Cycle Inhibiting Factors (Cifs): Cyclomodulins That Usurp the Ubiquitin-Dependent Degradation Pathway of Host Cells

Abstract
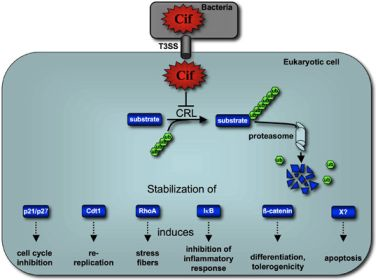
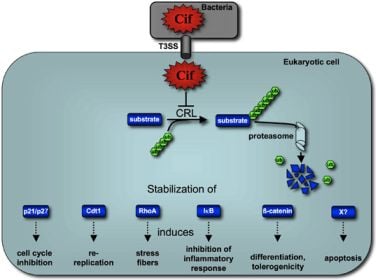
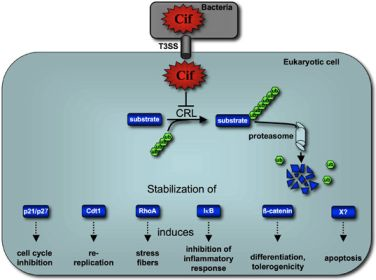
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Discovery and Distribution of the Cycle Inhibiting Factors (Cifs) That Triggers an Original Cytopathic Effect in Host Cells

2. Cif Proteins are Type III Effectors That Traffic to the Nucleus of the Host Cells
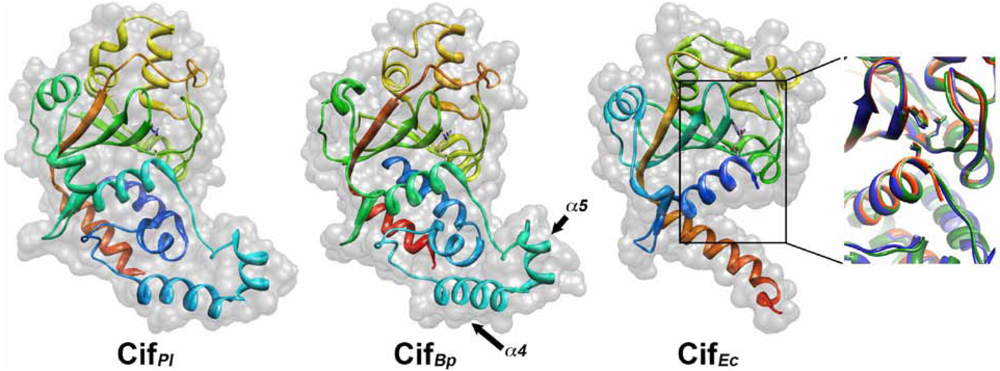
3. Crystal Structure Studies of Cif Homologs Reveals a Family of Proteins Sharing a Conserved Active Site

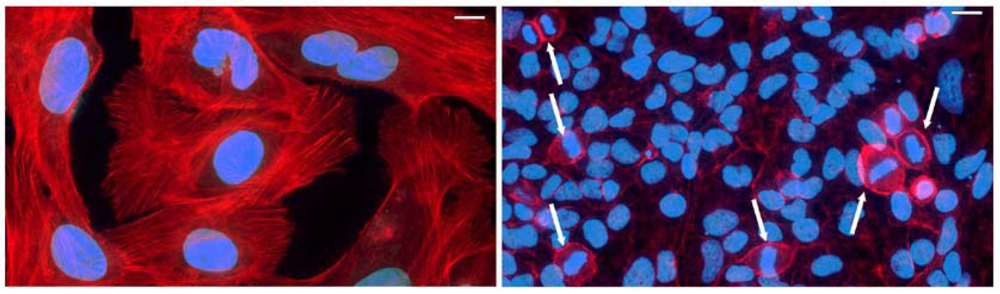
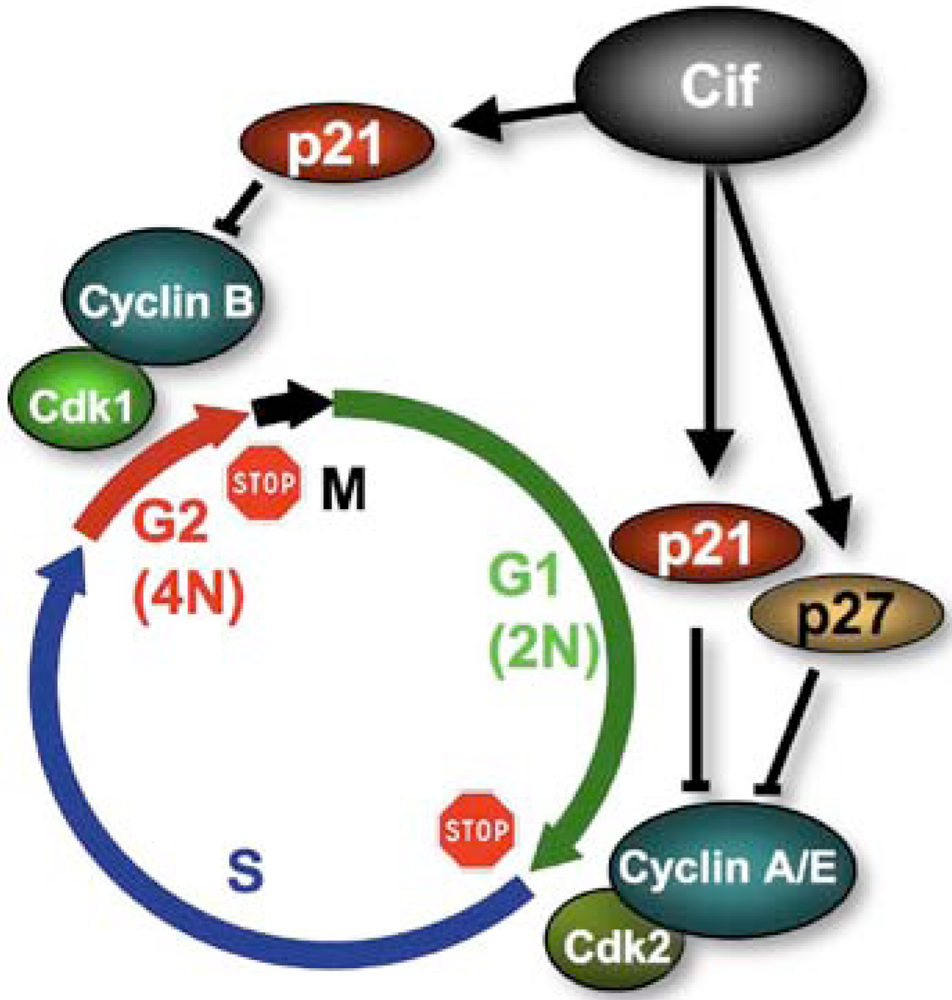
4. Cifs Are Cyclomodulins That Trigger Host Cell Cycle Arrest
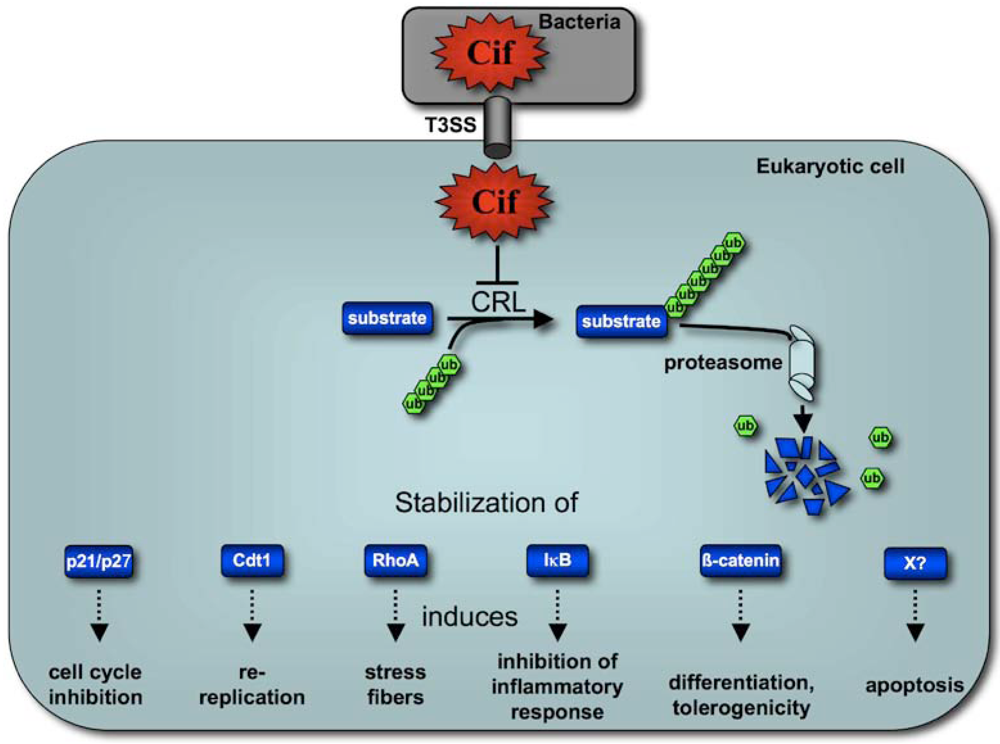
5. Cif Effector Proteins Target NEDD8 to Inhibit Specific Ubiquitin-Dependent Degradation Pathways
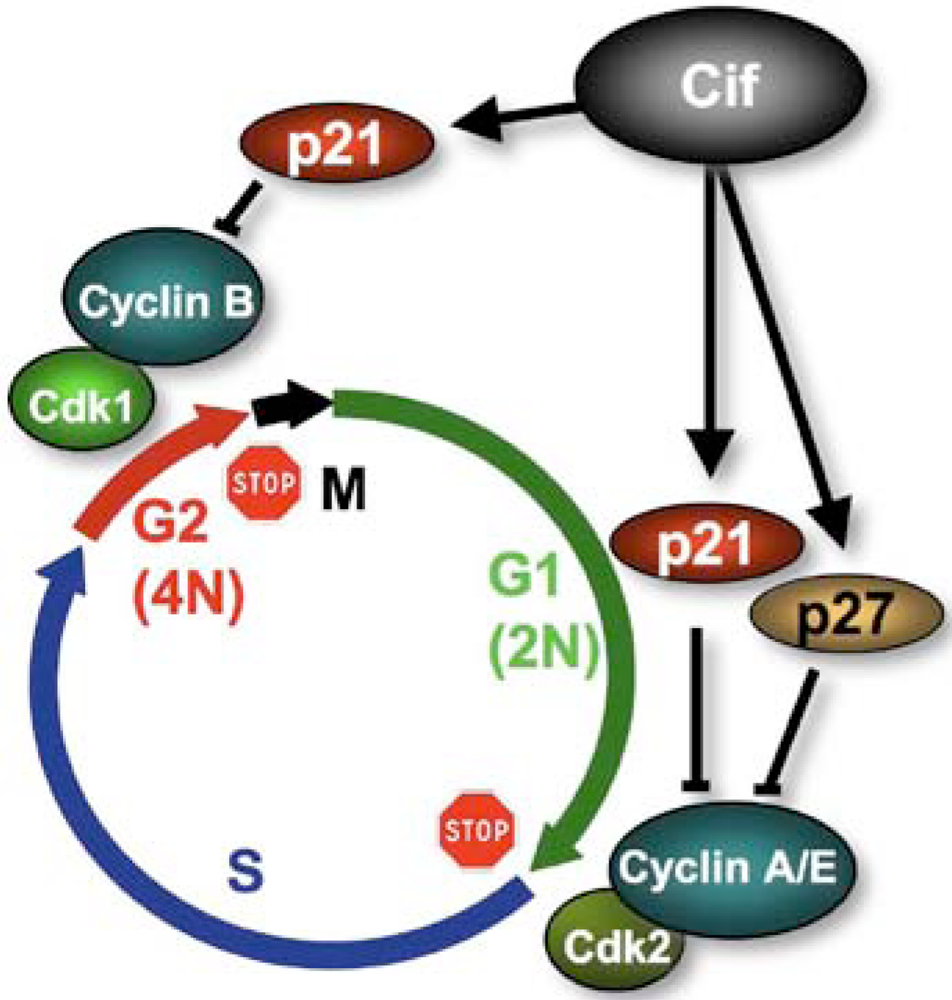
5.1. CifEc Interacts with Host Protein NEDD8
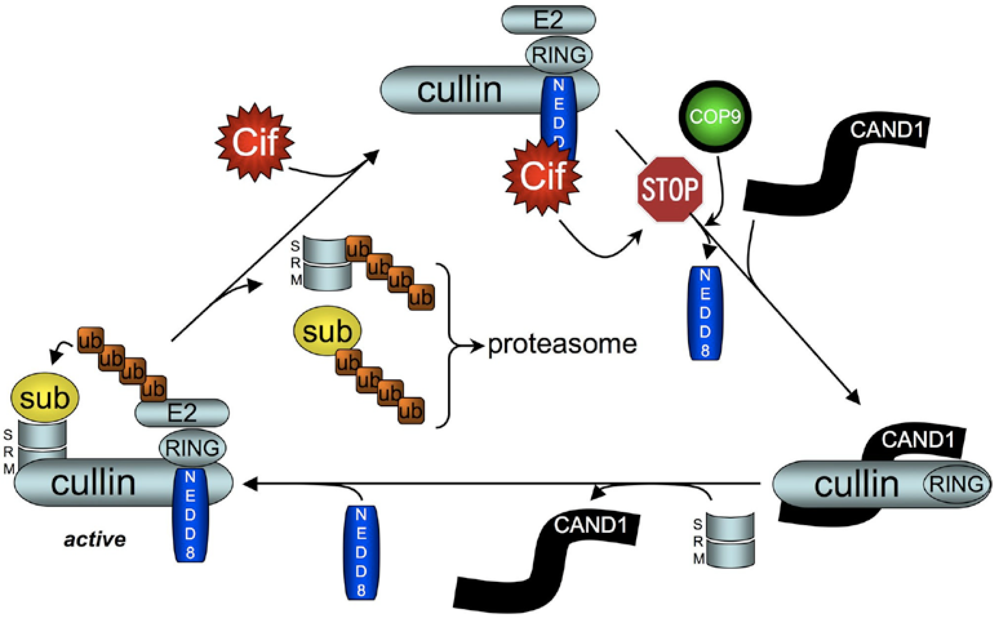
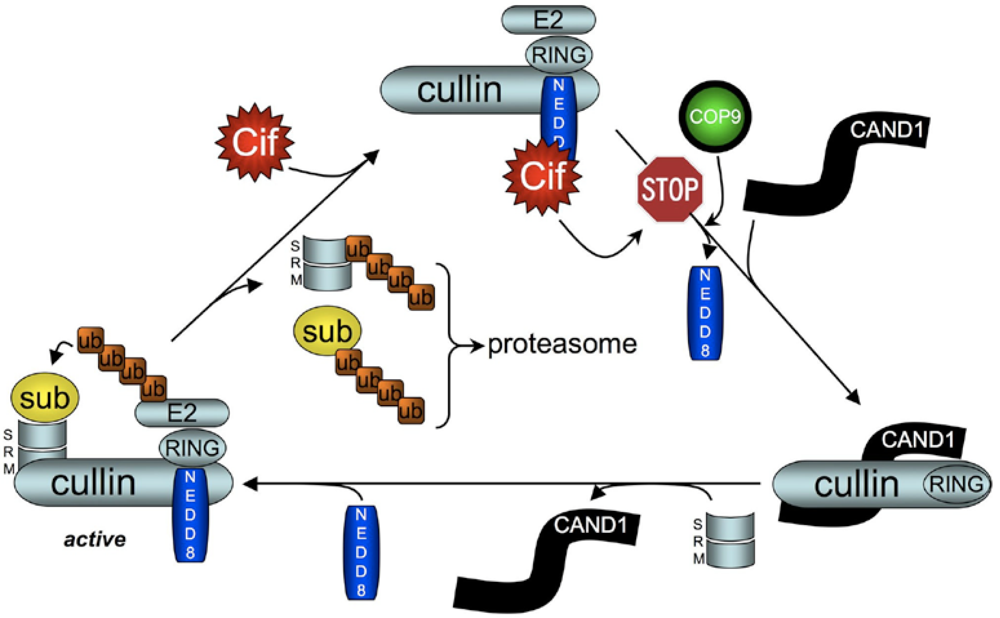
5.2. Cif Inhibits CRL Activity
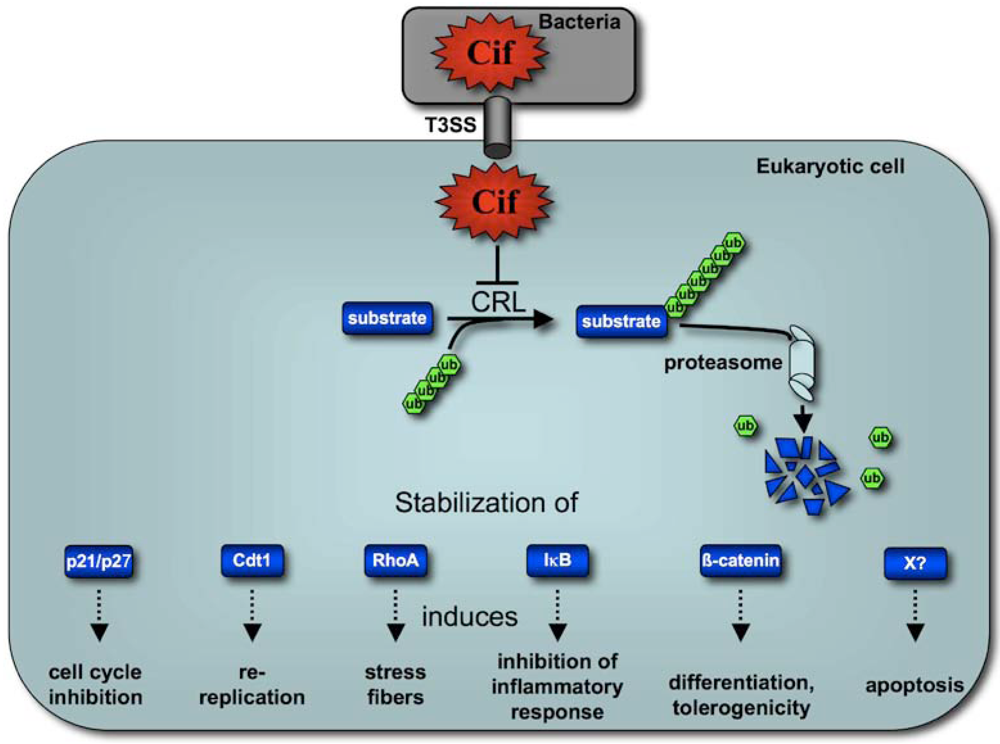
6. Concluding remarks and perspectives
References
- de Rycke, J.; Comtet, E.; Chalareng, C.; Boury, M.; Tasca, C.; Milon, A. Enteropathogenic Escherichia coli O103 from rabbit elicits actin stress fibers and focal adhesions in HeLa epithelial cells, cytopathic effects that are linked to an analog of the locus of enterocyte effacement. Infect. Immun. 1997, 65, 2555–2563. [Google Scholar] [PubMed]
- Nougayrede, J.P.; Boury, M.; Tasca, C.; Marches, O.; Milon, A.; Oswald, E.; de Rycke, J. Type III secretion-dependent cell cycle block caused in HeLa cells by enteropathogenic Escherichia coli O103. Infect. Immun. 2001, 69, 6785–6795. [Google Scholar] [PubMed]
