Shiga Toxin Interaction with Human Intestinal Epithelium

{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Regulation of Stx Production and Release
2.1. Iron
2.2. Phage Lytic Cycle
2.3. Outer Membrane Vesicles
2.4. Intestinal Environment
2.4.1. Oxygen
2.4.2. Microbiota
2.4.3. Host Cells
3. Gb3 Expression, Stx Uptake and Intracellular Trafficking
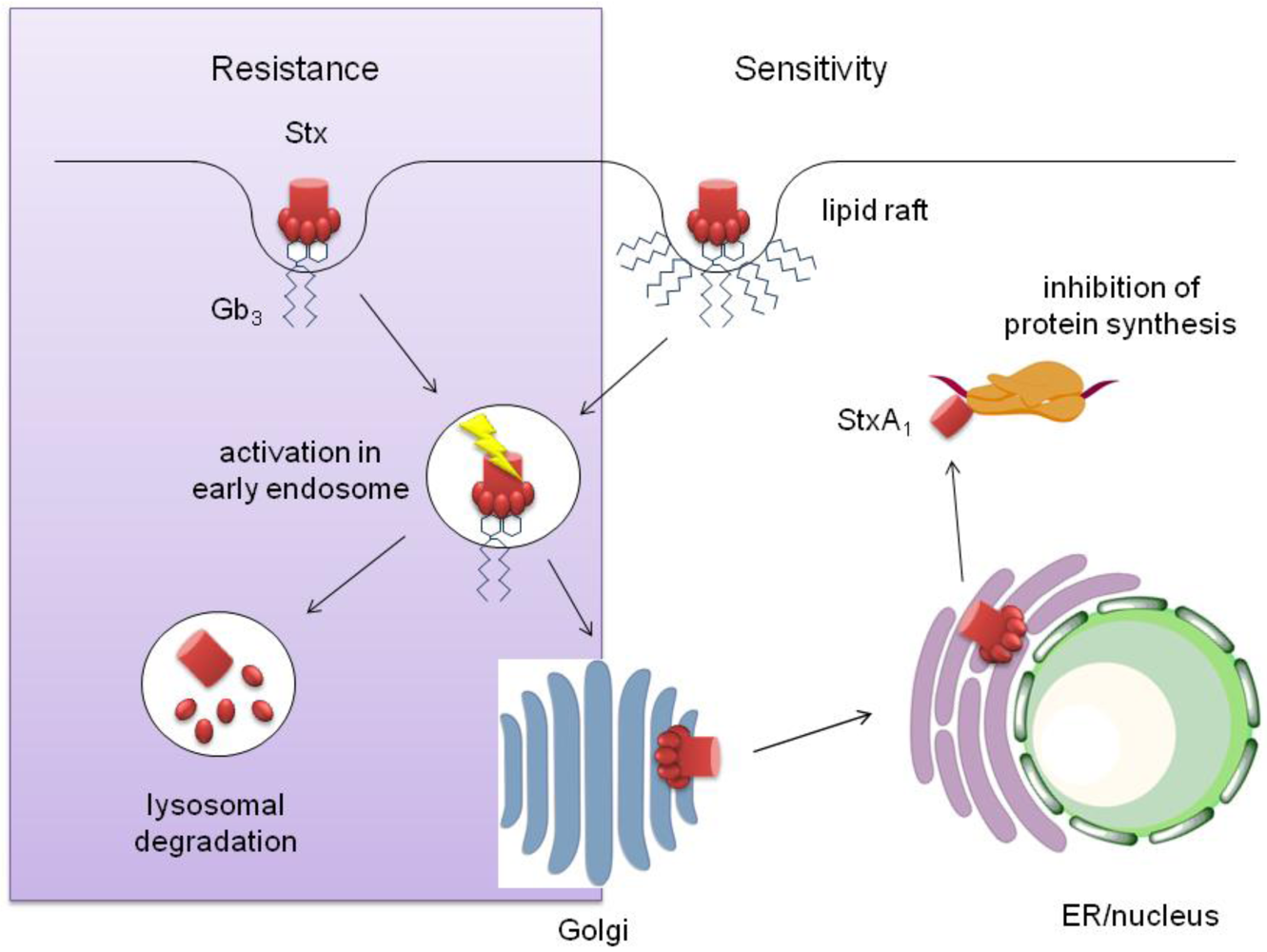
3.1. Stx Transport in Sensitive Cells
3.2. Stx Transport in Resistant Cells
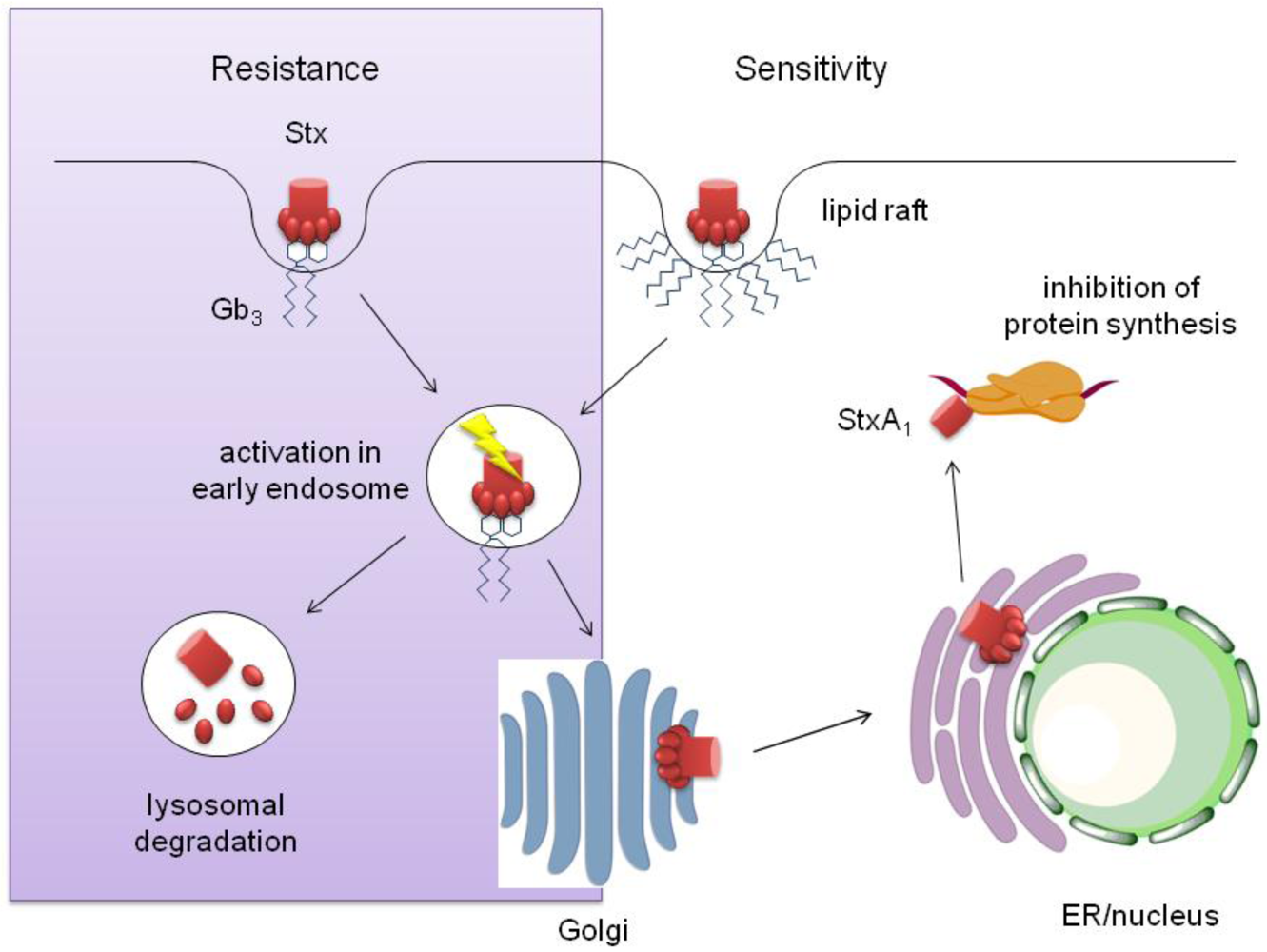
3.3. Stx Interaction with Human IEC and Intracellular Transport
4. Stx Translocation across Intestinal Epithelium
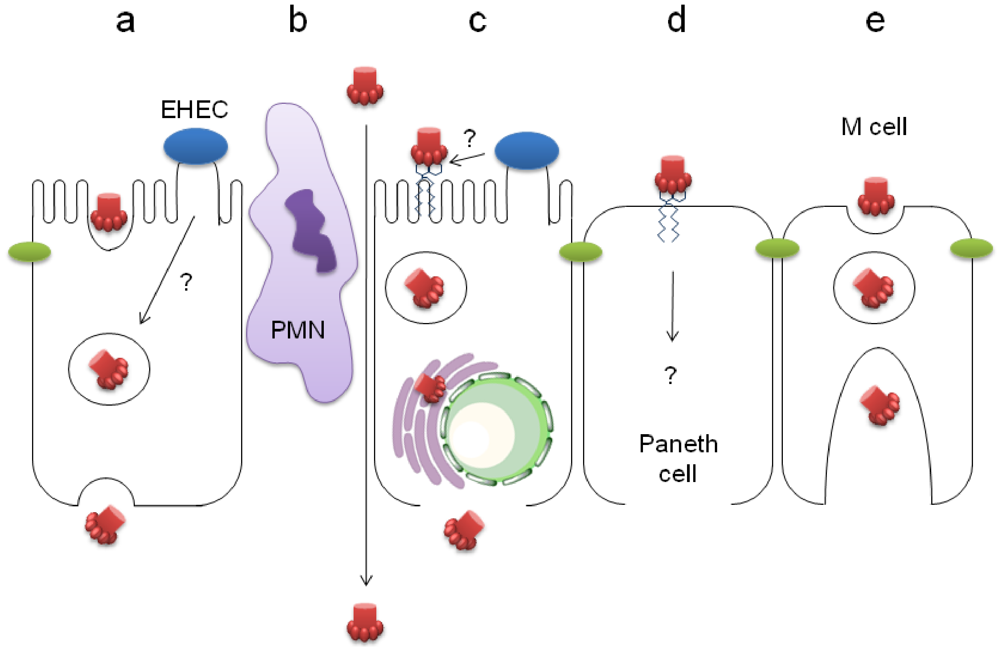
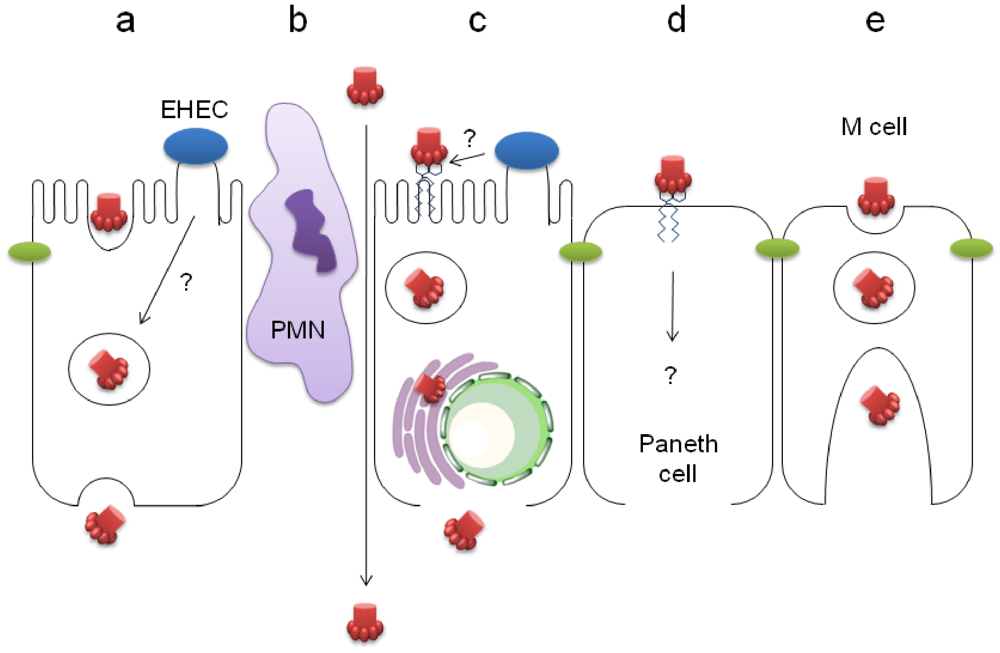
4.1. Stx Transcytosis by Gb3-Negative IEC
4.2. Paracellular Stx Translocation
4.3. Stx Uptake by Gb3-Positive IEC
4.4. Stx Translocation by M Cells
5. Conclusions
Acknowledgements
References
- Money, P.; Kelly, A.F.; Gould, S.W.; Denholm-Price, J.; Threlfall, E.J.; Fielder, M.D. Cattle, weather and water: Mapping Escherichia coli O157:H7 infections in humans in England and Scotland. Environ. Microbiol. 2010, 12, 2633–2644. [Google Scholar]
- Tarr, P.I.; Gordon, C.A.; Chandler, W.L. Shiga-toxin-producing Escherichia coli and haemolytic uraemic syndrome. Lancet 2005, 365, 1073–1086. [Google Scholar]
- Karmali, M.A.; Petric, M.; Lim, C.; Fleming, P.C.; Arbus, G.S.; Lior, H. The association between idiopathic hemolytic uremic syndrome and infection by verotoxin-producing Escherichia coli. J. Infect. Dis. 1985, 151, 775–782. [Google Scholar]
- Obrig, T.G.; Louise, C.B.; Lingwood, C.A.; Boyd, B.; Barley-Maloney, L.; Daniel, T.O. Endothelial heterogeneity in Shiga toxin receptors and responses. J. Biol. Chem. 1993, 268, 15484–15488. [Google Scholar]
- Lindberg, A.A.; Brown, J.E.; Stromberg, N.; Westling-Ryd, M.; Schultz, J.E.; Karlsson, K.A. Identification of the carbohydrate receptor for Shiga toxin produced by Shigella dysenteriae type 1. J. Biol. Chem. 1987, 262, 1779–1785. [Google Scholar]
- Strockbine, N.A.; Marques, L.R.; Newland, J.W.; Smith, H.W.; Holmes, R.K.; O'Brien, A.D. Two toxin-converting phages from Escherichia coli O157:H7 strain 933 encode antigenically distinct toxins with similar biologic activities. Infect. Immunol. 1986, 53, 135–140. [Google Scholar]
- Wagner, P.L.; Waldor, M.K. Bacteriophage control of bacterial virulence. Infect. Immunol. 2002, 70, 3985–3993. [Google Scholar]
- Calderwood, S.B.; Mekalanos, J.J. Iron regulation of Shiga-like toxin expression in Escherichia coli is mediated by the fur locus. J. Bacteriol. 1987, 169, 4759–4764. [Google Scholar]
- Ritchie, J.M.; Wagner, P.L.; Acheson, D.W.; Waldor, M.K. Comparison of Shiga toxin production by hemolytic-uremic syndrome-associated and bovine-associated Shiga toxin-producing Escherichia coli isolates. Appl. Environ. Microbiol. 2003, 69, 1059–1066. [Google Scholar]
