Calyculin A from Discodermia Calyx Is a Dual Action Toxin that Blocks Calcium Influx and Inhibits Protein Ser/Thr Phosphatases

Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results and Discussion
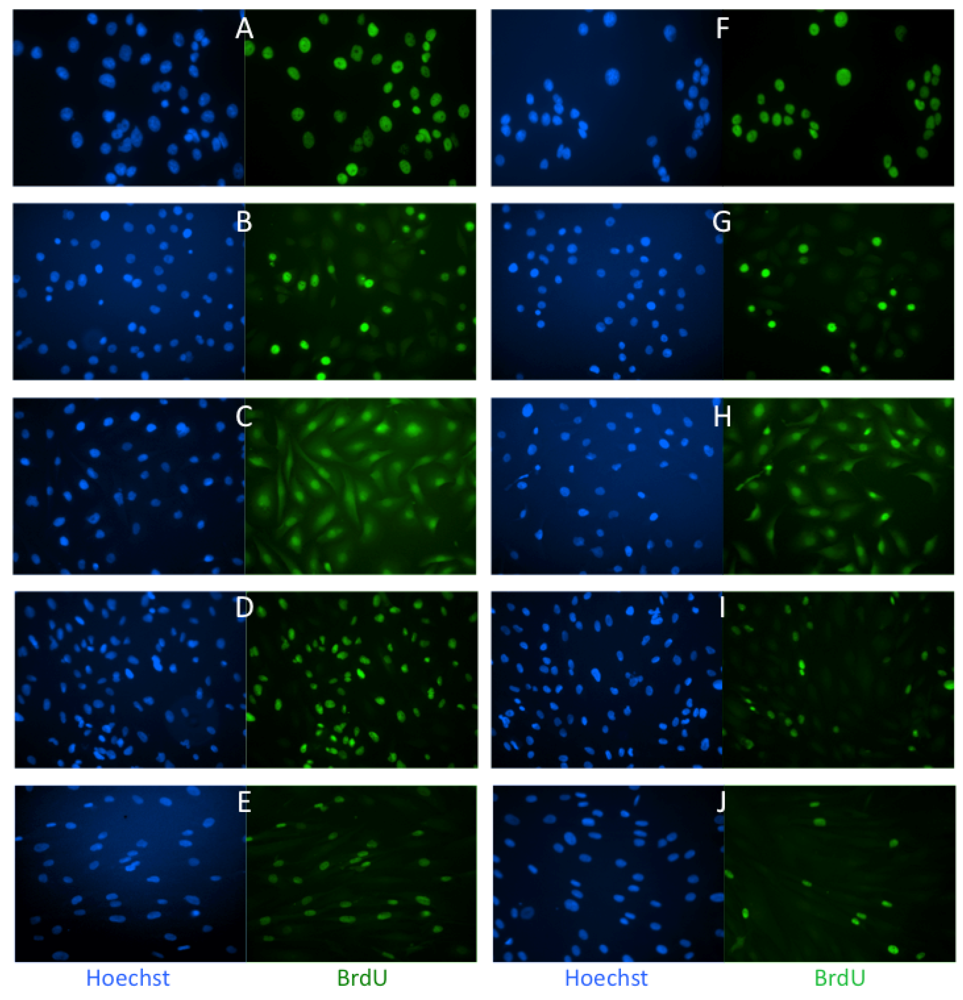
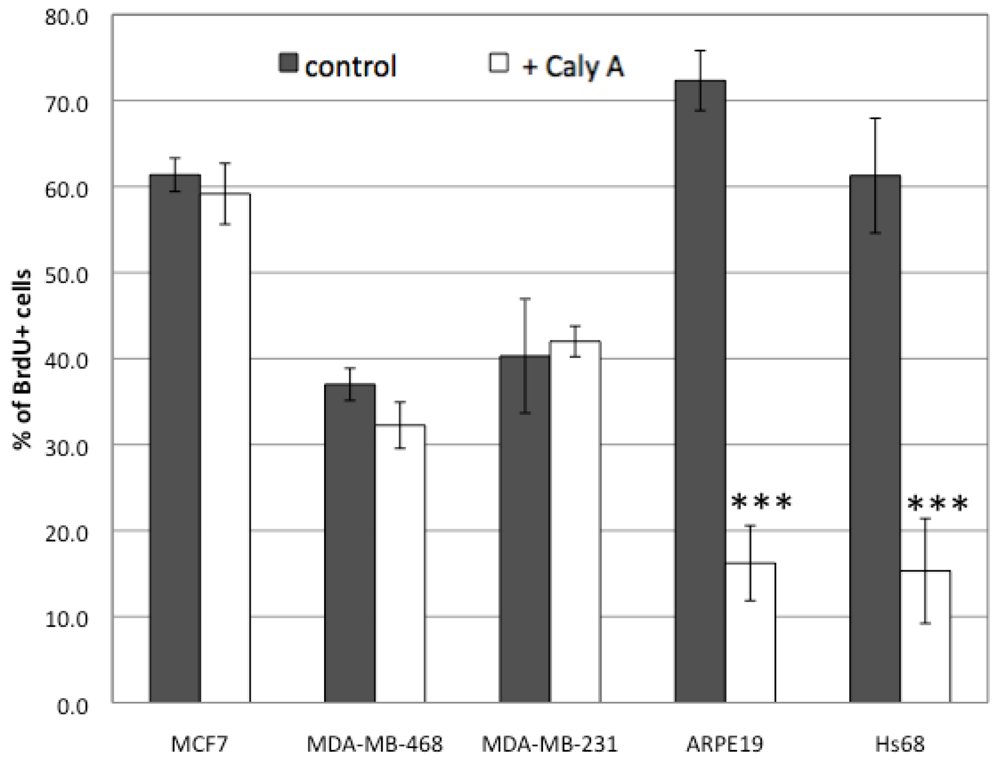
2.1. Effects of Low Dose Caly A on Cell Proliferation
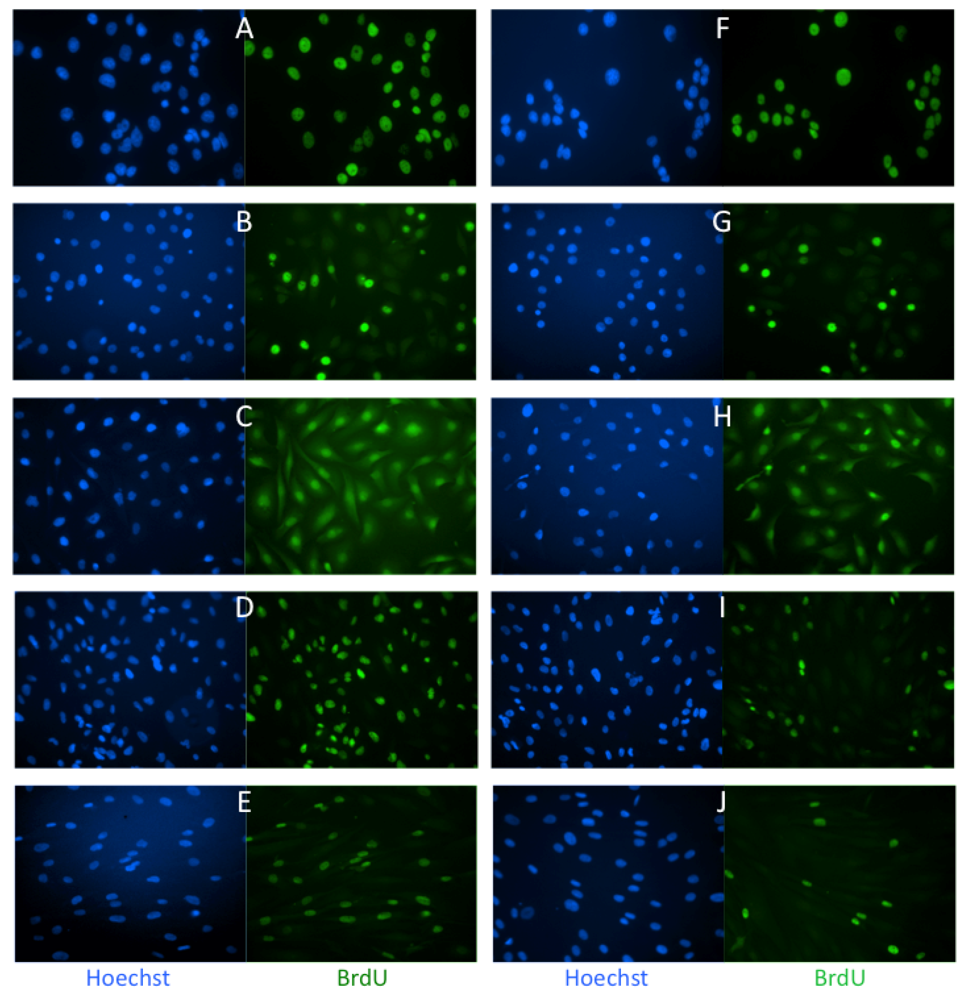
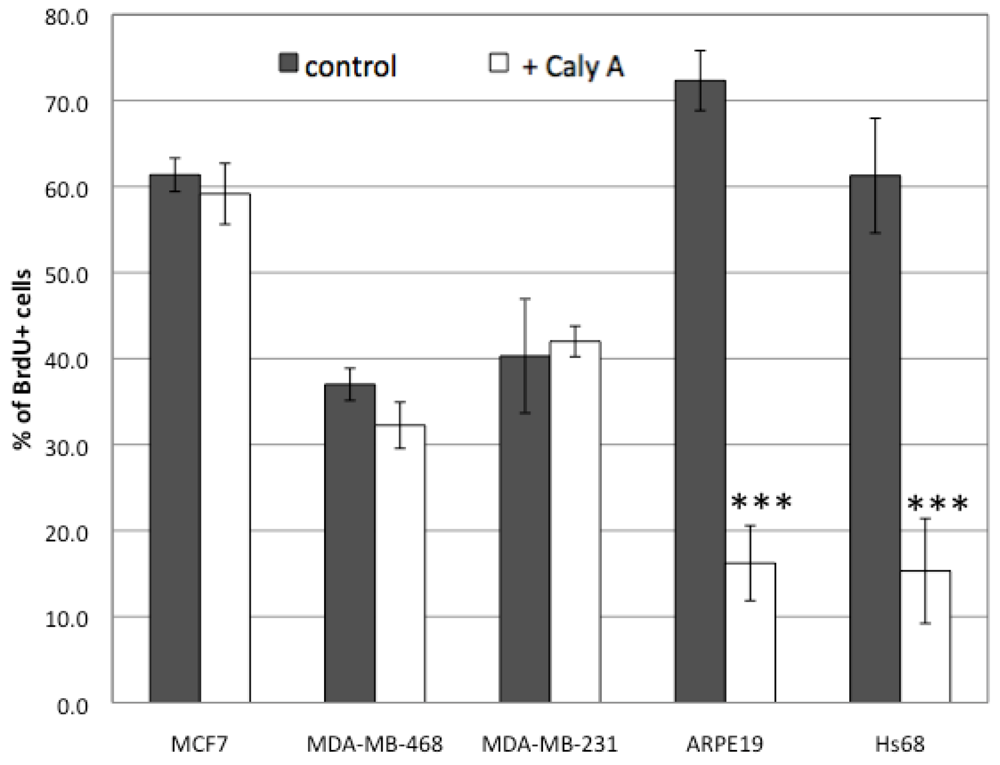
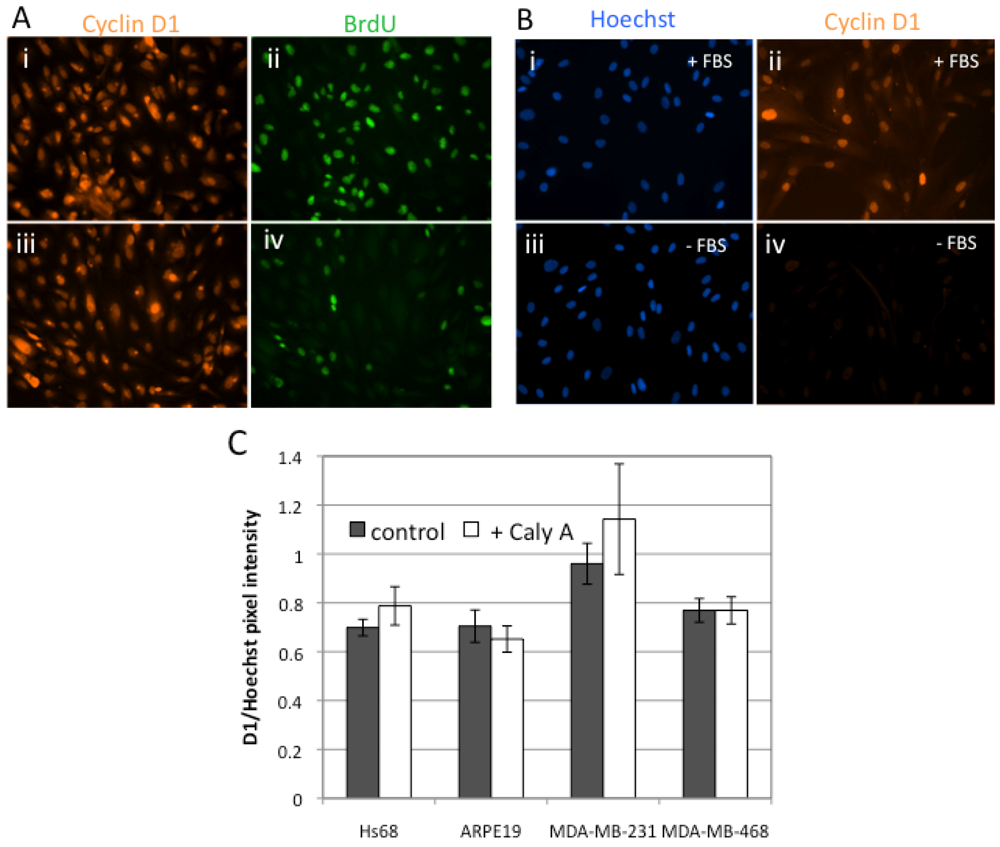
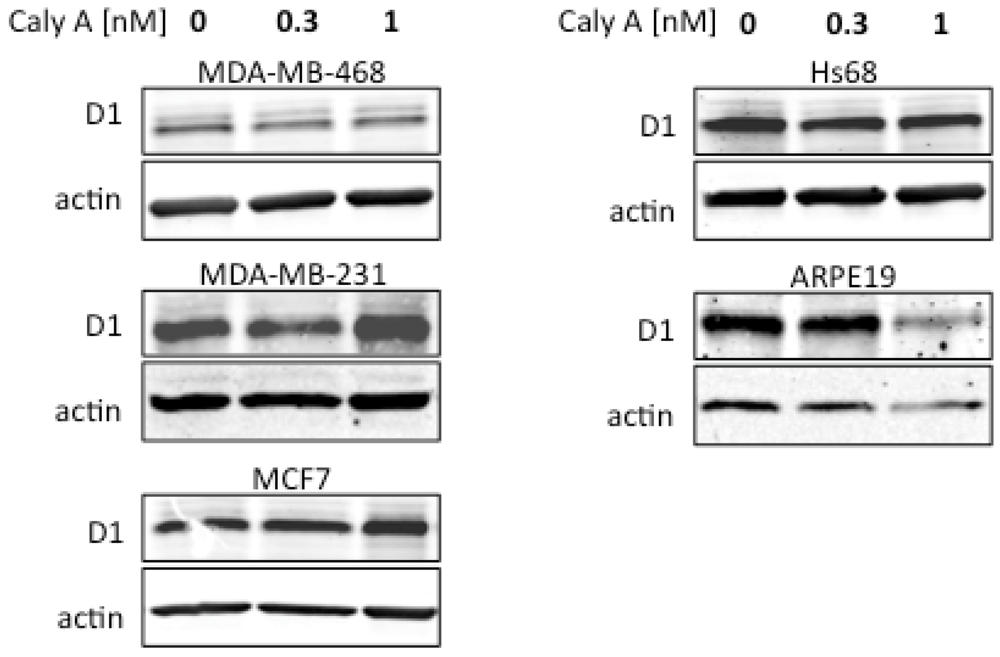
2.2. Caly A Arrests G1 to S Phase Progression without Cyclin D1 Ablation
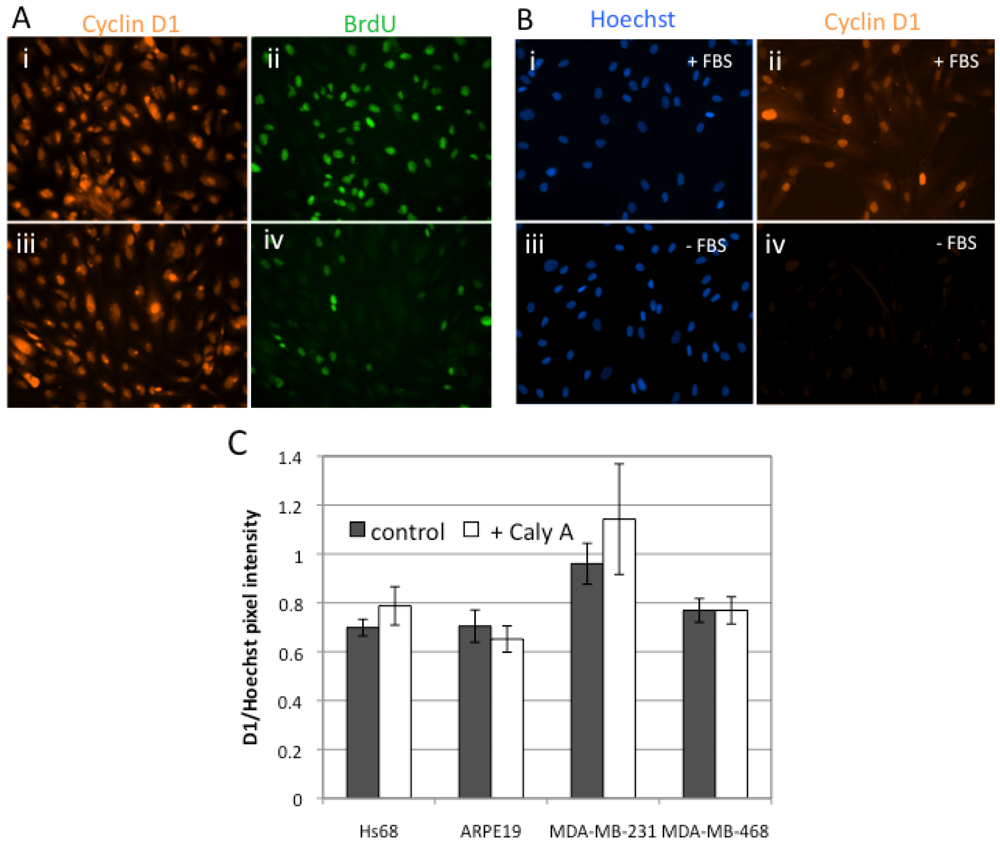
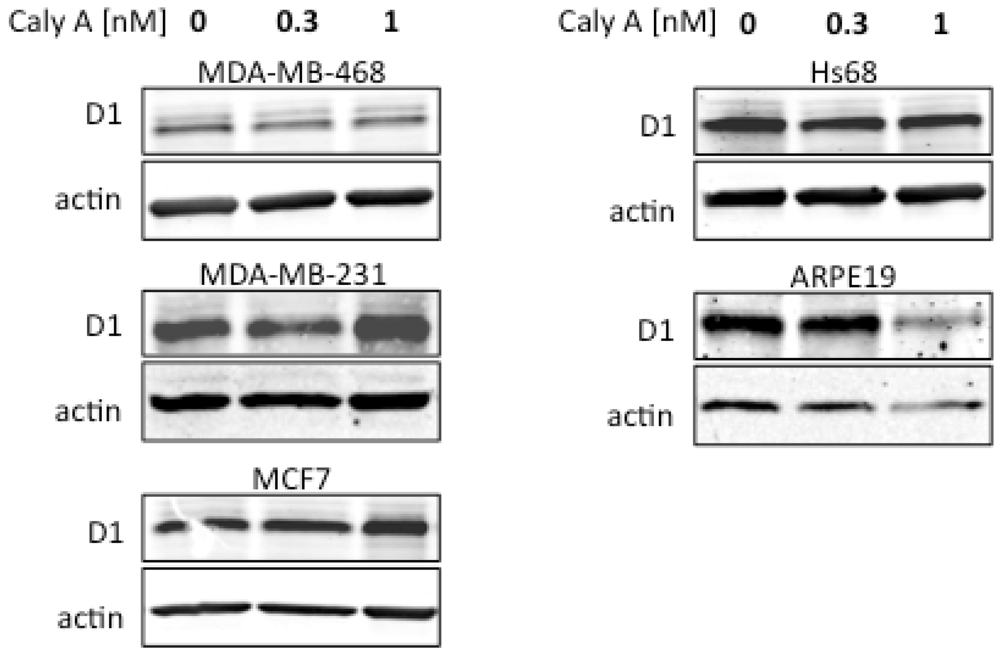
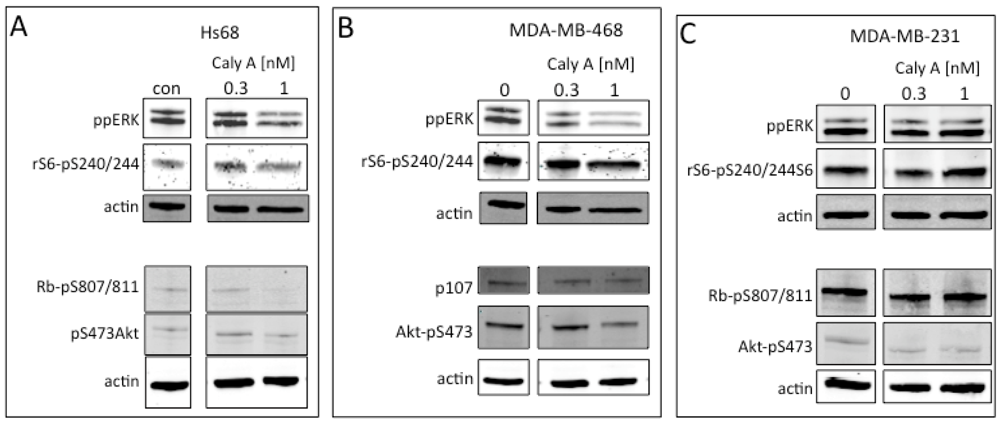
2.3. Protein Phosphorylation in Intact Cells Treated with Caly A
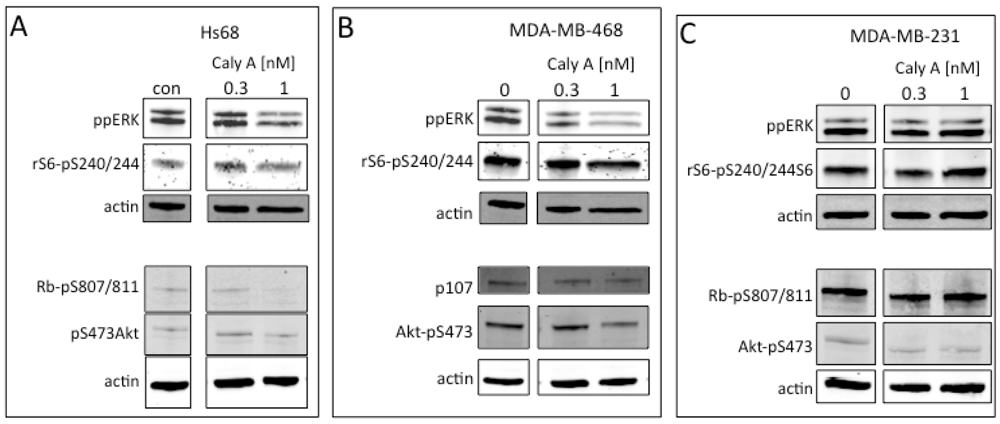
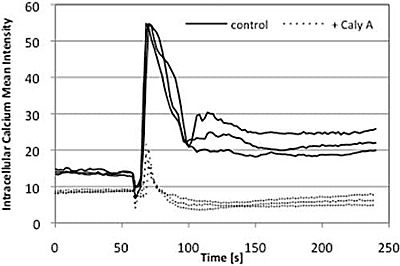
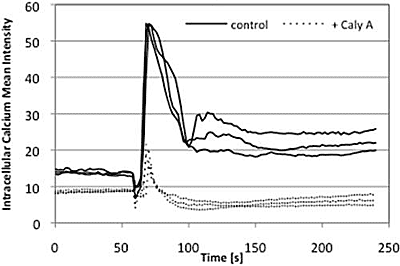
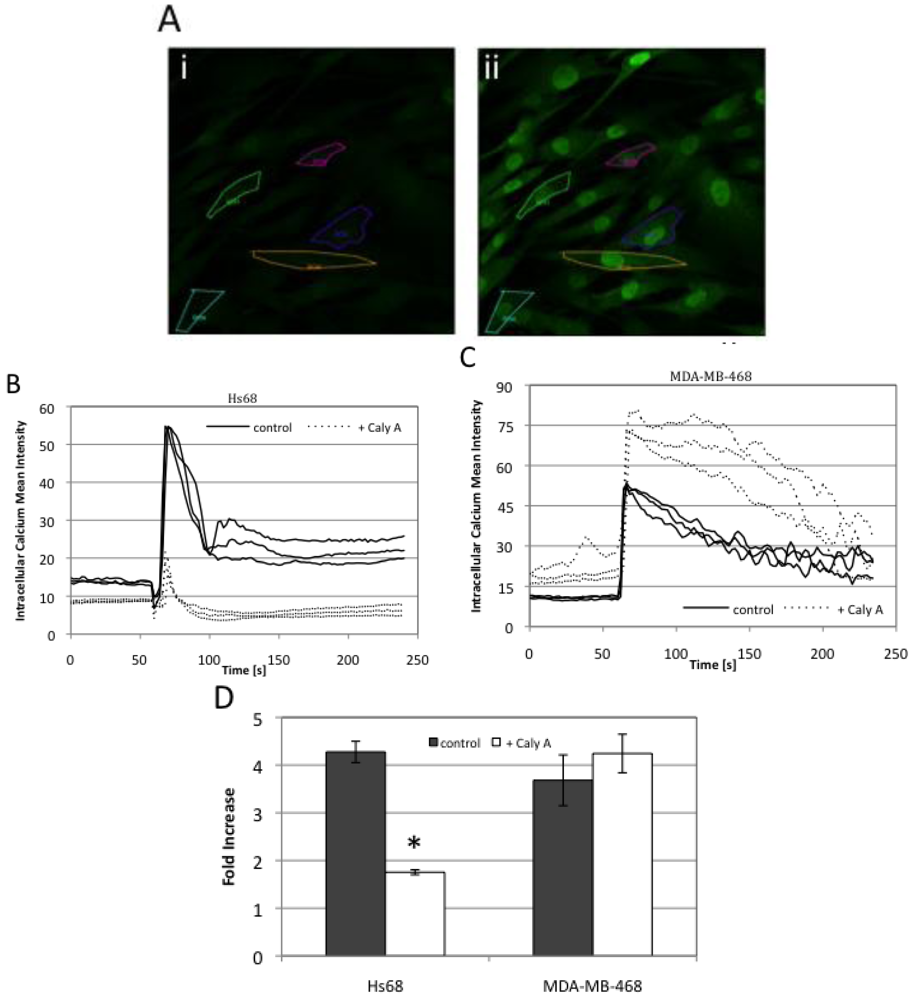
2.4. Caly A Blocks Serum-Stimulated Intracellular Calcium in Hs-68 Fibroblasts

2.5. Discussion
3. Experimental Section
3.1. Cell Culture and Reagents
