Shiga Toxins: Intracellular Trafficking to the ER Leading to Activation of Host Cell Stress Responses

{kind=link}
Abstract
:1. Introduction
2. Characterization of Shiga Toxin Intracellular Transport
2.1. Toxin Structure and Function
2.2. Toxin Interaction with Cell Surface Receptors
2.3. Intracellular Trafficking of the Toxins
2.3.1. Endocytosis of Shiga toxins
2.3.2. Endosome to Golgi Apparatus Transport of Shiga Toxins
2.3.3. Toxin Transport from the Trans-Golgi Network to the ER and Nuclear Envelope
2.3.4. Transport of Shiga Toxins from the ER to the Cytosol
3. Shiga Toxins Induce the Ribotoxic Stress Response
4. ER Stress Responses and Shiga Toxin-Induced Apoptosis
4.1. Shiga Toxins Induce Apoptosis
4.2. Shiga Toxins Induce the ER Stress Response Leading to Apoptosis
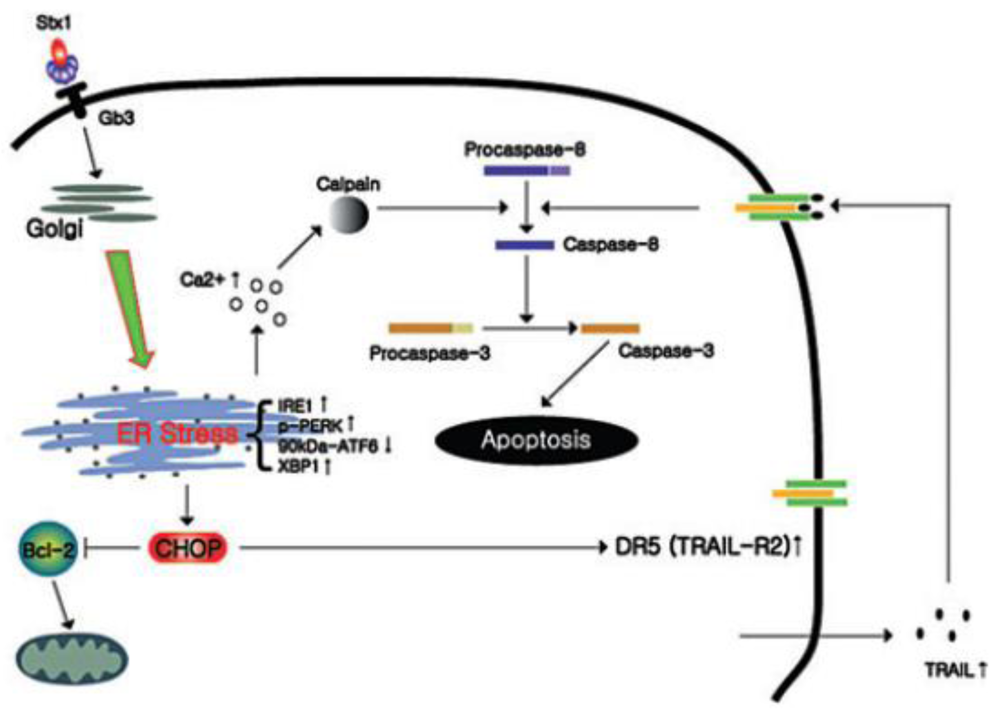
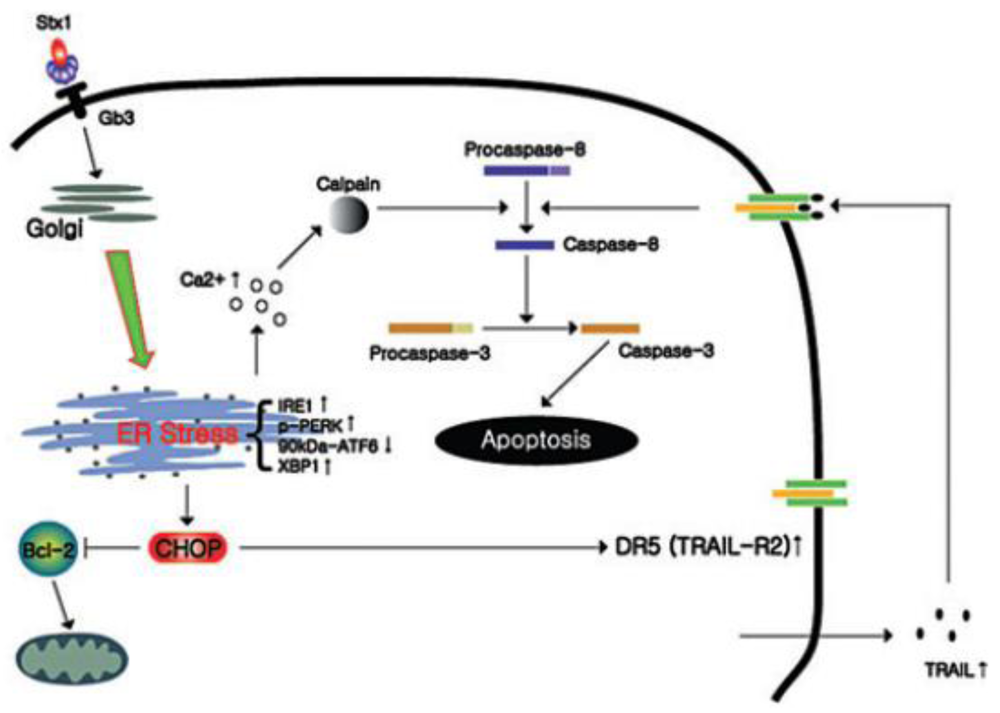
4.3. Calcium Involvement in Shiga Toxin-Induced Apoptosis
5. Shiga Toxin-Induced ER Stress: The Cell Death vs. Survival Decision
6. Conclusions
Acknowledgements
References
- Proulx, F.; Tesh, V.L. Renal diseases in the Pediatric Intensive Care Unit: Thrombotic microangiopathy, hemolytic uremic syndrome, and thrombotic thrombocytopenic purpura. In Pediatric Critical Care Medicine: Basic Science and Clinical Evidence; Wheeler, D.S., Wong, H.R., Shanley, T.P., Eds.; Springer Verlag: London, UK, 2007; pp. 1189–1204. [Google Scholar]
- Scheiring, J.; Andreoli, S.P.; Zimmerhackl, L.B. Treatment and outcome of Shiga-toxin-associated hemolytic uremic syndrome (HUS). Pediatr. Nephrol. 2008, 23, 1749–1760. [Google Scholar]
- Karch, H.; Tarr, P.I.; Bielaszewska, M. Enterohaemorrhagic Escherichia coli in human medicine. Int. J. Med. Microbiol. 2005, 295, 405–418. [Google Scholar]
- Bale, J.F.; Brasher, C.; Siegler, R.L. CNS manifestations of the hemolytic-uremic syndrome. Relationship to metabolic alterations and prognosis. Am. J. Dis. Child. 1980, 134, 869–872. [Google Scholar] [PubMed]
- Hurley, B.P.; Jacewicz, M.; Thorpe, C.M.; Lincicome, L.L.; King, A.J.; Keusch, G.T.; Acheson, D.W.K. Shiga toxins 1 and 2 translocate differently across polarized intestinal epithelial cells. Infect. Immun. 1999, 67, 6670–6677. [Google Scholar]
- Malyukova, I.; Murray, K.F.; Zhu, C.; Boedeker, E.; Kane, A.; Patterson, K.; Peterson, J.R.; Donowitz, M.; Kovbasnjuk, O. Macropinocytosis in Shiga toxin 1 uptake by human intestinal epithelial cells and transcellular transcytosis. Am. J. Physiol. Gastrointest. Liver Physiol. 2009, 296, G78–G92. [Google Scholar]
- Fontaine, A.; Arondel, J.; Sansonetti, P.J. Role of Shiga toxin in the pathogenesis of bacillary dysentery, studied by using a Tox− mutant of Shigella dysenteriae 1. Infect. Immun. 1988, 56, 3099–3109. [Google Scholar]
- Taylor, F.B.; Tesh, V.L.; DeBault, L.; Li, A.; Chang, A.C.K.; Kosanke, S.D.; Pysher, T.J.; Siegler, R.L. Characterization of the baboon responses to Shiga-like toxin: Descriptive study of a new primate model of toxic responses to Stx-1. Am. J. Path. 1999, 154, 1285–1299. [Google Scholar]
- te Loo, D.M.W.M.; Monnens, L.A.H.; van der Velden, T.J.A.N.; Vermeer, M.A.; Preyers, F.; Demacker, P.N.M.; van den Heuvel, L.P.W.J.; van Hinsbergh, V.W.M. Binding and transfer of verocytotoxin by polymorphonuclear leukocytes in hemolytic uremic syndrome. Blood 2000, 95, 3396–3402. [Google Scholar]
- Brigotti, M.; Carnicelli, D.; Ravanelli, E.; Barbieri, S.; Ricci, F.; Bontadini, A.; Tozzi, A.E.; Scavia, G.; Caprioli, A.; Tazzari, P.L. Interactions between Shiga toxins and human polymorphonuclear leukocytes. J. Leukoc. Biol. 2008, 84, 1019–1027. [Google Scholar]
- Lingwood, C.A. Shiga toxin receptor glycolipid binding. Pathology and utility. In Methods in Molecular Medicine. E. coli Shiga Toxin Methods and Protocols; Philpott, D., Ebel, F., Eds.; Humana Press, Inc.: Totowa, NJ, USA, 2003; Volume 73, pp. 165–186. [Google Scholar]
