The Subtle Balance between Lipolysis and Lipogenesis: A Critical Point in Metabolic Homeostasis



Abstract
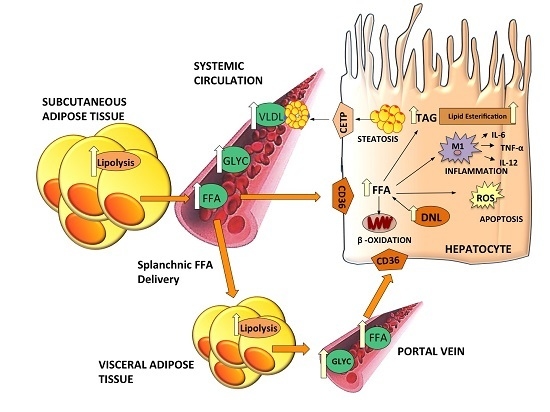
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Lipogenesis
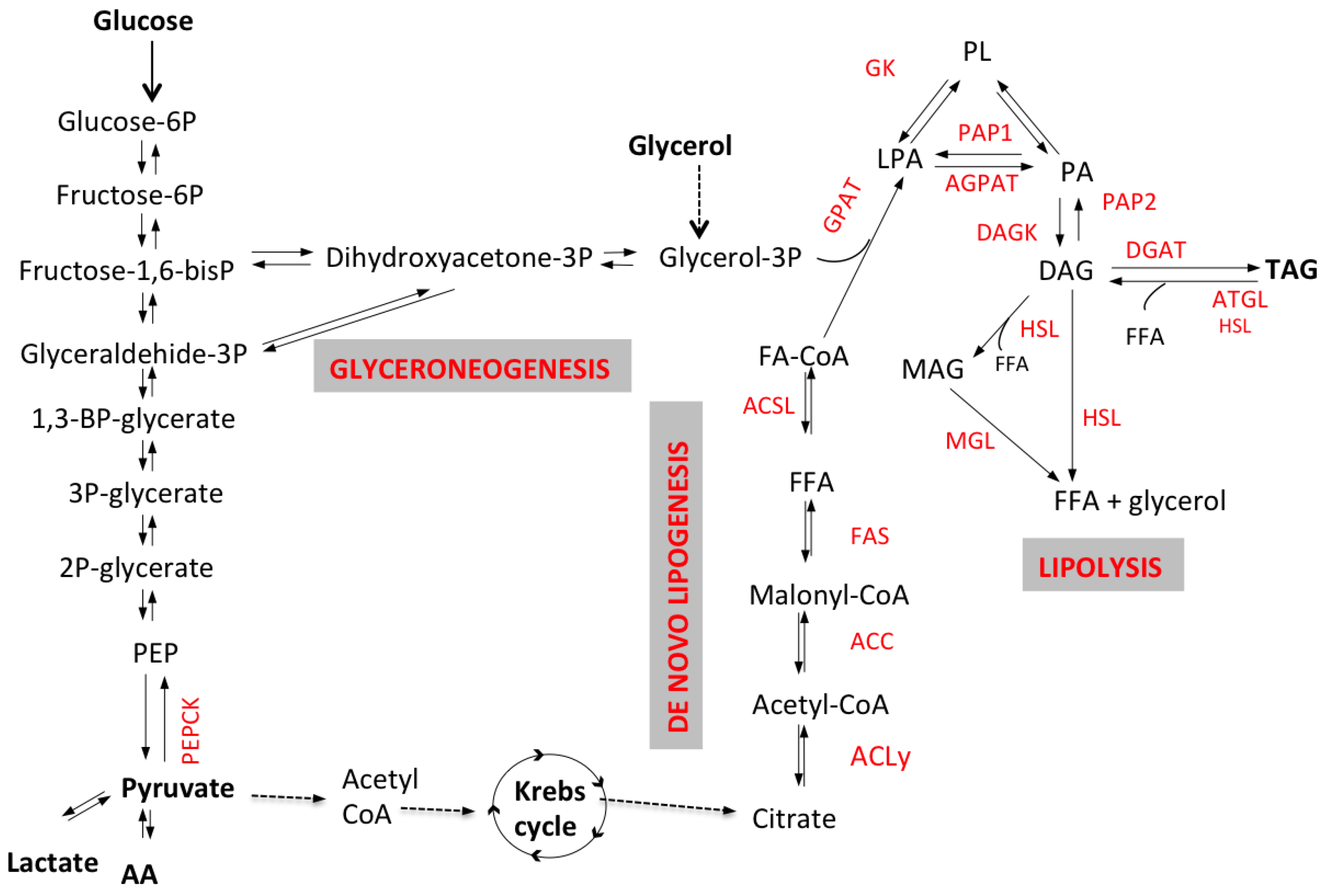
2.1. Glycerol-3-Phosphate (G3P) Synthesis and Glyceroneogenesis
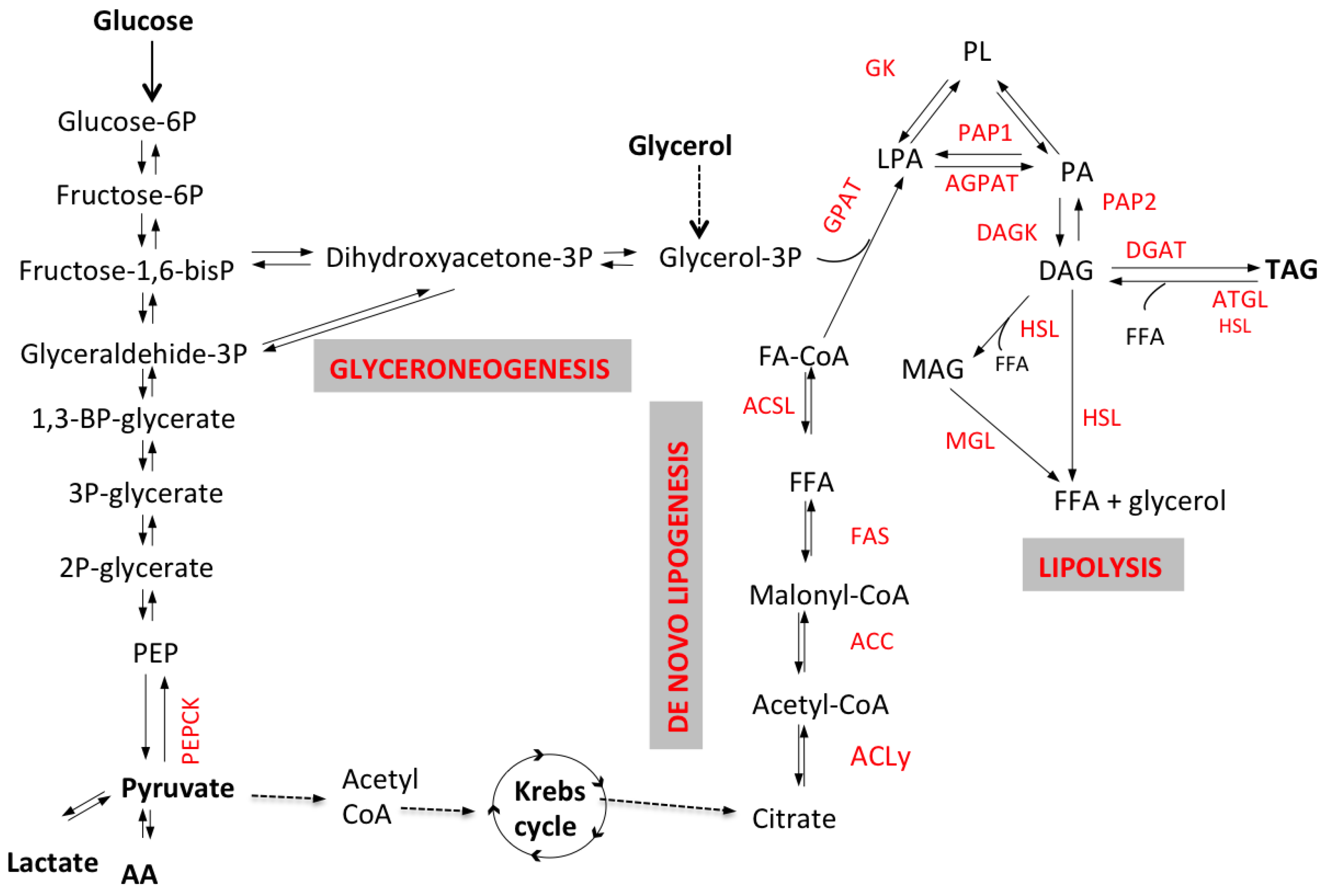
2.2. De Novo Lipogenesis
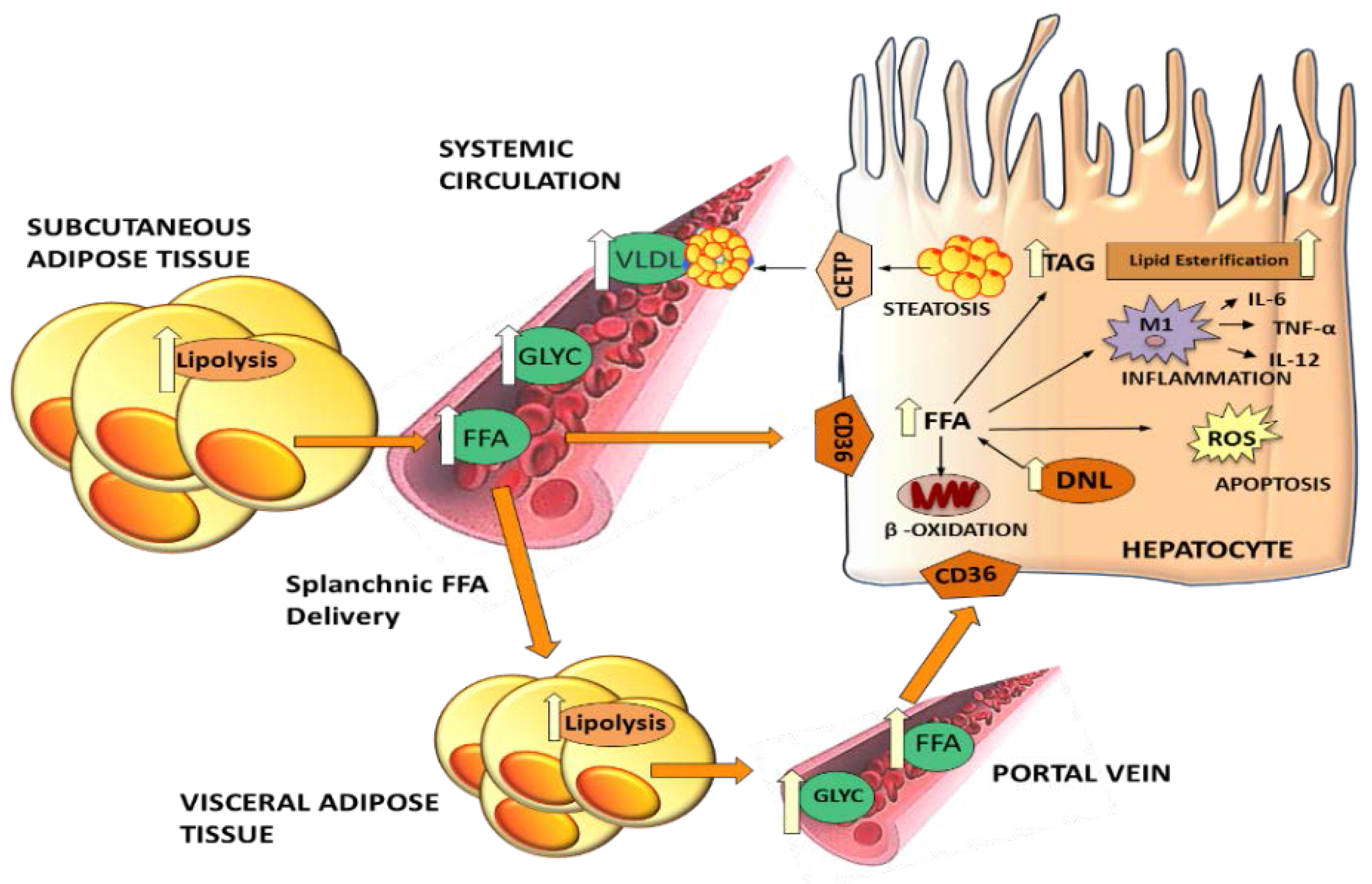
2.3. Hepatic TG Secretion as Very Low Density Lipoproteins (VLDL)
3. Lipolysis
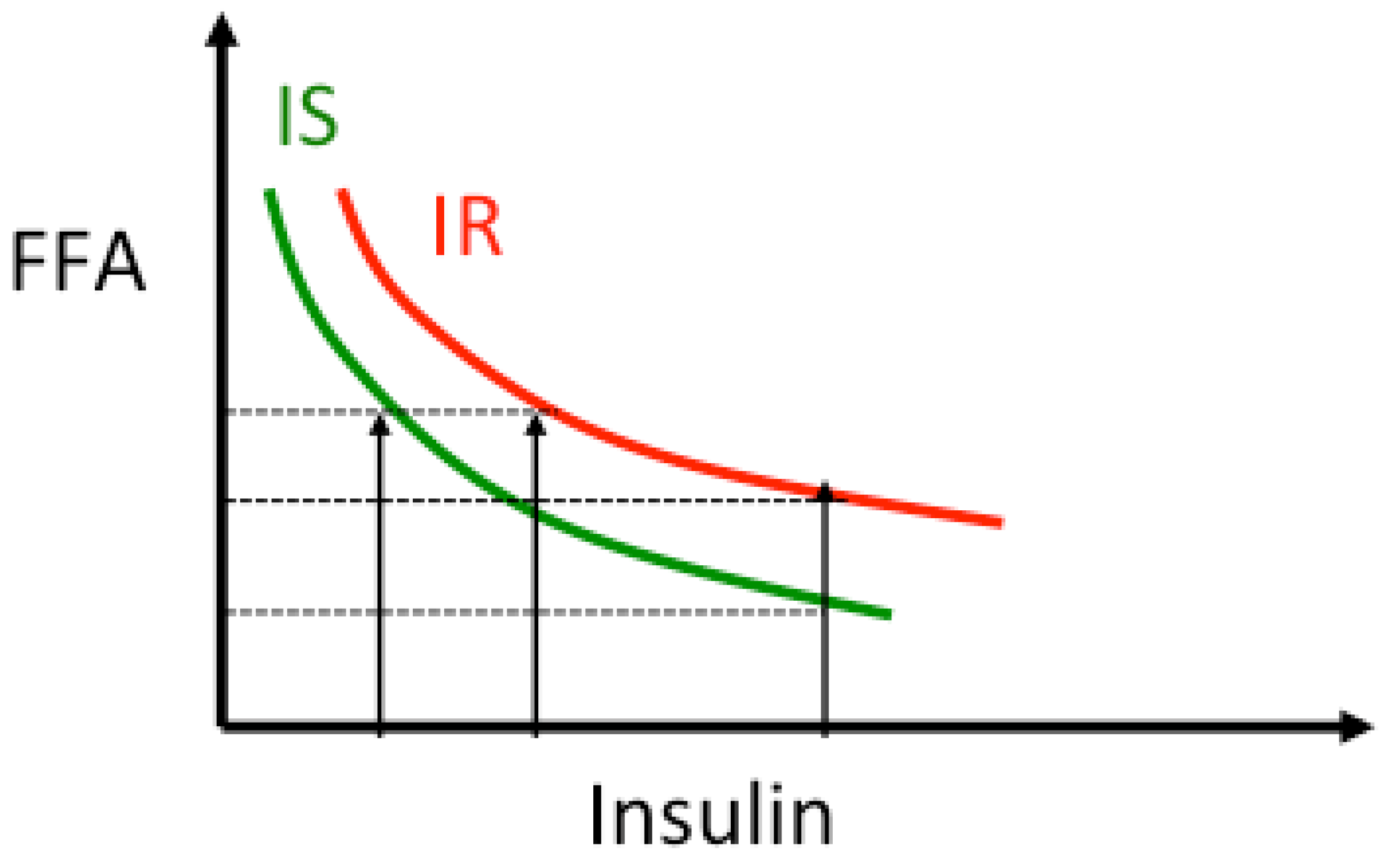
4. Insulin Resistance and Lipolysis

