The Role of Gluten in Celiac Disease and Type 1 Diabetes


Abstract
:1. Celiac Disease
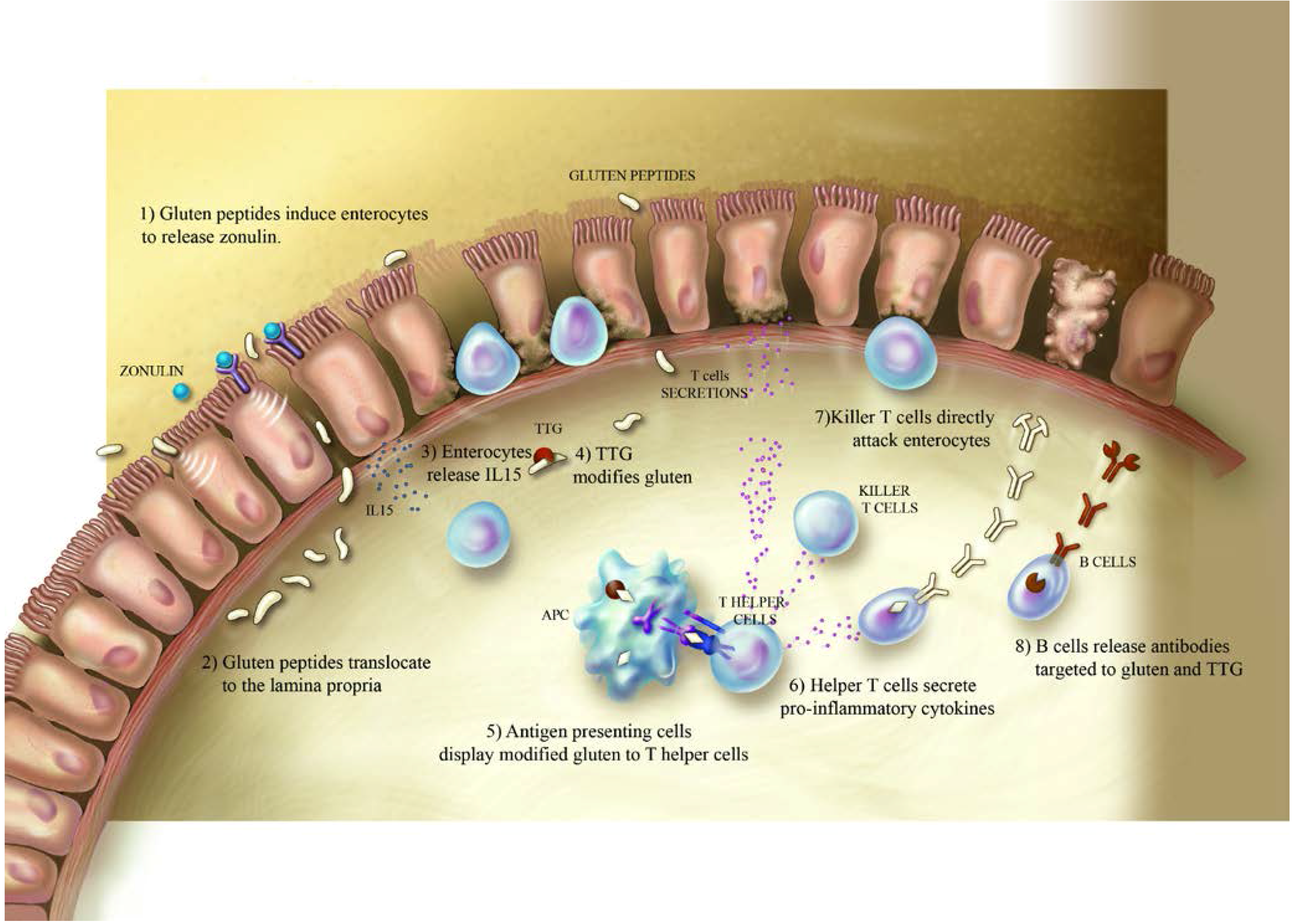
2. Celiac Disease and Gluten
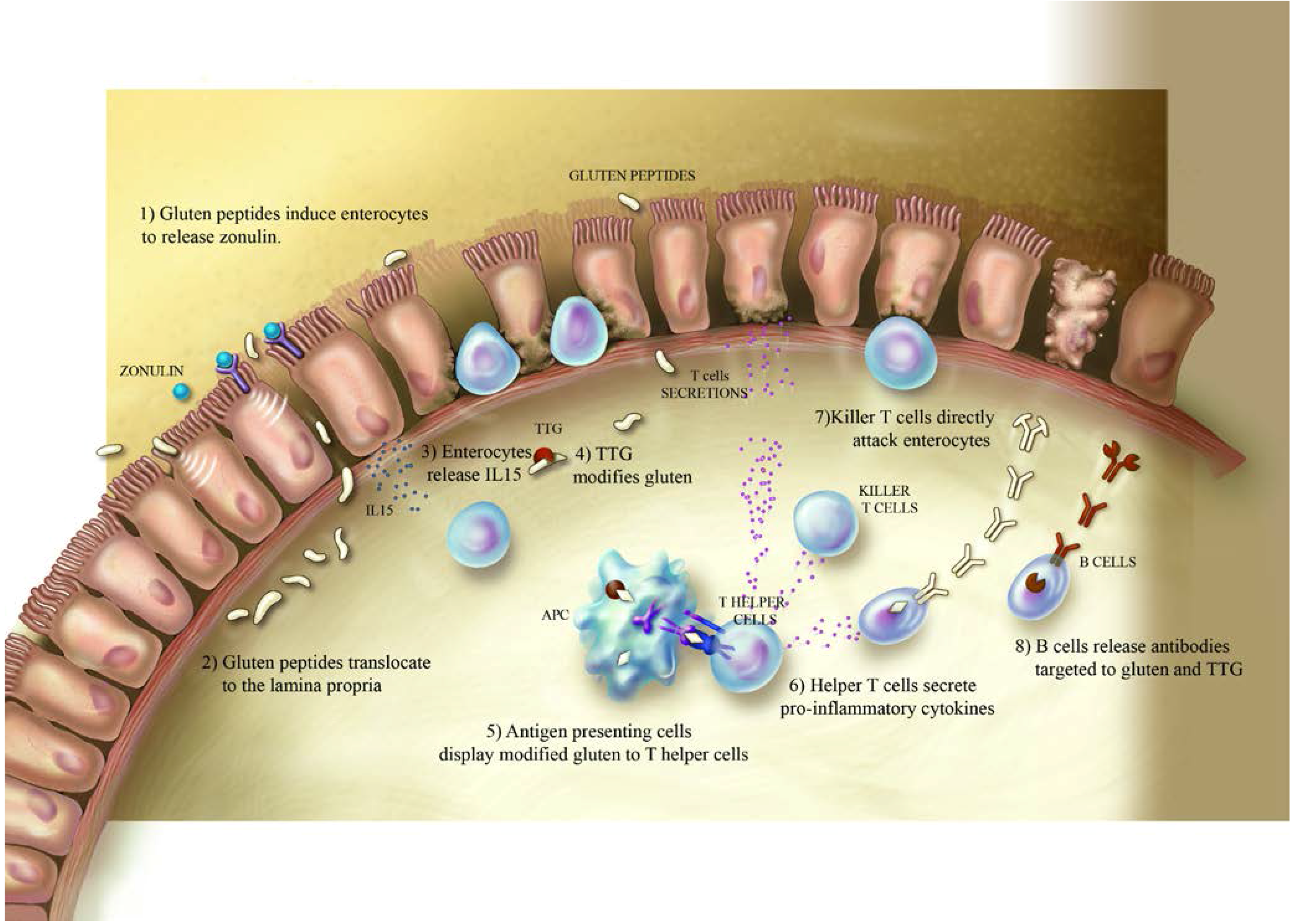
3. Type-1 Diabetes
4. Type 1 Diabetes and Gluten
- In vitro studies:
- In vivo studies:
- Incidence of T1D is reduced in offspring of NOD mice fed a GFD during pregnancy [87].
- Removal of gluten from the diet selectively protects NOD mice from developing T1D [96].
- FGD casein-based diet reduces incidence of hypoglycemia, delays onset of T1D and reduces IA titers in NOD mice [98].
- Gluten containing diet alters the composition of the innate immune system in BALB/c and NOD mice and it is correlated with an increased expression of dendritic cells activation markers in NOD mice [86].
- Box 1 summarizes the main findings about the correlation between gluten and the onset of T1D.
5. Comorbidity between Celiac Disease and Type-1 Diabetes

{kind=link}
| Feature | Celiac Disease | Type 1 Diabetes |
|---|---|---|
| Wordwide incidence | 0.6% *–1% | <1% |
| Contribution of HLA genes | HLA DQ2 (DQA1*05-DQB1*02) HLA DQ8 (DQA1*03-DQB1*03:02) | HLADQ2 and/or DQ8 (DRB1*0401-DQB1*03:02 and DRB1*0301-DQB1*0201) |
| Non-HLA candidate genes | CTLA4, PTPN22,CD28, ICOS, MYO9B | CTLA4, PTPN22, MIC-A |
| Symptoms | Diarrhea, steatorrhea, weight loss, failure to thrive, iron deficiency, abdominal pain, reduced bone density, chronic fatigue, growth failure. | Polyuria, polydipsia, extensive hunger, weight loss, chronic fatigue, reduced bone density, growth failure, hyperglycemia. |
| Diagnosis | Small intestinal biopsy, generally with supporting serological testing.Serologic tests: IgA anti-tTG, IgG anti-tTG, IgA anti-EMA, IgG DGP. | Blood test: Fasting blood glucose level, oral glucose tolerance test, A1C. Serologic tests: ICA, IAA, GADA, IA2 antibodies |
| Comorbidities | Type 1 diabetes, Down syndrome, Turner syndrome, William’s syndrome, vitiligo, Addison’s disease, hyperparathyroidism, neuropathy, IgA nephropathy, psoriasis. | Celiac disease, Grave’s disease, Hashimoto’s disease, Addison disease, vitiligo, autoimmune thyroid disease. |
| Pathogenesis | Enteropathy is due to dysregulation of the innate and adaptive immune system. Alteration of intestinal permeability. | Autoimmune destruction of pancreatic insulin-producing β-cells by an adaptive and innate immune response. Alteration of intestinal permeability. |
6. Conclusions
Conflicts of Interest
References
- Castillo, N.E.; Theethira, T.G.; Leffler, D.A. The present and the future in the diagnosis and management of celiac disease. Gastroenterol. Rep. 2015, 3, 3–11. [Google Scholar] [CrossRef] [PubMed]
- Lundin, K.E.; Sollid, L.M. Advances in coeliac disease. Curr. Opin. Gastroenterol. 2014, 30, 154–162. [Google Scholar] [CrossRef] [PubMed]
- Lionetti, E.; Castellaneta, S.; Francavilla, R.; Pulvirenti, A.; Tonutti, E.; Amarri, S.; Barbato, M.; Barbera, C.; Barera, G.; Bellantoni, A.; et al. Introduction of gluten, HLA status, and the risk of celiac disease in children. N. Engl. J. Med. 2014, 371, 1295–1303. [Google Scholar] [CrossRef] [PubMed]
- Gutierrez-Achury, J.; Zhernakova, A.; Pulit, S.L.; Trynka, G.; Hunt, K.A.; Romanos, J.; Raychaudhuri, S.; van Heel, D.A.; Wijmenga, C.; de Bakker, P.I. Fine mapping in the MHC region accounts for 18% additional genetic risk for celiac disease. Nat. Genet. 2015, 47, 577–578. [Google Scholar] [CrossRef] [PubMed]
- Collado, M.C.; Donat, E.; Ribes-Koninckx, C.; Calabuig, M.; Sanz, Y. Specific duodenal and faecal bacterial groups associated with paediatric coeliac disease. J. Clin. Pathol. 2009, 62, 264–269. [Google Scholar] [CrossRef] [PubMed]
- Sellitto, M.; Bai, G.; Serena, G.; Fricke, W.F.; Sturgeon, C.; Gajer, P.; White, J.R.; Koenig, S.S.; Sakamoto, J.; Boothe, D.; et al. Proof of concept of microbiome-metabolome analysis and delayed gluten exposure on celiac disease autoimmunity in genetically at-risk infants. PLoS ONE 2012, 7, e33387. [Google Scholar] [CrossRef] [PubMed]