- Sandvig, K.; Grimmer, S.; Lauvrak, S.U.; Torgersen, M.L.; Skretting, G.; van Deurs, B.; Iversen, T.G. Pathways followed by ricin and Shiga toxin into cells. Histochem. Cell Biol. 2002, 117, 131–141. [Google Scholar]
- Johannes, L.; Römer, W. Shiga toxins—From cell biology to biomedical applications. Nat. Rev. Microbiol. 2010, 8, 105–116. [Google Scholar]
- Armstrong, G.D.; Fodor, E.; Vanmaele, R. Investigation of Shiga-like toxin binding to chemically synthesized oligosaccharide sequences. J. Infect. Dis. 1991, 164, 1160–1167. [Google Scholar]
- Armstrong, G.D.; McLaine, P.N.; Rowe, P.C. Clinical trials of Synsorb-Pk in preventing hemolytic-uremic syndrome. In Escherichia coli O157:H7 and Other Shiga toxin-producing E. coli Strains; Kaper, J.B., O'Brien, A.D., Eds.; ASM Press: Washington, DC, USA, 1998; pp. 374–384. [Google Scholar]
- Trachtman, H.; Cnaan, A.; Christen, E.; Gibbs, K.; Zhao, S.; Acheson, D.W.K.; Weiss, R.; Kaskel, F.J.; Spitzer, A.; Hirschman, G.H. Effect of an oral Shiga toxin–binding agent on diarrhea-associated Hemolytic Uremic Syndrome in children: A randomized controlled trial. JAMA 2003, 290, 1337–1344. [Google Scholar]
- Nishikawa, K.; Matsuoka, K.; Kita, E.; Okabe, N.; Mizuguchi, M.; Hino, K.; Miyazawa, S.; Yamasaki, C.; Aoki, J.; Takashima, S.; Yamakawa, Y.; Nishijima, M.; Terunuma, D.; Kuzuhara, H.; Natori, Y. A therapeutic agent with oriented carbohydrates for treatment of infections by Shiga toxin-producing Escherichia coli O157:H7. Proc. Natl. Acad. Sci. USA 2002, 99, 7669–7674. [Google Scholar]
- Saenz, J.B.; Doggett, T.A.; Haslam, D.B. Identification and characterization of small molecules that inhibit intracellular toxin transport. Infect. Immun. 2007, 75, 4552–4561. [Google Scholar]
- Mukherjee, J.; Chios, K.; Fishwild, D.; Hudson, D.; O’Donnell, S.; Rich, S.M.; Donohue-Rolfe, A.; Tzipori, S. Production and characterization of protective human antibodies against Shiga toxin 1. Infect. Immun. 2002, 70, 5896–5899. [Google Scholar]
- Pinyon, R.A.; Paton, J.C.; Paton, A.W.; Botten, J.A.; Morona, R. Refinement of a therapeutic Shiga toxin—Binding probiotic for human trials. J. Infect. Dis. 2004, 189, 1547–1555. [Google Scholar]
- Strockbine, N.A.; Marques, L.R.M.; Newland, J.W.; Williams-Smith, H.; Holmes, R.K.; O'Brien, A.D. Two toxin-converting phages from Escherichia coli O157:H7 strain 933 encode antigenically distinct toxins with similar biologic activities. Infect. Immun. 1986, 53, 135–140. [Google Scholar]
- Mellmann, A.; Bielaszewska, M.; Kock, R.; Friedrich, A.W.; Fruth, A.; Middendorf, B.; Harmsen, D.; Schmidt, M.A.; Karch, H. Analysis of collection of hemolytic uremic syndrome-associated enterohemorrhagic Escherichia coli. Emerg. Infect. Dis. 2008, 14, 1287–1290. [Google Scholar]
- Jackson, M.P.; Newland, J.W.; Holmes, R.K.; O'Brien, A.D. Nucleotide sequence analysis of the structural genes for Shiga-like toxin I encoded by bacteriophage 933J from Escherichia coli. Microb. Pathog. 1987, 2, 147–153. [Google Scholar] [CrossRef] [PubMed]
- Stein, P.E.; Boodhoo, A.; Tyrrell, G.J.; Brunton, J.L.; Read, R.J. Crystal structure of the cell-binding B oligomer of verotoxin-1 from E coli. Nature 1992, 355, 748–750. [Google Scholar]
- Fraser, M.E.; Chernaia, M.M.; Kozlov, Y.V.; James, M.N.G. Crystal structure of the holotoxin from Shigella dysenteriae at 2.5 Å resolution. Nat. Struct. Biol. 1994, 1, 59–64. [Google Scholar] [CrossRef] [PubMed]
- Fraser, M.E.; Fujinaga, M.; Cherney, M.M.; Melton-Celsa, A.R.; Twiddy, E.M.; O'Brien, A.D.; James, M.N.G. Structure of Shiga toxin type 2 (Stx2) from Escherichia coli O157:H7. J. Biol. Chem. 2004, 279, 27511–27517. [Google Scholar]
- Endo, Y.; Tsurugi, K.; Yutsudo, T.; Takeda, Y.; Ogasawara, T.; Igarashi, K. Site of action of a Vero toxin (VT2) from Escherichia coli O157:H7 and of Shiga toxin on eukaryotic ribosomes. RNA N-glycosidase activity of the toxins. Eur. J. Biochem. 1988, 171, 45–50. [Google Scholar] [CrossRef] [PubMed]
