Insulin Protects Hepatic Lipotoxicity by Regulating ER Stress through the PI3K/Akt/p53 Involved Pathway Independently of Autophagy Inhibition

Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemicals
2.2. Animal and Experimental Protocol
2.3. Animal Sample Analysis
2.4. Cell Culture
2.5. Cell Death Assays
2.6. ROS Detection
2.7. Analysis of RFP-LC3 Puncta
2.8. Western Blot Analysis
2.9. RNA Interference
2.10. Statistical Analysis
3. Results
Subsection
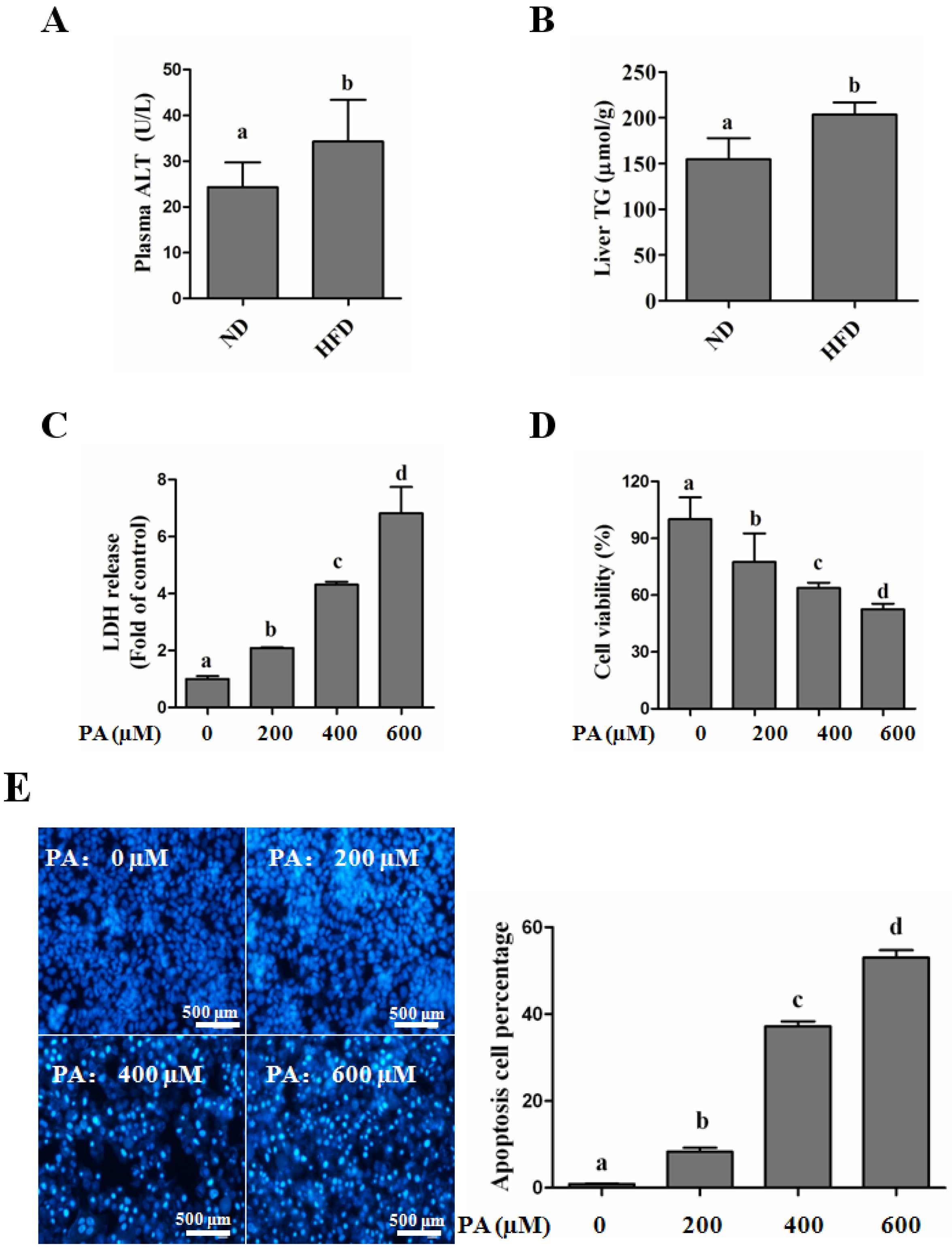
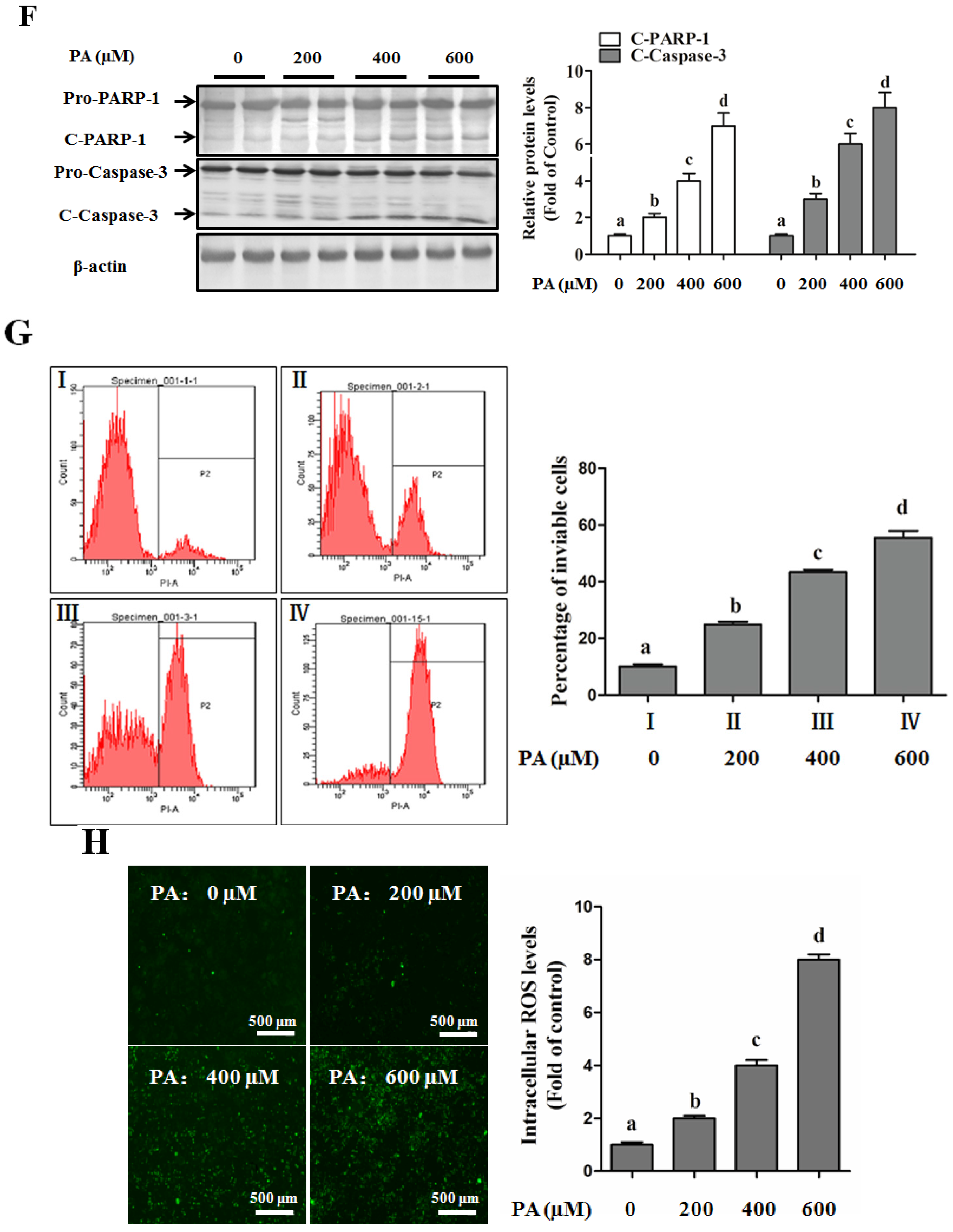
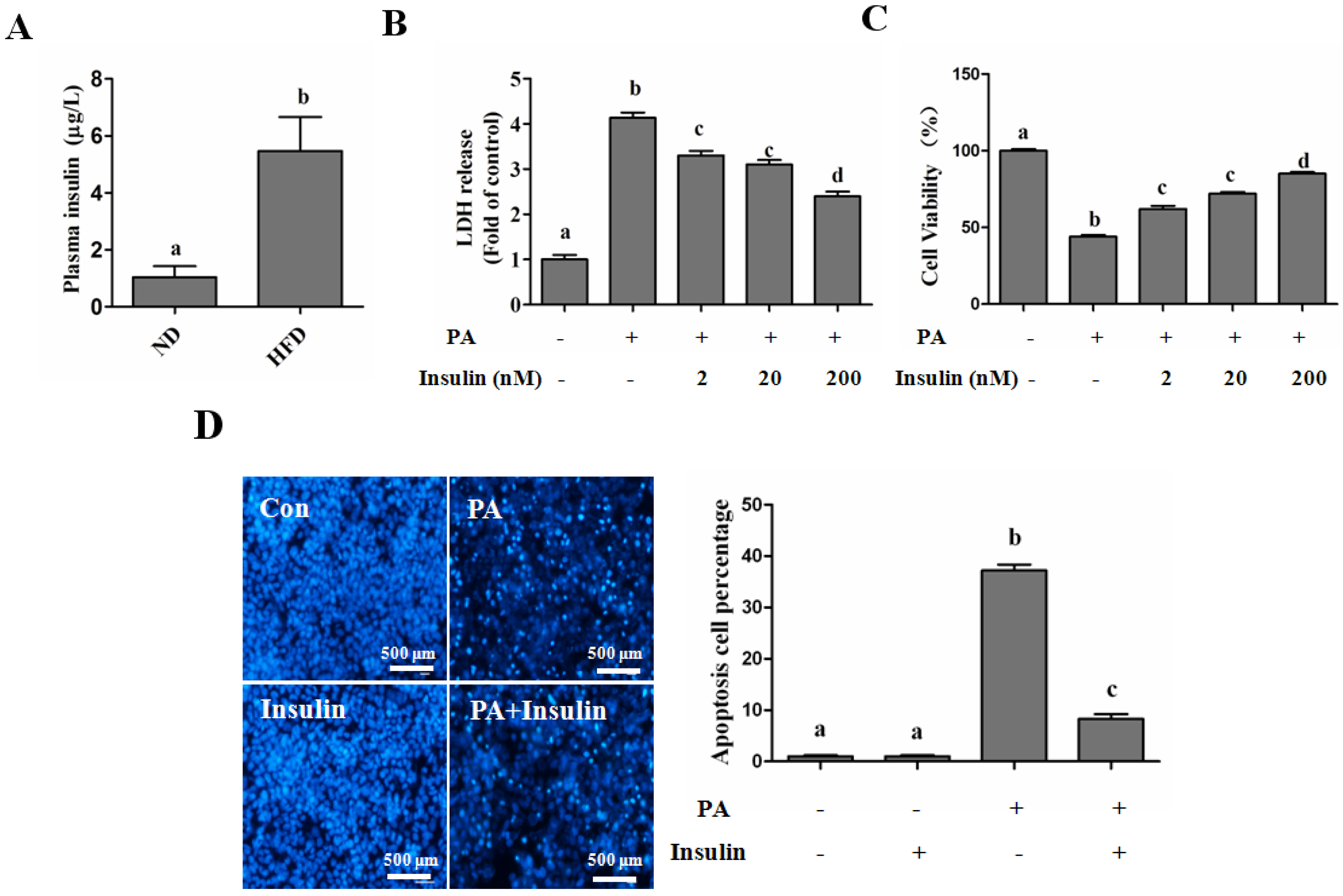
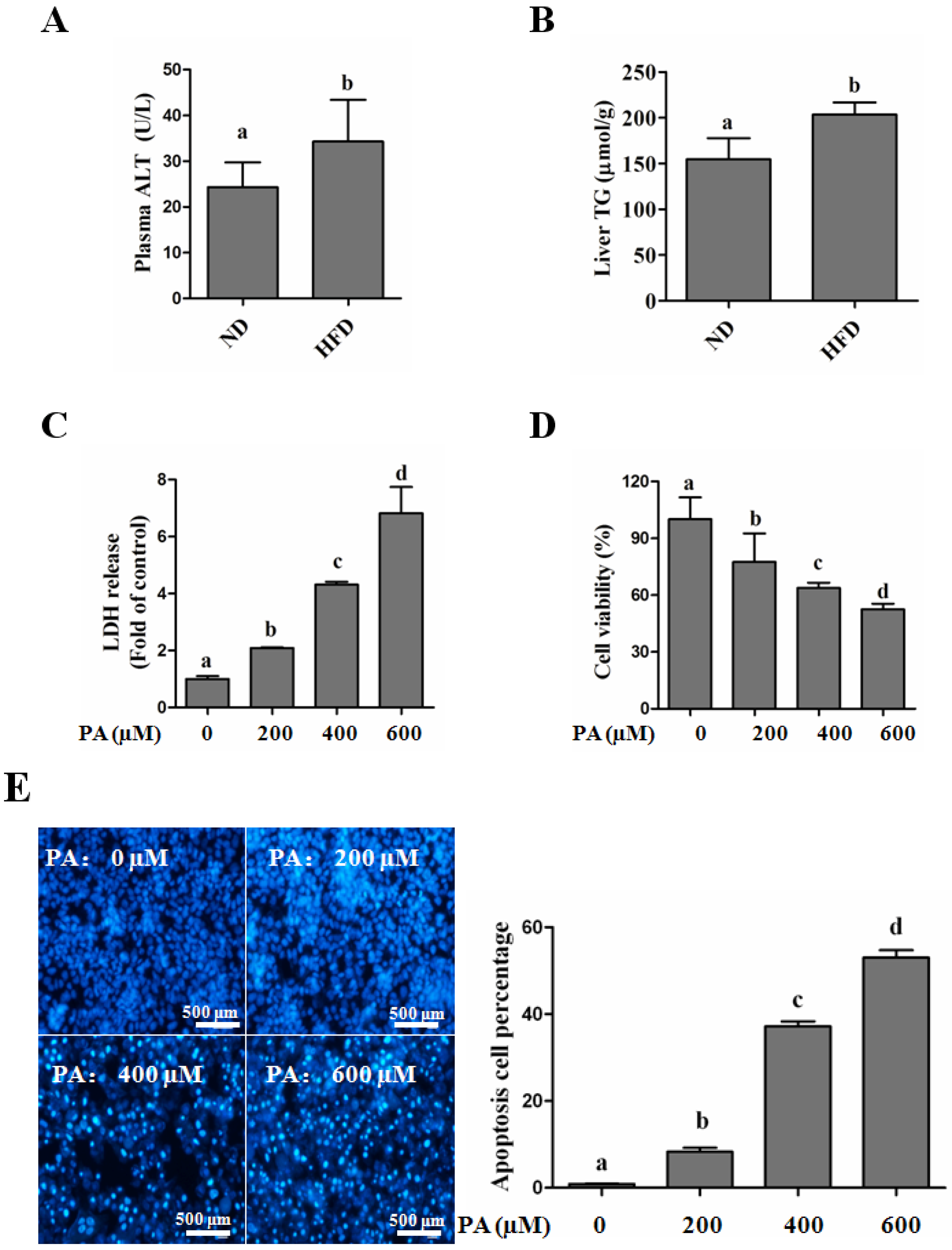
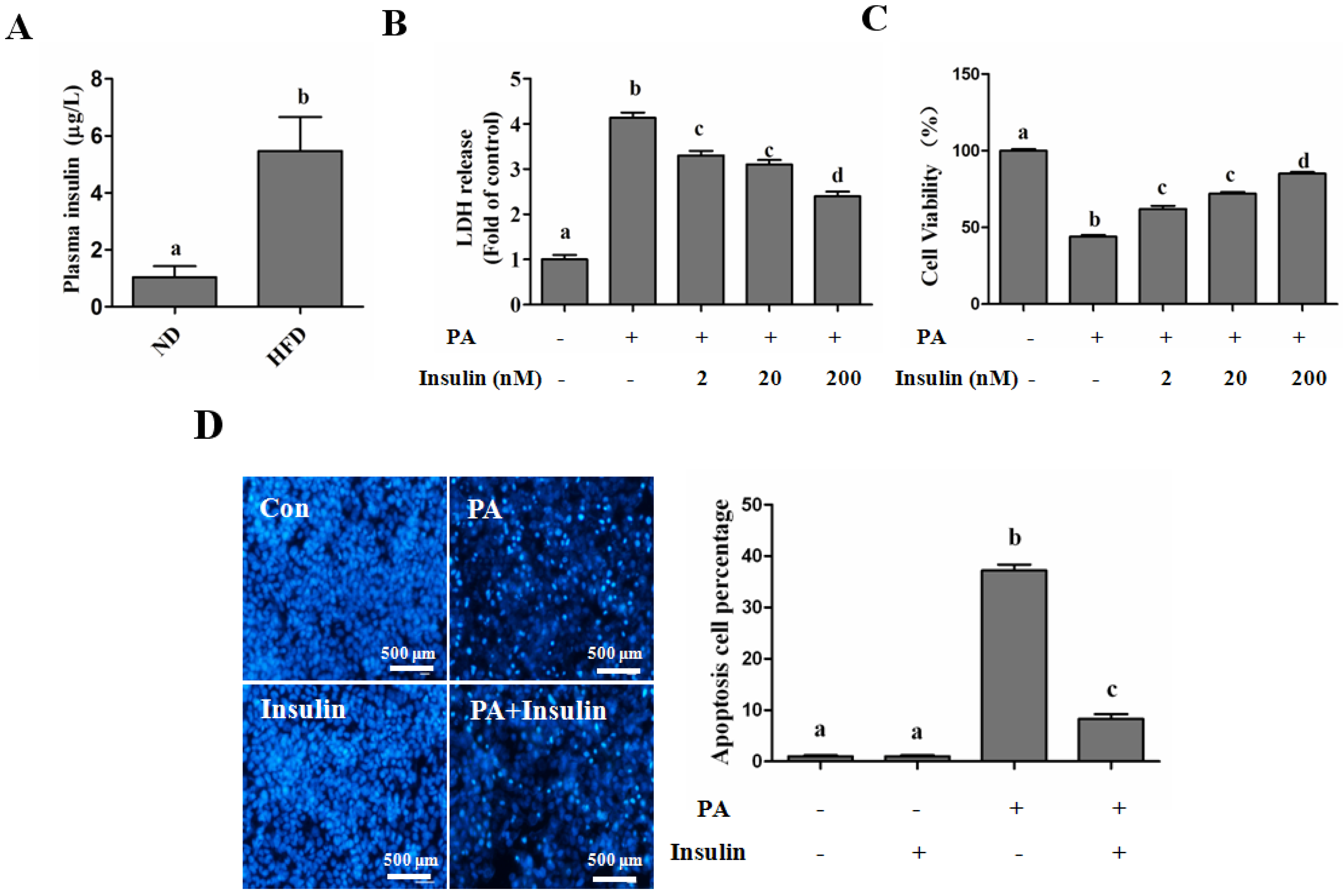
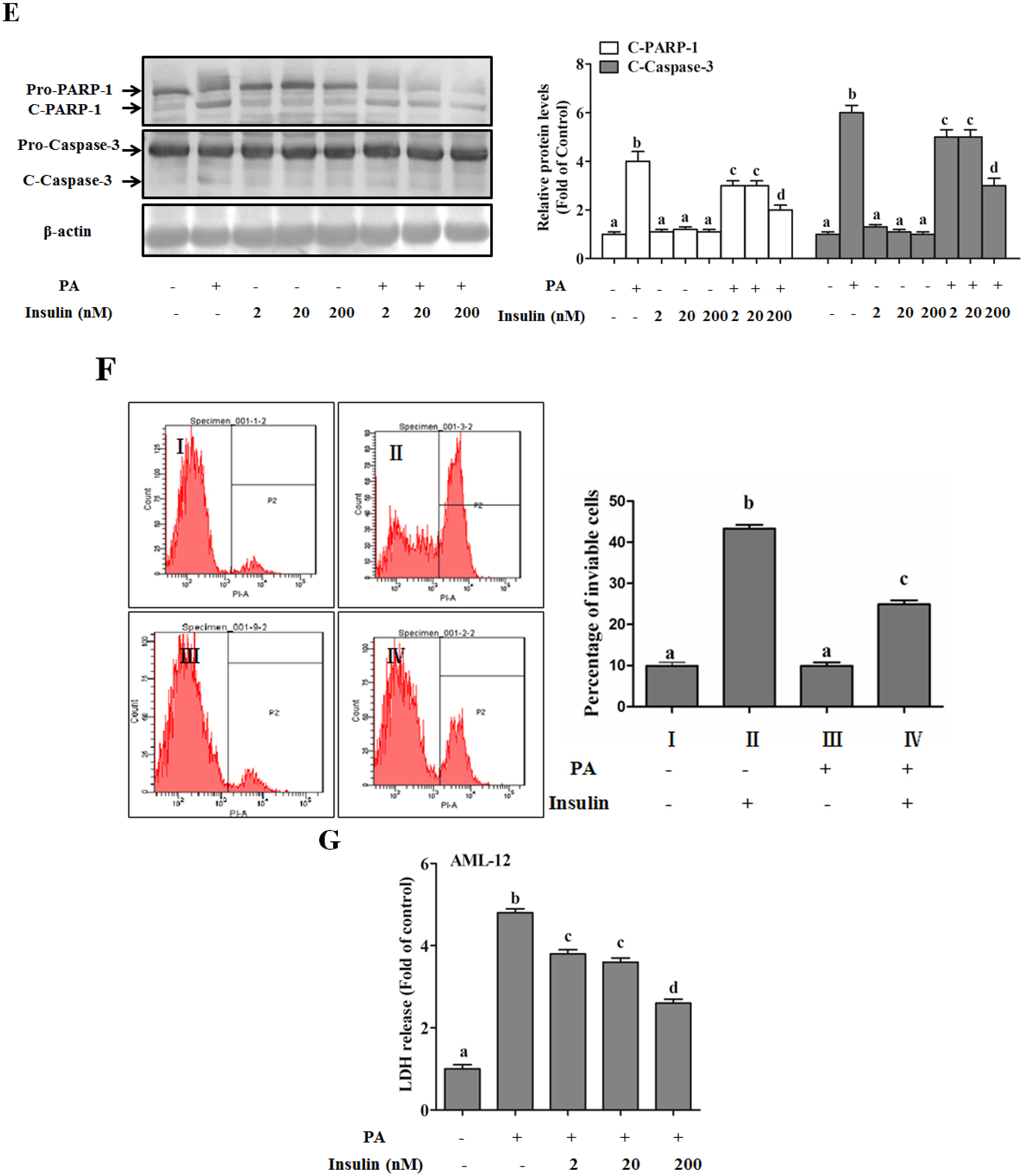
SFAs Induce Hepatotoxicity in Human Hepatocytes
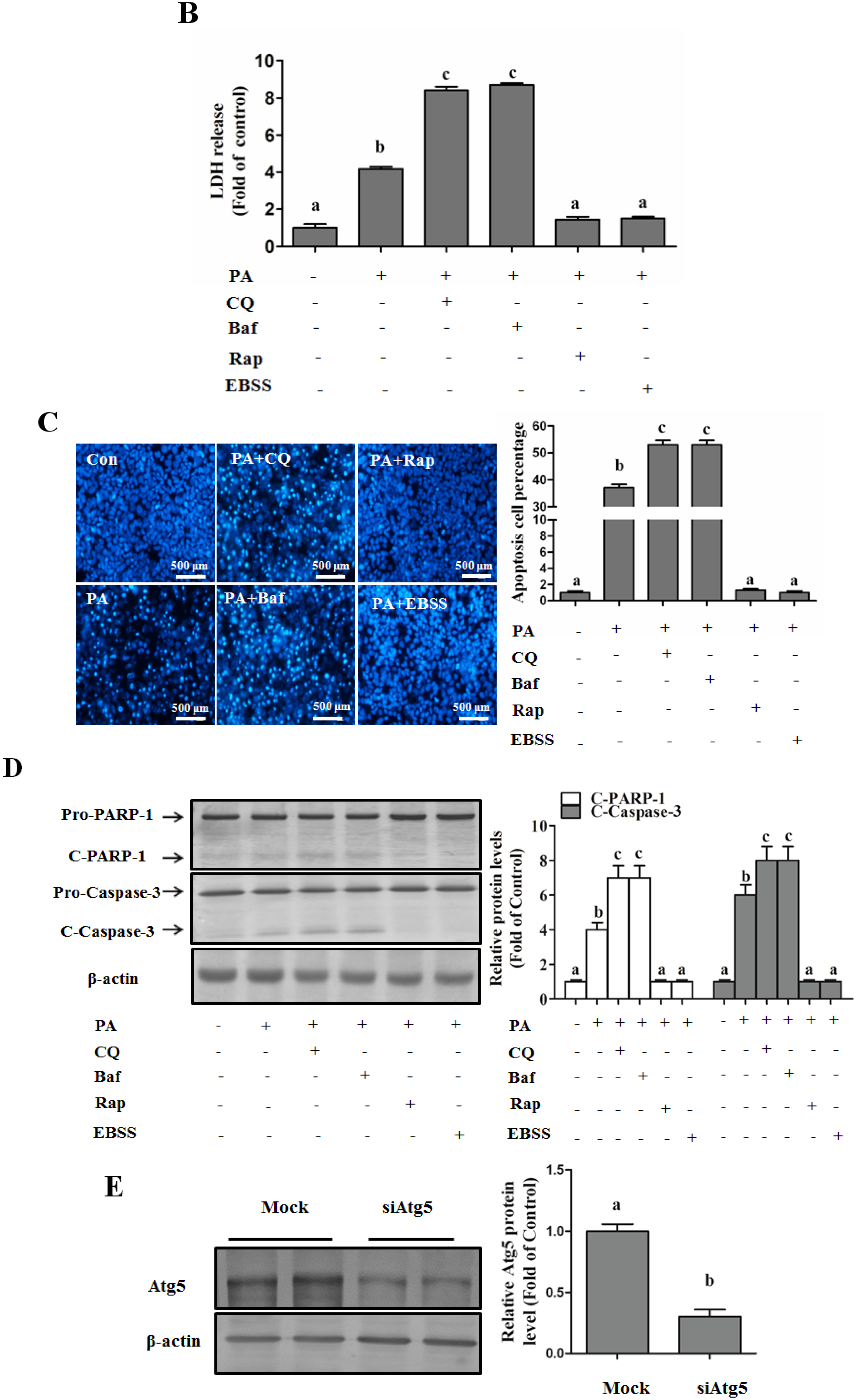
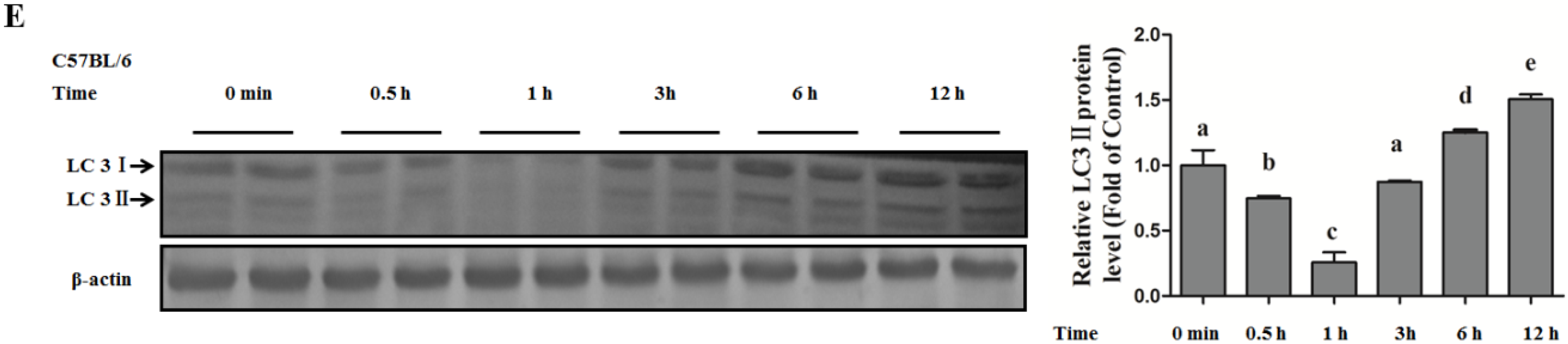
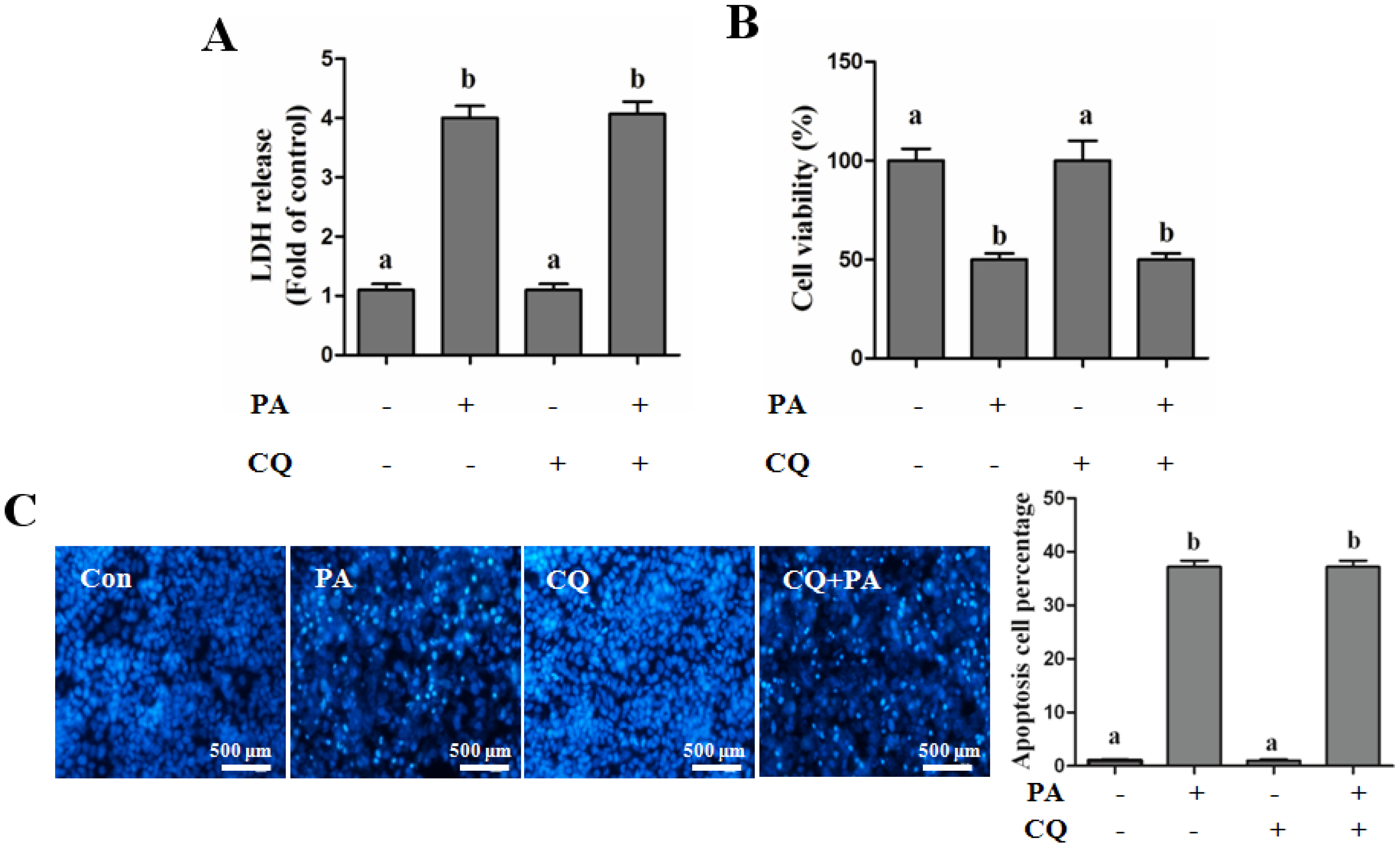
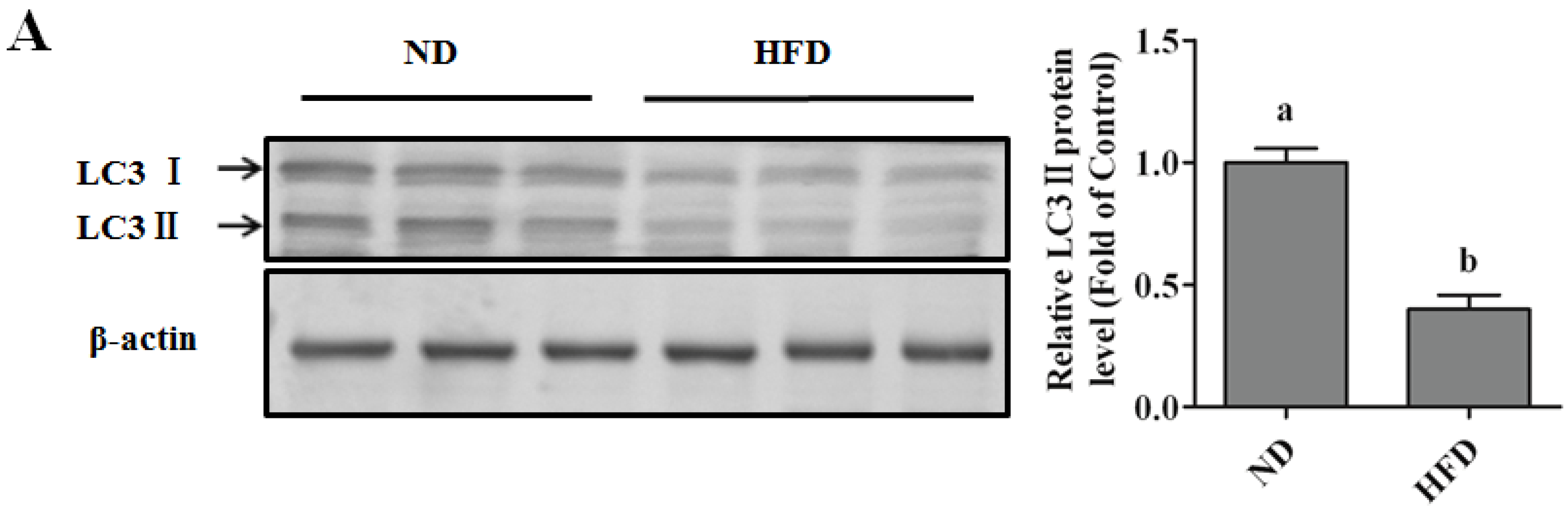
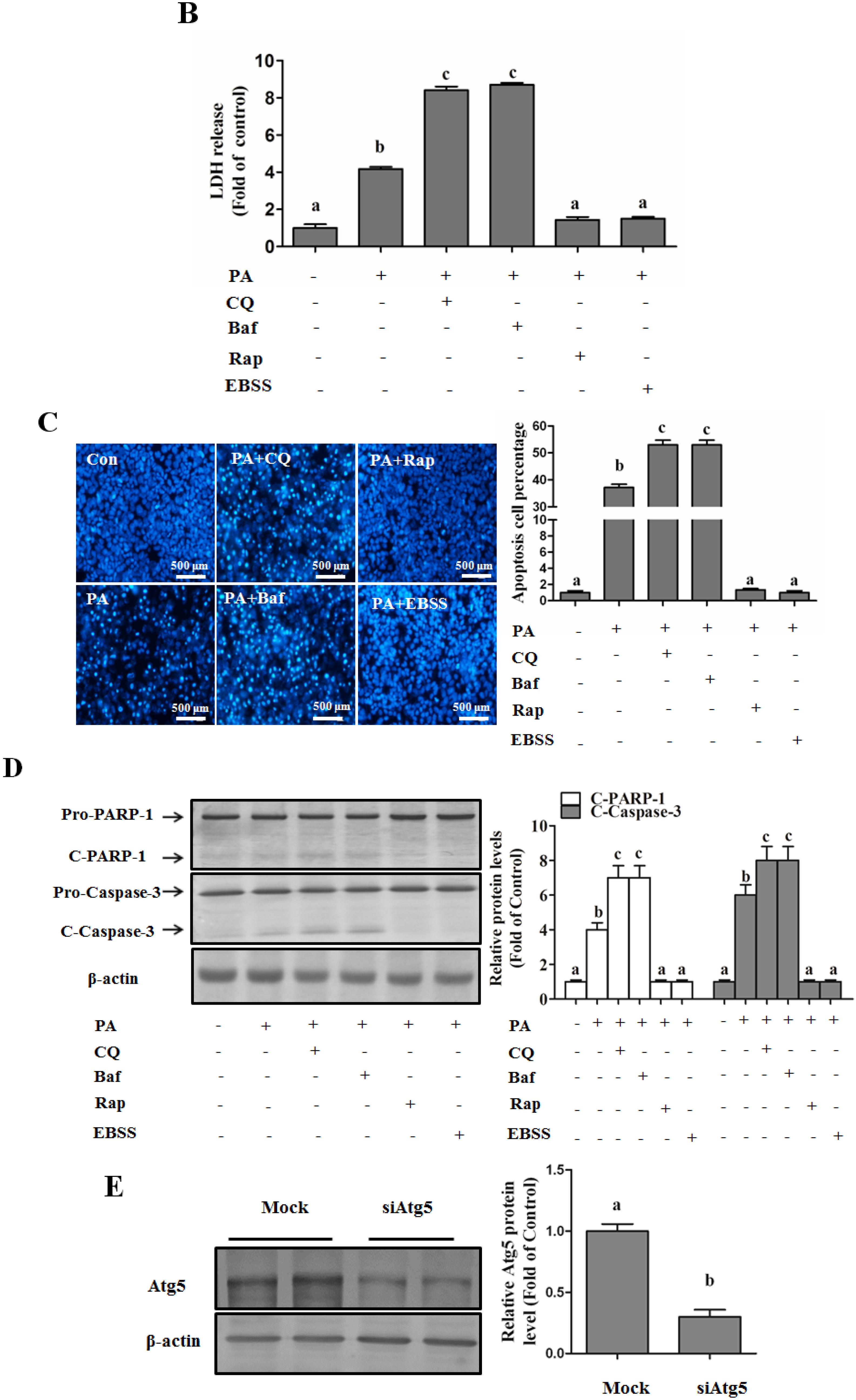
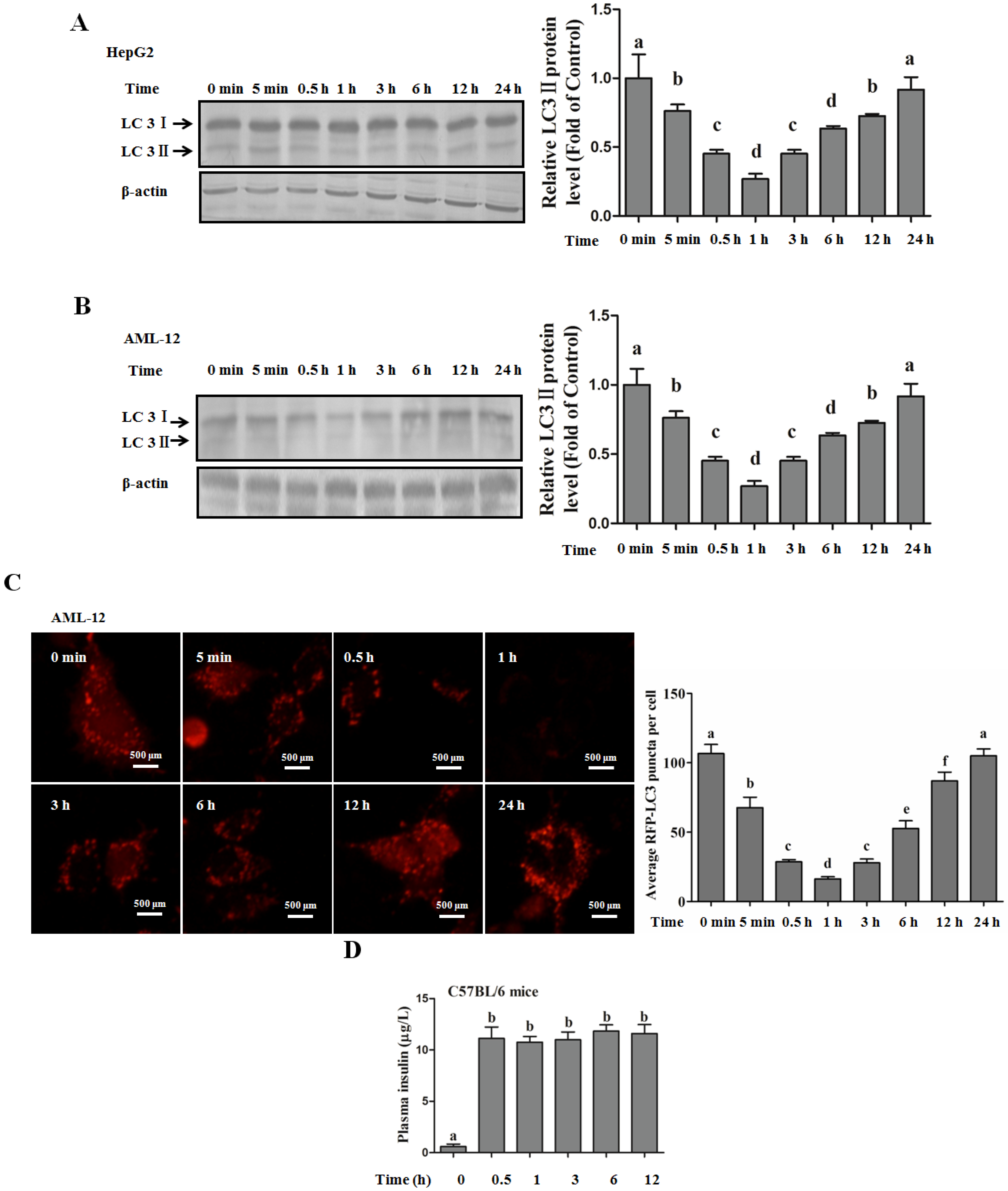
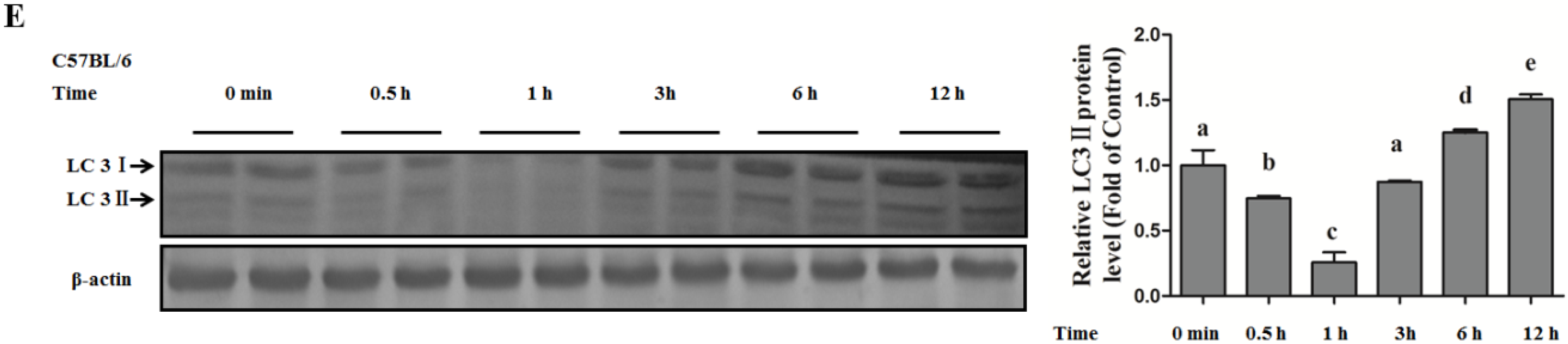
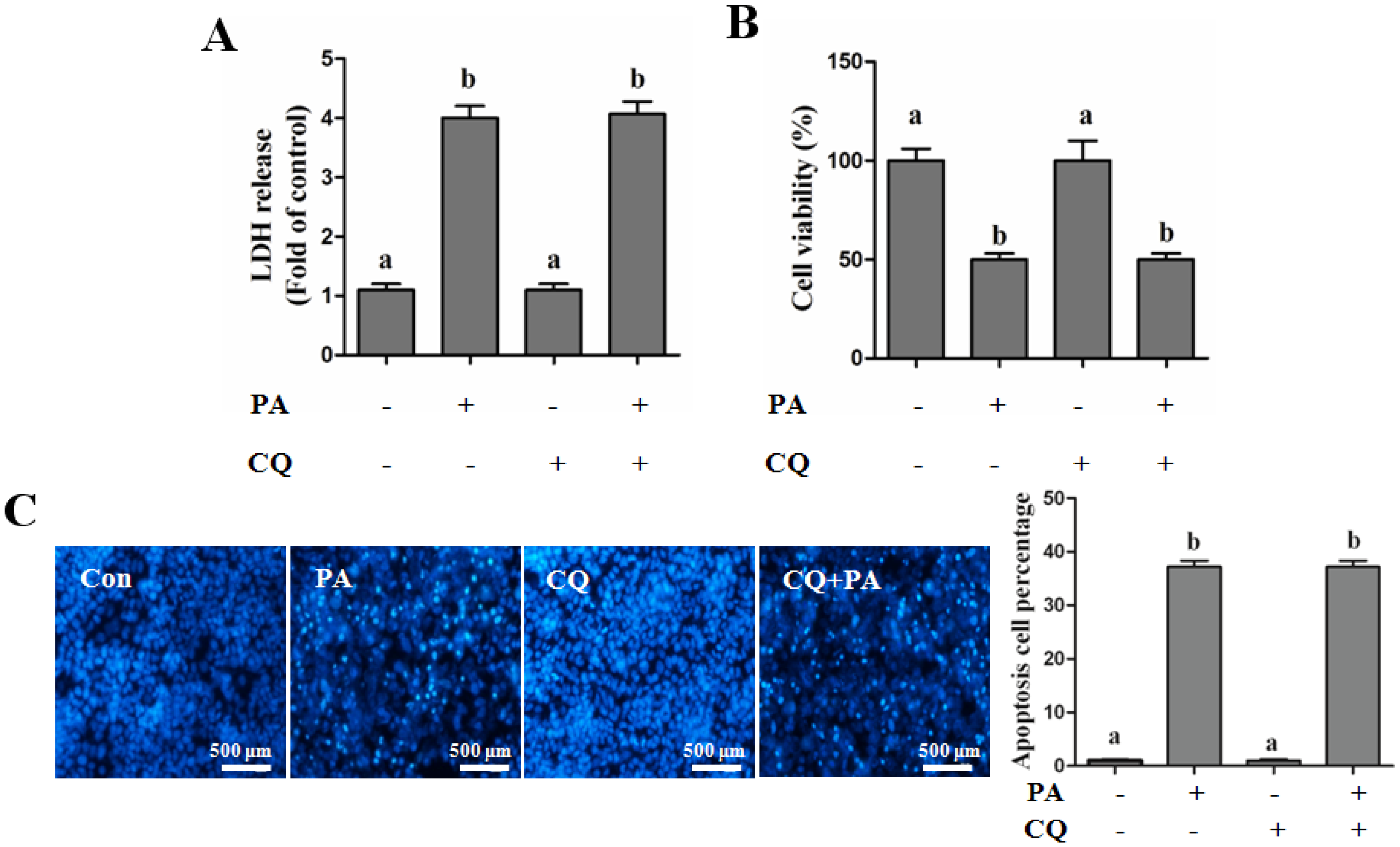
Autophagy Protects against Palmitate-Induced Lipotoxicity
Autophagy Protects against Palmitate-Induced Lipotoxicity
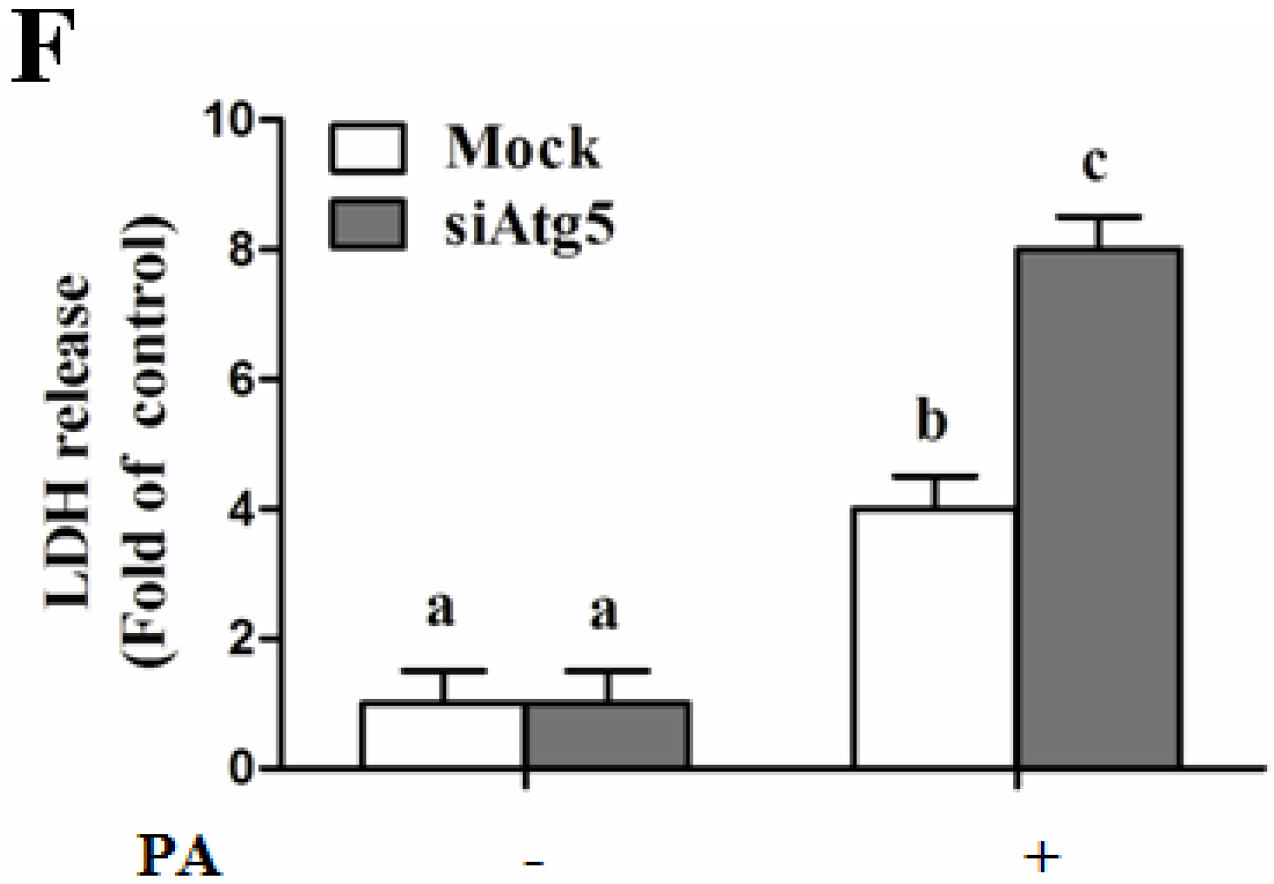
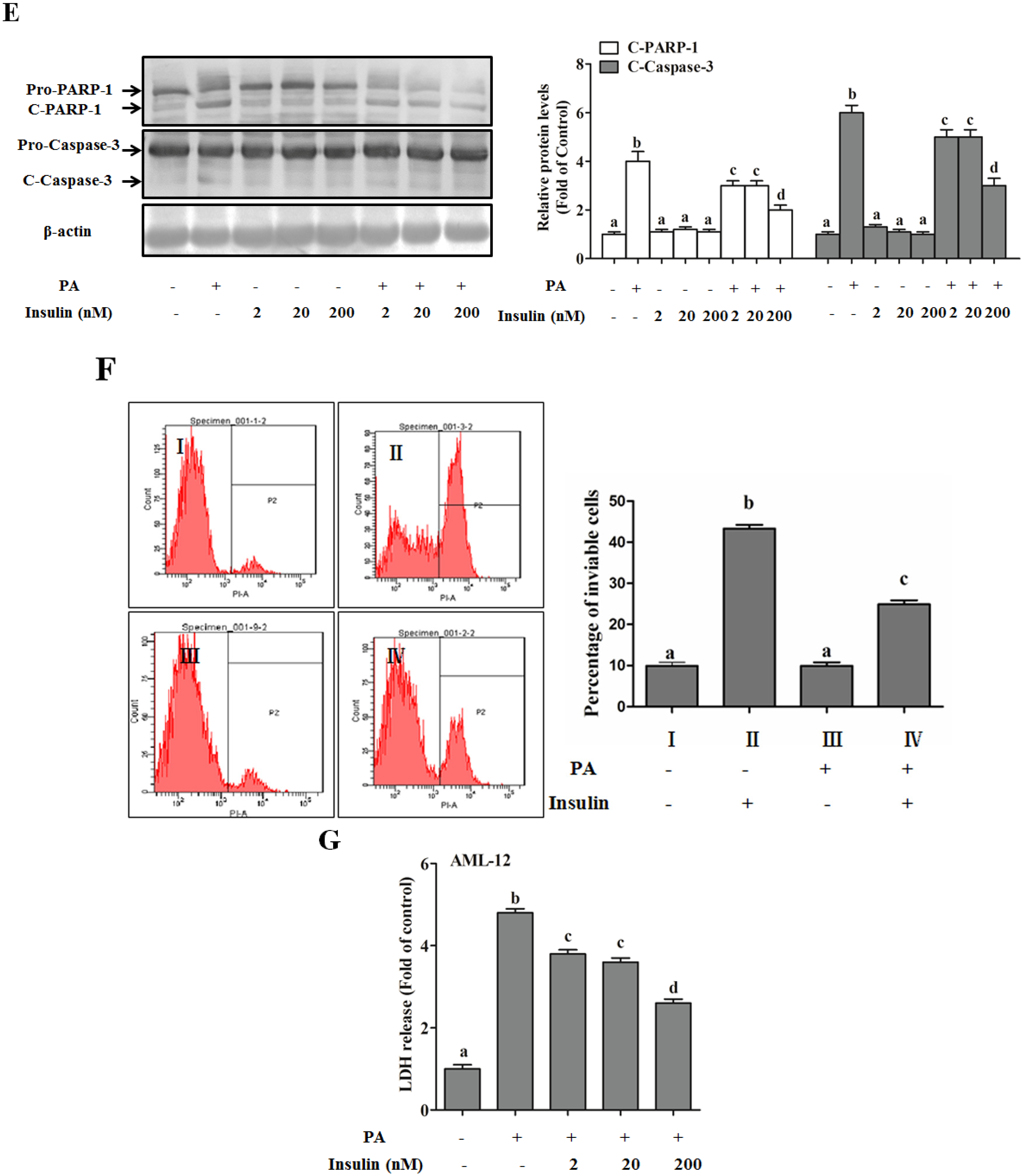
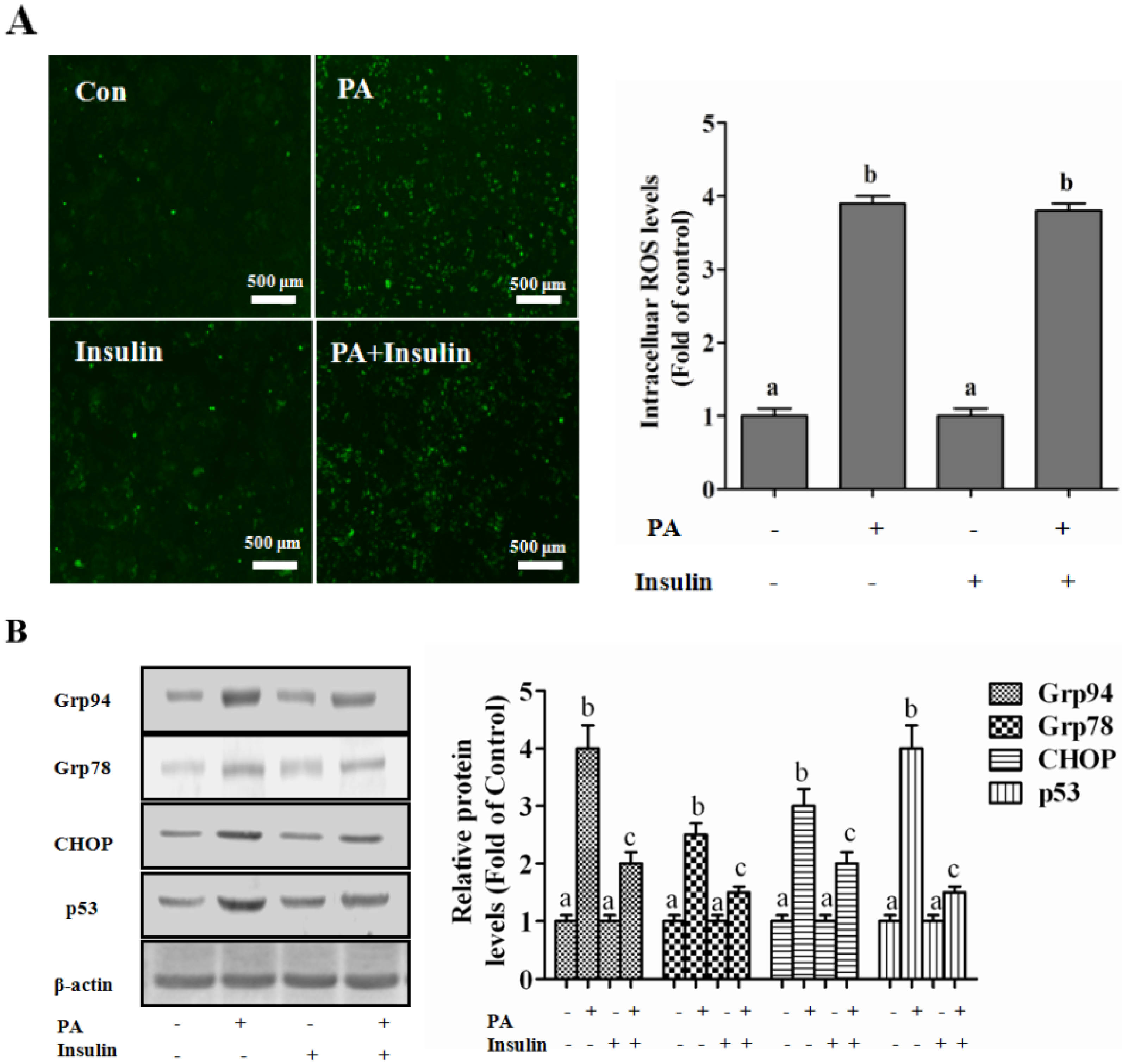
Autophagy Is Independent from Insulin-Protected Lipotoxicity in Hepatocytes
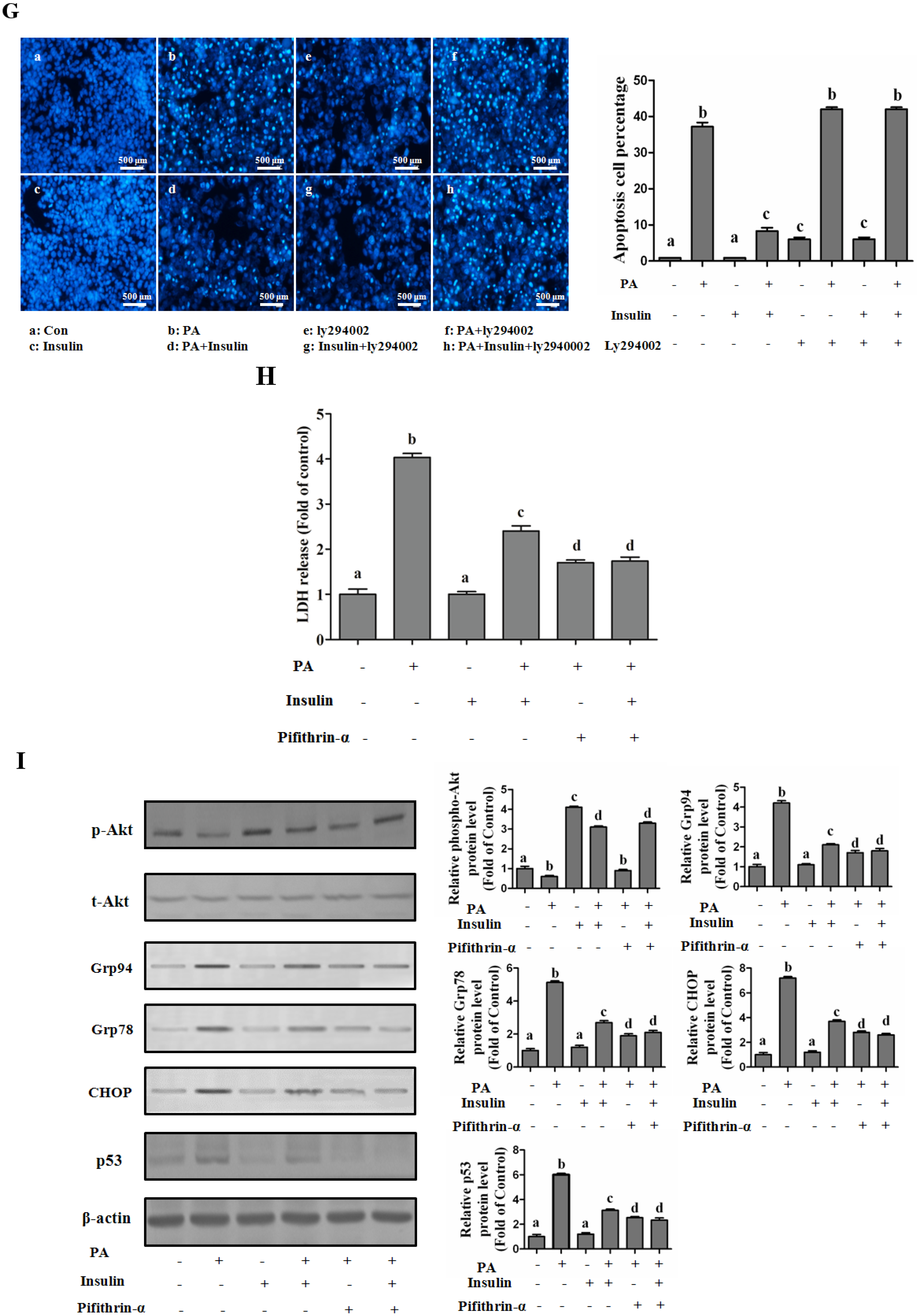
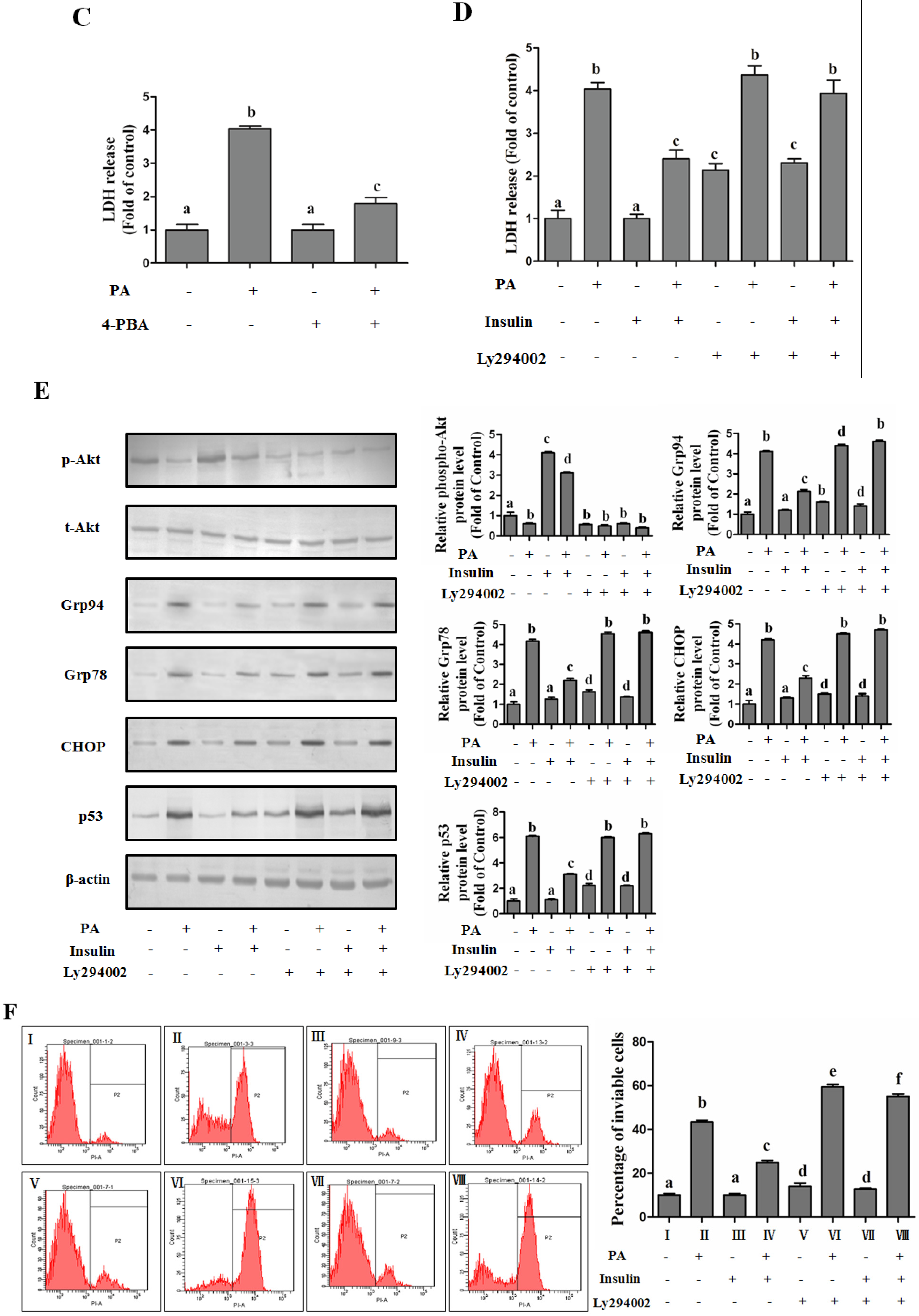
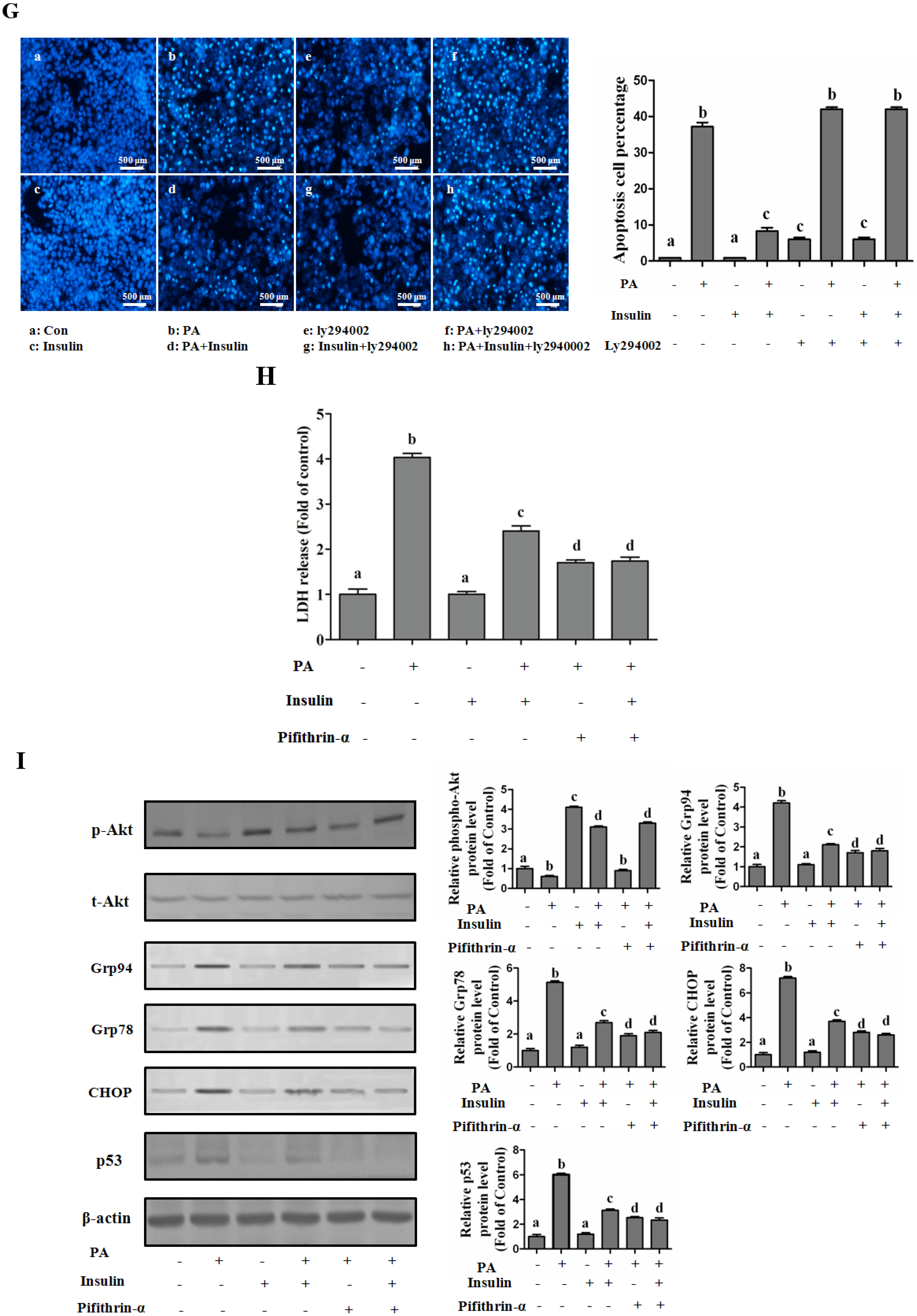
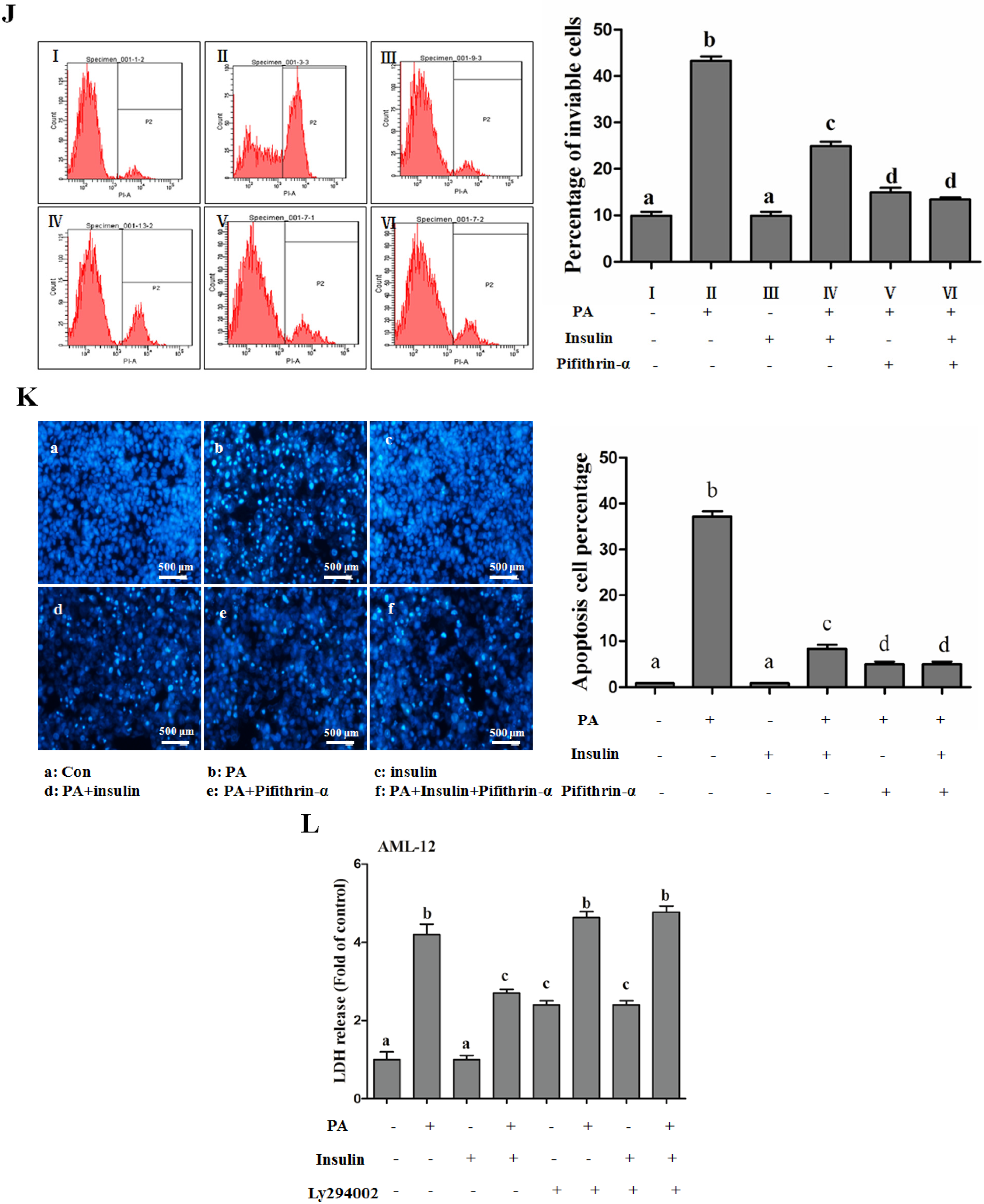
PI3K/Akt-Regulated p53 Pathway Contributes to Insulin-Protected Lipotoxicity
Promoting TG Synthesis Is Absent from the Hepatoprotective Role of Insulin
4. Discussion
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| AML | Alpha mouse liver |
| AMPK | AMP-activated protein kinase |
| Baf | Bafilomycin A1 |
| CQ | chloroquine |
| ER | Endoplasmic reticulum |
| EBSS | Earle’s Balanced Salt Solution |
| FFAs | Free fatty acids |
| HFD | High-fat diet group |
| HepG2 | Human liver carcinoma cell line |
| MUFAs | Monounsaturated fatty acids |