- Shimizu, T.; Ohta, Y.; Noda, M. Shiga toxin 2 is specifically released from bacterial cells by two different mechanisms. Infect. Immunol. 2009, 77, 2813–2823. [Google Scholar]
- Wagner, P.L.; Livny, J.; Neely, M.N.; Acheson, D.W.; Friedman, D.I.; Waldor, M.K. Bacteriophage control of Shiga toxin 1 production and release by Escherichia coli. Mol. Microbiol. 2002, 44, 957–970. [Google Scholar]
- Wagner, P.L.; Neely, M.N.; Zhang, X.; Acheson, D.W.; Waldor, M.K.; Friedman, D.I. Role for a phage promoter in Shiga toxin 2 expression from a pathogenic Escherichia coli strain. J. Bacteriol. 2001, 183, 2081–2085. [Google Scholar]
- McGannon, C.M.; Fuller, C.A.; Weiss, A.A. Different classes of antibiotics differentially influence Shiga toxin production. Antimicrob. Agents Chemother. 2010, 54, 3790–3798. [Google Scholar]
- Łoś, J.M.; Łoś, M.; Węgrzyn, G.; Węgrzyn, A. Differential efficiency of induction of various lambdoid prophages responsible for production of Shiga toxins in response to different induction agents. Microbiol. Pathog. 2009, 47, 289–298. [Google Scholar] [CrossRef]
- Mühldorfer, I.; Hacker, J.; Keusch, G.T.; Acheson, D.W.; Tschäpe, H.; Kane, A.V.; Ritter, A.; Ölschläger, T.; Donohue-Rolfe, A. Regulation of the Shiga-like toxin II operon in Escherichia coli. Infect. Immunol. 1996, 64, 495–502. [Google Scholar]
- Waldor, M.K.; Friedman, D.I. Phage regulatory circuits and virulence gene expression. Curr. Opin. Microbiol. 2005, 8, 459–465. [Google Scholar]
- Shimizu, T.; Ohta, Y.; Tsutsuki, H.; Noda, M. Construction of a novel bioluminescent reporter system for investigating Shiga toxin expression of enterohemorrhagic Escherichia coli. Gene 2011, 478, 1–10. [Google Scholar]
- Griffin, P.M.; Tauxe, R.V. The epidemiology of infections caused by Escherichia coli O157:H7, other enterohemorrhagic E. coli, and the associated hemolytic uremic syndrome. Epidemiol. Rev. 1991, 13, 60–98. [Google Scholar] [PubMed]
- Heuvelink, A.E.; van de Kar, N.C.; Meis, J.F.; Monnens, L.A.; Melchers, W.J. Characterization of verocytotoxin-producing Escherichia coli O157 isolates from patients with haemolytic uraemic syndrome in Western Europe. Epidemiol. Infect. 1995, 115, 1–14. [Google Scholar]
- Amano, A.; Takeuchi, H.; Furuta, N. Outer membrane vesicles function as offensive weapons in host-parasite interactions. Microbes Infect. 2010, 12, 791–798. [Google Scholar]
- Ellis, T.N.; Kuehn, M.J. Virulence and immunomodulatory roles of bacterial outer membrane vesicles. Microbiol. Mol. Biol. Rev. 2010, 74, 81–94. [Google Scholar]