- Saxena, S.K.; O'Brien, A.D.; Ackerman, E.J. Shiga toxin, Shiga-like toxin II variant, and ricin are all single-site RNA N-glycosidases of 28 S RNA when microinjected into Xenopus oocytes. J. Biol. Chem. 1989, 264, 596–601. [Google Scholar]
- Sandvig, K.; Olsnes, S.; Brown, J.E.; Petersen, O.W.; van Deurs, B. Endocytosis from coated pits of Shiga toxin: A glycolipid-binding protein from Shigella dysenteriae 1. J. Cell Biol. 1989, 108, 1331–1343. [Google Scholar]
- Sandvig, K.; Bergan, J.; Dyve, A.-B.; Skotland, T.; Torgersen, M.L. Endocytosis and retrograde transport of Shiga toxin. Toxicon 2009. [Google Scholar]
- DeGrandis, S.; Law, H.; Brunton, J.; Gyles, C.; Lingwood, C.A. Globotetraosylceramide is recognized by the pig edema disease toxin. J. Biol. Chem. 1989, 264, 12520–12525. [Google Scholar]
- Schweppe, C.H.; Bielaszewska, M.; Pohlentz, G.; Friedrich, A.W.; Büntemeyer, H.; Schmidt, M.A.; Kim, K.S.; Peter-Katalinić, J.; Karch, H.; Müthing, J. Glycosphingolipids in vascular endothelial cells: Relationship of heterogeneity in Gb3Cer/CD77 receptor expression with differential Shiga toxin 1 cytotoxicity. Glycoconj. J. 2008, 25, 291–304. [Google Scholar]
- Ling, H.; Boodhoo, A.; Hazes, B.; Cummings, M.D.; Armstrong, G.D.; Brunton, J.L.; Read, R.J. Structure of the Shiga-like toxin I B-pentamer complexed with an analogue of its receptor Gb3. Biochemistry 1998, 37, 1777–1788. [Google Scholar]
- Bast, D.J.; Banerjee, L.; Clark, C.; Read, R.J.; Brunton, J.L. The identification of three biologically relevant globotriaosyl ceramide receptor binding sites on the Verotoxin 1 B subunit. Mol. Microbiol. 1999, 32, 953–960. [Google Scholar]
- Römer, W.; Berland, L.; Chambon, V.; Gaus, K.; Windschiegl, B.; Tenza, D.; Aly, M.R.; Fraisier, V.; Florent, J.C.; Perrais, D.; Lamaze, C.; Raposo, G.; Steinem, C.; Sens, P.; Bassereau, P.; Johannes, L. Shiga toxin induces tubular membrane invaginations for its uptake into cells. Nature 2007, 450, 670–675. [Google Scholar]
- Okuda, T.; Tokuda, N.; Numata, S.; Ito, M.; Ohta, M.; Kawamura, K.; Wiels, J.; Urano, T.; Tajima, O.; Furukawa, K. Targeted disruption of Gb3/CD77 synthase gene resulted in the complete deletion of globo-series glycosphingolipids and loss of sensitivity to verotoxins. J. Biol. Chem. 2006, 281, 10230–10235. [Google Scholar]
- Saelinger, C.B. Receptors for bacterial toxins. In Bacterial Protein Toxins; Burns, D.L., Barbieri, J.T., Iglewski, B.H., Rappuoli, R., Eds.; ASM Press: Washington, DC, USA, 2003; pp. 131–148. [Google Scholar]
- Binnington, B.; Lingwood, D.; Nutikka, A.; Lingwood, C.A. Effect of globotriaosyl ceramide fatty acid alpha-hydroxylation on the binding by verotoxin 1 and verotoxin 2. Neurochem. Res. 2002, 27, 807–813. [Google Scholar]
- Römer, W.; Pontani, L.L.; Sorre, B.; Rentero, C.; Berland, L.; Chambon, V.; Lamaze, C.; Bassereau, P.; Sykes, C.; Gaus, K.; Johannes, L. Actin dynamics drive membrane reorganization and scission in clathrin-independent endocytosis. Cell 2010, 140, 540–553. [Google Scholar]
- Arab, S.; Lingwood, C.A. Influence of phospholipid chain length on verotoxin/globotriaosyl ceramide binding in model membranes: comparison of a supported bilayer film and liposomes. Glycoconj. J. 1996, 13, 159–166. [Google Scholar]
- Arab, S.; Lingwood, C.A. Intracellular targeting of the endoplasmic reticulum/nuclear envelope by retrograde transport may determine cell hypersensitivity to verotoxin via globotriaosyl ceramide fatty acid isoform traffic. J. Cell. Physiol. 1998, 177, 646–660. [Google Scholar]
- Schüller, S.; Frankel, G.; Phillips, A.D. Interaction of Shiga toxin from Escherichia coli with human intestinal epithelial cell lines and explants: Stx2 induces epithelial damage in organ culture. Cell. Microbiol. 2004, 6, 289–301. [Google Scholar]
- Griener, T.P.; Mulvey, G.L.; Marcato, P.; Armstrong, G.D. Differential binding of Shiga toxin 2 to human and murine neutrophils. J. Med. Microbiol. 2007, 56, 1423–1430. [Google Scholar]