| mTOR | Mammalian target of rapamycin |
| NAFLD | Nonalcoholic fatty liver disease |
| NASH | Nonalcoholic steatohepatitis |
| ND | Normal diet group |
| PI3K | Phosphatidylinositol 3-kinase |
| PPARα | Peroxisome proliferator-activated receptors α |
| Rap | rapamycin |
| SFAs | Saturated fatty acids |
| SCD-1 | Stearoyl-CoA desaturase-1 |
| TG | Triglyceride |
| USFAs | Unsaturated fatty acids |
References
- Clark, J.M.; Diehl, A.M. Nonalcoholic fatty liver disease: An underrecognized cause of cryptogenic cirrhosis. JAMA 2003, 289, 3000–3004. [Google Scholar] [CrossRef] [PubMed]
- Machado, M.; Marques-Vidal, P.; Cortez-Pinto, H. Hepatic histology in obese patients undergoing bariatric surgery. J. Hepatol. 2006, 45, 600–606. [Google Scholar] [CrossRef] [PubMed]
- Trauner, M.; Arrese, M.; Wagner, M. Fatty liver and lipotoxicity. Biochim. Biophys. Acta 2010, 1801, 299–310. [Google Scholar] [CrossRef] [PubMed]
- Malhi, H.; Gores, G.J. Molecular mechanisms of lipotoxicity in nonalcoholic fatty liver disease. Semin. Liver Dis. 2008, 28, 360–369. [Google Scholar] [CrossRef] [PubMed]
- Alkhouri, N.; Dixon, L.J.; Feldstein, A.E. Lipotoxicity in nonalcoholic fatty liver disease: Not all lipids are created equal. Expert Rev. Gastroenterol. Hepatol. 2009, 3, 445–451. [Google Scholar] [CrossRef] [PubMed]
- Dioufa, N.; Chatzistamou, I.; Farmaki, E.; Papavassiliou, A.G.; Kiaris, H. P53 antagonizes the unfolded protein response and inhibits ground glass hepatocyte development during endoplasmic reticulum stress. Exp. Biol. Med. 2012, 237, 1173–1180. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Li, X.; Li, Y.; Li, N.; Shi, X.; Ding, H.; Zhang, Y.; Li, X.; Liu, G.; Wang, Z. Non-esterified fatty acids activate the ROS-p38-p53/Nrf2 signaling pathway to induce bovine hepatocyte apoptosis in vitro. Apoptosis 2014, 19, 984–997. [Google Scholar] [CrossRef] [PubMed]
- Gastaldelli, A.; Cusi, K.; Pettiti, M.; Hardies, J.; Miyazaki, Y.; Berria, R.; Buzzigoli, E.; Sironi, A.M.; Cersosimo, E.; Ferrannini, E.; et al. Relationship between hepatic/visceral fat and hepatic insulin resistance in nondiabetic and type 2 diabetic subjects. Gastroenterology 2007, 133, 496–506. [Google Scholar] [CrossRef] [PubMed]
