J. Exp. Med. 1992, 176, 287–292. [Google Scholar] [CrossRef]
- Kadl, A.; Meher, A.K.; Sharma, P.R.; Lee, M.Y.; Doran, A.C.; Johnstone, S.R.; Elliott, M.R.; Gruber, F.; Han, J.; Chen, W.; et al. Identification of a novel macrophage phenotype that develops in response to atherogenic phospholipids via Nrf2. Circ. Res. 2010, 107, 737–746. [Google Scholar] [CrossRef]
- Boyle, J.J.; Harrington, H.A.; Piper, E.; Elderfield, K.; Stark, J.; Landis, R.C.; Haskard, D.O. Coronary intraplaque hemorrhage evokes a novel atheroprotective macrophage phenotype. Am. J. Pathol. 2009, 174, 1097–1108. [Google Scholar] [CrossRef]
- Boyle, J.J.; Johns, M.; Lo, J.; Chiodini, A.; Ambrose, N.; Evans, P.C.; Mason, J.C.; Haskard, D.O. Heme induces heme oxygenase 1 via Nrf2: role in the homeostatic macrophage response to intraplaque hemorrhage. Arter. Thromb. Vasc. Biol. 2011, 31, 2685–2691. [Google Scholar] [CrossRef]
- Gleissner, C.A.; Shaked, I.; Little, K.M.; Ley, K. CXC chemokine ligand 4 induces a unique transcriptome in monocyte-derived macrophages. J. Immunol. 2010, 184, 4810–4818. [Google Scholar]
- Grinberg, S.; Hasko, G.; Wu, D.; Leibovich, S.J. Suppression of PLCbeta2 by endotoxin plays a role in the adenosine A(2A) receptor-mediated switch of macrophages from an inflammatory to an angiogenic phenotype. Am. J. Pathol. 2009, 175, 2439–2453. [Google Scholar] [CrossRef]
- Loots, M.A.; Lamme, E.N.; Zeegelaar, J.; Mekkes, J.R.; Bos, J.D.; Middelkoop, E. Differences in cellular infiltrate and extracellular matrix of chronic diabetic and venous ulcers versus acute wounds. J. Invest. Dermatol. 1998, 111, 850–857. [Google Scholar]
- Eming, S.A.; Krieg, T.; Davidson, J.M. Inflammation in wound repair: Molecular and cellular mechanisms. J. Invest. Dermatol. 2007, 127, 514–525. [Google Scholar] [CrossRef]
- Ambarus, C.A.; Noordenbos, T.; de Hair, M.J.; Tak, P.P.; Baeten, D.L. Intimal lining layer macrophages but not synovial sublining macrophages display an IL-10 polarized-like phenotype in chronic synovitis. Arthritis Res. Ther. 2012, 14, R74. [Google Scholar] [CrossRef]
- Crielaard, B.J.; Lammers, T.; Morgan, M.E.; Chaabane, L.; Carboni, S.; Greco, B.; Zaratin, P.; Kraneveld, A.D.; Storm, G. Macrophages and liposomes in inflammatory disease: Friends or foes? Int. J. Pharm. 2011, 416, 499–506. [Google Scholar] [CrossRef]
- Lumeng, C.N.; Bodzin, J.L.; Saltiel, A.R. Obesity induces a phenotypic switch in adipose tissue macrophage polarization. J. Clin. Invest. 2007, 117, 175–184. [Google Scholar]
- Orme, J.; Mohan, C. Macrophage subpopulations in systemic lupus erythematosus. Discov. Med. 2012, 13, 151–158. [Google Scholar]
- Sindrilaru, A.; Peters, T.; Wieschalka, S.; Baican, C.; Baican, A.; Peter, H.; Hainzl, A.; Schatz, S.; Qi, Y.; Schlecht, A.; et al. An unrestrained proinflammatory M1 macrophage population induced by iron impairs wound healing in humans and mice. J. Clin. Invest. 2011, 121, 985–997. [Google Scholar] [CrossRef]
- Vandooren, B.; Noordenbos, T.; Ambarus, C.; Krausz, S.; Cantaert, T.; Yeremenko, N.; Boumans, M.; Lutter, R.; Tak, P.P.; Baeten, D. Absence of a classically activated macrophage cytokine signature in peripheral spondylarthritis, including psoriatic arthritis. Arthritis Rheum. 2009, 60, 966–975. [Google Scholar] [CrossRef]
- Singh, J.A.; Cameron, D.R. Summary of AHRQ’s comparative effectiveness review of drug therapy for rheumatoid arthritis (RA) in adults—an update. J. Manag. Care Pharm. 2012, 18 Suppl. C, 1–18. [Google Scholar]
- Hodgkins, P.; Yen, L.; Yarlas, A.; Karlstadt, R.; Solomon, D.; Kane, S. Impact of MMX(R) mesalamine on improvement and maintenance of health-related quality of life in patients with ulcerative colitis. Inflamm. Bowel Dis. 2012. [Google Scholar]
- Actis, G.C.; Rosina, F.; Pellicano, R.; Rizzetto, M. An aggressive medical approach for inflammatory bowel disease: Clinical challenges and therapeutic profiles in a retrospective hospital-based series. Curr. Clin. Pharmacol. 2012, 7, 209–213. [Google Scholar] [CrossRef]