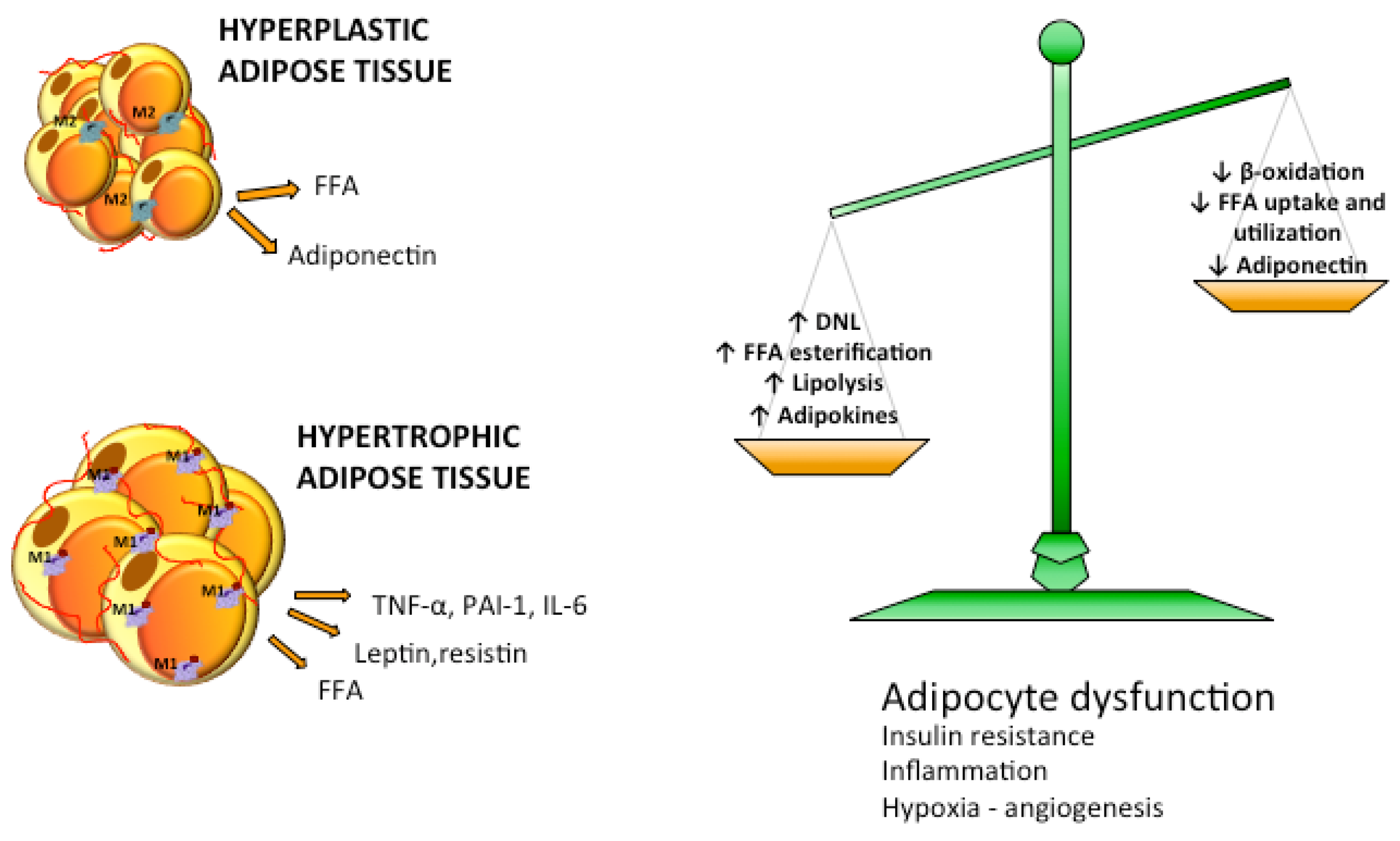
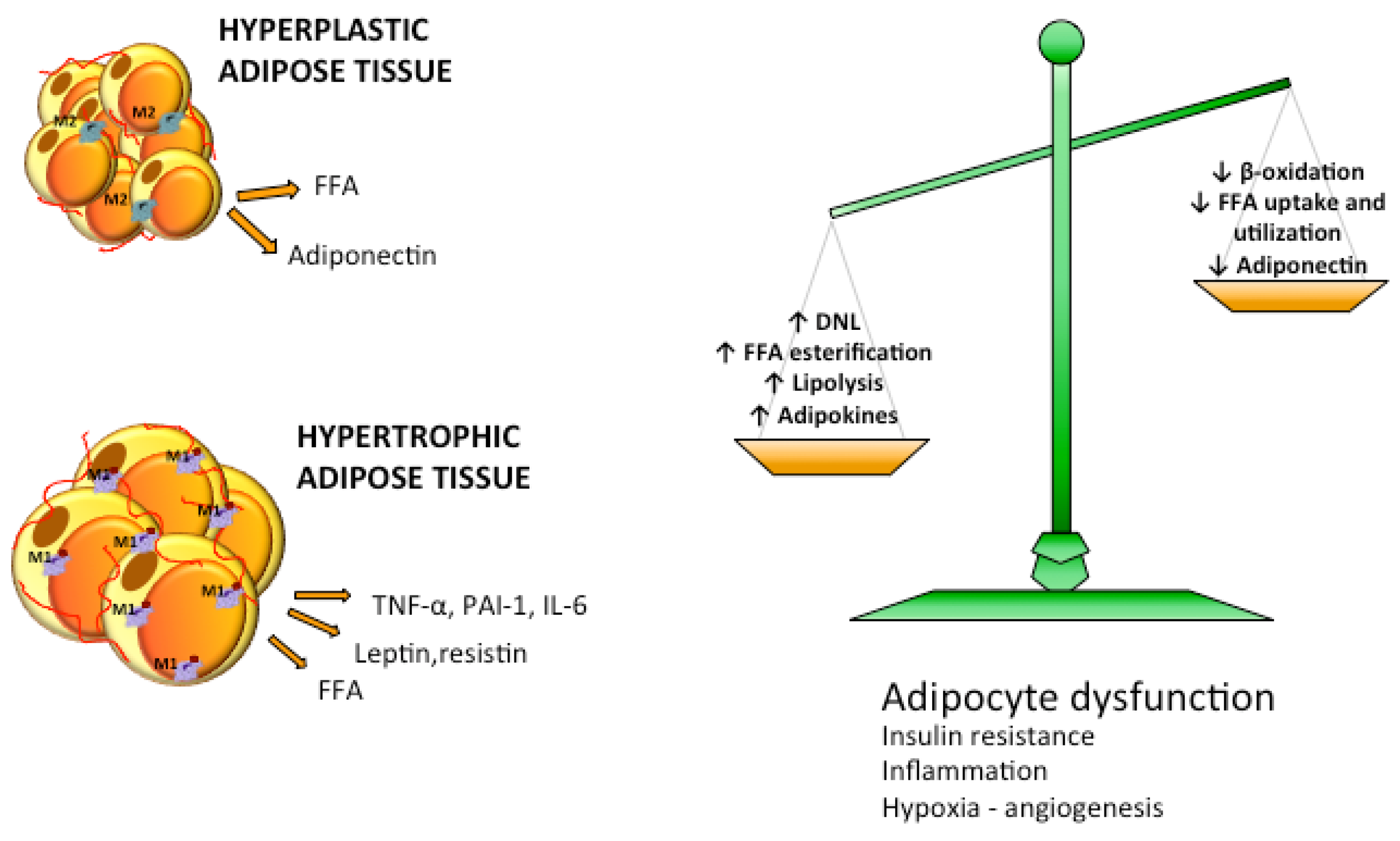
5. Dysfunctional Adipose Tissue: Accumulation and Remodeling
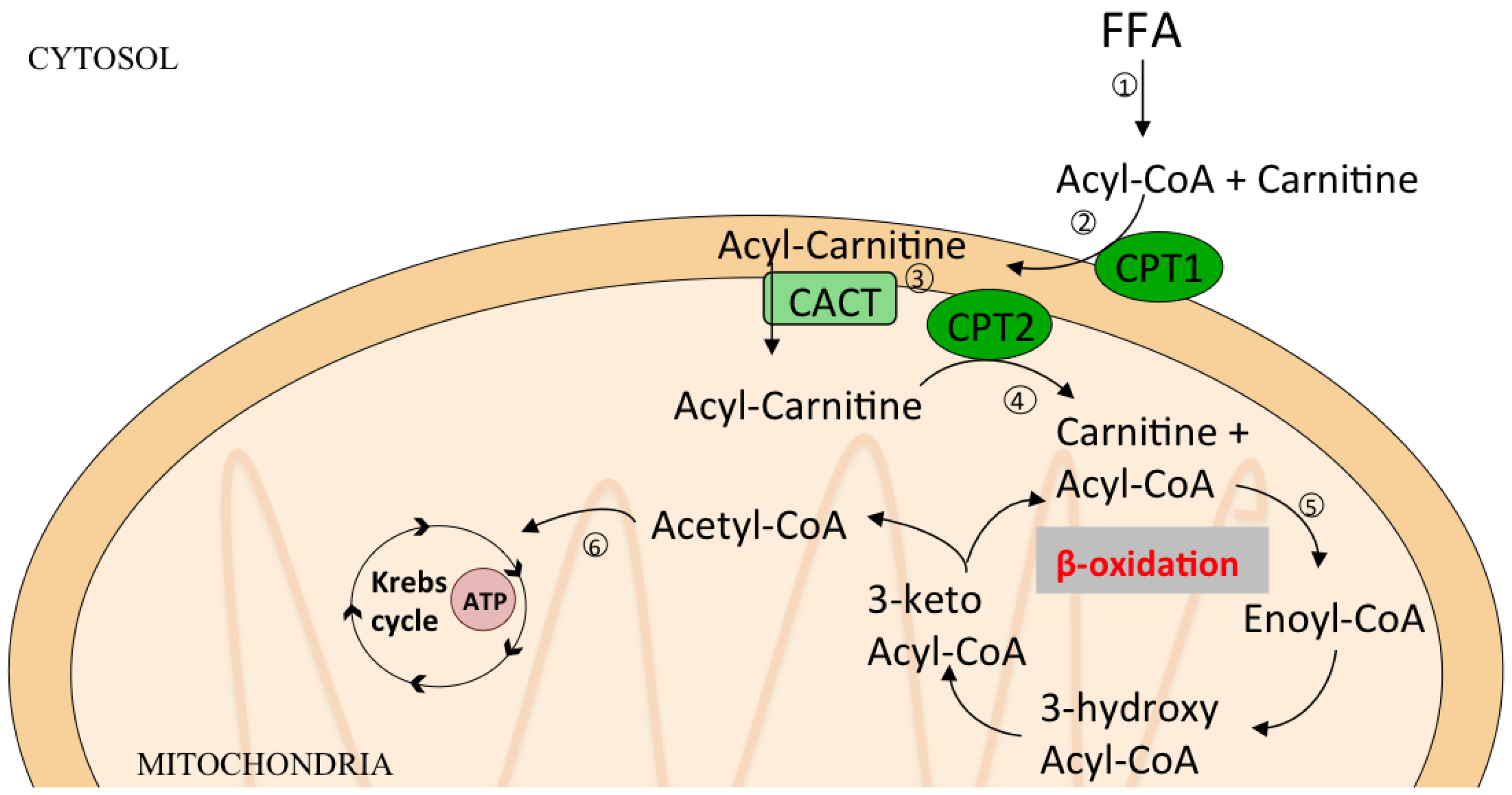
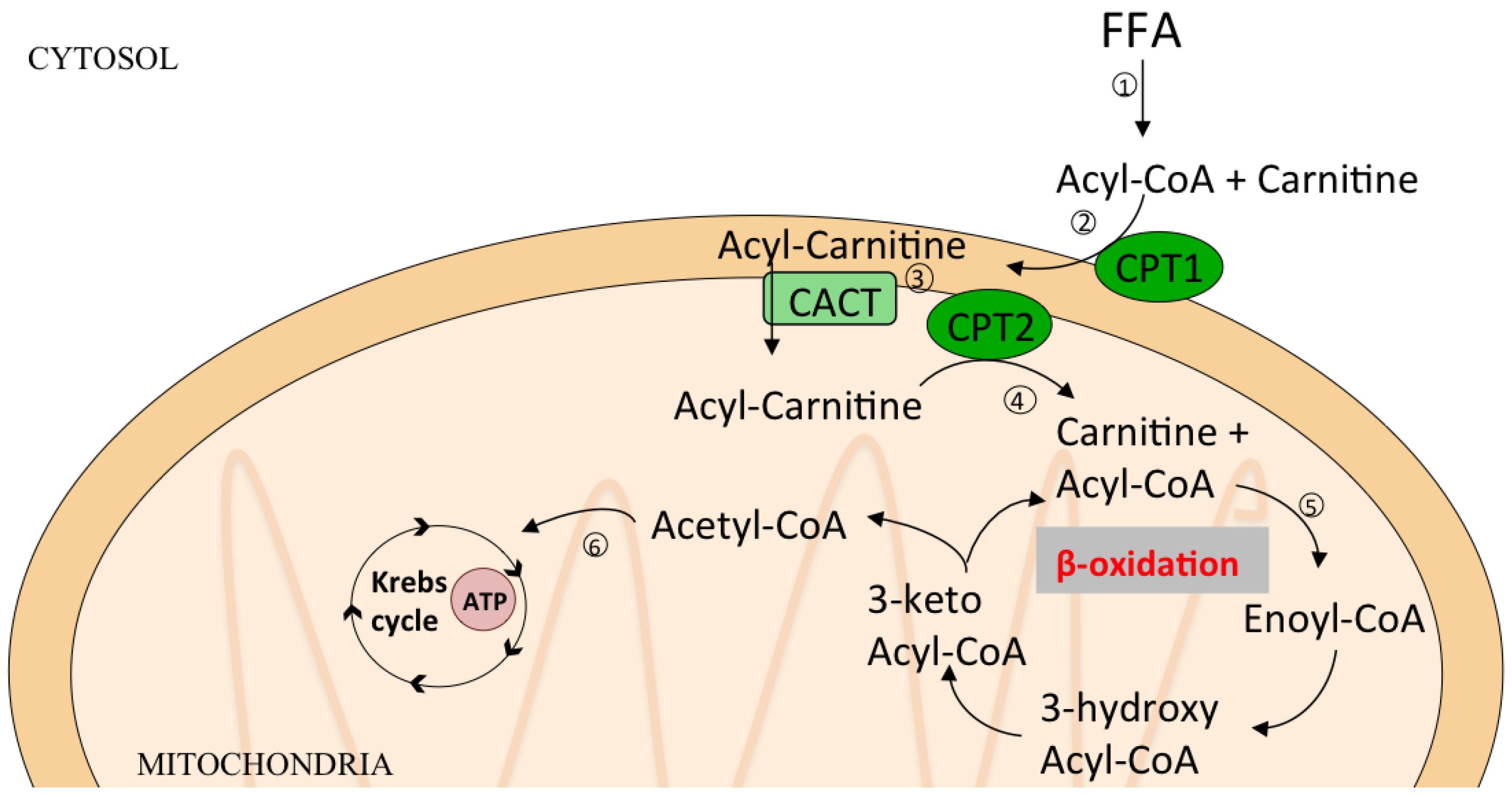
6. Lipid Oxidation
7. Saturated or Unsaturated Fat?
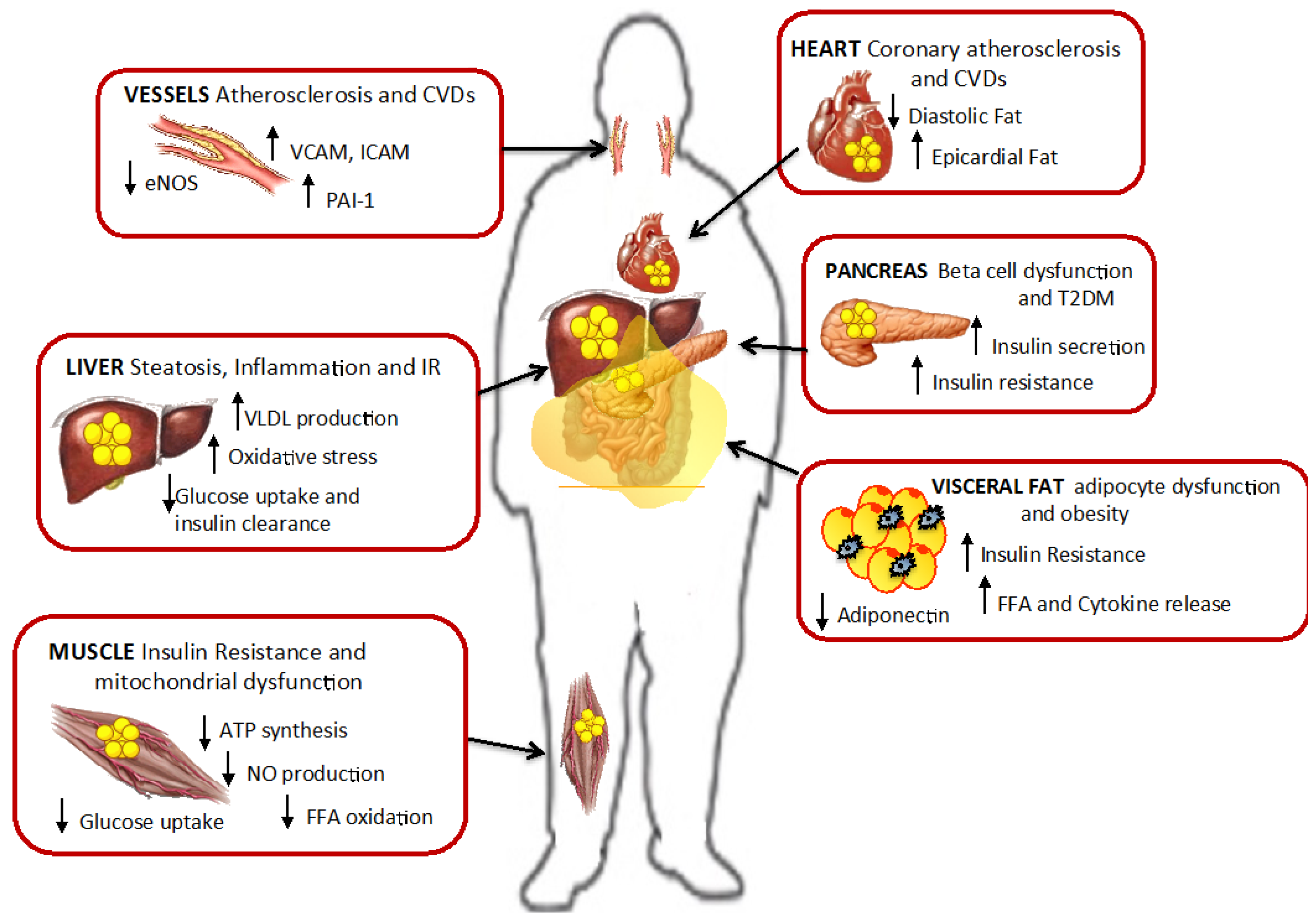
8. Lipotoxicity: Causes and Consequences
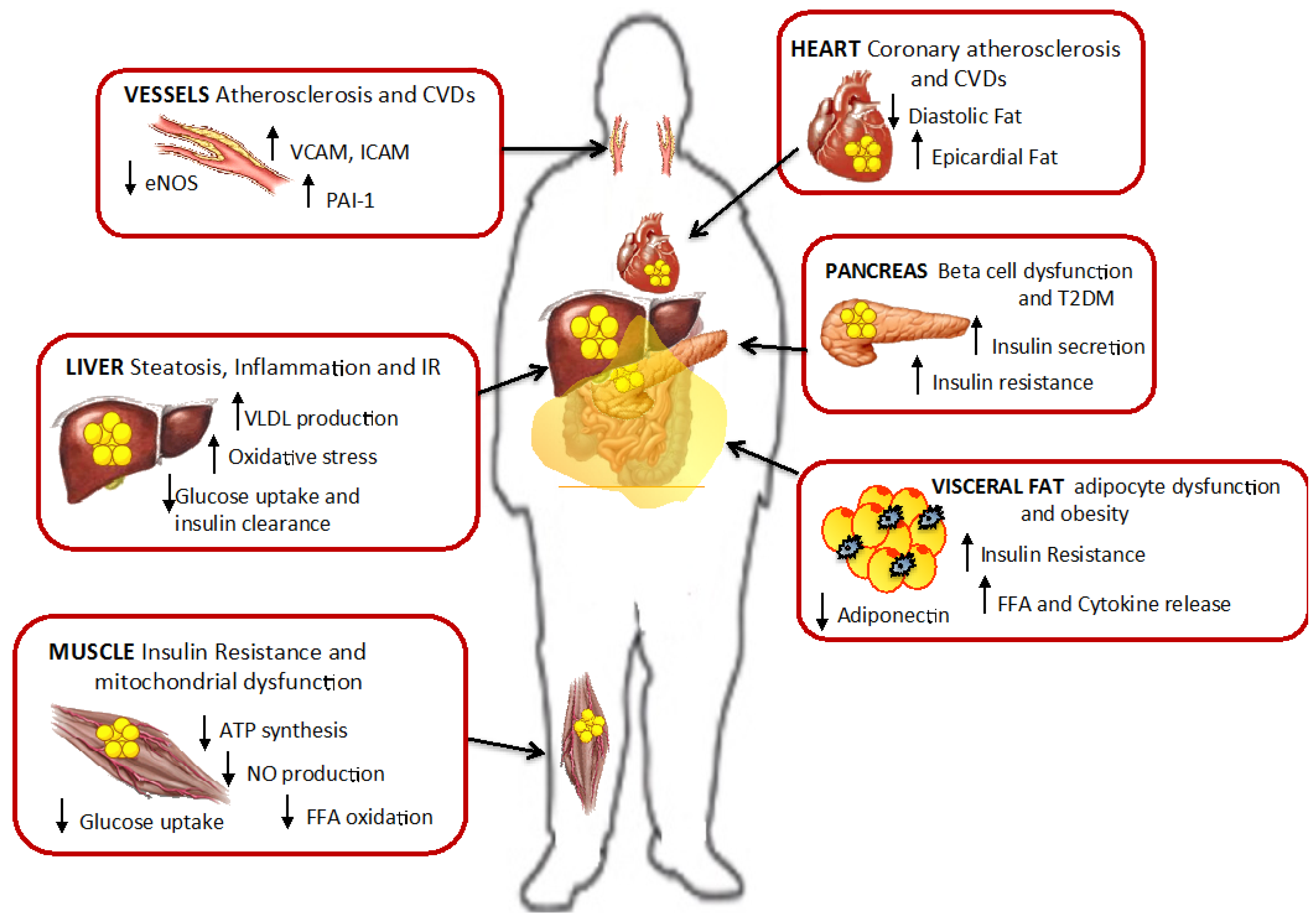
8.1. The Role of FFA in Lipotoxicity
8.2. Lipidomics and the Discovery of Harmful Lipids
9. Summary and Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
References
- Morelli, M.; Gaggini, M.; Daniele, G.; Marraccini, P.; Sicari, R.; Gastaldelli, A. Ectopic fat: The true culprit linking obesity and cardiovascular disease? Thromb. Haemost 2013, 110, 651–660. [Google Scholar] [CrossRef] [PubMed]
- Gaggini, M.; Saponaro, C.; Gastaldelli, A. Not all fats are created equal: Adipose vs. Ectopic fat, implication in cardiometabolic diseases. Horm. Mol. Biol. Clin. Investig. 2015, 22, 7–18. [Google Scholar] [CrossRef] [PubMed]
- Gaggini, M.; Morelli, M.; Buzzigoli, E.; DeFronzo, R.A.; Bugianesi, E.; Gastaldelli, A. Non-alcoholic fatty liver disease (NAFLD) and its connection with insulin resistance, dyslipidemia, atherosclerosis and coronary heart disease. Nutrients 2013, 5, 1544–1560. [Google Scholar] [CrossRef] [PubMed]
- Yki-Jarvinen, H. Non-alcoholic fatty liver disease as a cause and a consequence of metabolic syndrome. Lancet Diabetes Endocrinol. 2014, 2, 901–910. [Google Scholar] [CrossRef]
- Gastaldelli, A. Visceral adipose tissue and ectopic fat deposition. In Handbook of Obesity; Bray, G.A., Bouchard, C., Eds.; CRC Press: Boca Raton, FL, USA, 2014; Volume 1, pp. 237–248. [Google Scholar]
- Lim, E.L.; Hollingsworth, K.G.; Aribisala, B.S.; Chen, M.J.; Mathers, J.C.; Taylor, R. Reversal of type 2 diabetes: Normalisation of beta cell function in association with decreased pancreas and liver triacylglycerol. Diabetologia 2011, 54, 2506–2514. [Google Scholar] [CrossRef] [PubMed]
- Gastaldelli, A.; Morales, M.A.; Marraccini, P.; Sicari, R. The role of cardiac fat in insulin resistance. Curr. Opin. Clin. Nutr. Metab. Care 2012, 15, 523–528. [Google Scholar] [CrossRef] [PubMed]
- Gaborit, B.; Kober, F.; Jacquier, A.; Moro, P.J.; Cuisset, T.; Boullu, S.; Dadoun, F.; Alessi, M.C.; Morange, P.; Clement, K.; et al. Assessment of epicardial fat volume and myocardial triglyceride content in severely obese subjects: Relationship to metabolic profile, cardiac function and visceral fat. Int. J. Obes. 2012, 36, 422–430. [Google Scholar] [CrossRef] [PubMed]
- Boden, G.; Lebed, B.; Schatz, M.; Homko, C.; Lemieux, S. Effects of acute changes of plasma free fatty acids on intramyocellular fat content and insulin resistance in healthy subjects. Diabetes 2001, 50, 1612–1617. [Google Scholar] [CrossRef] [PubMed]
- Schrauwen, P.; Hesselink, M.K. Oxidative capacity, lipotoxicity, and mitochondrial damage in type 2 diabetes. Diabetes 2004, 53, 1412–1417. [Google Scholar] [CrossRef] [PubMed]
- Boden, G. Obesity, insulin resistance and free fatty acids. Curr. Opin. Endocrinol. Diabetes Obes. 2011, 18, 139–143. [Google Scholar] [CrossRef] [PubMed]
- Hanson, R.W.; Reshef, L. Glyceroneogenesis revisited. Biochimie 2003, 85, 1199–1205. [Google Scholar] [CrossRef] [PubMed]
- Kalhan, S.C.; Bugianesi, E.; McCullough, A.J.; Hanson, R.W.; Kelley, D.E. Estimates of hepatic glyceroneogenesis in type 2 diabetes mellitus in humans. Metabolism 2008, 57, 305–312. [Google Scholar] [CrossRef] [PubMed]
- Gastaldelli, A.; Cusi, K.; Pettiti, M.; Hardies, J.; Miyazaki, Y.; Berria, R.; Buzzigoli, E.; Sironi, A.M.; Cersosimo, E.; Ferrannini, E.; et al. Relationship between hepatic/visceral fat and hepatic insulin resistance in nondiabetic and type 2 diabetic subjects. Gastroenterology 2007, 133, 496–506. [Google Scholar] [CrossRef] [PubMed]
- Gastaldelli, A.; Miyazaki, Y.; Pettiti, M.; Santini, E.; Ciociaro, D.; Defronzo, R.A.; Ferrannini, E. The effect of rosiglitazone on the liver: Decreased gluconeogenesis in patients with type 2 diabetes. J. Clin. Endocrinol. Metab. 2006, 91, 806–812. [Google Scholar] [CrossRef] [PubMed]
- Belfort, R.; Harrison, S.A.; Brown, K.; Darland, C.; Finch, J.; Hardies, J.; Balas, B.; Gastaldelli, A.; Tio, F.; Pulcini, J.; et al. A placebo-controlled trial of pioglitazone in subjects with nonalcoholic steatohepatitis. N. Engl. J. Med. 2006, 355, 2297–2307. [Google Scholar] [CrossRef] [PubMed]
- Sanyal, A.J.; Chalasani, N.; Kowdley, K.V.; McCullough, A.; Diehl, A.M.; Bass, N.M.; Neuschwander-Tetri, B.A.; Lavine, J.E.; Tonascia, J.; Unalp, A.; et al. Pioglitazone, vitamin E, or placebo for nonalcoholic steatohepatitis. N. Engl. J. Med. 2010, 362, 1675–1685. [Google Scholar] [CrossRef] [PubMed]
- Tordjman, J.; Chauvet, G.; Quette, J.; Beale, E.G.; Forest, C.; Antoine, B. Thiazolidinediones block fatty acid release by inducing glyceroneogenesis in fat cells. J. Biol. Chem. 2003, 278, 18785–18790. [Google Scholar] [CrossRef] [PubMed]
- Hellerstein, M.K. De novo lipogenesis in humans: Metabolic and regulatory aspects. Eur. J. Clin. Nutr. 1999, 53 (Suppl. 1), S53–S65. [Google Scholar] [CrossRef] [PubMed]
- Donnelly, K.L.; Smith, C.I.; Schwarzenberg, S.J.; Jessurun, J.; Boldt, M.D.; Parks, E.J. Sources of fatty acids stored in liver and secreted via lipoproteins in patients with nonalcoholic fatty liver disease. J. Clin. Investig. 2005, 115, 1343–1351. [Google Scholar] [CrossRef] [PubMed]
- Timlin, M.T.; Parks, E.J. Temporal pattern of de novo lipogenesis in the postprandial state in healthy men. Am. J. Clin. Nutr. 2005, 81, 35–42. [Google Scholar] [PubMed]
- Flannery, C.; Dufour, S.; Rabol, R.; Shulman, G.I.; Petersen, K.F. Skeletal muscle insulin resistance promotes increased hepatic de novo lipogenesis, hyperlipidemia, and hepatic steatosis in the elderly. Diabetes 2012, 61, 2711–2717. [Google Scholar] [CrossRef] [PubMed]