- Taegtmeyer, H.; Stanley, W.C. Too much or not enough of a good thing? Cardiac glucolipotoxicity versus lipoprotection. J. Mol. Cell. Cardiol. 2011, 50, 2–5. [Google Scholar] [CrossRef] [PubMed]
- Sohn, J.; Siegelman, E.; Osiason, A. Unusual patterns of hepatic steatosis caused by the local effect of insulin revealed on chemical shift MR imaging. AJR 2001, 176, 471–474. [Google Scholar] [CrossRef] [PubMed]
- Frikke-Schmidt, H.; Pedersen, T.A.; Fledelius, C.; Olsen, G.S.; Hellerstein, M. Adipose weight gain during chronic insulin treatment of mice results from changes in lipid storage without affecting De Novo synthesis of palmitate. PLoS ONE 2013, 8, e76060. [Google Scholar]
- Kuma, A.; Hatano, M.; Matsui, M.; Yamamoto, A.; Nakaya, H.; Yoshimori, T.; Ohsumi, Y.; Tokuhisa, T.; Mizushima, N. The role of autophagy during the early neonatal starvation period. Nature 2004, 432, 1032–1036. [Google Scholar] [CrossRef] [PubMed]
- Meng, F.; Ning, H.; Sun, Z.; Huang, F.; Li, Y.; Chu, X.; Lu, H.; Sun, C.; Li, S. Ursolic acid protects hepatocytes against lipotoxicity through activating autophagy via an AMPK pathway. J. Funct. Foods 2015, 17, 172–182. [Google Scholar] [CrossRef]
- Sinha, R.A.; Farah, B.L.; Singh, B.K.; Siddique, M.M.; Li, Y.; Wu, Y.; Ilkayeva, O.R.; Gooding, J.; Ching, J.; Zhou, J.; et al. Caffeine stimulates hepatic lipid metabolism by the autophagy-lysosomal pathway in mice. Hepatology 2014, 59, 1366–1380. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.E.; Lee, S.M.; Lee, Y.J.; Li, L.J.; Lee, S.J.; Lee, J.H.; Kim, Y.; Jun, H.S.; Lee, K.W.; Kang, Y. Protective role of autophagy in palmitate-induced INS-1 beta-cell death. Endocrinology 2009, 150, 126–134. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Pan, X.Y.; Xu, Y.; Xiao, Y.; An, Y.; Tie, L.; Pan, Y.; Li, X.J. Curcumin induces autophagy to protect vascular endothelial cell survival from oxidative stress damage. Autophagy 2012, 8, 812–825. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Rodriguez, A.; Mayoral, R.; Agra, N.; Valdecantos, M.P.; Pardo, V.; Miquilena-Colina, M.E.; Vargas-Castrillon, J.; Lo Iacono, O.; Corazzari, M.; Fimia, G.M.; et al. Impaired autophagic flux is associated with increased endoplasmic reticulum stress during the development of nafld. Cell Death Dis. 2014, 5, e1179. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Li, J.; Shen, C.; Zhang, X.; Sun, S.; Cho, M.; Sun, C.; Song, Z. Tert-butylhydroquinone (tBHQ) protects hepatocytes against lipotoxicity via inducing autophagy independently of Nrf2 activation. Biochim. Biophys. Acta 2014, 1841, 22–33. [Google Scholar] [CrossRef] [PubMed]
- Ezaki, J.; Matsumoto, N.; Takeda-Ezaki, M.; Komatsu, M.; Takahashi, K.; Hiraoka, Y.; Taka, H.; Fujimura, T.; Takehana, K.; Yoshida, M.; et al. Liver autophagy contributes to the maintenance of blood glucose and amino acid levels. Autophagy 2011, 7, 727–736. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, J.L.; Basu, S.K.; Brown, M.S. Receptor-mediated endocytosis of low-density lipoprotein in cultured cells. Methods Enzymol. 1983, 98, 241–260. [Google Scholar] [PubMed]
- Liu, L.; Li, Y.; Guan, C.; Li, K.; Wang, C.; Feng, R.; Sun, C. Free fatty acid metabolic profile and biomarkers of isolated post-challenge diabetes and type 2 diabetes mellitus based on GC-MS and multivariate statistical analysis. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2010, 878, 2817–2825. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.J.; Youn, H.; Seong, K.M.; Yun, Y.J.; Kim, W.; Kim, Y.H.; Lee, J.Y.; Kim, C.S.; Jin, Y.W.; Youn, B. Psoralidin, a dual inhibitor of COX-2 and 5-LOX, regulates ionizing radiation (IR)-induced pulmonary inflammation. Biochem. Pharmacol. 2011, 82, 524–534. [Google Scholar] [CrossRef] [PubMed]
- Kimura, S.; Noda, T.; Yoshimori, T. Dissection of the autophagosome maturation process by a novel reporter protein, tandem fluorescent-tagged LC3. Autophagy 2007, 3, 452–460. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Li, Y.; Ning, H.; Na, L.; Niu, Y.; Wang, M.; Feng, R.; Liu, L.; Guo, F.; Hou, S.; et al. Calcium supplementation increases circulating cholesterol by reducing its catabolism via GPER and TRPC1-dependent pathway in estrogen deficient women. Int. J. Cardiol. 2013, 168, 2548–2560. [Google Scholar] [CrossRef] [PubMed]