- Kolling, G.L.; Matthews, K.R. Export of virulence genes and Shiga toxin by membrane vesicles of Escherichia coli O157:H7. Appl. Environ. Microbiol. 1999, 65, 1843–1848. [Google Scholar]
- Yokoyama, K.; Horii, T.; Yamashino, T.; Hashikawa, S.; Barua, S.; Hasegawa, T.; Watanabe, H.; Ohta, M. Production of Shiga toxin by Escherichia coli measured with reference to the membrane vesicle-associated toxins. FEMS Microbiol. Lett. 2000, 192, 139–144. [Google Scholar]
- Kesty, N.C.; Mason, K.M.; Reedy, M.; Miller, S.E.; Kuehn, M.J. Enterotoxigenic Escherichia coli vesicles target toxin delivery into mammalian cells. EMBO J. 2004, 23, 4538–4549. [Google Scholar]
- Marteyn, B.; Scorza, F.B.; Sansonetti, P.J.; Tang, C. Breathing life into pathogens: The influence of oxygen on bacterial virulence and host responses in the gastrointestinal tract. Cell. Microbiol. 2011, 13, 171–176. [Google Scholar]
- James, B.W.; Keevil, C.W. Influence of oxygen availability on physiology, verocytotoxin expression and adherence of Escherichia coli O157. J. Appl. Microbiol. 1999, 86, 117–124. [Google Scholar]
- Asahara, T.; Shimizu, K.; Nomoto, K.; Hamabata, T.; Ozawa, A.; Takeda, Y. Probiotic bifidobacteria protect mice from lethal infection with Shiga toxin-producing Escherichia coli O157:H7. Infect. Immunol. 2004, 72, 2240–2247. [Google Scholar]
- Carey, C.M.; Kostrzynska, M.; Ojha, S.; Thompson, S. The effect of probiotics and organic acids on Shiga-toxin 2 gene expression in enterohemorrhagic Escherichia coli O157:H7. J. Microbiol. Methods 2008, 73, 125–132. [Google Scholar]
- De Sablet, T.; Chassard, C.; Bernalier-Donadille, A.; Vareille, M.; Gobert, A.P.; Martin, C. Human microbiota-secreted factors inhibit Shiga toxin synthesis by enterohemorrhagic Escherichia coli O157:H7. Infect. Immunol. 2009, 77, 783–790. [Google Scholar]
- Gamage, S.D.; Strasser, J.E.; Chalk, C.L.; Weiss, A.A. Nonpathogenic Escherichia coli can contribute to the production of Shiga Toxin. Infect. Immunol. 2003, 71, 3107–3115. [Google Scholar]
- Vareille, M.; de Sablet, T.; Hindré, T.; Martin, C.; Gobert, A.P. Nitric oxide inhibits Shiga-toxin synthesis by enterohemorrhagic Escherichia coli. Proc. Natl. Acad. Sci. USA 2007, 104, 10199–10204. [Google Scholar]
- Wagner, P.L.; Acheson, D.W.; Waldor, M.K. Human neutrophils and their products induce Shiga toxin production by enterohemorrhagic Escherichia coli. Infect. Immunol. 2001, 69, 1934–1937. [Google Scholar]
- Phillips, A.D.; Navabpour, S.; Hicks, S.; Dougan, G.; Wallis, T.; Frankel, G. Enterohaemorrhagic Escherichia coli O157:H7 target Peyer’s patches in humans and cause attaching/effacing lesions in both human and bovine intestine. Gut 2000, 47, 377–381. [Google Scholar]