- Yap, T.W.; Chan, W.K.; Leow, A.H.; Azmi, A.N.; Loke, M.F.; Vadivelu, J.; Goh, K.L. Prevalence of serum celiac antibodies in a multiracial Asian population—A first study in the young Asian adult population of Malaysia. PLoS ONE 2015, 10, e0121908. [Google Scholar] [CrossRef] [PubMed]
- Emilsson, L.; Wijmenga, C.; Murray, J.A.; Ludvigsson, J.F. Autoimmune Disease in First-Degree Relatives and Spouses of Individuals With Celiac Disease. Clin. Gastroenterol. Hepatol. Off. Clin. Pract. J. Am. Gastroenterol. Assoc. 2015, 13, 1271–1277. [Google Scholar] [CrossRef] [PubMed]
- Catassi, C.; Kryszak, D.; Bhatti, B.; Sturgeon, C.; Helzlsouer, K.; Clipp, S.L.; Gelfond, D.; Puppa, E.; Sferruzza, A.; Fasano, A. Natural history of celiac disease autoimmunity in a USA cohort followed since 1974. Ann. Med. 2010, 42, 530–538. [Google Scholar] [CrossRef] [PubMed]
- Kelly, C.P.; Bai, J.C.; Liu, E.; Leffler, D.A. Advances in diagnosis and management of celiac disease. Gastroenterology 2015, 148, 1175–1186. [Google Scholar] [CrossRef] [PubMed]
- Dieterich, W.; Ehnis, T.; Bauer, M.; Donner, P.; Volta, U.; Riecken, E.O.; Schuppan, D. Identification of tissue transglutaminase as the autoantigen of celiac disease. Nat. Med. 1997, 3, 797–801. [Google Scholar] [CrossRef] [PubMed]
- Fasano, A.; Catassi, C. Clinical practice: Celiac disease. N. Engl. J. Med. 2012, 367, 2419–2426. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.J.; Andreson, Z.; Ryu, D. Gluten free contamination in foods labeles “Gluten free” in United States. J. Food Prot. 2014, 77, 1830–1833. [Google Scholar] [CrossRef] [PubMed]
- Fasano, A.; Not, T.; Wang, W.; Uzzau, S.; Berti, I.; Tommasini, A.; Goldblum, S.E. Zonulin, a newly discovered modulator of intestinal permeability, and its expression in coeliac disease. Lancet 2000, 355, 1518–1519. [Google Scholar] [CrossRef]
- Fasano, A. Surprises from celiac disease. Sci. Am. 2009, 301, 54–61. [Google Scholar] [CrossRef] [PubMed]
- Maiuri, L.; Ciacci, C.; Ricciardelli, I.; Vacca, L.; Raia, V.; Rispo, A.; Griffin, M.; Issekutz, T.; Quaratino, S.; Londei, M. Unexpected role of surface transglutaminase type II in celiac disease. Gastroenterology 2005, 129, 1400–1413. [Google Scholar] [CrossRef] [PubMed]
- Gianfrani, C.; Siciliano, R.A.; Facchiano, A.M.; Camarca, A.; Mazzeo, M.F.; Costantini, S.; Salvati, V.M.; Maurano, F.; Mazzarella, G.; Iaquinto, G.; et al. Transamidation of wheat flour inhibits the response to gliadin of intestinal T cells in celiac disease. Gastroenterology 2007, 133, 780–789. [Google Scholar] [CrossRef] [PubMed]
- Meresse, B.; Ripoche, J.; Heyman, M.; Cerf-Bensussan, N. Celiac disease: From oral tolerance to intestinal inflammation, autoimmunity and lymphomagenesis. Mucosal Immunol. 2009, 2, 8–23. [Google Scholar] [CrossRef] [PubMed]
- Nikulina, M.; Habich, C.; Flohe, S.B.; Scott, F.W.; Kolb, H. Wheat gluten causes dendritic cell maturation and chemokine secretion. J. Immunol. 2004, 173, 1925–1933. [Google Scholar] [CrossRef] [PubMed]
- Skovbjerg, H.; Koch, C.; Anthonsen, D.; Sjostrom, H. Deamidation and cross-linking of gliadin peptides by transglutaminases and the relation to celiac disease. Biochim. Biophys. Acta 2004, 1690, 220–230. [Google Scholar] [CrossRef] [PubMed]
- Castellanos-Rubio, A.; Santin, I.; Irastorza, I.; Castano, L.; Carlos Vitoria, J.; Ramon Bilbao, J. TH17 (and TH1) signatures of intestinal biopsies of CD patients in response to gliadin. Autoimmunity 2009, 42, 69–73. [Google Scholar] [CrossRef] [PubMed]
- Palova-Jelinkova, L.; Danova, K.; Drasarova, H.; Dvorak, M.; Funda, D.P.; Fundova, P.; Kotrbova-Kozak, A.; Cerna, M.; Kamanova, J.; Martin, S.F.; et al. Pepsin digest of wheat gliadin fraction increases production of IL-1β via TLR4/MyD88/TRIF/MAPK/NF-κB signaling pathway and an NLRP3 inflammasome activation. PLoS ONE 2013, 8, e62426. [Google Scholar] [CrossRef] [PubMed]