3.2. Cell Proliferation Assay and Immunofluorescence
3.3. Western Blotting
3.4. Live-Cell Imaging and Calcium Measurements
4. Conclusions
Acknowledgments
Conflict of Interest
References
- Kato, Y.; Fusetani, N.; Matsunaga, S.; Hashimoto, K. Calyculin A, a novel antitumor metabolite from the marine sponge Discodermia. Calyx. J. Am. Chem. Soc. 1986, 208, 2780–2781. [Google Scholar]
- Kato, Y.; Fusetani, N.; Matsunaga, S.; Hashimoto, K. Hashimoto, K. Calyculins, potent antitumor metabolites from the marine sponge Discodermia. Calyx: Biological activities. Drugs Exp. Clin. Res. 1988, 65, 723–728. [Google Scholar]
- Ishihara, H.; Ozaki, H.; Sato, K.; Hori, M.; Karaki, H.; Watabe, S.; Kato, Y.; Fusetani, N.; Hashimoto, K.; Uemura, D.; Hartshorne, D.J. Calcium-independent activation of contractile apparatus in smooth muscle by calyculin A. J. Pharmacol. Exp. Ther. 1989, 250, 388–396. [Google Scholar]
- Yabu, H.; Yoshino, M.; Usuki, T.; Someya, T.; Obara, K.; Ozaki, H.; Karaki, H. Modification by calyculin A of inward Ca currents in smooth muscle cells isolated from guinea pig taenia coli. Prog. Clin. Biol. Res. 1990, 327, 623–626. [Google Scholar]