- Kaper, J.B.; Nataro, J.P.; Mobley, H.L. Pathogenic Escherichia coli. Nat. Rev. Microbiol. 2004, 2, 123–140. [Google Scholar] [CrossRef] [PubMed]
- Nataro, J.P.; Kaper, J.B. Diarrheagenic Escherichia coli. Clin. Microbiol. Rev. 1998, 11, 142–201. [Google Scholar] [PubMed]
- Marches, O.; Ledger, T.N.; Boury, M.; Ohara, M.; Tu, X.; Goffaux, F.; Mainil, J.; Rosenshine, I.; Sugai, M.; de Rycke, J.; Oswald, E. Enteropathogenic and enterohaemorrhagic Escherichia coli deliver a novel effector called Cif, which blocks cell cycle G2/M transition. Mol. Microbiol. 2003, 50, 1553–1567. [Google Scholar] [CrossRef] [PubMed]
- Loukiadis, E.; Nobe, R.; Herold, S.; Tramuta, C.; Ogura, Y.; Ooka, T.; Morabito, S.; Kerouredan, M.; Brugere, H.; Schmidt, H.; Hayashi, T.; Oswald, E. Distribution, functional expression, and genetic organization of Cif, a phage-encoded type III-secreted effector from enteropathogenic and enterohemorrhagic Escherichia coli. J. Bacteriol. 2008, 190, 275–285. [Google Scholar] [PubMed]
- Creuzburg, K.; Middendorf, B.; Mellmann, A.; Martaler, T.; Holz, C.; Fruth, A.; Karch, H.; Schmidt, H. Evolutionary analysis and distribution of type III effector genes in pathogenic Escherichia coli from human, animal and food sources. Environ. Microbiol. 2011, 13, 439–452. [Google Scholar] [CrossRef] [PubMed]
- Jubelin, G.; Chavez, C.V.; Taieb, F.; Banfield, M.J.; Samba-Louaka, A.; Nobe, R.; Nougayrede, J.P.; Zumbihl, R.; Givaudan, A.; Escoubas, J.M.; Oswald, E. Cycle inhibiting factors (CIFs) are a growing family of functional cyclomodulins present in invertebrate and mammal bacterial pathogens. PLoS ONE 2009, 4, e4855. [Google Scholar] [PubMed]
- Konczy, P.; Ziebell, K.; Mascarenhas, M.; Choi, A.; Michaud, C.; Kropinski, A.M.; Whittam, T.S.; Wickham, M.; Finlay, B.; Karmali, M.A. Genomic O island 122, locus for enterocyte effacement, and the evolution of virulent verocytotoxin-producing Escherichia coli. J. Bacteriol. 2008, 190, 5832–5840. [Google Scholar] [CrossRef] [PubMed]