- Di Sabatino, A.; Ciccocioppo, R.; Cupelli, F.; Cinque, B.; Millimaggi, D.; Clarkson, M.M.; Paulli, M.; Cifone, M.G.; Corazza, G.R. Epithelium derived interleukin 15 regulates intraepithelial lymphocyte Th1 cytokine production, cytotoxicity, and survival in coeliac disease. Gut 2006, 55, 469–477. [Google Scholar] [CrossRef] [PubMed]
- Harris, K.M.; Fasano, A.; Mann, D.L. Monocytes differentiated with IL-15 support Th17 and Th1 responses to wheat gliadin: Implications for celiac disease. Clin. Immunol. 2010, 135, 430–439. [Google Scholar] [CrossRef] [PubMed]
- De Nitto, D.; Monteleone, I.; Franze, E.; Pallone, F.; Monteleone, G. Involvement of interleukin-15 and interleukin-21, two γ-chain-related cytokines, in celiac disease. World J. Gastroenterol. 2009, 15, 4609–4614. [Google Scholar] [CrossRef] [PubMed]
- Lammers, K.M.; Lu, R.; Brownley, J.; Lu, B.; Gerard, C.; Thomas, K.; Rallabhandi, P.; Shea-Donohue, T.; Tamiz, A.; Alkan, S.; et al. Gliadin induces an increase in intestinal permeability and zonulin release by binding to the chemokine receptor CXCR3. Gastroenterology 2008, 135, 194–204. [Google Scholar] [CrossRef] [PubMed]
- Lammers, K.M.; Khandelwal, S.; Chaudhry, F.; Kryszak, D.; Puppa, E.L.; Casolaro, V.; Fasano, A. Identification of a novel immunomodulatory gliadin peptide that causes interleukin-8 release in a chemokine receptor CXCR3-dependent manner only in patients with coeliac disease. Immunology 2011, 132, 432–440. [Google Scholar] [CrossRef] [PubMed]
- Drago, S.; El Asmar, R.; di Pierro, M.; Grazia Clemente, M.; Tripathi, A.; Sapone, A.; Thakar, M.; Iacono, G.; Carroccio, A.; D’Agate, C.; et al. Gliadin, zonulin and gut permeability: Effects on celiac and non-celiac intestinal mucosa and intestinal cell lines. Scand. J. Gastroenterol. 2006, 41, 408–419. [Google Scholar] [CrossRef] [PubMed]
- Fasano, A. Zonulin and its regulation of intestinal barrier function: The biological door to inflammation, autoimmunity, and cancer. Physiol. Rev. 2011, 91, 151–175. [Google Scholar] [CrossRef] [PubMed]
- Garrote, J.A.; Gomez-Gonzalez, E.; Bernardo, D.; Arranz, E.; Chirdo, F. Celiac disease pathogenesis: The proinflammatory cytokine network. J. Pediatr. Gastroenterol. Nutr. 2008, 47 (Suppl. 1), S27–S32. [Google Scholar] [CrossRef] [PubMed]
- Thomas, K.E.; Sapone, A.; Fasano, A.; Vogel, S.N. Gliadin stimulation of murine macrophage inflammatory gene expression and intestinal permeability are MyD88-dependent: Role of the innate immune response in Celiac disease. J. Immunol. 2006, 176, 2512–2521. [Google Scholar] [CrossRef] [PubMed]
- Ortega, C.; Fernandez, S.; Estevez, O.A.; Aguado, R.; Molina, I.J.; Santamaria, M. IL-17 producing T cells in celiac disease: Angels or devils? Int. Rev. Immunol. 2013, 32, 534–543. [Google Scholar] [CrossRef] [PubMed]
- Anderson, R.P.; Degano, P.; Godkin, A.J.; Jewell, D.P.; Hill, A.V. In vivo antigen challenge in celiac disease identifies a single transglutaminase-modified peptide as the dominant A-gliadin T-cell epitope. Nat. Med. 2000, 6, 337–342. [Google Scholar] [CrossRef] [PubMed]
- Shan, L.; Molberg, O.; Parrot, I.; Hausch, F.; Filiz, F.; Gray, G.M.; Sollid, L.M.; Khosla, C. Structural basis for gluten intolerance in celiac sprue. Science 2002, 297, 2275–2279. [Google Scholar] [CrossRef] [PubMed]
- Camarca, M.E.; Mozzillo, E.; Nugnes, R.; Zito, E.; Falco, M.; Fattorusso, V.; Mobilia, S.; Buono, P.; Valerio, G.; Troncone, R.; et al. Celiac disease in type 1 diabetes mellitus. Ital. J. Pediatr. 2012, 38, 10. [Google Scholar] [CrossRef] [PubMed]
- Rubio-Tapia, A.; Kyle, R.A.; Kaplan, E.L.; Johnson, D.R.; Page, W.; Erdtmann, F.; Brantner, T.L.; Kim, W.R.; Phelps, T.K.; Lahr, B.D.; et al. Increased prevalence and mortality in undiagnosed celiac disease. Gastroenterology 2009, 137, 88–93. [Google Scholar] [CrossRef] [PubMed]
- Vilppula, A.; Kaukinen, K.; Luostarinen, L.; Krekela, I.; Patrikainen, H.; Valve, R.; Maki, M.; Collin, P. Increasing prevalence and high incidence of celiac disease in elderly people: A population-based study. BMC Gastroenterol. 2009, 9, 49. [Google Scholar] [CrossRef] [PubMed]
