Diet-Gene Interactions and PUFA Metabolism: A Potential Contributor to Health Disparities and Human Diseases

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Review
2.1. A Dramatic Change in the PUFA Content in Our Diet
2.2. A Debate about the Health Impact of n-6 PUFAs
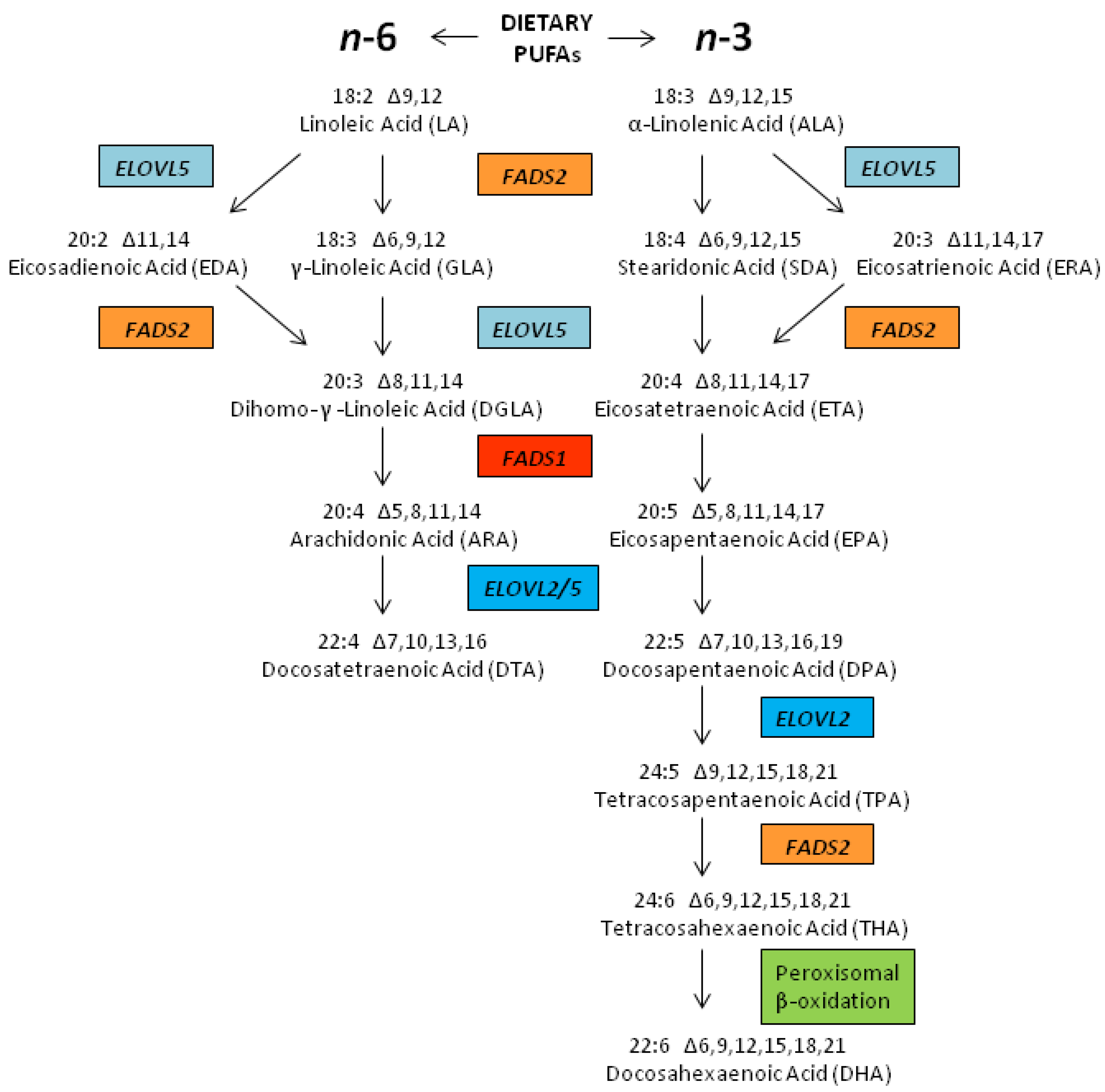