- Coombes, B.K.; Wickham, M.E.; Mascarenhas, M.; Gruenheid, S.; Finlay, B.B.; Karmali, M.A. Molecular analysis as an aid to assess the public health risk of non-O157 Shiga toxin-producing Escherichia coli strains. Appl. Environ. Microbiol. 2008, 74, 2153–2160. [Google Scholar] [CrossRef] [PubMed]
- Chavez, C.V.; Jubelin, G.; Courties, G.; Gomard, A.; Ginibre, N.; Pages, S.; Taieb, F.; Girard, P.A.; Oswald, E.; Givaudan, A.; Zumbihl, R.; Escoubas, J.M. The cyclomodulin Cif of Photorhabdus luminescens inhibits insect cell proliferation and triggers host cell death by apoptosis. Microbes Infect. 2010, 12, 1208–1218. [Google Scholar] [CrossRef] [PubMed]
- Cui, J.; Yao, Q.; Li, S.; Ding, X.; Lu, Q.; Mao, H.; Liu, L.; Zheng, N.; Chen, S.; Shao, F. Glutamine Deamidation and Dysfunction of Ubiquitin/NEDD8 Induced by a Bacterial Effector Family. Science 2010, 329, 1215–1218. [Google Scholar] [CrossRef] [PubMed]
- Nougayrede, J.P.; Marches, O.; Boury, M.; Mainil, J.; Charlier, G.; Pohl, P.; de Rycke, J.; Milon, A.; Oswald, E. The long-term cytoskeletal rearrangement induced by rabbit enteropathogenic Escherichia coli is Esp dependent but intimin independent. Mol. Microbiol. 1999, 31, 19–30. [Google Scholar] [CrossRef] [PubMed]
- Frankel, G.; Phillips, A.D.; Rosenshine, I.; Dougan, G.; Kaper, J.B.; Knutton, S. Enteropathogenic and enterohaemorrhagic Escherichia coli: more subversive elements. Mol. Microbiol. 1998, 30, 911–921. [Google Scholar] [CrossRef] [PubMed]
- Charpentier, X.; Oswald, E. Identification of the secretion and translocation domain of the enteropathogenic and enterohemorrhagic Escherichia coli effector Cif, using TEM-1 beta-lactamase as a new fluorescence-based reporter. J. Bacteriol. 2004, 186, 5486–5495. [Google Scholar] [CrossRef]