- LeMaistre, C.F. DAB(389)IL-2 (denileukin diftitox, ONTAK): other potential applications. Clin. Lymphoma 2000, 1 Suppl. 1, S37–S40. [Google Scholar] [CrossRef]
- Jia, Y.; Li, H.; Chen, W.; Li, M.; Lv, M.; Feng, P.; Hu, H.; Zhang, L. Prevention of murine experimental autoimmune encephalomyelitis by in vivo expression of a novel recombinant immunotoxin DT390-RANTES. Gene Ther. 2006, 13, 1351–1359. [Google Scholar] [CrossRef]
- Bruhl, H.; Cihak, J.; Stangassinger, M.; Schlondorff, D.; Mack, M. Depletion of CCR5-expressing cells with bispecific antibodies and chemokine toxins: A new strategy in the treatment of chronic inflammatory diseases and HIV. J. Immunol. 2001, 166, 2420–2426. [Google Scholar]
- Ravetch, J.V.; Kinet, J.P. Fc receptors. Annu. Rev. Immunol. 1991, 9, 457–492. [Google Scholar] [CrossRef]
- Van de Winkel, J.G.; Anderson, C.L. Biology of human immunoglobulin G Fc receptors. J. Leukoc. Biol. 1991, 49, 511–524. [Google Scholar]
- Daeron, M. Structural bases of Fc gamma R functions. Int. Rev. Immunol. 1997, 1, 1–27. [Google Scholar] [CrossRef]
- Daeron, M. Fc receptor biology. Annu. Rev. Immunol. 1997, 1, 203–234. [Google Scholar] [CrossRef]
- Thepen, T.; van Vuuren, A.J.; Kiekens, R.C.; Damen, C.A.; Vooijs, W.C.; van de Winkel, J.G. Resolution of cutaneous inflammation after local elimination of macrophages. Nat. Biotechnol. 2000, 18, 48–51. [Google Scholar]
- Van Vuuren, A.J.; van Roon, J.A.; Walraven, V.; Stuij, I.; Harmsen, M.C.; McLaughlin, P.M.; van de Winkel, J.G.; Thepen, T. CD64-directed immunotoxin inhibits arthritis in a novel CD64 transgenic rat model. J. Immunol. 2006, 176, 5833–5838. [Google Scholar]
- Fet, N.G.; Fiebeler, A.; Klinge, U.; Park, J.K.; Barth, S.; Thepen, T.; Tolba, R.H. Reduction of activated macrophages after ischaemia-reperfusion injury diminishes oxidative stress and ameliorates renal damage. Nephrol. Dial. Transplant. 2012, 2, 3149–3155. [Google Scholar]
- Leemans, J.C.; Thepen, T.; Weijer, S.; Florquin, S.; van Rooijen, N.; van de Winkel, J.G.; van der Poll, T. Macrophages play a dual role during pulmonary tuberculosis in mice. J. Infect. Dis. 2005, 191, 65–74. [Google Scholar] [CrossRef]
- Stahnke, B.; Thepen, T.; Stocker, M.; Rosinke, R.; Jost, E.; Fischer, R.; Tur, M.K.; Barth, S. Granzyme B-H22(scFv), a human immunotoxin targeting CD64 in acute myeloid leukemia of monocytic subtypes. Mol. Cancer Ther. 2008, 7, 2924–2932. [Google Scholar] [CrossRef]
- Van de Winkel, J.G.; Capel, P.J. Human IgG Fc receptor heterogeneity: molecular aspects and clinical implications. Immunol. Today 1993, 14, 215–221. [Google Scholar] [CrossRef]
- Ribbert, T.; Thepen, T.; Tur, M.K.; Fischer, R.; Huhn, M.; Barth, S. Recombinant, ETA’-based CD64 immunotoxins: Improved efficacy by increased valency, both in vitro and in vivo in a chronic cutaneous inflammation model in human CD64 transgenic mice. Br. J. Dermatol. 2010, 163, 279–286. [Google Scholar] [CrossRef]
- Hetzel, C.; Bachran, C.; Tur, M.K.; Fuchs, H.; Stocker, M. Improved immunotoxins with novel functional elements. Curr. Pharm. Des. 2009, 15, 2700–2711. [Google Scholar]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Hristodorov, D.; Mladenov, R.; Huhn, M.; Barth, S.; Thepen, T. Macrophage-Targeted Therapy: CD64-Based Immunotoxins for Treatment of Chronic Inflammatory Diseases. Toxins 2012, 4, 676-694. https://doi.org/10.3390/toxins4090676
Hristodorov D, Mladenov R, Huhn M, Barth S, Thepen T. Macrophage-Targeted Therapy: CD64-Based Immunotoxins for Treatment of Chronic Inflammatory Diseases. Toxins. 2012; 4(9):676-694. https://doi.org/10.3390/toxins4090676
Chicago/Turabian StyleHristodorov, Dmitrij, Radoslav Mladenov, Michael Huhn, Stefan Barth, and Theo Thepen. 2012. "Macrophage-Targeted Therapy: CD64-Based Immunotoxins for Treatment of Chronic Inflammatory Diseases" Toxins 4, no. 9: 676-694. https://doi.org/10.3390/toxins4090676