- Gaemers, I.C.; Stallen, J.M.; Kunne, C.; Wallner, C.; van Werven, J.; Nederveen, A.; Lamers, W.H. Lipotoxicity and steatohepatitis in an overfed mouse model for non-alcoholic fatty liver disease. Biochim. Biophys. Acta 2011, 1812, 447–458. [Google Scholar] [CrossRef] [PubMed]
- Zeng, M.D. Glucotoxicity, lipotoxicity and the nonalcoholic fatty liver disease. Zhonghua Gan Zang Bing Za Zhi 2005, 13, 81–82. [Google Scholar] [PubMed]
- Browning, J.D.; Horton, J.D. Molecular mediators of hepatic steatosis and liver injury. J. Clin. Investig. 2004, 114, 147–152. [Google Scholar] [CrossRef] [PubMed]
- Hardwick, J.P.; Osei-Hyiaman, D.; Wiland, H.; Abdelmegeed, M.A.; Song, B.J. PPAR/RXR regulation of fatty acid metabolism and fatty acid omega-hydroxylase (CYP4) isozymes: Implications for prevention of lipotoxicity in fatty liver disease. PPAR Res. 2009, 2009, 952734. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N. The pleiotropic role of autophagy: From protein metabolism to bactericide. Cell Death Differ. 2005, 12 (Suppl. 2), 1535–1541. [Google Scholar] [CrossRef] [PubMed]
- Bergamini, E.; Cavallini, G.; Donati, A.; Gori, Z. The role of autophagy in aging: Its essential part in the anti-aging mechanism of caloric restriction. Ann. N. Y. Acad. Sci. 2007, 1114, 69–78. [Google Scholar] [CrossRef] [PubMed]
- Hansen, T.E.; Johansen, T. Following autophagy step by step. BMC Biol. 2011, 9, 39. [Google Scholar] [CrossRef] [PubMed]
- Cai, N.; Zhao, X.; Jing, Y.; Sun, K.; Jiao, S.; Chen, X.; Yang, H.; Zhou, Y.; Wei, L. Autophagy protects against palmitate-induced apoptosis in hepatocytes. Cell Biosci. 2014, 4, 28. [Google Scholar] [CrossRef] [PubMed]
- Eltahir, E.M.; El Ghazali, G.; TM, A.E.; IE, A.E.; Elbashir, M.I.; Giha, H.A. Raised plasma insulin level and homeostasis model assessment (HOMA) score in cerebral malaria: Evidence for insulin resistance and marker of virulence. Acta Biochim. Pol. 2010, 57, 513–520. [Google Scholar] [PubMed]