- Ishihara, H.; Martin, B.L.; Brautigan, D.L.; Karaki, H.; Ozaki, H.; Kato, Y.; Fusetani, N.; Watabe, S.; Hashimoto, K.; Uemura, D.; et al. Calyculin A and okadaic acid: Inhibitors of protein phosphatase activity. Biochem. Biophys. Res. Commun. 1989, 159, 871–877. [Google Scholar] [CrossRef]
- Suganuma, M.; Fujiki, H.; Okabe, S.; Nishiwaki, S.; Brautigan, D.; Ingebritsen, T.S.; Rosner, M.R. Structurally different members of the okadaic acid class selectively inhibit protein serine/threonine but not tyrosine phosphatase activity. Toxicon 1992, 30, 873–878. [Google Scholar] [CrossRef]
- Brautigan, D.L. Protein Ser/Thr phosphatases-The ugly ducklings of cell signaling. FEBS J. 2012. [Google Scholar] [CrossRef]
- Wakimoto, T.; Matsunaga, S.; Takai, A.; Fusetani, N. Insight into binding of calyculin A to protein phosphatase 1. Isolation of hemicalyculin A and chemical transformation of calyculin A. Chem. Biol. 2002, 9, 309–319. [Google Scholar] [CrossRef]
- Kita, A.; Matsunaga, S.; Takai, A.; Kataiwa, H.; Wakimoto, T.; Fusetani, N.; Isobe, M.; Miki, K. Crystal structure of the complex between calyculin A and the catalytic subunit of protein phosphatase 1. Structure 2002, 10, 715–724. [Google Scholar] [CrossRef]
- Favre, B.; Turowski, P.; Hemmings, B.A. Differential inhibition and posttranslational modification of protein phosphatase 1 and 2A in MCF7 cells treated with calyculin-A, okadaic acid, and tautomycin. J. Biol. Chem. 1997, 272, 13856–13863. [Google Scholar]
- Namboodiripad, A.N.; Jennings, M.L. Permeability characteristics of erythrocyte membrane to okadaic acid and calyculin A. Am. J. Physiol. 1996, 270, C449–C456. [Google Scholar]
- Edelson, J.R.; Brautigan, D.L. The Discodermia calyx Toxin Calyculin A Enhances Cyclin D1 Phosphorylation and Degradation, and Arrests Cell Cycle Progression in Human Breast Cancer Cells. Toxins 2011, 3, 105–119. [Google Scholar] [CrossRef]
- Baldin, V.; Lukas, J.; Marcote, M.J.; Pagano, M.; and Draetta, C. Cyclin D 1 is a nuclear protein required for cell cycle progression in G1. Genes Dev. 1993, 7, 812–821. [Google Scholar] [CrossRef]