- Taieb, F.; Nougayrede, J.P.; Watrin, C.; Samba-Louaka, A.; Oswald, E. Escherichia coli cyclomodulin Cif induces G2 arrest of the host cell cycle without activation of the DNA-damage checkpoint-signalling pathway. Cell Microbiol. 2006, 8, 1910–1921. [Google Scholar] [CrossRef] [PubMed]
- Jubelin, G.; Taieb, F.; Duda, D.M.; Hsu, Y.; Samba-Louaka, A.; Nobe, R.; Penary, M.; Watrin, C.; Nougayrede, J.P.; Schulman, B.A.; Stebbins, C.E.; Oswald, E. Pathogenic Bacteria Target NEDD8-Conjugated Cullins to Hijack Host-Cell Signaling Pathways. PLoS Pathog. 2010, 6, e1001128. [Google Scholar] [CrossRef] [PubMed]
- Crow, A.; Race, P.R.; Jubelin, G.; Varela Chavez, C.; Escoubas, J.M.; Oswald, E.; Banfield, M.J. Crystal structures of Cif from bacterial pathogens Photorhabdus luminescens and Burkholderia pseudomallei. PLoS ONE 2009, 4, e5582. [Google Scholar] [PubMed]
- Hsu, Y.; Jubelin, G.; Taieb, F.; Nougayrede, J.P.; Oswald, E.; Stebbins, C.E. Structure of the cyclomodulin Cif from pathogenic Escherichia coli. J. Mol. Biol. 2008, 384, 465–477. [Google Scholar] [CrossRef] [PubMed]
- Yao, Q.; Cui, J.; Zhu, Y.; Wang, G.; Hu, L.; Long, C.; Cao, R.; Liu, X.; Huang, N.; Chen, S.; Liu, L.; Shao, F. A bacterial type III effector family uses the papain-like hydrolytic activity to arrest the host cell cycle. Proc. Natl. Acad. Sci. USA 2009, 106, 3716–3721. [Google Scholar]
- Samba-Louaka, A.; Taieb, F.; Nougayrede, J.P.; Oswald, E. Cif type III effector protein: A smart hijacker of the host cell cycle. Future Microbiol. 2009, 4, 867–877. [Google Scholar] [CrossRef] [PubMed]
- Nougayrede, J.P.; Taieb, F.; de Rycke, J.; Oswald, E. Cyclomodulins: Bacterial effectors that modulate the eukaryotic cell cycle. Trends Microbiol. 2005, 13, 103–110. [Google Scholar] [CrossRef] [PubMed]
- Oswald, E.; Nougayrede, J.P.; Taieb, F.; Sugai, M. Bacterial toxins that modulate host cell-cycle progression. Curr. Opin. Microbiol. 2005, 8, 83–91. [Google Scholar] [CrossRef] [PubMed]