- Kasarda, D.D. Can an increase in celiac disease be attributed to an increase in the gluten content of wheat as a consequence of wheat breeding? J. Agric. Food Chem. 2013, 61, 1155–1159. [Google Scholar] [CrossRef] [PubMed]
- Hogberg, L.; Falth-Magnusson, K.; Grodzinsky, E.; Stenhammar, L. Familial prevalence of coeliac disease: A twenty-year follow-up study. Scand. J. Gastroenterol. 2003, 38, 61–65. [Google Scholar] [CrossRef] [PubMed]
- Ivarsson, A.; Persson, L.A.; Nystrom, L.; Ascher, H.; Cavell, B.; Danielsson, L.; Dannaeus, A.; Lindberg, T.; Lindquist, B.; Stenhammar, L.; et al. Epidemic of coeliac disease in Swedish children. Acta Paediatr. 2000, 89, 165–171. [Google Scholar] [CrossRef] [PubMed]
- Ivarsson, A.; Myleus, A.; Norstrom, F.; van der Pals, M.; Rosen, A.; Hogberg, L.; Danielsson, L.; Halvarsson, B.; Hammarroth, S.; Hernell, O.; et al. Prevalence of childhood celiac disease and changes in infant feeding. Pediatrics 2013, 131, e687–e694. [Google Scholar] [CrossRef] [PubMed]
- Liu, E.; Lee, H.S.; Aronsson, C.A.; Hagopian, W.A.; Koletzko, S.; Rewers, M.J.; Eisenbarth, G.S.; Bingley, P.J.; Bonifacio, E.; Simell, V.; et al. Risk of pediatric celiac disease according to HLA haplotype and country. N. Engl. J. Med. 2014, 371, 42–49. [Google Scholar] [CrossRef] [PubMed]
- Hosea Blewett, H.J.; Cicalo, M.C.; Holland, C.D.; Field, C.J. The immunological components of human milk. Adv. Food Nutr. Res. 2008, 54, 45–80. [Google Scholar] [PubMed]
- Newburg, D.S.; Walker, W.A. Protection of the neonate by the innate immune system of developing gut and of human milk. Pediatr. Res. 2007, 61, 2–8. [Google Scholar] [CrossRef] [PubMed]
- Ivarsson, A.; Hernell, O.; Stenlund, H.; Persson, L.A. Breast-feeding protects against celiac disease. Am. J. Clin. Nutr. 2002, 75, 914–921. [Google Scholar] [PubMed]
- Vriezinga, S.L.; Auricchio, R.; Bravi, E.; Castillejo, G.; Chmielewska, A.; Crespo Escobar, P.; Kolacek, S.; Koletzko, S.; Korponay-Szabo, I.R.; Mummert, E.; et al. Randomized feeding intervention in infants at high risk for celiac disease. N. Engl. J. Med. 2014, 371, 1304–1315. [Google Scholar] [CrossRef] [PubMed]
- Nadal, I.; Donat, E.; Ribes-Koninckx, C.; Calabuig, M.; Sanz, Y. Imbalance in the composition of the duodenal microbiota of children with coeliac disease. J. Med. Microbiol. 2007, 56, 1669–1674. [Google Scholar] [CrossRef] [PubMed]
- Tjellstrom, B.; Hogberg, L.; Stenhammar, L.; Falth-Magnusson, K.; Magnusson, K.E.; Norin, E.; Sundqvist, T.; Midtvedt, T. Faecal short-chain fatty acid pattern in childhood coeliac disease is normalised after more than one year’s gluten-free diet. Microb. Ecol. Health Dis. 2013, 24. [Google Scholar] [CrossRef] [PubMed]
- Krumbhaar, E.B. Spontaneous Diabetes in a Dog. J. Exp. Med. 1916, 24, 361–365. [Google Scholar] [CrossRef] [PubMed]
- Patterson, C.C.; Gyurus, E.; Rosenbauer, J.; Cinek, O.; Neu, A.; Schober, E.; Parslow, R.C.; Joner, G.; Svensson, J.; Castell, C.; et al. Trends in childhood type 1 diabetes incidence in Europe during 1989–2008: Evidence of non-uniformity over time in rates of increase. Diabetologia 2012, 55, 2142–2147. [Google Scholar] [CrossRef] [PubMed]
- Patterson, C.C.; Dahlquist, G.G.; Gyurus, E.; Green, A.; Soltesz, G.; Group, E.S. Incidence trends for childhood type 1 diabetes in Europe during 1989–2003 and predicted new cases 2005–20: A multicentre prospective registration study. Lancet 2009, 373, 2027–2033. [Google Scholar] [CrossRef]
- Harjutsalo, V.; Sjoberg, L.; Tuomilehto, J. Time trends in the incidence of type 1 diabetes in Finnish children: A cohort study. Lancet 2008, 371, 1777–1782. [Google Scholar] [CrossRef]
- Tuomilehto, J. The emerging global epidemic of type 1 diabetes. Curr. Diab. Rep. 2013, 13, 795–804. [Google Scholar] [CrossRef] [PubMed]
- Hagopian, W.A.; Lernmark, A.; Rewers, M.J.; Simell, O.G.; She, J.X.; Ziegler, A.G.; Krischer, J.P.; Akolkar, B. TEDDY—The Environmental Determinants of Diabetes in the Young: An observational clinical trial. Ann. N. Y. Acad. Sci. 2006, 1079, 320–326. [Google Scholar] [CrossRef] [PubMed]