- Ma, W.; Wu, J.H.; Wang, Q.; Lemaitre, R.N.; Mukamal, K.J.; Djousse, L.; King, I.B.; Song, X.; Biggs, M.L.; Delaney, J.A.; et al. Prospective association of fatty acids in the de novo lipogenesis pathway with risk of type 2 diabetes: The cardiovascular health study. Am. J. Clin. Nutr. 2015, 101, 153–163. [Google Scholar] [CrossRef] [PubMed]
- Cai, D.; Yuan, M.; Frantz, D.F.; Melendez, P.A.; Hansen, L.; Lee, J.; Shoelson, S.E. Local and systemic insulin resistance resulting from hepatic activation of ikk-beta and nf-kappab. Nat. Med. 2005, 11, 183–190. [Google Scholar] [CrossRef] [PubMed]
- Marques-Lopes, I.; Ansorena, D.; Astiasaran, I.; Forga, L.; Martinez, J.A. Postprandial de novo lipogenesis and metabolic changes induced by a high-carbohydrate, low-fat meal in lean and overweight men. Am. J. Clin. Nutr. 2001, 73, 253–261. [Google Scholar] [PubMed]
- Bechmann, L.P.; Hannivoort, R.A.; Gerken, G.; Hotamisligil, G.S.; Trauner, M.; Canbay, A. The interaction of hepatic lipid and glucose metabolism in liver diseases. J. Hepatol. 2012, 56, 952–964. [Google Scholar] [CrossRef] [PubMed]
- Shimano, H.; Yahagi, N.; Amemiya-Kudo, M.; Hasty, A.H.; Osuga, J.; Tamura, Y.; Shionoiri, F.; Iizuka, Y.; Ohashi, K.; Harada, K.; et al. Sterol regulatory element-binding protein-1 as a key transcription factor for nutritional induction of lipogenic enzyme genes. J. Biol. Chem. 1999, 274, 35832–35839. [Google Scholar] [CrossRef] [PubMed]
- Dentin, R.; Pegorier, J.P.; Benhamed, F.; Foufelle, F.; Ferre, P.; Fauveau, V.; Magnuson, M.A.; Girard, J.; Postic, C. Hepatic glucokinase is required for the synergistic action of chrebp and SREBP-1c on glycolytic and lipogenic gene expression. J. Biol. Chem. 2004, 279, 20314–20326. [Google Scholar] [CrossRef] [PubMed]
- Herman, M.A.; Peroni, O.D.; Villoria, J.; Schon, M.R.; Abumrad, N.A.; Bluher, M.; Klein, S.; Kahn, B.B. A novel chrebp isoform in adipose tissue regulates systemic glucose metabolism. Nature 2012, 484, 333–338. [Google Scholar] [CrossRef] [PubMed]
- Musso, G.; Gambino, R.; Cassader, M. Recent insights into hepatic lipid metabolism in non-alcoholic fatty liver disease (NAFLD). Prog. Lipid. Res. 2009, 48, 1–26. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, J.M.; Linfoot, P.; Dare, D.; Aghajanian, K. Hepatic de novo lipogenesis in normoinsulinemic and hyperinsulinemic subjects consuming high-fat, low-carbohydrate and low-fat, high-carbohydrate isoenergetic diets. Am. J. Clin. Nutr. 2003, 77, 43–50. [Google Scholar] [PubMed]
- Shimomura, I.; Matsuda, M.; Hammer, R.E.; Bashmakov, Y.; Brown, M.S.; Goldstein, J.L. Decreased IRS-2 and increased SREBP-1c lead to mixed insulin resistance and sensitivity in livers of lipodystrophic and ob/ob mice. Mol. Cell 2000, 6, 77–86. [Google Scholar] [CrossRef]
- Boden, G.; Salehi, S.; Cheung, P.; Homko, C.; Song, W.; Loveland-Jones, C.; Jayarajan, S. Comparison of in vivo effects of insulin on SREBP-1c activation and INSIG-1/2 in rat liver and human and rat adipose tissue. Obesity 2013, 21, 1208–1214. [Google Scholar] [CrossRef] [PubMed]
- Guo, Z.K.; Cella, L.K.; Baum, C.; Ravussin, E.; Schoeller, D.A. De novo lipogenesis in adipose tissue of lean and obese women: Application of deuterated water and isotope ratio mass spectrometry. Int. J. Obes. Relat. Metab. Disord. 2000, 24, 932–937. [Google Scholar] [CrossRef] [PubMed]
- Garrido-Sanchez, L.; Vendrell, J.; Fernandez-Garcia, D.; Ceperuelo-Mallafre, V.; Chacon, M.R.; Ocana-Wilhelmi, L.; Alcaide, J.; Tinahones, F.J.; Garcia-Fuentes, E. De novo lipogenesis in adipose tissue is associated with course of morbid obesity after bariatric surgery. PLoS ONE 2012, 7, e31280. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, M.J.; Hull, D.; Guo, K.; Barton, D.; Hazlehurst, J.M.; Gathercole, L.L.; Nasiri, M.; Yu, J.; Gough, S.C.; Newsome, P.N.; et al. Glucagon-like peptide 1 decreases lipotoxicity in non-alcoholic steatophepatitis. J. Hepatol. 2015. [Google Scholar] [CrossRef] [PubMed]
- Beysen, C.; Murphy, E.J.; Nagaraja, H.; Decaris, M.; Riiff, T.; Fong, A.; Hellerstein, M.K.; Boyle, P.J. A pilot study of the effects of pioglitazone and rosiglitazone on de novo lipogenesis in type 2 diabetes. J. Lipid. Res. 2008, 49, 2657–2663. [Google Scholar] [CrossRef] [PubMed]
- Krauss, R.M. Lipids and lipoproteins in patients with type 2 diabetes. Diabetes Care 2004, 27, 1496–1504. [Google Scholar] [CrossRef] [PubMed]
- Van de Weijer, T.; Schrauwen-Hinderling, V.B.; Schrauwen, P. Lipotoxicity in type 2 diabetic cardiomyopathy. Cardiovasc Res. 2011, 92, 10–18. [Google Scholar] [CrossRef] [PubMed]
- Zammit, V.A. Hepatic triacylglycerol synthesis and secretion: DGAT2 as the link between glycaemia and triglyceridaemia. Biochem. J. 2013, 451, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Yen, C.L.; Nelson, D.W.; Yen, M.I. Intestinal triacylglycerol synthesis in fat absorption and systemic energy metabolism. J. Lipid Res. 2015, 56, 489–501. [Google Scholar] [CrossRef] [PubMed]
- Perry, R.J.; Samuel, V.T.; Petersen, K.F.; Shulman, G.I. The role of hepatic lipids in hepatic insulin resistance and type 2 diabetes. Nature 2014, 510, 84–91. [Google Scholar] [CrossRef] [PubMed]
- Adiels, M.; Taskinen, M.R.; Packard, C.; Caslake, M.J.; Soro-Paavonen, A.; Westerbacka, J.; Vehkavaara, S.; Hakkinen, A.; Olofsson, S.O.; Yki-Jarvinen, H.; et al. Overproduction of large vldl particles is driven by increased liver fat content in man. Diabetologia 2006, 49, 755–765. [Google Scholar] [CrossRef] [PubMed]
- Fabbrini, E.; Mohammed, B.S.; Magkos, F.; Korenblat, K.M.; Patterson, B.W.; Klein, S. Alterations in adipose tissue and hepatic lipid kinetics in obese men and women with nonalcoholic fatty liver disease. Gastroenterology 2008, 134, 424–431. [Google Scholar] [CrossRef] [PubMed]
- Wree, A.; Schlattjan, M.; Bechmann, L.P.; Claudel, T.; Sowa, J.P.; Stojakovic, T.; Scharnagl, H.; Kofeler, H.; Baba, H.A.; Gerken, G.; et al. Adipocyte cell size, free fatty acids and apolipoproteins are associated with non-alcoholic liver injury progression in severely obese patients. Metabolism 2014, 63, 1542–1552. [Google Scholar] [CrossRef] [PubMed]