- Poirier, K.; Faucher, S.P.; Beland, M.; Brousseau, R.; Gannon, V.; Martin, C.; Harel, J.; Daigle, F. Escherichia coli O157:H7 survives within human macrophages: Global gene expression profile and involvement of the Shiga toxins. Infect. Immunol. 2008, 76, 4814–4822. [Google Scholar]
- Jacewicz, M.S.; Mobassaleh, M.; Gross, S.K.; Balasubramanian, K.A.; Daniel, P.F.; Raghavan, S.; McCluer, R.H.; Keusch, G.T. Pathogenesis of Shigella diarrhea: XVII. A mammalian cell membrane glycolipid, Gb3, is required but not sufficient to confer sensitivity to Shiga toxin. J. Infect. Dis. 1994, 169, 538–546. [Google Scholar] [CrossRef] [PubMed]
- Waddell, T.; Cohen, A.; Lingwood, C.A. Induction of verotoxin sensitivity in receptor-deficient cell lines using the receptor glycolipid globotriosylceramide. Proc. Natl. Acad. Sci. USA 1990, 87, 7898–7901. [Google Scholar]
- Kovbasnjuk, O.; Edidin, M.; Donowitz, M. Role of lipid rafts in Shiga toxin 1 interaction with the apical surface of Caco-2 cells. J. Cell Sci. 2001, 114, 4025–4031. [Google Scholar]
- Falguières, T.; Mallard, F.; Baron, C.; Hanau, D.; Lingwood, C.; Goud, B.; Salamero, J.; Johannes, L. Targeting of Shiga toxin B-subunit to retrograde transport route in association with detergent-resistant membranes. Mol. Biol. Cell 2001, 12, 2453–2468. [Google Scholar]
- Hanashima, T.; Miyake, M.; Yahiro, K.; Iwamaru, Y.; Ando, A.; Morinaga, N.; Noda, M. Effect of Gb3 in lipid rafts in resistance to Shiga-like toxin of mutant Vero cells. Microbiol. Pathog. 2008, 45, 124–133. [Google Scholar]
- Kiarash, A.; Boyd, B.; Lingwood, C.A. Glycosphingolipid receptor function is modified by fatty acid content. Verotoxin 1 and verotoxin 2c preferentially recognize different globotriaosyl ceramide fatty acid homologues. J. Biol. Chem. 1994, 269, 11138–11146. [Google Scholar] [PubMed]
- Binnington, B.; Lingwood, D.; Nutikka, A.; Lingwood, C.A. Effect of globotriaosyl ceramide fatty acid alpha-hydroxylation on the binding by verotoxin 1 and verotoxin 2. Neurochem. Res. 2002, 27, 807–813. [Google Scholar]
- Arab, S.; Lingwood, C.A. Intracellular targeting of the endoplasmic reticulum/nuclear envelope by retrograde transport may determine cell hypersensitivity to verotoxin via globotriaosyl ceramide fatty acid isoform traffic. J. Cell. Physiol. 1998, 177, 646–660. [Google Scholar]
- Fraser, M.E.; Fujinaga, M.; Cherney, M.M.; Melton-Celsa, A.R.; Twiddy, E.M.; O'Brien, A.D.; James, M.N. Structure of Shiga toxin type 2 (Stx2) from Escherichia coli O157:H7. J. Biol. Chem. 2004, 279, 27511–27517. [Google Scholar]
- Garred, O.; van Deurs, B.; Sandvig, K. Furin-induced cleavage and activation of Shiga toxin. J. Biol. Chem. 1995, 270, 10817–10821. [Google Scholar]
- Sandvig, K.; Garred, O.; Prydz, K.; Kozlov, J.V.; Hansen, S.H.; van Deurs, B. Retrograde transport of endocytosed Shiga toxin to the endoplasmic reticulum. Nature 1992, 358, 510–512. [Google Scholar]