- Rewers, M.; Bugawan, T.L.; Norris, J.M.; Blair, A.; Beaty, B.; Hoffman, M.; McDuffie, R.S., Jr.; Hamman, R.F.; Klingensmith, G.; Eisenbarth, G.S.; et al. Newborn screening for HLA markers associated with IDDM: Diabetes autoimmunity study in the young (DAISY). Diabetologia 1996, 39, 807–812. [Google Scholar] [CrossRef] [PubMed]
- Skyler, J.S.; Greenbaum, C.J.; Lachin, J.M.; Leschek, E.; Rafkin-Mervis, L.; Savage, P.; Spain, L.; Type 1 Diabetes TrialNet Study Group. Type 1 Diabetes TrialNet—An international collaborative clinical trials network. Ann. N. Y. Acad. Sci. 2008, 1150, 14–24. [Google Scholar] [CrossRef] [PubMed]
- Foster, B. Diabetic Coma: Acetonaemia. Br. Med. J. 1878, 1, 78–81. [Google Scholar] [CrossRef] [PubMed]
- American Diabetes Association. Standards of medical care in diabetes—2012. Diabetes Care 2012, 35 (Suppl. 1), S11–S63. [Google Scholar]
- Bottazzo, G.F.; Florin-Christensen, A.; Doniach, D. Islet-cell antibodies in diabetes mellitus with autoimmune polyendocrine deficiencies. Lancet 1974, 2, 1279–1283. [Google Scholar] [CrossRef]
- Palmer, J.P.; Asplin, C.M.; Clemons, P.; Lyen, K.; Tatpati, O.; Raghu, P.K.; Paquette, T.L. Insulin antibodies in insulin-dependent diabetics before insulin treatment. Science 1983, 222, 1337–1339. [Google Scholar] [CrossRef] [PubMed]
- Baekkeskov, S.; Aanstoot, H.J.; Christgau, S.; Reetz, A.; Solimena, M.; Cascalho, M.; Folli, F.; Richter-Olesen, H.; de Camilli, P. Identification of the 64K autoantigen in insulin-dependent diabetes as the GABA-synthesizing enzyme glutamic acid decarboxylase. Nature 1990, 347, 151–156. [Google Scholar] [CrossRef] [PubMed]
- Atkinson, M.A.; Eisenbarth, G.S.; Michels, A.W. Type 1 diabetes. Lancet 2014, 383, 69–82. [Google Scholar] [CrossRef]
- Stankov, K.; Benc, D.; Draskovic, D. Genetic and epigenetic factors in etiology of diabetes mellitus type 1. Pediatrics 2013, 132, 1112–1122. [Google Scholar] [CrossRef] [PubMed]
- Svejgaard, A.; Ryder, L.P. HLA and insulin-dependent diabetes: An overview. Genet. Epidemiol. 1989, 6, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Vella, A.; Cooper, J.D.; Lowe, C.E.; Walker, N.; Nutland, S.; Widmer, B.; Jones, R.; Ring, S.M.; McArdle, W.; Pembrey, M.E.; et al. Localization of a type 1 diabetes locus in the IL2RA/CD25 region by use of tag single-nucleotide polymorphisms. Am. J. Hum. Genet. 2005, 76, 773–779. [Google Scholar] [CrossRef] [PubMed]
- Nistico, L.; Buzzetti, R.; Pritchard, L.E.; van der Auwera, B.; Giovannini, C.; Bosi, E.; Larrad, M.T.; Rios, M.S.; Chow, C.C.; Cockram, C.S.; et al. The CTLA-4 gene region of chromosome 2q33 is linked to, and associated with, type 1 diabetes. Belgian Diabetes Registry. Hum. Mol. Genet. 1996, 5, 1075–1080. [Google Scholar] [CrossRef] [PubMed]
- Bottini, N.; Musumeci, L.; Alonso, A.; Rahmouni, S.; Nika, K.; Rostamkhani, M.; MacMurray, J.; Meloni, G.F.; Lucarelli, P.; Pellecchia, M.; et al. A functional variant of lymphoid tyrosine phosphatase is associated with type I diabetes. Nat. Genet. 2004, 36, 337–338. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, M.; Obayashi, H.; Maruya, E.; Ohta, M.; Tegoshi, H.; Fukui, M.; Hasegawa, G.; Shigeta, H.; Kitagawa, Y.; Nakano, K.; et al. Association between type 1 diabetes age-at-onset and intercellular adhesion molecule-1 (ICAM-1) gene polymorphism. Hum. Immunol. 2000, 61, 507–510. [Google Scholar] [CrossRef]
- Bell, G.I.; Horita, S.; Karam, J.H. A polymorphic locus near the human insulin gene is associated with insulin-dependent diabetes mellitus. Diabetes 1984, 33, 176–183. [Google Scholar] [CrossRef] [PubMed]
- Sadeharju, K.; Hamalainen, A.M.; Knip, M.; Lonnrot, M.; Koskela, P.; Virtanen, S.M.; Ilonen, J.; Akerblom, H.K.; Hyoty, H.; Finnish, T.S.G. Enterovirus infections as a risk factor for type I diabetes: Virus analyses in a dietary intervention trial. Clin. Exp. Immunol. 2003, 132, 271–277. [Google Scholar] [CrossRef] [PubMed]