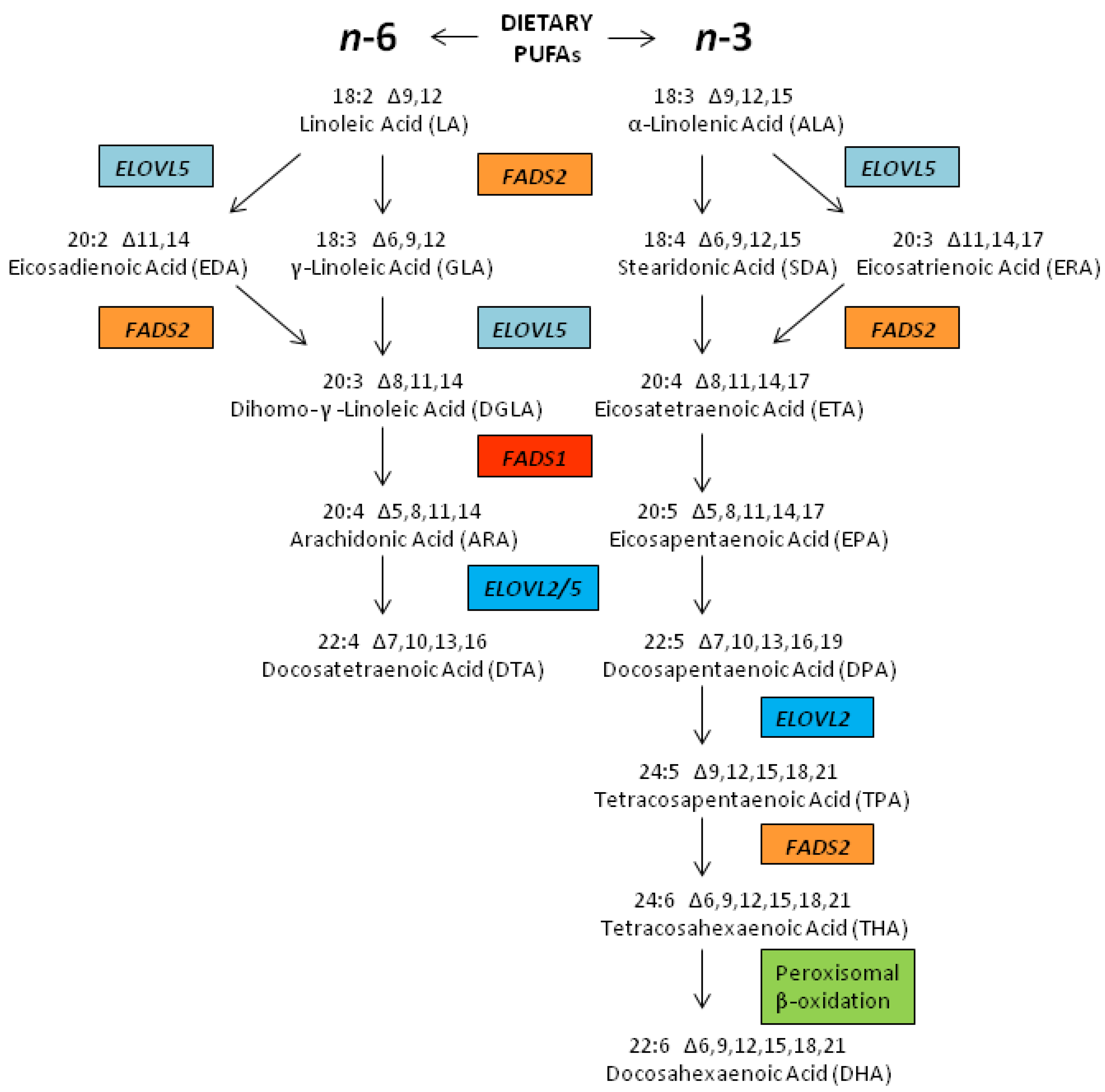
2.3. Biosynthesis of LCPUFAs
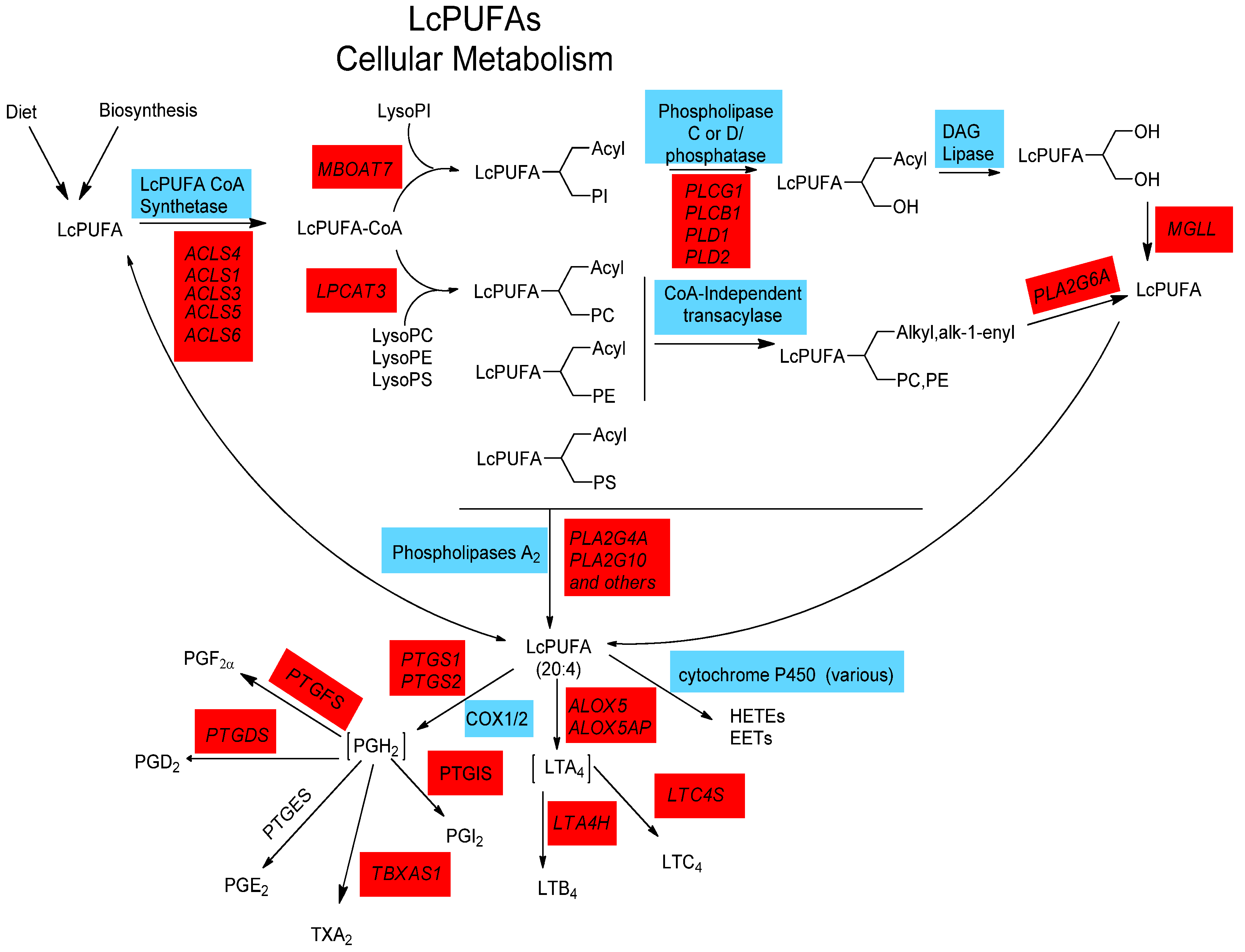
2.4. LCPUFA Cellular Metabolism