- Richardson, S.E.; Karmali, M.A.; Becker, L.E.; Smith, C.R. The histopathology of the hemolytic uremic syndrome associated with verocytotoxin-producing Escherichia coli infections. Hum. Pathol. 1988, 19, 1102–1108. [Google Scholar]
- Habib, R. Pathology of the hemolytic uremic syndrome. In Hemolytic Uremic Syndrome and Thrombotic Thrombocytopenic Purpura; Kaplan, B.S., Trompeter, R.S., Moake, J.L., Eds.; Decker: New York, NY, USA, 1992; pp. 315–353. [Google Scholar]
- Obrig, T.G.; Del Vecchio, P.J.; Brown, J.E.; Moran, T.P.; Rowland, B.M.; Judge, T.K.; Rothman, S.W. Direct cytotoxic action of Shiga toxin on human vascular endothelial cells. Infect. Immun. 1988, 56, 2373–2378. [Google Scholar]
- Fujii, J.; Wood, K.; Matsuda, F.; Carneiro-Filho, B.A.; Schlegel, K.H.; Yutsudo, T.; Binnington-Boyd, B.; Lingwood, C.A.; Obata, F.; Kim, K.S.; Yoshida, S.; Obrig, T. Shiga toxin 2 causes apoptosis in human brain microvascular endothelial cells via C/EBP homologous protein. Infect. Immun. 2008, 76, 3679–3689. [Google Scholar]
- Obrig, T.G.; Louise, C.B.; Lingwood, C.A.; Boyd, B.; Barley-Maloney, L.; Daniel, T.O. Endothelial heterogeneity in Shiga toxin receptors and responses. J. Biol. Chem. 1993, 268, 15484–15488. [Google Scholar]
- Müthing, J.; Schweppe, C.H.; Karch, H.; Friedrich, A.W. Shiga toxins, glycosphingolipid diversity, and endothelial cell injury. Thromb. Haemost. 2009, 101, 252–264. [Google Scholar]
- Sandvig, K. Shiga toxins. Toxicon 2001, 39, 1629–1635. [Google Scholar]
- Sandvig, K.; Torgersen, M.L.; Engedal, N.; Skotland, T.; Iversen, T.G. Protein toxins from plants and bacteria: Probes for intracellular transport and tools in medicine. FEBS Lett. 2010. [Google Scholar]
- Garred, Ø.; Dubinina, E.; Holm, P.K.; Olsnes, S.; van Deurs, B.; Kozlov, J.V.; Sandvig, K. Role of processing and intracellular transport for optimal toxicity of Shiga toxin and toxin mutants. Exp. Cell Res. 1995, 218, 39–49. [Google Scholar]
- Garred, Ø.; van Deurs, B.; Sandvig, K. Furin-induced cleavage and activation of Shiga toxin. J. Biol. Chem. 1995, 270, 10817–10821. [Google Scholar]
- Sandvig, K.; Prydz, K.; Ryd, M.; van Deurs, B. Endocytosis and intracellular transport of the glycolipid-binding ligand Shiga toxin in polarized MDCK cells. J. Cell Biol. 1991, 113, 553–562. [Google Scholar]
- Lauvrak, S.U.; Wälchli, S.; Iversen, T.G.; Slagsvold, H.H.; Torgersen, M.L.; Spilsberg, B.; Sandvig, K. Shiga toxin regulates its entry in a Syk-dependent manner. Mol. Biol. Cell 2006, 17, 1096–1109. [Google Scholar]
- Lauvrak, S.U.; Torgersen, M.L.; Sandvig, K. Efficient endosome-to-Golgi transport of Shiga toxin is dependent on dynamin and clathrin. J. Cell Sci. 2004, 117, 2321–2331. [Google Scholar]
- Falguières, T.; Mallard, F.; Baron, C.; Hanau, D.; Lingwood, C.; Goud, B.; Salamero, J.; Johannes, L. Targeting of Shiga toxin B-subunit to retrograde transport route in association with detergent-resistant membranes. Mol. Biol. Cell 2001, 12, 2453–2468. [Google Scholar]
- Mallard, F.; Antony, C.; Tenza, D.; Salamero, J.; Goud, B.; Johannes, L. Direct pathway from early/recycling endosomes to the Golgi apparatus revealed through the study of Shiga toxin B-fragment transport. J. Cell Biol. 1998, 143, 973–990. [Google Scholar]
- Natarajan, R.; Linstedt, A.D. A cycling cis-Golgi protein mediates endosome-to-Golgi traffic. Mol. Biol. Cell 2004, 15, 4798–4806. [Google Scholar]
- Lombardi, P.; Mulder, M.; van der Boom, H.; Frants, R.R.; Havekes, L.M. Inefficient degradation of triglyceride-rich lipoprotein by HepG2 cells is due to a retarded transport to the lysosomal compartment. J. Biol. Chem. 1993, 268, 26113–26119. [Google Scholar]
- Riederer, M.A.; Soldati, T.; Shapiro, A.D.; Lin, J.; Pfeffer, S.R. Lysosome biogenesis requires Rab9 function and receptor recycling from endosomes to the trans-Golgi network. J. Cell Biol. 1994, 125, 573–582. [Google Scholar]