- Mahdessian, H.; Taxiarchis, A.; Popov, S.; Silveira, A.; Franco-Cereceda, A.; Hamsten, A.; Eriksson, P.; van’t Hooft, F. TM6SF2 is a regulator of liver fat metabolism influencing triglyceride secretion and hepatic lipid droplet content. Proc. Natl. Acad. Sci. USA 2014, 111, 8913–8918. [Google Scholar] [CrossRef] [PubMed]
- Dongiovanni, P.; Petta, S.; Maglio, C.; Fracanzani, A.L.; Pipitone, R.; Mozzi, E.; Motta, B.M.; Kaminska, D.; Rametta, R.; Grimaudo, S.; et al. Transmembrane 6 superfamily member 2 gene variant disentangles nonalcoholic steatohepatitis from cardiovascular disease. Hepatology 2015, 61, 506–514. [Google Scholar] [CrossRef] [PubMed]
- Mittendorfer, B.; Magkos, F.; Fabbrini, E.; Mohammed, B.S.; Klein, S. Relationship between body fat mass and free fatty acid kinetics in men and women. Obesity 2009, 17, 1872–1877. [Google Scholar] [CrossRef] [PubMed]
- Smirnova, E.; Goldberg, E.B.; Makarova, K.S.; Lin, L.; Brown, W.J.; Jackson, C.L. ATGL has a key role in lipid droplet/adiposome degradation in mammalian cells. EMBO Rep. 2006, 7, 106–113. [Google Scholar] [CrossRef] [PubMed]
- Haemmerle, G.; Lass, A.; Zimmermann, R.; Gorkiewicz, G.; Meyer, C.; Rozman, J.; Heldmaier, G.; Maier, R.; Theussl, C.; Eder, S.; et al. Defective lipolysis and altered energy metabolism in mice lacking adipose triglyceride lipase. Science 2006, 312, 734–737. [Google Scholar] [CrossRef] [PubMed]
- Shaw, C.S.; Clark, J.A.; Shepherd, S.O. HSL and ATGL: The movers and shakers of muscle lipolysis. J. Physiol. 2013, 591, 6137–6138. [Google Scholar] [CrossRef] [PubMed]
- Huijsman, E.; van de Par, C.; Economou, C.; van der Poel, C.; Lynch, G.S.; Schoiswohl, G.; Haemmerle, G.; Zechner, R.; Watt, M.J. Adipose triacylglycerol lipase deletion alters whole body energy metabolism and impairs exercise performance in mice. Am. J. Physiol. Endocrinol. Metab. 2009, 297, E505–E513. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.M.; Betters, J.L.; Lord, C.; Ma, Y.; Han, X.; Yang, K.; Alger, H.M.; Melchior, J.; Sawyer, J.; Shah, R.; et al. CGI-58 knockdown in mice causes hepatic steatosis but prevents diet-induced obesity and glucose intolerance. J. Lipid Res. 2010, 51, 3306–3315. [Google Scholar] [CrossRef] [PubMed]
- Ong, K.T.; Mashek, M.T.; Bu, S.Y.; Greenberg, A.S.; Mashek, D.G. Adipose triglyceride lipase is a major hepatic lipase that regulates triacylglycerol turnover and fatty acid signaling and partitioning. Hepatology 2010, 53, 116–126. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.W.; Wang, S.P.; Alvarez, F.; Casavant, S.; Gauthier, N.; Abed, L.; Soni, K.G.; Yang, G.; Mitchell, G.A. Deficiency of liver adipose triglyceride lipase in mice causes progressive hepatic steatosis. Hepatology 2011, 54, 122–132. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, C.D.; Claudel, T.; Kumari, P.; Haemmerle, G.; Pollheimer, M.J.; Stojakovic, T.; Scharnagl, H.; Halilbasic, E.; Gumhold, J.; Silbert, D.; et al. Absence of adipose triglyceride lipase protects from hepatic endoplasmic reticulum stress in mice. Hepatology 2012, 56, 270–280. [Google Scholar] [CrossRef] [PubMed]
- Pulinilkunnil, T.; Kienesberger, P.C.; Nagendran, J.; Sharma, N.; Young, M.E.; Dyck, J.R. Cardiac-specific adipose triglyceride lipase overexpression protects from cardiac steatosis and dilated cardiomyopathy following diet-induced obesity. Int. J. Obes. 2013, 38, 205–215. [Google Scholar] [CrossRef] [PubMed]
- Kraemer, F.B.; Shen, W.J. Hormone-sensitive lipase: Control of intracellular tri-(di-)acylglycerol and cholesteryl ester hydrolysis. J. Lipid. Res. 2002, 43, 1585–1594. [Google Scholar] [CrossRef] [PubMed]
- Osuga, J.; Ishibashi, S.; Oka, T.; Yagyu, H.; Tozawa, R.; Fujimoto, A.; Shionoiri, F.; Yahagi, N.; Kraemer, F.B.; Tsutsumi, O.; et al. Targeted disruption of hormone-sensitive lipase results in male sterility and adipocyte hypertrophy, but not in obesity. Proc. Natl. Acad. Sci. USA 2000, 97, 787–792. [Google Scholar] [CrossRef] [PubMed]
- Haemmerle, G.; Zimmermann, R.; Hayn, M.; Theussl, C.; Waeg, G.; Wagner, E.; Sattler, W.; Magin, T.M.; Wagner, E.F.; Zechner, R. Hormone-sensitive lipase deficiency in mice causes diglyceride accumulation in adipose tissue, muscle, and testis. J. Biol. Chem. 2002, 277, 4806–4815. [Google Scholar] [CrossRef] [PubMed]
- Gastaldelli, A.; Coggan, A.R.; Wolfe, R.R. Assessment of methods for improving tracer estimation of non-steady-state rate of appearance. J. Appl. Physiol. 1999, 87, 1813–1822. [Google Scholar] [PubMed]
- Natali, A.; Gastaldelli, A.; Galvan, A.Q.; Sironi, A.M.; Ciociaro, D.; Sanna, G.; Rosenzweig, P.; Ferrannini, E. Effects of acute alpha 2-blockade on insulin action and secretion in humans. Am. J. Physiol. 1998, 274, E57–E64. [Google Scholar] [PubMed]
- Karpe, F.; Dickmann, J.R.; Frayn, K.N. Fatty acids, obesity, and insulin resistance: Time for a reevaluation. Diabetes 2011, 60, 2441–2449. [Google Scholar] [CrossRef] [PubMed]
- Gastaldelli, A. Role of beta-cell dysfunction, ectopic fat accumulation and insulin resistance in the pathogenesis of type 2 diabetes mellitus. Diabetes Res. Clin. Pract. 2011, 93 (Suppl. 1), S60–S65. [Google Scholar] [CrossRef]
- Groop, L.C.; Bonadonna, R.C.; DelPrato, S.; Ratheiser, K.; Zyck, K.; Ferrannini, E.; DeFronzo, R.A. Glucose and free fatty acid metabolism in non-insulin-dependent diabetes mellitus. Evidence for multiple sites of insulin resistance. J. Clin. Investig. 1989, 84, 205–213. [Google Scholar] [CrossRef] [PubMed]
- Brehm, A.; Krssak, M.; Schmid, A.I.; Nowotny, P.; Waldhausl, W.; Roden, M. Increased lipid availability impairs insulin-stimulated ATP synthesis in human skeletal muscle. Diabetes 2006, 55, 136–140. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.L.; Zhang, L.; Youker, K.; Zhang, M.X.; Wang, J.; LeMaire, S.A.; Coselli, J.S.; Shen, Y.H. Free fatty acids inhibit insulin signaling-stimulated endothelial nitric oxide synthase activation through upregulating Pten or inhibiting Akt kinase. Diabetes 2006, 55, 2301–2310. [Google Scholar] [CrossRef] [PubMed]