- Endo, Y.; Tsurugi, K.; Yutsudo, T.; Takeda, Y.; Ogasawara, T.; Igarashi, K. Site of action of a Vero toxin (VT2) from Escherichia coli O157:H7 and of Shiga toxin on eukaryotic ribosomes. RNA N-glycosidase activity of the toxins. Eur. J. Biochem. 1988, 171, 45–50. [Google Scholar] [CrossRef] [PubMed]
- Smith, W.E.; Kane, A.V.; Campbell, S.T.; Acheson, D.W.; Cochran, B.H.; Thorpe, C.M. Shiga toxin 1 triggers a ribotoxic stress response leading to p38 and JNK activation and induction of apoptosis in intestinal epithelial cells. Infect. Immunol. 2003, 71, 1497–1504. [Google Scholar]
- Hoey, D.E.; Sharp, L.; Currie, C.; Lingwood, C.A.; Gally, D.L.; Smith, D.G. Verotoxin 1 binding to intestinal crypt epithelial cells results in localization to lysosomes and abrogation of toxicity. Cell. Microbiol. 2003, 5, 85–97. [Google Scholar]
- Naylor, S.W.; Low, J.C.; Besser, T.E.; Mahajan, A.; Gunn, G.J.; Pearce, M.C.; McKendrick, I.J.; Smith, D.G.; Gally, D.L. Lymphoid follicle-dense mucosa at the terminal rectum is the principal site of colonization of enterohemorrhagic Escherichia coli O157:H7 in the bovine host. Infect. Immunol. 2003, 71, 1505–1512. [Google Scholar]
- Pruimboom-Brees, I.M.; Morgan, T.W.; Ackermann, M.R.; Nystrom, E.D.; Samuel, J.E.; Cornick, N.A.; Moon, H.W. Cattle lack vascular receptors for Escherichia coli O157:H7 Shiga toxins. Proc. Natl. Acad. Sci. USA 2000, 97, 10325–10329. [Google Scholar]
- Sandvig, K.; Ryd, M.; Garred, O.; Schweda, E.; Holm, P.K.; van Deurs, B. Retrograde transport from the Golgi complex to the ER of both Shiga toxin and the nontoxic Shiga B-fragment is regulated by butyric acid and cAMP. J. Cell Biol. 1994, 126, 53–64. [Google Scholar]
- Björk, S.; Breimer, M.E.; Hansson, G.C.; Karlsson, K.A.; Leffler, H. Structures of blood group glycosphingolipids of human small intestine. A relation between the expression of fucolipids of epithelial cells and the ABO, Le and Se phenotype of the donor. J. Biol. Chem. 1987, 262, 6758–6765. [Google Scholar] [PubMed]
- Holgersson, J.; Jovall, P.A.; Breimer, M.E. Glycosphingolipids of human large intestine: Detailed structural characterization with special reference to blood group compounds and bacterial receptor structures. J. Biochem. 1991, 110, 120–131. [Google Scholar]
- Schüller, S.; Frankel, G.; Phillips, A.D. Interaction of Shiga toxin from Escherichia coli with human intestinal epithelial cell lines and explants: Stx2 induces epithelial damage in organ culture. Cell. Microbiol. 2004, 6, 289–301. [Google Scholar]
- Miyamoto, Y.; Iimura, M.; Kaper, J.B.; Torres, A.G.; Kagnoff, M.F. Role of Shiga toxin versus H7 flagellin in enterohaemorrhagic Escherichia coli signalling of human colon epithelium in vivo. Cell. Microbiol. 2006, 8, 869–879. [Google Scholar]