- Miwako, I.; Yamamoto, A.; Kitamura, T.; Nagayama, K.; Ohashi, M. Cholesterol requirement for cation-independent mannose 6-phosphate receptor exit from multivesicular late endosomes to the Golgi. J. Cell Sci. 2001, 114, 1765–1776. [Google Scholar]
- Sandvig, K. Transport of toxins across intracellular membranes. In Bacterial Protein Toxins; Burns, D.L., Barbieri, J.T., Iglewski, B.H., Rappuoli, R., Eds.; ASM Press: Washington, DC, USA, 2003; pp. 157–172. [Google Scholar]
- Saint-Pol, A.; Yelamos, B.; Amessou, M.; Mills, I.G.; Dugast, M.; Tenza, D.; Schu, P.; Antony, C.; McMahon, H.T.; Lamaze, C.; Johannes, L. Clathrin adaptor epsinR is required for retrograde sorting on early endosomal membranes. Dev. Cell 2004, 6, 525–538. [Google Scholar]
- Chen, Y.A.; Scheller, R.H. SNARE-mediated membrane fusion. Nat. Rev. Mol. Cell Biol. 2001, 2, 98–106. [Google Scholar]
- Johannes, L.; Goud, B. Facing inward from compartment shores: How many pathways were we looking for? Traffic 2000, 1, 119–123. [Google Scholar] [CrossRef] [PubMed]
- Del Nery, E.; Miserey-Lenkei, S.; Falguières, T.; Nizak, C.; Johannes, L.; Perez, F.; Goud, B. Rab6A and Rab6A' GTPases play non-overlapping roles in membrane trafficking. Traffic 2006, 7, 394–407. [Google Scholar]
- Mallard, F.; Tang, B.L.; Galli, T.; Tenza, D.; Saint-Pol, A.; Yue, X.; Antony, C.; Hong, W.; Goud, B.; Johannes, L. Early/recycling endosomes-to-TGN transport involves two SNARE complexes and a Rab6 isoform. J. Cell Biol. 2002, 156, 653–664. [Google Scholar]
- Popoff, V.; Mardones, G.A.; Tenza, D.; Rojas, R.; Lamaze, C.; Bonifacino, J.S.; Raposo, G.; Johannes, L. The retromer complex and clathrin define an early endosomal retrograde exit site. J. Cell Sci. 2007, 120, 2022–2031. [Google Scholar]
- Bujny, M.V.; Popoff, V.; Johannes, L.; Cullen, P.J. The retromer component sorting nexin-1 is required for efficient retrograde transport of Shiga toxin from early endosome to the trans Golgi network. J. Cell Sci. 2007, 120, 2010–2021. [Google Scholar]
- Utskarpen, A.; Slagsvold, H.H.; Dyve, A.B.; Skanland, S.S.; Sandvig, K. SNX1 and SNX2 mediate retrograde transport of Shiga toxin. Biochem. Biophys. Res. Commun. 2007, 358, 566–570. [Google Scholar]
- Jackson, M.E.; Simpson, J.C.; Girod, A.; Pepperkok, R.; Roberts, L.M.; Lord, J.M. The KDEL retrieval system is exploited by Pseudomonas exotoxin A, but not by Shiga-like toxin-1, during retrograde transport from the Golgi complex to the endoplasmic reticulum. J. Cell Sci. 1999, 112, 467–475. [Google Scholar]
- White, J.; Johannes, L.; Mallard, F.; Girod, A.; Grill, S.; Reinsch, S.; Keller, P.; Tzschaschel, B.; Echard, A.; Goud, B.; Stelzer, E.H. Rab6 coordinates a novel Golgi to ER retrograde transport pathway in live cells. J. Cell Biol. 1999, 147, 743–760. [Google Scholar]
- Girod, A.; Storrie, B.; Simpson, J.C.; Johannes, L.; Goud, B.; Roberts, L.M.; Lord, J.M.; Nilsson, T.; Pepperkok, R. Evidence for a COP-I-independent transport route from the Golgi complex to the endoplasmic reticulum. Nat. Cell Biol. 1999, 1, 423–430. [Google Scholar]
- Sandvig, K.; Garred, Ø.; Prydz, K.; Kozlov, J.V.; Hansen, S.H.; van Deurs, B. Retrograde transport of endocytosed Shiga toxin to the endoplasmic reticulum. Nature 1992, 358, 510–512. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, A.; Doi, H.; Matsuzawa, F.; Aikawa, S.; Takiguchi, K.; Kawano, H.; Hayashida, M.; Ohno, S. Bcl-2 antiapoptotic protein mediates verotoxin II-induced cell death: possible association between Bcl-2 and tissue failure by E. coli O157:H7. Genes Dev. 2000, 14, 1734–1740. [Google Scholar] [PubMed]
- Brigotti, M.; Alfieri, R.; Sestili, P.; Bonelli, M.; Petronini, P.G.; Guidarelli, A.; Barbieri, L.; Stirpe, F.; Sperti, S. Damage to nuclear DNA induced by Shiga toxin 1 and ricin in human endothelial cells. FASEB J. 2002, 16, 365–372. [Google Scholar]
- Schiavo, G.; van der Goot, F.G. The bacterial toxin toolkit. Nat. Rev. Mol. Cell Biol. 2001, 2, 530–537. [Google Scholar]