- Itani, S.I.; Ruderman, N.B.; Schmieder, F.; Boden, G. Lipid-induced insulin resistance in human muscle is associated with changes in diacylglycerol, protein kinase C, and IκB-α. Diabetes 2002, 51, 2005–2011. [Google Scholar] [CrossRef] [PubMed]
- Bugianesi, E.; Gastaldelli, A.; Vanni, E.; Gambino, R.; Cassader, M.; Baldi, S.; Ponti, V.; Pagano, G.; Ferrannini, E.; Rizzetto, M. Insulin resistance in non-diabetic patients with non-alcoholic fatty liver disease: Sites and mechanisms. Diabetologia 2005, 48, 634–642. [Google Scholar] [CrossRef] [PubMed]
- Fabbrini, E.; Magkos, F.; Conte, C.; Mittendorfer, B.; Patterson, B.W.; Okunade, A.L.; Klein, S. Validation of a novel index to assess insulin resistance of adipose tissue lipolytic activity in obese subjects. J. Lipid Res. 2012, 53, 321–324. [Google Scholar] [CrossRef] [PubMed]
- Fabbrini, E.; Yoshino, J.; Yoshino, M.; Magkos, F.; Luecking, C.T.; Samovski, D.; Fraterrigo, G.; Okunade, A.L.; Patterson, B.W.; Klein, S. Metabolically normal obese people are protected from adverse effects following weight gain. J. Clin. Investig. 2015, 125, 787–795. [Google Scholar] [CrossRef] [PubMed]
- Kashyap, S.; Belfort, R.; Gastaldelli, A.; Pratipanawatr, T.; Berria, R.; Pratipanawatr, W.; Bajaj, M.; Mandarino, L.; DeFronzo, R.; Cusi, K. A sustained increase in plasma free fatty acids impairs insulin secretion in nondiabetic subjects genetically predisposed to develop type 2 diabetes. Diabetes 2003, 52, 2461–2474. [Google Scholar] [CrossRef] [PubMed]
- Lupi, R.; Dotta, F.; Marselli, L.; Del Guerra, S.; Masini, M.; Santangelo, C.; Patane, G.; Boggi, U.; Piro, S.; Anello, M.; et al. Prolonged exposure to free fatty acids has cytostatic and pro-apoptotic effects on human pancreatic islets: Evidence that beta-cell death is caspase mediated, partially dependent on ceramide pathway, and Bcl-2 regulated. Diabetes 2002, 51, 1437–1442. [Google Scholar] [CrossRef] [PubMed]
- Marques, B.G.; Hausman, D.B.; Martin, R.J. Association of fat cell size and paracrine growth factors in development of hyperplastic obesity. Am. J. Physiol. 1998, 275, R1898–R1908. [Google Scholar] [PubMed]
- Thorne, A.; Lofgren, P.; Hoffstedt, J. Increased visceral adipocyte lipolysis—A pathogenic role in nonalcoholic fatty liver disease? J. Clin. Endocrinol. Metab. 2010, 95, E209–E213. [Google Scholar] [CrossRef] [PubMed]
- Jensen, M.D.; Haymond, M.W.; Rizza, R.A.; Cryer, P.E.; Miles, J.M. Influence of body fat distribution on free fatty acid metabolism in obesity. J. Clin. Investig. 1989, 83, 1168–1173. [Google Scholar] [CrossRef] [PubMed]
- Spalding, K.L.; Arner, E.; Westermark, P.O.; Bernard, S.; Buchholz, B.A.; Bergmann, O.; Blomqvist, L.; Hoffstedt, J.; Naslund, E.; Britton, T.; et al. Dynamics of fat cell turnover in humans. Nature 2008, 453, 783–787. [Google Scholar] [CrossRef] [PubMed]
- Ye, R.; Scherer, P.E. Adiponectin, driver or passenger on the road to insulin sensitivity? Mol. Metab. 2013, 2, 133–141. [Google Scholar] [CrossRef] [PubMed]
- Arner, E.; Westermark, P.O.; Spalding, K.L.; Britton, T.; Ryden, M.; Frisen, J.; Bernard, S.; Arner, P. Adipocyte turnover: Relevance to human adipose tissue morphology. Diabetes 2010, 59, 105–109. [Google Scholar] [CrossRef] [PubMed]
- Sainz, N.; Barrenetxe, J.; Moreno-Aliaga, M.J.; Martinez, J.A. Leptin resistance and diet-induced obesity: Central and peripheral actions of leptin. Metabolism 2015, 64, 35–46. [Google Scholar] [CrossRef] [PubMed]
- Skurk, T.; Alberti-Huber, C.; Herder, C.; Hauner, H. Relationship between adipocyte size and adipokine expression and secretion. J. Clin. Endocrinol. Metab. 2007, 92, 1023–1033. [Google Scholar] [CrossRef] [PubMed]
- Vargas, G.; Chandalia, M.; Jiang, Y.; Davila, H.; Motamedi, M.; Abate, N. Heterogeneity in subcutaneous adipose tissue morphology and metabolic complications in overweight and obese women. Metab. Syndr. Relat. Disord. 2013, 11, 276–282. [Google Scholar] [CrossRef] [PubMed]
- Alligier, M.; Gabert, L.; Meugnier, E.; Lambert-Porcheron, S.; Chanseaume, E.; Pilleul, F.; Debard, C.; Sauvinet, V.; Morio, B.; Vidal-Puig, A.; et al. Visceral fat accumulation during lipid overfeeding is related to subcutaneous adipose tissue characteristics in healthy men. J. Clin. Endocrinol. Metab. 2013, 98, 802–810. [Google Scholar] [CrossRef] [PubMed]
- Samocha-Bonet, D.; Dixit, V.D.; Kahn, C.R.; Leibel, R.L.; Lin, X.; Nieuwdorp, M.; Pietilainen, K.H.; Rabasa-Lhoret, R.; Roden, M.; Scherer, P.E.; et al. Metabolically healthy and unhealthy obese—The 2013 stock conference report. Obes. Rev. 2014, 15, 697–708. [Google Scholar] [CrossRef] [PubMed]
- McLaughlin, T.; Lamendola, C.; Coghlan, N.; Liu, T.C.; Lerner, K.; Sherman, A.; Cushman, S.W. Subcutaneous adipose cell size and distribution: Relationship to insulin resistance and body fat. Obesity 2014, 22, 673–680. [Google Scholar] [CrossRef] [PubMed]
- Johannsen, D.L.; Tchoukalova, Y.; Tam, C.S.; Covington, J.D.; Xie, W.; Schwarz, J.M.; Bajpeyi, S.; Ravussin, E. Effect of 8 weeks of overfeeding on ectopic fat deposition and insulin sensitivity: Testing the "adipose tissue expandability" hypothesis. Diabetes Care 2014, 37, 2789–2797. [Google Scholar] [CrossRef] [PubMed]
- McLaughlin, T.; Abbasi, F.; Lamendola, C.; Reaven, G. Heterogeneity in the prevalence of risk factors for cardiovascular disease and type 2 diabetes mellitus in obese individuals: Effect of differences in insulin sensitivity. Arch. Intern. Med. 2007, 167, 642–648. [Google Scholar] [CrossRef] [PubMed]
- Scherer, P.E. Adipose tissue: From lipid storage compartment to endocrine organ. Diabetes 2006, 55, 1537–1545. [Google Scholar] [CrossRef] [PubMed]
- Arner, P.; Arner, E.; Hammarstedt, A.; Smith, U. Genetic predisposition for type 2 diabetes, but not for overweight/obesity, is associated with a restricted adipogenesis. PLoS ONE 2011, 6, e18284. [Google Scholar] [CrossRef] [PubMed]
- Arner, P. The adipocyte in insulin resistance: Key molecules and the impact of the thiazolidinediones. Trends Endocrinol. Metab. 2003, 14, 137–145. [Google Scholar] [CrossRef]