- Pfutzner, A.; Raz, I.; Bitton, G.; Klonoff, D.; Nagar, R.; Hermanns, N.; Haak, T. Improved insulin absorption by means of standardized injection site modulation results in a safer and more efficient prandial insulin treatment. A review of the existing clinical data. J. Diabetes Sci. Technol. 2015, 9, 116–122. [Google Scholar] [CrossRef] [PubMed]
- Goldman, J.; White, J.R., Jr. New insulin glargine 300 U/mL for the treatment of type 1 and type 2 diabetes mellitus. Ann. Pharmacother. 2015, 49, 1153–1161. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.T.; Tan, H.L.; Huang, Q.; Ong, C.N.; Shen, H.M. Activation of the PI3K-Akt-mTOR signaling pathway promotes necrotic cell death via suppression of autophagy. Autophagy 2009, 5, 824–834. [Google Scholar] [CrossRef] [PubMed]
- Jia, G.; Cheng, G.; Gangahar, D.M.; Agrawal, D.K. Insulin-like growth factor-1 and TNF-alpha regulate autophagy through c-jun N-terminal kinase and AKt pathways in human atherosclerotic vascular smooth cells. Immunol. Cell Biol. 2006, 84, 448–454. [Google Scholar] [CrossRef] [PubMed]
- Naito, T.; Kuma, A.; Mizushima, N. Differential contribution of insulin and amino acids to the mTORC1-autophagy pathway in the liver and muscle. J. Biol. Chem. 2013, 288, 21074–21081. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.Y.; Han, J.; Cao, S.Y.; Hong, T.; Zhuo, D.; Shi, J.; Liu, Z.; Cao, W. Hepatic autophagy is suppressed in the presence of insulin resistance and hyperinsulinemia: Inhibition of FoxO1-dependent expression of key autophagy genes by insulin. J. Biol. Chem. 2009, 284, 31484–31492. [Google Scholar] [CrossRef] [PubMed]
- Sun, W.; Bi, Y.; Liang, H.; Cai, M.; Chen, X.; Zhu, Y.; Li, M.; Xu, F.; Yu, Q.; He, X.; et al. Inhibition of obesity-induced hepatic er stress by early insulin therapy in obese diabetic rats. Endocrine 2011, 39, 235–241. [Google Scholar] [CrossRef] [PubMed]
- Boden, G.; Cheung, P.; Salehi, S.; Homko, C.; Loveland-Jones, C.; Jayarajan, S.; Stein, T.; Williams, K.; Liu, M.; Barrero, C. Insulin regulates the unfolded protein response in human adipose tissue. Diabetes 2014, 63, 912–922. [Google Scholar] [CrossRef] [PubMed]