- Roy, C.R. Exploitation of the endoplasmic reticulum by bacterial pathogens. Trends Microbiol. 2002, 10, 418–424. [Google Scholar]
- Wernick, N.L.B.; Chinnapen, D.J.-F; Cho, J.A.; Lencer, W.I. Cholera toxin: an intracellular journey into the cytosol by way of the endoplasmic reticulum. Toxins 2010, 2, 310–325. [Google Scholar] [CrossRef]
- Kurmanova, A.; Llorente, A.; Polesskaya, A.; Garred, Ø.; Olsnes, S.; Kozlov, J.; Sandvig, K. Structural requirements for furin-induced cleavage and activation of Shiga toxin. Biochem. Biophys. Res. Commun. 2007, 357, 144–149. [Google Scholar] [CrossRef] [PubMed]
- Yu, M.; Haslam, D.B. Shiga toxin is transported from the endoplasmic reticulum following interaction with the luminal chaperone HEDJ/ERdj3. Infect. Immun. 2005, 73, 2524–2532. [Google Scholar]
- Falguières, T.; Johannes, L. Shiga toxin B-subunit binds to the chaperone BiP and the nucleolar protein B23. Biol. Cell 2006, 98, 125–134. [Google Scholar]
- Tam, P.J.; Lingwood, C.A. Membrane cytosolic translocation of verotoxin A1 subunit in target cells. Microbiology 2007, 153, 2700–2710. [Google Scholar]
- Iordanov, M.S.; Pribnow, D.; Magun, J.L.; Dinh, T.-H.; Pearson, J.A.; Chen, S.L.; Magun, B.E. Ribotoxic stress response: Activation of the stress-activated protein kinase JNK1 by inhibitors of the peptidyl transferase reaction and by sequence-specific RNA damage to the alpha-sarcin/ricin loop in the 28S rRNA. Mol. Cell. Biol. 1997, 17, 3373–3381. [Google Scholar]
- Cherla, R.P.; Lee, S.-Y.; Mees, P.L.; Tesh, V.L. Shiga toxin 1-induced cytokine production is mediated by MAP kinase pathways and translation initiation factor eIF4E in the macrophage-like THP-1 cell line. J. Leukoc. Biol. 2006, 79, 397–407. [Google Scholar]
- Smith, W.E.; Kane, A.V.; Campbell, S.T.; Acheson, D.W.K.; Cochran, B.H.; Thorpe, C.M. Shiga toxin 1 triggers a ribotoxic stress response leading to p38 and JNK activation and induction of apoptosis in intestinal epithelial cells. Infect. Immun. 2003, 71, 1497–1504. [Google Scholar]
- Foster, G.H.; Tesh, V.L. Shiga toxin 1-induced activation of c-Jun NH(2)-terminal kinase and p38 in the human monocytic cell line THP-1: Possible involvement in the production of TNF-alpha. J. Leukoc. Biol. 2002, 71, 107–114. [Google Scholar]
- Zhou, H.R.; Lau, A.S.; Pestka, J.J. Role of double-stranded RNA-activated protein kinase R (PKR) in deoxynivalenol-induced ribotoxic stress response. Toxicol. Sci. 2003, 74, 335–344. [Google Scholar]
- Jandhyala, D.M.; Ahluwalia, A.; Obrig, T.; Thorpe, C.M. ZAK: A MAP3Kinase that transduces Shiga toxin- and ricin-induced proinflammatory cytokine expression. Cell. Microbiol. 2008, 10, 1468–1477. [Google Scholar]
- Deng, X.; Xiao, L.; Lang, W.; Gao, F.; Ruvolo, P.; May, W.S. Novel role for JNK as a stress-activated Bcl2 kinase. J. Biol. Chem. 2001, 276, 23681–23688. [Google Scholar]
- De Chiara, G.; Marcocci, M.E.; Torcia, M.; Lucibello, M.; Rosini, P.; Bonini, P.; Higashimoto, Y.; Damonte, G.; Armirotti, A.; Amodei, S.; Palamara, A.T.; Russo, T.; Garaci, E.; Cozzolino, F. Bcl-2 Phosphorylation by p38 MAPK: Identification of target sites and biologic consequences. J. Biol. Chem. 2006, 281, 21353–21361. [Google Scholar]
- Lee, S.-Y.; Cherla, R.P.; Caliskan, I.; Tesh, V.L. Shiga toxin 1 induces apoptosis in the human myelogenous leukemia cell line THP-1 by a caspase-8-dependent, tumor necrosis factor receptor-independent mechanism. Infect. Immun. 2005, 73, 5115–5126. [Google Scholar]
- Tesh, V.L. Induction of apoptosis by Shiga toxins. Future Microbiol. 2010, 5, 431–453. [Google Scholar]
- Cameron, P.; Smith, S.J.; Giembycz, M.A.; Rotondo, D.; Plevin, R. Verotoxin activates mitogen-activated protein kinase in human peripheral blood monocytes: role in apoptosis and proinflammatory cytokine release. Br. J. Pharmacol. 2003, 140, 1320–1330. [Google Scholar]