- Kanda, H.; Tateya, S.; Tamori, Y.; Kotani, K.; Hiasa, K.; Kitazawa, R.; Kitazawa, S.; Miyachi, H.; Maeda, S.; Egashira, K.; et al. Mcp-1 contributes to macrophage infiltration into adipose tissue, insulin resistance, and hepatic steatosis in obesity. J. Clin. Investig. 2006, 116, 1494–1505. [Google Scholar] [CrossRef] [PubMed]
- Bohm, A.; Halama, A.; Meile, T.; Zdichavsky, M.; Lehmann, R.; Weigert, C.; Fritsche, A.; Stefan, N.; Konigsrainer, A.; Haring, H.U.; et al. Metabolic signatures of cultured human adipocytes from metabolically healthy versus unhealthy obese individuals. PLoS ONE 2014, 9, e93148. [Google Scholar] [CrossRef] [PubMed]
- Sidossis, L.S.; Wolfe, R.R. Glucose and insulin-induced inhibition of fatty acid oxidation: The glucose-fatty acid cycle reversed. Am. J. Physiol. 1996, 270, E733–E738. [Google Scholar] [PubMed]
- Ricchi, M.; Odoardi, M.R.; Carulli, L.; Anzivino, C.; Ballestri, S.; Pinetti, A.; Fantoni, L.I.; Marra, F.; Bertolotti, M.; Banni, S.; et al. Differential effect of oleic and palmitic acid on lipid accumulation and apoptosis in cultured hepatocytes. J. Gastroenterol Hepatol. 2009, 24, 830–840. [Google Scholar] [CrossRef] [PubMed]
- Siriwardhana, N.; Kalupahana, N.S.; Cekanova, M.; LeMieux, M.; Greer, B.; Moustaid-Moussa, N. Modulation of adipose tissue inflammation by bioactive food compounds. J. Nutr. Biochem. 2013, 24, 613–623. [Google Scholar] [CrossRef] [PubMed]
- Zhao, G.; Etherton, T.D.; Martin, K.R.; Vanden Heuvel, J.P.; Gillies, P.J.; West, S.G.; Kris-Etherton, P.M. Anti-inflammatory effects of polyunsaturated fatty acids in THP-1 cells. Biochem. Biophys. Res. Commun. 2005, 336, 909–917. [Google Scholar] [CrossRef] [PubMed]
- Calder, P.C. Polyunsaturated fatty acids and inflammation. Prostaglandins Leukot. Essent. Fatty Acids. 2006, 75, 197–202. [Google Scholar] [CrossRef] [PubMed]
- Mei, S.; Ni, H.M.; Manley, S.; Bockus, A.; Kassel, K.M.; Luyendyk, J.P.; Copple, B.L.; Ding, W.X. Differential roles of unsaturated and saturated fatty acids on autophagy and apoptosis in hepatocytes. J. Pharmacol. Exp. Ther. 2011, 339, 487–498. [Google Scholar] [CrossRef] [PubMed]
- Haus, J.M.; Kashyap, S.R.; Kasumov, T.; Zhang, R.; Kelly, K.R.; Defronzo, R.A.; Kirwan, J.P. Plasma ceramides are elevated in obese subjects with type 2 diabetes and correlate with the severity of insulin resistance. Diabetes 2009, 58, 337–343. [Google Scholar] [CrossRef] [PubMed]
- Kang, M.J.; Shin, M.S.; Park, J.N.; Lee, S.S. The effects of polyunsaturated:Saturated fatty acids ratios and peroxidisability index values of dietary fats on serum lipid profiles and hepatic enzyme activities in rats. Br. J. Nutr. 2005, 94, 526–532. [Google Scholar] [CrossRef] [PubMed]
- Liao, F.H.; Liou, T.H.; Shieh, M.J.; Chien, Y.W. Effects of different ratios of monounsaturated and polyunsaturated fatty acids to saturated fatty acids on regulating body fat deposition in hamsters. Nutrition 2010, 26, 811–817. [Google Scholar] [CrossRef] [PubMed]
- Summers, L.K.; Fielding, B.A.; Bradshaw, H.A.; Ilic, V.; Beysen, C.; Clark, M.L.; Moore, N.R.; Frayn, K.N. Substituting dietary saturated fat with polyunsaturated fat changes abdominal fat distribution and improves insulin sensitivity. Diabetologia 2002, 45, 369–377. [Google Scholar] [CrossRef] [PubMed]
- Hudgins, L.C.; Hellerstein, M.; Seidman, C.; Neese, R.; Diakun, J.; Hirsch, J. Human fatty acid synthesis is stimulated by a eucaloric low fat, high carbohydrate diet. J. Clin. Investig. 1996, 97, 2081–2091. [Google Scholar] [CrossRef] [PubMed]
- Chong, M.F.; Hodson, L.; Bickerton, A.S.; Roberts, R.; Neville, M.; Karpe, F.; Frayn, K.N.; Fielding, B.A. Parallel activation of de novo lipogenesis and stearoyl-CoA desaturase activity after 3 d of high-carbohydrate feeding. Am. J. Clin. Nutr. 2008, 87, 817–823. [Google Scholar] [PubMed]
- Flowers, M.T. The Delta9 fatty acid desaturation index as a predictor of metabolic disease. Clin. Chem. 2009, 55, 2071–2073. [Google Scholar] [CrossRef] [PubMed]
- Warensjo, E.; Rosell, M.; Hellenius, M.L.; Vessby, B.; De Faire, U.; Riserus, U. Associations between estimated fatty acid desaturase activities in serum lipids and adipose tissue in humans: Links to obesity and insulin resistance. Lipids Health. Dis. 2009, 8, 37. [Google Scholar] [CrossRef] [PubMed]
- Mar-Heyming, R.; Miyazaki, M.; Weissglas-Volkov, D.; Kolaitis, N.A.; Sadaat, N.; Plaisier, C.; Pajukanta, P.; Cantor, R.M.; de Bruin, T.W.; Ntambi, J.M.; et al. Association of stearoyl-CoA desaturase 1 activity with familial combined hyperlipidemia. Arterioscler. Thromb. Vasc. Biol. 2008, 28, 1193–1199. [Google Scholar] [CrossRef] [PubMed]
- Riserus, U.; Tan, G.D.; Fielding, B.A.; Neville, M.J.; Currie, J.; Savage, D.B.; Chatterjee, V.K.; Frayn, K.N.; O’Rahilly, S.; Karpe, F. Rosiglitazone increases indexes of stearoyl-CoA desaturase activity in humans: Link to insulin sensitization and the role of dominant-negative mutation in peroxisome proliferator-activated receptor-gamma. Diabetes 2005, 54, 1379–1384. [Google Scholar] [CrossRef] [PubMed]
- Peter, A.; Cegan, A.; Wagner, S.; Lehmann, R.; Stefan, N.; Konigsrainer, A.; Konigsrainer, I.; Haring, H.U.; Schleicher, E. Hepatic lipid composition and stearoyl-coenzyme a desaturase 1 mRNA expression can be estimated from plasma VLDL fatty acid ratios. Clin. Chem. 2009, 55, 2113–2120. [Google Scholar] [CrossRef] [PubMed]
- Attie, A.D.; Krauss, R.M.; Gray-Keller, M.P.; Brownlie, A.; Miyazaki, M.; Kastelein, J.J.; Lusis, A.J.; Stalenhoef, A.F.; Stoehr, J.P.; Hayden, M.R.; et al. Relationship between stearoyl-CoA desaturase activity and plasma triglycerides in human and mouse hypertriglyceridemia. J. Lipid Res. 2002, 43, 1899–1907. [Google Scholar] [CrossRef] [PubMed]
- DeFronzo, R.A. Insulin resistance, lipotoxicity, type 2 diabetes and atherosclerosis: The missing links. The claude bernard lecture 2009. Diabetologia 2010, 53, 1270–1287. [Google Scholar] [CrossRef] [PubMed]