- Greineisen, W.E.; Maaetoft-Udsen, K.; Speck, M.; Balajadia, J.; Shimoda, L.M.; Sung, C.; Turner, H. Chronic insulin exposure induces ER stress and lipid body accumulation in mast cells at the expense of their secretory degranulation response. PLoS ONE 2015, 10, e0130198. [Google Scholar] [CrossRef] [PubMed]
- Pagliassotti, M.J.; Wei, Y.; Wang, D. Insulin protects liver cells from saturated fatty acid-induced apoptosis via inhibition of c-jun NH2 terminal kinase activity. Endocrinology 2007, 148, 3338–3345. [Google Scholar] [CrossRef] [PubMed]
- Ricchi, M.; Odoardi, M.R.; Carulli, L.; Anzivino, C.; Ballestri, S.; Pinetti, A.; Fantoni, L.I.; Marra, F.; Bertolotti, M.; Banni, S.; et al. Differential effect of oleic and palmitic acid on lipid accumulation and apoptosis in cultured hepatocytes. J. Gastroenterol. Hepatol. 2009, 24, 830–840. [Google Scholar] [CrossRef] [PubMed]
- Enoch, H.G.; Catala, A.; Strittmatter, P. Mechanism of rat liver microsomal stearyl-CoA desaturase. Studies of the substrate specificity, enzyme-substrate interactions, and the function of lipid. J. Biol. Chem. 1976, 251, 5095–5103. [Google Scholar] [PubMed]
- Li, Z.Z.; Berk, M.; McIntyre, T.M.; Feldstein, A.E. Hepatic lipid partitioning and liver damage in nonalcoholic fatty liver disease: Role of stearoyl-CoA desaturase. J. Biol. Chem. 2009, 284, 5637–5644. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Free Fatty Acids | ND (μmol/L) | HFD (μmol/L) |
|---|---|---|
| C14:0, MA (Myristic acid) | 6.76 ± 0.32 | 5.07 ± 0.08 |
| C16:0, PA (Palmitic acid) | 580.88 ± 0.88 | 730.11 ± 20.11 * |
| C16:1, PLA (Palmitoleic acid) | 73.14 ± 3.95 | 49.30 ± 1.39 * |
| C18:0, SA (Stearic acid) | 211.88 ± 4.16 | 383.58 ± 13.59 ** |
| C18:1, OA (Oleic acid) | 185.58 ± 8.47 | 461.37 ± 13.67 ** |
| C18:2, LA (Linoleic acid) | 716.22 ± 14.16 | 919.71 ± 28.19 * |
| γ-C18:3, γ-LNA (γ-Linolenic acid) | 23.18 ± 0.75 | 22.46 ± 0.81 |
| C18:3, LNA (Linoleic acid) | 7.60 ± 0.29 | 5.54 ± 0.18 * |
| C20:2, EDA (Eicosadienoic acid) | 5.04 ± 0.18 | 8.08 ± 0.26 |