- Samba-Louaka, A.; Nougayrede, J.P.; Watrin, C.; Jubelin, G.; Oswald, E.; Taieb, F. Bacterial cyclomodulin Cif blocks the host cell cycle by stabilizing the cyclin dependent kinase inhibitors p21(waf1) and p27(kip1). Cell Microbiol. 2008, 10, 2496–2508. [Google Scholar] [CrossRef] [PubMed]
- Besson, A.; Dowdy, S.F.; Roberts, J.M. CDK inhibitors: Cell cycle regulators and beyond. Dev. Cell 2008, 14, 159–169. [Google Scholar] [CrossRef] [PubMed]
- Samba-Louaka, A.; Nougayrede, J.P.; Watrin, C.; Oswald, E.; Taieb, F. The enteropathogenic Escherichia coli effector Cif induces delayed apoptosis in epithelial cells. Infect Immun. 2009, 77, 5471–5477. [Google Scholar] [CrossRef] [PubMed]
- Morikawa, H.; Kim, M.; Mimuro, H.; Punginelli, C.; Koyama, T.; Nagai, S.; Miyawaki, A.; Iwai, K.; Sasakawa, C. The bacterial effector Cif interferes with SCF ubiquitin ligase function by inhibiting deneddylation of Cullin1. Biochem. Biophys. Res. Commun. 2010, 401, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Kitagawa, K.; Kotake, Y.; Kitagawa, M. Ubiquitin-mediated control of oncogene and tumor suppressor gene products. Cancer Sci. 2009, 100, 1374–1381. [Google Scholar] [CrossRef] [PubMed]
- Ciechanover, A.; Finley, D.; Varshavsky, A. Ubiquitin dependence of selective protein degradation demonstrated in the mammalian cell cycle mutant ts85. Cell 1984, 37, 57–66. [Google Scholar] [CrossRef] [PubMed]
- Hershko, A.; Ciechanover, A. The ubiquitin system for protein degradation. Annu. Rev. Biochem. 1992, 61, 761–807. [Google Scholar] [CrossRef] [PubMed]
- Hershko, A.; Ciechanover, A. The ubiquitin system. Annu. Rev. Biochem. 1998, 67, 425–479. [Google Scholar] [PubMed]