- Belfort, R.; Mandarino, L.; Kashyap, S.; Wirfel, K.; Pratipanawatr, T.; Berria, R.; Defronzo, R.A.; Cusi, K. Dose-response effect of elevated plasma free fatty acid on insulin signaling. Diabetes 2005, 54, 1640–1648. [Google Scholar] [CrossRef] [PubMed]
- Chinen, I.; Shimabukuro, M.; Yamakawa, K.; Higa, N.; Matsuzaki, T.; Noguchi, K.; Ueda, S.; Sakanashi, M.; Takasu, N. Vascular lipotoxicity: Endothelial dysfunction via fatty-acid-induced reactive oxygen species overproduction in obese zucker diabetic fatty rats. Endocrinology 2007, 148, 160–165. [Google Scholar] [CrossRef] [PubMed]
- Inoguchi, T.; Li, P.; Umeda, F.; Yu, H.Y.; Kakimoto, M.; Imamura, M.; Aoki, T.; Etoh, T.; Hashimoto, T.; Naruse, M.; et al. High glucose level and free fatty acid stimulate reactive oxygen species production through protein kinase C—Dependent activation of NAD(P)H oxidase in cultured vascular cells. Diabetes 2000, 49, 1939–1945. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, S.; Tagawa, T.; Yamakawa, K.; Shimabukuro, M.; Ueda, S. Inhibition of the renin-angiotensin system prevents free fatty acid-induced acute endothelial dysfunction in humans. Arterioscler Thromb. Vasc. Biol. 2005, 25, 2376–2380. [Google Scholar] [CrossRef] [PubMed]
- Egan, B.M.; Hennes, M.M.; Stepniakowski, K.T.; O'Shaughnessy, I.M.; Kissebah, A.H.; Goodfriend, T.L. Obesity hypertension is related more to insulin's fatty acid than glucose action. Hypertension 1996, 27, 723–728. [Google Scholar] [CrossRef] [PubMed]
- DeFronzo, R.A.; Tripathy, D. Skeletal muscle insulin resistance is the primary defect in type 2 diabetes. Diabetes Care 2009, 32 Suppl. 2, S157–S163. [Google Scholar] [CrossRef] [PubMed]
- Perseghin, G.; Scifo, P.; De Cobelli, F.; Pagliato, E.; Battezzati, A.; Arcelloni, C.; Vanzulli, A.; Testolin, G.; Pozza, G.; Del Maschio, A.; et al. Intramyocellular triglyceride content is a determinant of in vivo insulin resistance in humans: A 1H-13C nuclear magnetic resonance spectroscopy assessment in offspring of type 2 diabetic parents. Diabetes 1999, 48, 1600–1606. [Google Scholar] [CrossRef] [PubMed]
- Szendroedi, J.; Roden, M. Mitochondrial fitness and insulin sensitivity in humans. Diabetologia 2008, 51, 2155–2167. [Google Scholar] [CrossRef] [PubMed]
- Jensen, M.D. Role of body fat distribution and the metabolic complications of obesity. J. Clin. Endocrinol. Metab. 2008, 93, S57–S63. [Google Scholar] [CrossRef] [PubMed]
- Rahman, R.; Hammoud, G.M.; Almashhrawi, A.A.; Ahmed, K.T.; Ibdah, J.A. Primary hepatocellular carcinoma and metabolic syndrome: An update. World J. Gastrointest Oncol. 2013, 5, 186–194. [Google Scholar] [CrossRef] [PubMed]
- Marra, F.; Gastaldelli, A.; Svegliati Baroni, G.; Tell, G.; Tiribelli, C. Molecular basis and mechanisms of progression of non-alcoholic steatohepatitis. Trends Mol. Med. 2008, 14, 72–81. [Google Scholar] [CrossRef] [PubMed]
- Park, J.W.; Chen, M.; Colombo, M.; Roberts, L.R.; Schwartz, M.; Chen, P.J.; Kudo, M.; Johnson, P.; Wagner, S.; Orsini, L.S.; et al. Global patterns of hepatocellular carcinoma management from diagnosis to death: The bridge study. Liver Int. 2015, 35, 2155–2166. [Google Scholar] [CrossRef] [PubMed]
- De Minicis, S.; Agostinelli, L.; Rychlicki, C.; Sorice, G.P.; Saccomanno, S.; Candelaresi, C.; Giaccari, A.; Trozzi, L.; Pierantonelli, I.; Mingarelli, E.; et al. HCC development is associated to peripheral insulin resistance in a mouse model of NASH. PLoS ONE 2014, 9, e97136. [Google Scholar] [CrossRef] [PubMed]
- Chalasani, N.; Younossi, Z.; Lavine, J.E.; Diehl, A.M.; Brunt, E.M.; Cusi, K.; Charlton, M.; Sanyal, A.J. The diagnosis and management of non-alcoholic fatty liver disease: Practice guideline by the American association for the study of liver diseases, American college of gastroenterology, and the American gastroenterological association. Hepatology 2012, 55, 2005–2023. [Google Scholar] [CrossRef] [PubMed]
- Ertle, J.; Dechene, A.; Sowa, J.P.; Penndorf, V.; Herzer, K.; Kaiser, G.; Schlaak, J.F.; Gerken, G.; Syn, W.K.; Canbay, A. Non-alcoholic fatty liver disease progresses to hepatocellular carcinoma in the absence of apparent cirrhosis. Int. J. Cancer 2011, 128, 2436–2443. [Google Scholar] [CrossRef] [PubMed]
- Dongiovanni, P.; Romeo, S.; Valenti, L. Hepatocellular carcinoma in nonalcoholic fatty liver: Role of environmental and genetic factors. World J. Gastroenterol. 2014, 20, 12945–12955. [Google Scholar] [CrossRef] [PubMed]
- Wong, R.J.; Cheung, R.; Ahmed, A. Nonalcoholic steatohepatitis is the most rapidly growing indication for liver transplantation in patients with hepatocellular carcinoma in the U.S. Hepatology 2014, 59, 2188–2195. [Google Scholar] [CrossRef] [PubMed]
- Basta, G.; Navarra, T.; De Simone, P.; Del Turco, S.; Gastaldelli, A.; Filipponi, F. What is the role of the receptor for advanced glycation end products-ligand axis in liver injury? Liver Transpl. 2011, 17, 633–640. [Google Scholar] [CrossRef] [PubMed]
- Kasumov, T.; Li, L.; Li, M.; Gulshan, K.; Kirwan, J.P.; Liu, X.; Previs, S.; Willard, B.; Smith, J.D.; McCullough, A. Ceramide as a mediator of non-alcoholic fatty liver disease and associated atherosclerosis. PLoS ONE 2015, 10, e0126910. [Google Scholar] [CrossRef] [PubMed]
- Lomonaco, R.; Ortiz-Lopez, C.; Orsak, B.; Webb, A.; Hardies, J.; Darland, C.; Finch, J.; Gastaldelli, A.; Harrison, S.; Tio, F.; et al. Effect of adipose tissue insulin resistance on metabolic parameters and liver histology in obese patients with non-alcoholic fatty liver disease. Hepatology 2012, 55, 1389–1397. [Google Scholar] [CrossRef] [PubMed]
- Sommerfeld, A.; Reinehr, R.; Haussinger, D. Free fatty acids shift insulin-induced hepatocyte proliferation towards CD95-dependent apoptosis. J. Biol. Chem. 2015, 290, 4398–4409. [Google Scholar] [CrossRef] [PubMed]
- Clarke, J.D.; Novak, P.; Lake, A.D.; Shipkova, P.; Aranibar, N.; Robertson, D.; Severson, P.L.; Reily, M.D.; Futscher, B.W.; Lehman-McKeeman, L.D.; et al. Characterization of hepatocellular carcinoma related genes and metabolites in human nonalcoholic fatty liver disease. Dig. Dis. Sci. 2014, 59, 365–374. [Google Scholar] [CrossRef] [PubMed]