- Harrison, L.M.; Cherla, R.P.; van den Hoogen, C.; van Haaften, W.C.E.; Lee, S.-Y.; Tesh, V.L. Comparative evaluation of apoptosis induced by Shiga toxin 1 and/or lipopolysaccharides in human monocytic and macrophage-like cells. Microb. Pathog. 2005, 38, 63–76. [Google Scholar]
- Jimbo, A.; Fujita, E.; Kouroku, Y.; Ohnishi, J.; Inohara, N.; Kuida, K.; Sakamaki, K.; Yonehara, S.; Momoi, T. ER stress induces caspase-8 activation, stimulating cytochrome c release and caspase-9 activation. Exp. Cell Res. 2003, 283, 156–166. [Google Scholar]
- Gething, M.J.; Sambrook, J. Protein folding in the cell. Nature 1992, 355, 33–45. [Google Scholar]
- Stirling, P.C.; Lundin, V.F.; Leroux, M.R. Getting a grip on non-native proteins. EMBO Rep. 2003, 4, 565–570. [Google Scholar]
- Feldman, D.E.; Frydman, J. Protein folding in vivo: The importance of molecular chaperones. Curr. Opin. Struct. Biol. 2000, 10, 26–33. [Google Scholar]
- Hatahet, F.; Ruddock, L.W.; Ahn, K.; Benham, A.; Craik, D.; Ellgaard, L.; Ferrari, D.; Ventura, S. Protein disulfide isomerase: A critical evaluation of its function in disulfide bond formation. Antioxid. Redox Signal. 2009, 11, 2807–2850. [Google Scholar]
- Ellgaard, L.; Helenius, A. Quality control in the endoplasmic reticulum. Nat. Rev. Mol. Cell Biol. 2003, 4, 181–191. [Google Scholar]
- Ron, D.; Walter, P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat. Rev. Mol. Cell Biol. 2007, 8, 519–529. [Google Scholar]
- Hendershot, L.M. The ER chaperone BiP is a master regulator of ER function. Mt. Sinai J. Med. 2004, 71, 289–297. [Google Scholar]
- Boyce, M.; Yuan, J. Cellular response to endoplasmic reticulum stress: A matter of life or death. Cell Death Differ. 2006, 13, 363–373. [Google Scholar]
- Schroder, M.; Kaufman, R.J. The mammalian unfolded protein response. Annu. Rev. Biochem. 2005, 74, 739–789. [Google Scholar]
- Szegezdi, E.; Logue, S.E.; Gorman, A.M.; Samali, A. Mediators of endoplasmic reticulum stress-induced apoptosis. EMBO Rep. 2006, 7, 880–885. [Google Scholar]
- Yoshida, H.; Matsui, T.; Yamamoto, A.; Okada, T.; Mori, K. XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell 2001, 107, 881–891. [Google Scholar]
- Calfon, M.; Zeng, H.; Urano, F.; Till, J.H.; Hubbard, S.R.; Harding, H.P.; Clark, S.G.; Ron, D. IRE1 couples endoplasmic reticulum load to secretory capacity by processing the XBP-1 mRNA. Nature 2002, 415, 92–96. [Google Scholar]
- Ye, J.; Rawson, R.B.; Komuro, R.; Chen, X.; Dave, U.P.; Prywes, R.; Brown, M.S.; Goldstein, J.L. ER stress induces cleavage of membrane-bound ATF6 by the same proteases that process SREBPs. Mol. Cell 2000, 6, 1355–1364. [Google Scholar]
- Kadowaki, H.; Nishitoh, H.; Ichijo, H. Survival and apoptosis signals in ER stress: The role of protein kinases. J. Chem. Neuroanat. 2004, 28, 93–100. [Google Scholar]
- Joyce, M.A.; Walters, K.A.; Lamb, S.E.; Yeh, M.M.; Zhu, L.F.; Kneteman, N.; Doyle, J.S.; Katze, M.G.; Tyrrell, D.L. HCV induces oxidative and ER stress, and sensitizes infected cells to apoptosis in SCID/Alb-uPA mice. PLoS Pathog. 2009, 5, e1000291. [Google Scholar]
- Lee, S.-Y.; Lee, M.-S.; Cherla, R.P.; Tesh, V.L. Shiga toxin 1 induces apoptosis through the endoplasmic reticulum stress response in human monocytic cells. Cell. Microbiol. 2008, 10, 770–780. [Google Scholar]
- Marciniak, S.J.; Yun, C.Y.; Oyadomari, S.; Novoa, I.; Zhang, Y.; Jungreis, R.; Nagata, K.; Harding, H.P.; Ron, D. CHOP induces death by promoting protein synthesis and oxidation in the stressed endoplasmic reticulum. Genes Dev. 2004, 18, 3066–3077. [Google Scholar]
- Yamaguchi, H.; Wang, H.G. CHOP is involved in endoplasmic reticulum stress-induced apoptosis by enhancing DR5 expression in human carcinoma cells. J. Biol. Chem. 2004, 279, 45495–45502. [Google Scholar]
- McCullough, K.D.; Martindale, J.L.; Klotz, L.O.; Aw, T.Y.; Holbrook, N.J. Gadd153 sensitizes cells to endoplasmic reticulum stress by down-regulating Bcl2 and perturbing the cellular redox state. Mol. Cell. Biol. 2001, 21, 1249–1259. [Google Scholar]