- Huang, D.T.; Ayrault, O.; Hunt, H.W.; Taherbhoy, A.M.; Duda, D.M.; Scott, D.C.; Borg, L.A.; Neale, G.; Murray, P.J.; Roussel, M.F.; Schulman, B.A. E2-RING expansion of the NEDD8 cascade confers specificity to cullin modification. Mol. Cell 2009, 33, 483–495. [Google Scholar] [PubMed]
- Saha, A.; Deshaies, R.J. Multimodal activation of the ubiquitin ligase SCF by Nedd8 conjugation. Mol. Cell 2008, 32, 21–31. [Google Scholar] [CrossRef] [PubMed]
- Duda, D.M.; Borg, L.A.; Scott, D.C.; Hunt, H.W.; Hammel, M.; Schulman, B.A. Structural insights into NEDD8 activation of cullin-RING ligases: Conformational control of conjugation. Cell 2008, 134, 995–1006. [Google Scholar] [CrossRef] [PubMed]
- Merlet, J.; Burger, J.; Gomes, J.E.; Pintard, L. Regulation of cullin-RING E3 ubiquitin-ligases by neddylation and dimerization. Cell Mol. Life Sci. 2009, 66, 1924–1938. [Google Scholar] [CrossRef] [PubMed]
- Bosu, D.R.; Kipreos, E.T. Cullin-RING ubiquitin ligases: Global regulation and activation cycles. Cell Div. 2008, 3, 7. [Google Scholar] [CrossRef] [PubMed]
- Tomoda, K.; Yoneda-Kato, N.; Fukumoto, A.; Yamanaka, S.; Kato, J.Y. Multiple functions of Jab1 are required for early embryonic development and growth potential in mice. J. Biol. Chem. 2004, 279, 43013–43018. [Google Scholar] [PubMed]
- Cerda-Maira, F.A.; Pearce, M.J.; Fuortes, M.; Bishai, W.R.; Hubbard, S.R.; Darwin, K.H. Molecular analysis of the prokaryotic ubiquitin-like protein (Pup) conjugation pathway in Mycobacterium tuberculosis. Mol. Microbiol. 2010, 77, 1123–1135. [Google Scholar] [CrossRef] [PubMed]
- Striebel, F.; Imkamp, F.; Sutter, M.; Steiner, M.; Mamedov, A.; Weber-Ban, E. Bacterial ubiquitin-like modifier Pup is deamidated and conjugated to substrates by distinct but homologous enzymes. Nat. Struct. Mol. Biol. 2009, 16, 647–651. [Google Scholar] [CrossRef] [PubMed]
- Flatau, G.; Lemichez, E.; Gauthier, M.; Chardin, P.; Paris, S.; Fiorentini, C.; Boquet, P. Toxin-induced activation of the G protein p21 Rho by deamidation of glutamine. Nature 1997, 387, 729–733. [Google Scholar] [PubMed]