- Yang, K.; Hitomi, M.; Stacey, D.W. Variations in cyclin D1 levels through the cell cycle determine the proliferative fate of a cell. CellDiv. 2006, 1, 32. [Google Scholar]
- Perrais, M.; Chen, X.; Perez-Moreno, M.; Gumbiner, B.M. E-cadherin homophilic ligation inhibits cell growth and epidermal growth factor receptor signaling independently of other cell interactions. Mol. Biol. Cell 2007, 18, 2013–2025. [Google Scholar] [CrossRef]
- Ward, S.M.; Vogalis, F.; Blondfield, D.P.; Ozaki, H.; Fusetani, N.; Uemura, D.; Publicover, N.G.; Sanders, K.M. Inhibition of electrical slow waves and Ca2+ currents of gastric and colonic smooth muscle by phosphatase inhibitors. Am. J. Physiol. 1991, 261, C64–C70. [Google Scholar]
- Obara, K.; Usuki, T.; Yabu, H. Calyculin A modulates voltage-dependent outward current in guinea-pig taenia coli smooth muscle cells. Adv. Exp. Med. Biol. 1992, 311, 373–374. [Google Scholar]
- Kahl, C.R.; Means, A.R. Regulation of cell cycle progression by calcium/calmodulin-dependent pathways. Endocr. Rev. 2003, 24, 719–736. [Google Scholar] [CrossRef]