- Lonnrot, M.; Knip, M.; Roivainen, M.; Koskela, P.; Akerblom, H.K.; Hyoty, H. Onset of type 1 diabetes mellitus in infancy after enterovirus infections. Diabet. Med. 1998, 15, 431–434. [Google Scholar] [CrossRef]
- Hyoty, H.; Hiltunen, M.; Knip, M.; Laakkonen, M.; Vahasalo, P.; Karjalainen, J.; Koskela, P.; Roivainen, M.; Leinikki, P.; Hovi, T.; et al. A prospective study of the role of coxsackie B and other enterovirus infections in the pathogenesis of IDDM. Diabetes 1995, 44, 652–657. [Google Scholar] [CrossRef] [PubMed]
- Muir, P.; Singh, N.B.; Banatvala, J.E. Enterovirus-specific serum IgA antibody responses in patients with acute infections, chronic cardiac disease, and recently diagnosed insulin-dependent diabetes mellitus. J. Med. Virol. 1990, 32, 236–242. [Google Scholar] [CrossRef] [PubMed]
- Honeyman, M.C.; Stone, N.L.; Harrison, L.C. T-cell epitopes in type 1 diabetes autoantigen tyrosine phosphatase IA-2: Potential for mimicry with rotavirus and other environmental agents. Mol. Med. 1998, 4, 231–239. [Google Scholar] [PubMed]
- Honeyman, M.C.; Coulson, B.S.; Stone, N.L.; Gellert, S.A.; Goldwater, P.N.; Steele, C.E.; Couper, J.J.; Tait, B.D.; Colman, P.G.; Harrison, L.C. Association between rotavirus infection and pancreatic islet autoimmunity in children at risk of developing type 1 diabetes. Diabetes 2000, 49, 1319–1324. [Google Scholar] [CrossRef] [PubMed]
- Okada, H.; Kuhn, C.; Feillet, H.; Bach, J.F. The “hygiene hypothesis” for autoimmune and allergic diseases: An update. Clin. Exp. Immunol. 2010, 160, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Hu, C.; Wong, F.S.; Wen, L. Type 1 diabetes and gut microbiota: Friend or foe? Pharmacol. Res. 2015, 98, 9–15. [Google Scholar] [CrossRef] [PubMed]
- Wen, L.; Ley, R.E.; Volchkov, P.Y.; Stranges, P.B.; Avanesyan, L.; Stonebraker, A.C.; Hu, C.; Wong, F.S.; Szot, G.L.; Bluestone, J.A.; et al. Innate immunity and intestinal microbiota in the development of Type 1 diabetes. Nature 2008, 455, 1109–1113. [Google Scholar] [CrossRef] [PubMed]
- De Goffau, M.C.; Fuentes, S.; van den Bogert, B.; Honkanen, H.; de vos, W.M.; Welling, G.W.; Hyoty, H.; Harmsen, H.J. Aberrant gut microbiota composition at the onset of type 1 diabetes in young children. Diabetologia 2014, 57, 1569–1577. [Google Scholar] [CrossRef] [PubMed]
- De Goffau, M.C.; Luopajarvi, K.; Knip, M.; Ilonen, J.; Ruohtula, T.; Harkonen, T.; Orivuori, L.; Hakala, S.; Welling, G.W.; Harmsen, H.J.; et al. Fecal microbiota composition differs between children with β-cell autoimmunity and those without. Diabetes 2013, 62, 1238–1244. [Google Scholar] [CrossRef] [PubMed]
- Simmons, K.M.; Michels, A.W. Type 1 diabetes: A predictable disease. World J. Diabetes 2015, 6, 380–390. [Google Scholar] [CrossRef] [PubMed]
- Mojibian, M.; Chakir, H.; Lefebvre, D.E.; Crookshank, J.A.; Sonier, B.; Keely, E.; Scott, F.W. Diabetes-specific HLA-DR-restricted proinflammatory T-cell response to wheat polypeptides in tissue transglutaminase antibody-negative patients with type 1 diabetes. Diabetes 2009, 58, 1789–1796. [Google Scholar] [CrossRef] [PubMed]
- Auricchio, R.; Paparo, F.; Maglio, M.; Franzese, A.; Lombardi, F.; Valerio, G.; Nardone, G.; Percopo, S.; Greco, L.; Troncone, R. In vitro-deranged intestinal immune response to gliadin in type 1 diabetes. Diabetes 2004, 53, 1680–1683. [Google Scholar] [CrossRef] [PubMed]
- Troncone, R.; Franzese, A.; Mazzarella, G.; Paparo, F.; Auricchio, R.; Coto, I.; Mayer, M.; Greco, L. Gluten sensitivity in a subset of children with insulin dependent diabetes mellitus. Am. J. Gastroenterol. 2003, 98, 590–595. [Google Scholar] [CrossRef] [PubMed]
- Klemetti, P.; Savilahti, E.; Ilonen, J.; Akerblom, H.K.; Vaarala, O. T-cell reactivity to wheat gluten in patients with insulin-dependent diabetes mellitus. Scand. J. Immunol. 1998, 47, 48–53. [Google Scholar] [CrossRef] [PubMed]