- Paton, A.W.; Beddoe, T.; Thorpe, C.M.; Whisstock, J.C.; Wilce, M.C.; Rossjohn, J.; Talbot, U.M.; Paton, J.C. AB5 subtilase cytotoxin inactivates the endoplasmic reticulum chaperone BiP. Nature 2006, 443, 548–552. [Google Scholar]
- Wolfson, J.J.; May, K.L.; Thorpe, C.M.; Jandhyala, D.M.; Paton, J.C.; Paton, A.W. Subtilase cytotoxin activates PERK, IRE1 and ATF6 endoplasmic reticulum stress-signalling pathways. Cell. Microbiol. 2008, 10, 1775–1786. [Google Scholar]
- Shi, Y.; Porter, K.; Parameswaran, N.; Bae, H.K.; Pestka, J.J. Role of GRP78/BiP degradation and ER stress in deoxynivalenol–induced interleukin-6 upregulation in the macrophage. Toxicol. Sci. 2009, 109, 247–255. [Google Scholar]
- McConkey, D.J.; Orrenius, S. The role of calcium in the regulation of apoptosis. Biochem. Biophys. Res. Commun. 1997, 239, 357–366. [Google Scholar]
- Pinton, P.; Giorgi, C.; Siviero, R.; Zecchini, E.; Rizzuto, R. Calcium and apoptosis: ER-mitochondria Ca2+ transfer in the control of apoptosis. Oncogene 2008, 27, 6407–6418. [Google Scholar]
- Filippin, L.; Magalhaes, P.J.; Di Benedetto, G.; Colella, M.; Pozzan, T. Stable interactions between mitochondria and endoplasmic reticulum allow rapid accumulation of calcium in a subpopulation of mitochondria. J. Biol. Chem. 2003, 278, 39224–39234. [Google Scholar]
- Boya, P.; Cohen, I.; Zamzami, N.; Vieira, H.L.; Kroemer, G. Endoplasmic reticulum stress-induced cell death requires mitochondrial membrane permeabilization. Cell Death Differ. 2002, 9, 465–467. [Google Scholar]
- Gil-Parrado, S.; Fernandez-Montalvan, A.; Assfalg-Machleidt, I.; Popp, O.; Bestvater, F.; Holloschi, A.; Knoch, T.A.; Auerswald, E.A.; Welsh, K.; Reed, J.C.; Fritz, H.; Fuentes-Prior, P.; Spiess, E.; Salvesen, G.S.; Machleidt, W. Ionomycin-activated calpain triggers apoptosis. A probable role for Bcl-2 family members. J. Biol. Chem. 2002, 277, 27217–27226. [Google Scholar] [PubMed]
- Cherla, R.P.; Lee, S.-Y.; Mulder, R.A.; Lee, M.-S.; Tesh, V.L. Shiga toxin 1-induced proinflammatory cytokine production is regulated by the phosphatidylinositol 3-kinase/Akt/mammalian target of rapamycin signaling pathway. Infect. Immun. 2009, 77, 3919–3931. [Google Scholar]
- Lee, S.-Y.; Cherla, R.P.; Tesh, V.L. Simultaneous induction of apoptotic and survival signaling pathways in macrophage-like THP-1 cells by Shiga toxin 1. Infect. Immun. 2007, 75, 1291–1302. [Google Scholar]
- Lee, M.-S.; Cherla, R.P.; Leyva-Illades, D.; Tesh, V.L. Bcl-2 regulates the onset of Shiga toxin 1-induced apoptosis in THP-1 cells. Infect. Immun. 2009, 77, 5233–5344. [Google Scholar]
- Youle, R.J.; Strasser, A. The BCL-2 protein family: Opposing activities that mediate cell death. Nat. Rev. Mol. Cell Biol. 2008, 9, 47–59. [Google Scholar]
- Giam, M.; Huang, D.C.; Bouillet, P. BH3-only proteins and their roles in programmed cell death. Oncogene 2008, 27 (Suppl. 1), S128–S136. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.-S.; Cherla, R.P.; Lentz, E.K.; Leyva-Illades, D.; Tesh, V.L. Signaling through C/EBP homology protein and death receptor 5, and calpain activation differentially regulates THP-1 cell maturation-dependent apoptosis induced by Shiga toxin type 1. Infect. Immun. 2010. [Google Scholar]
© 2010 by the authors; licensee MDPI, Basel, Switzerland This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Lee, M.-S.; Cherla, R.P.; Tesh, V.L. Shiga Toxins: Intracellular Trafficking to the ER Leading to Activation of Host Cell Stress Responses. Toxins 2010, 2, 1515-1535. https://doi.org/10.3390/toxins2061515
Lee M-S, Cherla RP, Tesh VL. Shiga Toxins: Intracellular Trafficking to the ER Leading to Activation of Host Cell Stress Responses. Toxins. 2010; 2(6):1515-1535. https://doi.org/10.3390/toxins2061515
Chicago/Turabian StyleLee, Moo-Seung, Rama P. Cherla, and Vernon L. Tesh. 2010. "Shiga Toxins: Intracellular Trafficking to the ER Leading to Activation of Host Cell Stress Responses" Toxins 2, no. 6: 1515-1535. https://doi.org/10.3390/toxins2061515