- Muir, K.; Hazim, A.; He, Y.; Peyressatre, M.; Kim, D.Y.; Song, X.; Beretta, L. Proteomic and lipidomic signatures of lipid metabolism in NASH-associated hepatocellular carcinoma. Cancer Res. 2013, 73, 4722–4731. [Google Scholar] [CrossRef] [PubMed]
- Sironi, A.M.; Petz, R.; De Marchi, D.; Buzzigoli, E.; Ciociaro, D.; Positano, V.; Lombardi, M.; Ferrannini, E.; Gastaldelli, A. Impact of increased visceral and cardiac fat on cardiometabolic risk and disease. Diabet Med. 2012, 29, 622–627. [Google Scholar] [CrossRef] [PubMed]
- Djaberi, R.; Schuijf, J.D.; van Werkhoven, J.M.; Nucifora, G.; Jukema, J.W.; Bax, J.J. Relation of epicardial adipose tissue to coronary atherosclerosis. Am. J. Cardiol. 2008, 102, 1602–1607. [Google Scholar] [CrossRef] [PubMed]
- Ding, J.; Kritchevsky, S.B.; Harris, T.B.; Burke, G.L.; Detrano, R.C.; Szklo, M.; Jeffrey Carr, J.; Multi-Ethnic Study of Atherosclerosis. The association of pericardial fat with calcified coronary plaque. Obesity 2008, 16, 1914–1919. [Google Scholar] [CrossRef] [PubMed]
- Floegel, A.; Stefan, N.; Yu, Z.; Muhlenbruch, K.; Drogan, D.; Joost, H.G.; Fritsche, A.; Haring, H.U.; Hrabe de Angelis, M.; Peters, A.; et al. Identification of serum metabolites associated with risk of type 2 diabetes using a targeted metabolomic approach. Diabetes 2013, 62, 639–648. [Google Scholar] [CrossRef] [PubMed]
- Wang-Sattler, R.; Yu, Z.; Herder, C.; Messias, A.C.; Floegel, A.; He, Y.; Heim, K.; Campillos, M.; Holzapfel, C.; Thorand, B.; et al. Novel biomarkers for pre-diabetes identified by metabolomics. Mol. Syst. Biol. 2012, 8, 615. [Google Scholar] [CrossRef] [PubMed]
- Ferrannini, E.; Natali, A.; Camastra, S.; Nannipieri, M.; Mari, A.; Adam, K.P.; Milburn, M.V.; Kastenmuller, G.; Adamski, J.; Tuomi, T.; et al. Early metabolic markers of the development of dysglycemia and type 2 diabetes and their physiological significance. Diabetes 2013, 62, 1730–1737. [Google Scholar] [CrossRef] [PubMed]
- Rhee, E.P.; Cheng, S.; Larson, M.G.; Walford, G.A.; Lewis, G.D.; McCabe, E.; Yang, E.; Farrell, L.; Fox, C.S.; O'Donnell, C.J.; et al. Lipid profiling identifies a triacylglycerol signature of insulin resistance and improves diabetes prediction in humans. J. Clin. Investig. 2011, 121, 1402–1411. [Google Scholar] [CrossRef] [PubMed]
- Sala-Vila, A.; Cofan, M.; Mateo-Gallego, R.; Cenarro, A.; Civeira, F.; Ortega, E.; Ros, E. Inverse association between serum phospholipid oleic acid and insulin resistance in subjects with primary dyslipidaemia. Clin. Nutr. 2011, 30, 590–592. [Google Scholar] [CrossRef] [PubMed]
- Hodge, A.M.; English, D.R.; O’Dea, K.; Sinclair, A.J.; Makrides, M.; Gibson, R.A.; Giles, G.G. Plasma phospholipid and dietary fatty acids as predictors of type 2 diabetes: Interpreting the role of linoleic acid. Am. J. Clin. Nutr. 2007, 86, 189–197. [Google Scholar] [PubMed]
- Nestel, P.J.; Straznicky, N.; Mellett, N.A.; Wong, G.; De Souza, D.P.; Tull, D.L.; Barlow, C.K.; Grima, M.T.; Meikle, P.J. Specific plasma lipid classes and phospholipid fatty acids indicative of dairy food consumption associate with insulin sensitivity. Am. J. Clin. Nutr. 2014, 99, 46–53. [Google Scholar] [CrossRef] [PubMed]
- Fox, T.E.; Bewley, M.C.; Unrath, K.A.; Pedersen, M.M.; Anderson, R.E.; Jung, D.Y.; Jefferson, L.S.; Kim, J.K.; Bronson, S.K.; Flanagan, J.M.; et al. Circulating sphingolipid biomarkers in models of type 1 diabetes. J. Lipid Res. 2011, 52, 509–517. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.C.; Kim, B.R.; Lee, S.Y.; Park, T.S. Sphingolipid metabolism and obesity-induced inflammation. Front. Endocrinol. 2013, 4, 67. [Google Scholar] [CrossRef] [PubMed]
- Samad, F.; Hester, K.D.; Yang, G.; Hannun, Y.A.; Bielawski, J. Altered adipose and plasma sphingolipid metabolism in obesity: A potential mechanism for cardiovascular and metabolic risk. Diabetes 2006, 55, 2579–2587. [Google Scholar] [CrossRef] [PubMed]
- Jornayvaz, F.R.; Shulman, G.I. Diacylglycerol activation of protein kinase cepsilon and hepatic insulin resistance. Cell Metab. 2012, 15, 574–584. [Google Scholar] [CrossRef] [PubMed]
- Kotronen, A.; Seppanen-Laakso, T.; Westerbacka, J.; Kiviluoto, T.; Arola, J.; Ruskeepaa, A.L.; Oresic, M.; Yki-Jarvinen, H. Hepatic stearoyl-CoA desaturase (SCD)-1 activity and diacylglycerol but not ceramide concentrations are increased in the nonalcoholic human fatty liver. Diabetes 2009, 58, 203–208. [Google Scholar] [CrossRef] [PubMed]
- Magkos, F.; Su, X.; Bradley, D.; Fabbrini, E.; Conte, C.; Eagon, J.C.; Varela, J.E.; Brunt, E.M.; Patterson, B.W.; Klein, S. Intrahepatic diacylglycerol content is associated with hepatic insulin resistance in obese subjects. Gastroenterology 2012, 142, 1444–1446. [Google Scholar] [CrossRef] [PubMed]
- Gorden, D.L.; Ivanova, P.T.; Myers, D.S.; McIntyre, J.O.; VanSaun, M.N.; Wright, J.K.; Matrisian, L.M.; Brown, H.A. Increased diacylglycerols characterize hepatic lipid changes in progression of human nonalcoholic fatty liver disease; comparison to a murine model. PLoS ONE 2011, 6, e22775. [Google Scholar] [CrossRef] [PubMed]
- Pagadala, M.; Kasumov, T.; McCullough, A.J.; Zein, N.N.; Kirwan, J.P. Role of ceramides in nonalcoholic fatty liver disease. Trends Endocrinol. Metab. 2012, 23, 365–371. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Saponaro, C.; Gaggini, M.; Carli, F.; Gastaldelli, A. The Subtle Balance between Lipolysis and Lipogenesis: A Critical Point in Metabolic Homeostasis. Nutrients 2015, 7, 9453-9474. https://doi.org/10.3390/nu7115475
Saponaro C, Gaggini M, Carli F, Gastaldelli A. The Subtle Balance between Lipolysis and Lipogenesis: A Critical Point in Metabolic Homeostasis. Nutrients. 2015; 7(11):9453-9474. https://doi.org/10.3390/nu7115475
Chicago/Turabian StyleSaponaro, Chiara, Melania Gaggini, Fabrizia Carli, and Amalia Gastaldelli. 2015. "The Subtle Balance between Lipolysis and Lipogenesis: A Critical Point in Metabolic Homeostasis" Nutrients 7, no. 11: 9453-9474. https://doi.org/10.3390/nu7115475