- Engedal, N.; Skotland, T.; Torgersen, M.L.; Sandvig, K. Shiga toxin and its use in targeted cancer therapy and imaging. Microbiol. Biotechnol. 2010, 4, 32–46. [Google Scholar]
- Falguières, T.; Maak, M.; von Weyhern, C.; Sarr, M.; Sastre, X.; Poupon, M.F.; Robine, S.; Johannes, L.; Janssen, K.P. Human colorectal tumors and metastases express Gb3 and can be targeted by an intestinal pathogen-based delivery tool. Mol. Cancer Ther. 2008, 7, 2498–2508. [Google Scholar]
- Kovbasnjuk, O.; Mourtazina, R.; Baibakov, B.; Wang, T.; Elowsky, C.; Choti, M.A.; Kane, A.; Donowitz, M. The glycosphingolipid globotriaosylceramide in the metastatic transformation of colon cancer. Proc. Natl. Acad. Sci. USA 2005, 102, 19087–19092. [Google Scholar]
- Jacewicz, M.S.; Acheson, D.W.; Mobassaleh, M.; Donohue-Rolfe, A.; Balasubramanian, K.A.; Keusch, G.T. Maturational regulation of globotriaosylceramide, the Shiga-like toxin 1 receptor, in cultured human gut epithelial cells. J. Clin. Invest. 1995, 96, 1328–1335. [Google Scholar]
- Zumbrun, S.D.; Hanson, L.; Sinclair, J.F.; Freedy, J.; Melton-Celsa, A.R.; Rodriguez-Canales, J.; Hanson, J.C.; O’Brien, A.D. Human intestinal tissue and cultured colonic cells contain globotriaosylceramide synthase mRNA and the alternate Shiga toxin receptor globotetraosylceramide. Infect. Immunol. 2010, 78, 4488–4499. [Google Scholar]
- Philpott, D.J.; Ackerley, C.A.; Kiliaan, A.J.; Karmali, M.A.; Perdue, M.H.; Sherman, P.M. Translocation of verotoxin-1 across T84 monolayers: Mechanism of bacterial toxin penetration of epithelium. Am. J. Physiol. 1997, 273, 1349–1358. [Google Scholar]
- Tam, P.J.; Lingwood, C.A. Membrane cytosolic translocation of verotoxin A1 subunit in target cells. Microbiology 2007, 153, 2700–2710. [Google Scholar]
- Laiko, M.; Murtazina, R.; Malyukova, I.; Zhu, C.; Boedeker, E.C.; Gutsal, O.; O’Malley, R.; Cole, R.N.; Tarr, P.I.; Murray, K.F.; et al. Shiga toxin 1 interaction with enterocytes causes apical protein mistargeting through the depletion of intracellular galectin-3. Exp. Cell Res. 2010, 316, 657–666. [Google Scholar] [CrossRef] [PubMed]
- Schüller, S.; Heuschkel, R.; Torrente, F.; Kaper, J.B.; Phillips, A.D. Shiga toxin binding in normal and inflamed human intestinal mucosa. Microbes Infect. 2007, 9, 35–39. [Google Scholar]
- Brigotti, M.; Tazzari, P.L.; Ravanelli, E.; Carnicelli, D.; Rocchi, L.; Arfilli, V.; Scavia, G.; Minelli, F.; Ricci, F.; Pagliaro, P.; et al. Clinical Relevance of Shiga Toxin Concentrations in the Blood of Patients With Hemolytic Uremic Syndrome. Pediatr. Infect. Dis. J. 2011, 30, 486–490. [Google Scholar] [PubMed]