- Schmidt, G.; Sehr, P.; Wilm, M.; Selzer, J.; Mann, M.; Aktories, K. Gln 63 of Rho is deamidated by Escherichia coli cytotoxic necrotizing factor-1. Nature 1997, 387, 725–729. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Yang, Z.; Meng, M.; Zhao, Y.; Dong, N.; Yan, H.; Liu, L.; Ding, M.; Peng, H.B.; Shao, F. Cullin mediates degradation of RhoA through evolutionarily conserved BTB adaptors to control actin cytoskeleton structure and cell movement. Mol. Cell 2009, 35, 841–855. [Google Scholar] [CrossRef] [PubMed]
- Xouri, G.; Dimaki, M.; Bastiaens, P.I.; Lygerou, Z. Cdt1 interactions in the licensing process: A model for dynamic spatiotemporal control of licensing. Cell Cycle 2007, 6, 1549–1552. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Kipreos, E.T. The Caenorhabditis elegans replication licensing factor CDT-1 is targeted for degradation by the CUL-4/DDB-1 complex. Mol. Cell Biol. 2007, 27, 1394–1406. [Google Scholar] [CrossRef] [PubMed]
- Nishitani, H.; Sugimoto, N.; Roukos, V.; Nakanishi, Y.; Saijo, M.; Obuse, C.; Tsurimoto, T.; Nakayama, K.I.; Nakayama, K.; Fujita, M.; Lygerou, Z.; Nishimoto, T. Two E3 ubiquitin ligases, SCF-Skp2 and DDB1-Cul4, target human Cdt1 for proteolysis. EMBO J. 2006, 25, 1126–1136. [Google Scholar] [PubMed]
- Collier-Hyams, L.S.; Sloane, V.; Batten, B.C.; Neish, A.S. Cutting edge: Bacterial modulation of epithelial signaling via changes in neddylation of cullin-1. J. Immunol. 2005, 175, 4194–4198. [Google Scholar] [PubMed]
- Kumar, A.; Wu, H.; Collier-Hyams, L.S.; Hansen, J.M.; Li, T.; Yamoah, K.; Pan, Z.Q.; Jones, D.P.; Neish, A.S. Commensal bacteria modulate cullin-dependent signaling via generation of reactive oxygen species. EMBO J. 2007, 26, 4457–4466. [Google Scholar] [CrossRef] [PubMed]