- Larsen, J.; Weile, C.; Antvorskov, J.C.; Engkilde, K.; Nielsen, S.M.; Josefsen, K.; Buschard, K. Effect of dietary gluten on dendritic cells and innate immune subsets in BALB/c and NOD mice. PLoS ONE 2015, 10, e0118618. [Google Scholar] [CrossRef] [PubMed]
- Hansen, C.H.; Krych, L.; Buschard, K.; Metzdorff, S.B.; Nellemann, C.; Hansen, L.H.; Nielsen, D.S.; Frokiaer, H.; Skov, S.; Hansen, A.K. A maternal gluten-free diet reduces inflammation and diabetes incidence in the offspring of NOD mice. Diabetes 2014, 63, 2821–2832. [Google Scholar] [CrossRef] [PubMed]
- Schmid, S.; Koczwara, K.; Schwinghammer, S.; Lampasona, V.; Ziegler, A.G.; Bonifacio, E. Delayed exposure to wheat and barley proteins reduces diabetes incidence in non-obese diabetic mice. Clin. Immunol. 2004, 111, 108–118. [Google Scholar] [CrossRef] [PubMed]
- Lamb, M.M.; Myers, M.A.; Barriga, K.; Zimmet, P.Z.; Rewers, M.; Norris, J.M. Maternal diet during pregnancy and islet autoimmunity in offspring. Pediatr. Diabetes 2008, 9, 135–141. [Google Scholar] [CrossRef] [PubMed]
- Chmiel, R.; Beyerlein, A.; Knopff, A.; Hummel, S.; Ziegler, A.G.; Winkler, C. Early infant feeding and risk of developing islet autoimmunity and type 1 diabetes. Acta Diabetol. 2014, 52, 621–624. [Google Scholar] [CrossRef] [PubMed]
- Ziegler, A.G.; Schmid, S.; Huber, D.; Hummel, M.; Bonifacio, E. Early infant feeding and risk of developing type 1 diabetes-associated autoantibodies. JAMA 2003, 290, 1721–1728. [Google Scholar] [CrossRef] [PubMed]
- Elenberg, Y.; Shaoul, R. The role of infant nutrition in the prevention of future disease. Front. Pediatr. 2014, 2, 73. [Google Scholar] [CrossRef] [PubMed]
- Wahlberg, J.; Vaarala, O.; Ludvigsson, J.; ABIS-Study Group. Dietary risk factors for the emergence of type 1 diabetes-related autoantibodies in 21/2 year-old Swedish children. Br. J. Nutr. 2006, 95, 603–608. [Google Scholar] [CrossRef] [PubMed]
- Hummel, S.; Pfluger, M.; Hummel, M.; Bonifacio, E.; Ziegler, A.G. Primary dietary intervention study to reduce the risk of islet autoimmunity in children at increased risk for type 1 diabetes: The BABYDIET study. Diabetes Care 2011, 34, 1301–1305. [Google Scholar] [CrossRef] [PubMed]
- Lamb, M.M.; Simpson, M.D.; Seifert, J.; Scott, F.W.; Rewers, M.; Norris, J.M. The association between IgG4 antibodies to dietary factors, islet autoimmunity and type 1 diabetes: The Diabetes Autoimmunity Study in the Young. PLoS ONE 2013, 8, e57936. [Google Scholar] [CrossRef] [PubMed]
- Marietta, E.V.; Gomez, A.M.; Yeoman, C.; Tilahun, A.Y.; Clark, C.R.; Luckey, D.H.; Murray, J.A.; White, B.A.; Kudva, Y.C.; Rajagopalan, G. Low incidence of spontaneous type 1 diabetes in non-obese diabetic mice raised on gluten-free diets is associated with changes in the intestinal microbiome. PLoS ONE 2013, 8, e78687. [Google Scholar] [CrossRef] [PubMed]
- Sildorf, S.M.; Fredheim, S.; Svensson, J.; Buschard, K. Remission without insulin therapy on gluten-free diet in a 6-year old boy with type 1 diabetes mellitus. BMJ Case Rep 2012, 2012. [Google Scholar] [CrossRef] [PubMed]
- Patrick, C.; Wang, G.S.; Lefebvre, D.E.; Crookshank, J.A.; Sonier, B.; Eberhard, C.; Mojibian, M.; Kennedy, C.R.; Brooks, S.P.; Kalmokoff, M.L.; et al. Promotion of autoimmune diabetes by cereal diet in the presence or absence of microbes associated with gut immune activation, regulatory imbalance, and altered cathelicidin antimicrobial Peptide. Diabetes 2013, 62, 2036–2047. [Google Scholar] [CrossRef] [PubMed]
- Watts, T.; Berti, I.; Sapone, A.; Gerarduzzi, T.; Not, T.; Zielke, R.; Fasano, A. Role of the intestinal tight junction modulator zonulin in the pathogenesis of type I diabetes in BB diabetic-prone rats. Proc. Natl. Acad. Sci. USA 2005, 102, 2916–2921. [Google Scholar] [CrossRef] [PubMed]
- Mackinder, M.; Allison, G.; Svolos, V.; Buchanan, E.; Johnston, A.; Cardigan, T.; Laird, N.; Duncan, H.; Fraser, K.; Edwards, C.A.; et al. Nutritional status, growth and disease management in children with single and dual diagnosis of type 1 diabetes mellitus and coeliac disease. BMC Gastroenterol. 2014, 14, 99. [Google Scholar] [CrossRef] [PubMed]