| C20:4, AA (Arachidonic acid) | 383.16 ± 9.88 | 748.75 ± 24.72 ** |
| C20:5, EPA (Eicosapentaenoic acid) | 15.18 ± 0.75 | 17.87 ± 0.75 |
| C22:5, DPA (Docosapentaenoic acid) | 17.48 ± 0.37 | 23.93 ± 0.47 ** |
| C22:6, DHA (Docosahexaenoic acid) | 576.08 ± 12.48 | 957.23 ± 26.60 ** |
| C24:0, TA (Tetracosanoic acid) | 15.24 ± 1.27 | 17.32 ± 0.81 |
| C24:1, SOA (Selacholeic acid) | 17.16 ± 0.47 | 18.02 ± 0.67 |
| Total fatty acids | 2834.59 ± 58.42 | 4368.35 ± 132.21 ** |
| Saturated fatty acid | 814.79 ± 6.61 | 1136.77 ± 34.44 ** |
| Monounsaturated fatty acids | 275.86 ± 12.91 | 528.68 ± 15.72 |
| Polyunsaturated fatty acids | 1743.92 ± 38.84 | 2703.56 ± 81.97 ** |
| n-3 fatty acids | 616.32 ± 13.87 | 1004.56 ± 28.01 ** |
| n-6 fatty acids | 1127.61 ± 24.95 | 1698.99 ±53.97 ** |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ning, H.; Sun, Z.; Liu, Y.; Liu, L.; Hao, L.; Ye, Y.; Feng, R.; Li, J.; Li, Y.; Chu, X.; et al. Insulin Protects Hepatic Lipotoxicity by Regulating ER Stress through the PI3K/Akt/p53 Involved Pathway Independently of Autophagy Inhibition. Nutrients 2016, 8, 227. https://doi.org/10.3390/nu8040227
Ning H, Sun Z, Liu Y, Liu L, Hao L, Ye Y, Feng R, Li J, Li Y, Chu X, et al. Insulin Protects Hepatic Lipotoxicity by Regulating ER Stress through the PI3K/Akt/p53 Involved Pathway Independently of Autophagy Inhibition. Nutrients. 2016; 8(4):227. https://doi.org/10.3390/nu8040227
Chicago/Turabian StyleNing, Hua, Zongxiang Sun, Yunyun Liu, Lei Liu, Liuyi Hao, Yaxin Ye, Rennan Feng, Jie Li, Ying Li, Xia Chu, and et al. 2016. "Insulin Protects Hepatic Lipotoxicity by Regulating ER Stress through the PI3K/Akt/p53 Involved Pathway Independently of Autophagy Inhibition" Nutrients 8, no. 4: 227. https://doi.org/10.3390/nu8040227
APA StyleNing, H., Sun, Z., Liu, Y., Liu, L., Hao, L., Ye, Y., Feng, R., Li, J., Li, Y., Chu, X., Li, S., & Sun, C. (2016). Insulin Protects Hepatic Lipotoxicity by Regulating ER Stress through the PI3K/Akt/p53 Involved Pathway Independently of Autophagy Inhibition. Nutrients, 8(4), 227. https://doi.org/10.3390/nu8040227