- Imai, Y.; Fukui, T.; Kurohane, K.; Miyamoto, D.; Suzuki, Y.; Ishikawa, T.; Ono, Y.; Miyake, M. Restricted expression of Shiga toxin binding sites on mucosal epithelium of mouse distal colon. Infect. Immunol. 2003, 71, 985–990. [Google Scholar]
- Mobassaleh, M.; Gross, S.K.; McCluer, R.H.; Donohue-Rolfe, A.; Keusch, G.T. Quantitation of the rabbit intestinal glycolipid receptor for Shiga toxin. Further evidence for the developmental regulation of globotriaosylceramide in microvillus membranes. Gastroenterol 1989, 97, 384–391. [Google Scholar]
- Golan, L.; Gonen, E.; Yagel, S.; Rosenshine, I.; Shpigel, N.Y. Enterohemorrhagic Escherichia coli induce attaching and effacing lesions and hemorrhagic colitis in human and bovine intestinal xenograft models. Dis. Models Mech. 2011, 4, 86–94. [Google Scholar]
- Shigeno, T.; Akamatsu, T.; Fujimori, K.; Nakatsuji, Y.; Nagata, A. The clinical significance of colonoscopy in hemorrhagic colitis due to enterohemorrhagic Escherichia coli O157:H7 infection. Endoscopy 2002, 34, 311–314. [Google Scholar]
- Acheson, D.W.; Moore, R.; De Breucker, S.; Lincicome, L.; Jacewicz, M.; Skutelsky, E.; Keusch, G.T. Translocation of Shiga toxin across polarized intestinal cells in tissue culture. Infect. Immunol. 1996, 64, 3294–3300. [Google Scholar]
- Hurley, B.P.; Jacewicz, M.; Thorpe, C.M.; Lincicome, L.L.; King, A.J.; Keusch, G.T.; Acheson, D.W. Shiga toxins 1 and 2 translocate differently across polarized intestinal epithelial cells. Infect. Immunol. 1999, 67, 6670–6677. [Google Scholar]
- Hurley, B.P.; Thorpe, C.M.; Acheson, D.W. Shiga toxin translocation across intestinal epithelial cells is enhanced by neutrophil transmigration. Infect. Immunol. 2001, 69, 6148–6155. [Google Scholar]
- Maluykova, I.; Gutsal, O.; Laiko, M.; Kane, A.; Donowitz, M.; Kovbasnjuk, O. Latrunculin B facilitates Shiga toxin 1 transcellular transcytosis across T84 intestinal epithelial cells. Biochim. Biophys. Acta 2008, 1782, 370–377. [Google Scholar]
- Malyukova, I.; Murray, K.F.; Zhu, C.; Boedeker, E.; Kane, A.; Patterson, K.; Peterson, J.R.; Donowitz, M.; Kovbasnjuk, O. Macropinocytosis in Shiga toxin 1 uptake by human intestinal epithelial cells and transcellular transcytosis. Am. J. Physiol. Gastrointest. Liver Physiol. 2009, 296, G78–G92. [Google Scholar]
- Viswanathan, V.K.; Koutsouris, A.; Lukic, S.; Pilkinton, M.; Simonovic, I.; Simonovic, M.; Hecht, G. Comparative analysis of EspF from enteropathogenic and enterohemorrhagic Escherichia coli in alteration of epithelial barrier function. Infect. Immunol. 2004, 72, 3218–3227. [Google Scholar]
- Philpott, D.J.; McKay, D.M.; Mak, W.; Perdue, M.H.; Sherman, P.M. Signal transduction pathways involved in enterohemorrhagic Escherichia coli-induced alterations in T84 epithelial permeability. Infect. Immunol. 1998, 66, 1680–1687. [Google Scholar]