- Tsouka, A.; Mahmud, F.H.; Marcon, M.A. Celiac Disease Associated with Type 1 Diabetes and Celiac Disease Alone: Are these patients different? J. Pediatr. Gastroenterol. Nutr. 2015. [Google Scholar] [CrossRef] [PubMed]
- Castellaneta, S.; Piccinno, E.; Oliva, M.; Cristofori, F.; Vendemiale, M.; Ortolani, F.; Papadia, F.; Catassi, C.; Cavallo, L.; Francavilla, R. High rate of spontaneous normalization of celiac serology in a cohort of 446 children with type 1 diabetes: A prospective study. Diabetes Care 2015, 38, 760–766. [Google Scholar] [CrossRef] [PubMed]
- Rohrer, T.R.; Wolf, J.; Liptay, S.; Zimmer, K.P.; Frohlich-Reiterer, E.; Scheuing, N.; Marg, W.; Stern, M.; Kapellen, T.M.; Hauffa, B.P.; et al. Microvascular Complications in Childhood-Onset Type 1 Diabetes and Celiac Disease: A Multicenter Longitudinal Analysis of 56,514 Patients From the German-Austrian DPV Database. Diabetes Care 2015, 38, 801–807. [Google Scholar] [CrossRef] [PubMed]
- Larizza, D.; Calcaterra, V.; Klersy, C.; Badulli, C.; Caramagna, C.; Ricci, A.; Brambilla, P.; Salvaneschi, L.; Martinetti, M. Common immunogenetic profile in children with multiple autoimmune diseases: The signature of HLA-DQ pleiotropic genes. Autoimmunity 2012, 45, 470–475. [Google Scholar] [CrossRef] [PubMed]
- Bakker, S.F.; Tushuizen, M.E.; Stokvis-Brantsma, W.H.; Aanstoot, H.J.; Winterdijk, P.; van Setten, P.A.; von Blomberg, B.M.; Mulder, C.J.; Simsek, S. Frequent delay of coeliac disease diagnosis in symptomatic patients with type 1 diabetes mellitus: Clinical and genetic characteristics. Eur. J. Intern. Med. 2013, 24, 456–460. [Google Scholar] [CrossRef] [PubMed]
- Bakker, S.F.; Tushuizen, M.E.; von Blomberg, M.E.; Mulder, C.J.; Simsek, S. Type 1 diabetes and celiac disease in adults: Glycemic control and diabetic complications. Acta Diabetol. 2013, 50, 319–324. [Google Scholar] [CrossRef] [PubMed]
- Narula, P.; Porter, L.; Langton, J.; Rao, V.; Davies, P.; Cummins, C.; Kirk, J.; Barrett, T.; Protheroe, S. Gastrointestinal symptoms in children with type 1 diabetes screened for celiac disease. Pediatrics 2009, 124, e489–e495. [Google Scholar] [CrossRef] [PubMed]
- Warncke, K.; Liptay, S.; Frohlich-Reiterer, E.; Scheuing, N.; Schebek, M.; Wolf, J.; Rohrer, T.R.; Meissner, T.; Holl, R.W. Vascular risk factors in children, adolescents, and young adults with type 1 diabetes complicated by celiac disease: Results from the DPV initiative. Pediatr. Diabetes 2015. [Google Scholar] [CrossRef] [PubMed]
- Adlercreutz, E.H.; Wingren, C.J.; Vincente, R.P.; Merlo, J.; Agardh, D. Perinatal risk factors increase the risk of being affected by both type 1 diabetes and coeliac disease. Acta Paediatr. 2015, 104, 178–184. [Google Scholar] [CrossRef] [PubMed]
- Setty-Shah, N.; Maranda, L.; Nwosu, B.U. Increased risk for vitamin d deficiency in obese children with both celiac disease and type 1 diabetes. Gastroenterol. Res. Pract. 2014, 2014. [Google Scholar] [CrossRef] [PubMed]
- Joshi, A.S.; Varthakavi, P.K.; Bhagwat, N.M.; Chadha, M.D.; Mittal, S.S. Coeliac autoimmunity in type I diabetes mellitus. Arab. J. Gastroenterol. 2014, 15, 53–57. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Serena, G.; Camhi, S.; Sturgeon, C.; Yan, S.; Fasano, A. The Role of Gluten in Celiac Disease and Type 1 Diabetes. Nutrients 2015, 7, 7143-7162. https://doi.org/10.3390/nu7095329
Serena G, Camhi S, Sturgeon C, Yan S, Fasano A. The Role of Gluten in Celiac Disease and Type 1 Diabetes. Nutrients. 2015; 7(9):7143-7162. https://doi.org/10.3390/nu7095329
Chicago/Turabian StyleSerena, Gloria, Stephanie Camhi, Craig Sturgeon, Shu Yan, and Alessio Fasano. 2015. "The Role of Gluten in Celiac Disease and Type 1 Diabetes" Nutrients 7, no. 9: 7143-7162. https://doi.org/10.3390/nu7095329
APA StyleSerena, G., Camhi, S., Sturgeon, C., Yan, S., & Fasano, A. (2015). The Role of Gluten in Celiac Disease and Type 1 Diabetes. Nutrients, 7(9), 7143-7162. https://doi.org/10.3390/nu7095329