- Manicassamy, S.; Reizis, B.; Ravindran, R.; Nakaya, H.; Salazar-Gonzalez, R.M.; Wang, Y.C.; Pulendran, B. Activation of beta-catenin in dendritic cells regulates immunity versus tolerance in the intestine. Science 2010, 329, 849–853. [Google Scholar] [CrossRef] [PubMed]
- Munro, P.; Flatau, G.; Lemichez, E. Bacteria and the ubiquitin pathway. Curr. Opin. Microbiol. 2007, 10, 39–46. [Google Scholar] [CrossRef] [PubMed]
- Rytkonen, A.; Holden, D.W. Bacterial interference of ubiquitination and deubiquitination. Cell Host Microbe 2007, 1, 13–22. [Google Scholar] [CrossRef] [PubMed]
- Angot, A.; Vergunst, A.; Genin, S.; Peeters, N. Exploitation of eukaryotic ubiquitin signaling pathways by effectors translocated by bacterial type III and type IV secretion systems. PLoS Pathog. 2007, 3, e3. [Google Scholar] [CrossRef] [PubMed]
- Soucy, T.A.; Smith, P.G.; Milhollen, M.A.; Berger, A.J.; Gavin, J.M.; Adhikari, S.; Brownell, J.E.; Burke, K.E.; Cardin, D.P.; Critchley, S.; et al. An inhibitor of NEDD8-activating enzyme as a new approach to treat cancer. Nature 2009, 458, 732–736. [Google Scholar] [PubMed]
© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Taieb, F.; Nougayrède, J.-P.; Oswald, E. Cycle Inhibiting Factors (Cifs): Cyclomodulins That Usurp the Ubiquitin-Dependent Degradation Pathway of Host Cells. Toxins 2011, 3, 356-368. https://doi.org/10.3390/toxins3040356
Taieb F, Nougayrède J-P, Oswald E. Cycle Inhibiting Factors (Cifs): Cyclomodulins That Usurp the Ubiquitin-Dependent Degradation Pathway of Host Cells. Toxins. 2011; 3(4):356-368. https://doi.org/10.3390/toxins3040356
Chicago/Turabian StyleTaieb, Frédéric, Jean-Philippe Nougayrède, and Eric Oswald. 2011. "Cycle Inhibiting Factors (Cifs): Cyclomodulins That Usurp the Ubiquitin-Dependent Degradation Pathway of Host Cells" Toxins 3, no. 4: 356-368. https://doi.org/10.3390/toxins3040356