- Takuwa, N.; Zhou, W.; Takuwa, Y. Calcium, calmodulin and cell cycle progression. Cell Signal. 1994, 7, 93–104. [Google Scholar]
- Takuwa, N.; Zhou, W.; Kumada, M.; Takuwa, Y. Involvement of intact inositol-1,4,5-trisphosphate-sensitive Ca2+ stores in cell cycle progression at the G1/S boundary in serum-stimulated human fibroblasts. FEBS Lett. 1995, 360, 173–176. [Google Scholar] [CrossRef]
- Hazelton, B.; Mitchell, B.; Tupper, J. Calcium, magnesium, and growth control in the WI-38 human fibroblast cell. J. Cell Biol. 1979, 83, 487–498. [Google Scholar] [CrossRef]
- Sée, V.; Rajala, N.K.; Spiller, D.G.; White, M.R. Calcium-dependent regulation of the cell cycle via a novel MAPK-NF-kappaB pathway in Swiss 3T3 cells. J. Cell Biol. 2004, 166, 661–672. [Google Scholar] [CrossRef]
- Cobbold, P.H.; Goyns, M.H. Measurements of the free calcium concentration of single quiescent human fibroblasts before and after serum addition. Biosci. Rep. 1983, 3, 79–86. [Google Scholar] [CrossRef]
- Goad, D.L.; Meurer, E.A.; Voelkel, E.F.; Petrou, C.P.; Tashjian, A.H., Jr. Protein phosphatase inhibitors and bone resorption: inhibition by okadaic acid and biphasic actions of calyculin-A. Endocrinology 1992, 130, 3402–3410. [Google Scholar] [CrossRef]