- Klein, E.J.; Stapp, J.R.; Clausen, C.R.; Boster, D.R.; Wells, J.G.; Qin, X.; Swerdlow, D.L.; Tarr, P.I. Shiga toxin-producing Escherichia coli in children with diarrhea: A prospective point-of-care study. J. Pediatr. 2002, 141, 172–177. [Google Scholar]
- Slutsker, L.; Ries, A.A.; Greene, K.D.; Wells, J.G.; Hutwagner, L.; Griffin, P.M. Escherichia coli O157:H7 diarrhea in the United States: Clinical and epidemiologic features. Ann. Intern. Med. 1997, 126, 505–513. [Google Scholar]
- Griffin, P.M.; Olmstead, L.C.; Petras, R.E. Escherichia coli 0157:H7-associated colitis. A clinical and histological study of 11 cases. Gastroenterol 1990, 99, 142–149. [Google Scholar]
- Moon, H.W. Comparative histopathology of intestinal infections. Adv. Exp. Med. Biol. 1997, 412, 1–19. [Google Scholar]
- van de Kar, N.C.; Monnens, L.A.; Karmali, M.A.; van Hinsbergh, V.W. Tumor necrosis factor and interleukin-1 induce expression of the verocytotoxin receptor globotriaosylceramide on human endothelial cells: Implications for the pathogenesis of the hemolytic uremic syndrome. Blood 1992, 80, 2755–2764. [Google Scholar]
- Keusch, G.T.; Acheson, D.W.; Aaldering, L.; Erban, J.; Jacewicz, M.S. Comparison of the effects of Shiga-like toxin 1 on cytokine- and butyrate-treated human umbilical and saphenous vein endothelial cells. J. Infect. Dis. 1996, 173, 1164–1170. [Google Scholar]
- Louise, C.B.; Kaye, S.A.; Boyd, B.; Lingwood, C.A.; Obrig, T.G. Shiga toxin-associated hemolytic uremic syndrome: Effect of sodium butyrate on sensitivity of human umbilical vein endothelial cells to Shiga toxin. Infect. Immunol. 1995, 63, 2766–2769. [Google Scholar]
- Ohmi, K.; Kiyokawa, N.; Takeda, T.; Fujimoto, J. Human microvascular endothelial cells are strongly sensitive to Shiga toxins. Biochem. Biophys. Res. Commun. 1998, 251, 137–141. [Google Scholar]
- Sansonetti, P.J.; Phalipon, A. M cells as ports of entry for enteroinvasive pathogens: Mechanisms of interaction, consequences for the disease process. Semin. Immunol. 1999, 11, 193–203. [Google Scholar]
- Maresca, M.; Dumay, E.; Fantini, J.; Caporiccio, B. Selective transport of staphylococcal enterotoxin A through in vitro generated human M cells. Microbes Infect. 2007, 9, 1507–1510. [Google Scholar]
© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Schüller, S. Shiga Toxin Interaction with Human Intestinal Epithelium. Toxins 2011, 3, 626-639. https://doi.org/10.3390/toxins3060626
Schüller S. Shiga Toxin Interaction with Human Intestinal Epithelium. Toxins. 2011; 3(6):626-639. https://doi.org/10.3390/toxins3060626
Chicago/Turabian StyleSchüller, Stephanie. 2011. "Shiga Toxin Interaction with Human Intestinal Epithelium" Toxins 3, no. 6: 626-639. https://doi.org/10.3390/toxins3060626