- Barbiero, G.; Munaron, L.; Antoniotti, S.; Baccino, F.M.; Bonelli, G.; Lovisolo, D. Role of mitogen-induced calcium influx in the control of the cell cycle in Balb-c 3T3 fibroblasts. Cell Calcium 1995, 18, 542–556. [Google Scholar] [CrossRef]
- Sakagami, K.; Wu, D.M.; Puro, D.G. Physiology of rat retinal pericytes: modulation of ion channel activity by serum-derived molecules. J. Physiol. 1999, 521, 637–650. [Google Scholar] [CrossRef]
- Kusaka, S.; Kapousta-Bruneau, N.; Green, D.G.; Puro, D.G. Serum-induced changes in the physiology of mammalian retinal glial cells: role of lysophosphatidic acid. J. Physiol. 1998, 506, 445–458. [Google Scholar] [CrossRef]
- Peña, F.; Ordaz, B. Non-selective cation channel blockers: potential use in nervous system basic research and therapeutics. Mini Rev. Med. Chem. 2008, 8, 812–819. [Google Scholar] [CrossRef]
- Taylor, J.T.; Zeng, X.B.; Pottle, J.E.; Lee, K.; Wang, A.R.; Yi, S.G.; Scruggs, J.A.; Sikka, S.S.; Li, M. Calcium signaling and T-type calcium channels in cancer cell cycling. World J. Gastroenterol. 2008, 14, 4984–4991. [Google Scholar] [CrossRef]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Holy, M.; Brautigan, D.L. Calyculin A from Discodermia Calyx Is a Dual Action Toxin that Blocks Calcium Influx and Inhibits Protein Ser/Thr Phosphatases. Toxins 2012, 4, 940-954. https://doi.org/10.3390/toxins4100940
Holy M, Brautigan DL. Calyculin A from Discodermia Calyx Is a Dual Action Toxin that Blocks Calcium Influx and Inhibits Protein Ser/Thr Phosphatases. Toxins. 2012; 4(10):940-954. https://doi.org/10.3390/toxins4100940
Chicago/Turabian StyleHoly, Maja, and David L. Brautigan. 2012. "Calyculin A from Discodermia Calyx Is a Dual Action Toxin that Blocks Calcium Influx and Inhibits Protein Ser/Thr Phosphatases" Toxins 4, no. 10: 940-954. https://doi.org/10.3390/toxins4100940
APA StyleHoly, M., & Brautigan, D. L. (2012). Calyculin A from Discodermia Calyx Is a Dual Action Toxin that Blocks Calcium Influx and Inhibits Protein Ser/Thr Phosphatases. Toxins, 4(10), 940-954. https://doi.org/10.3390/toxins4100940