2.5. Biological Roles for LCPUFAs Such as ARA, EPA and DHA
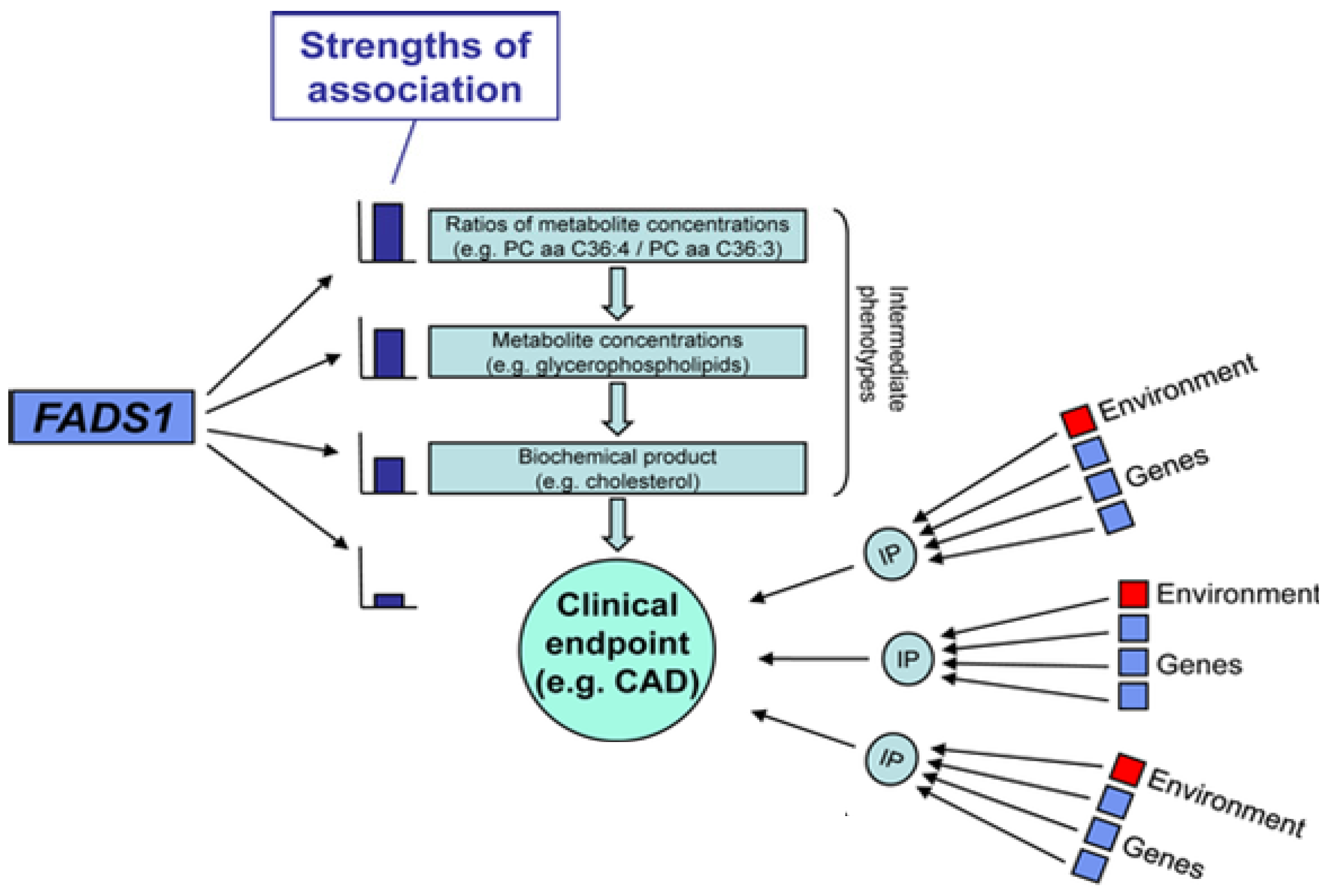
2.6. Genetic Variations within the FADS Gene Cluster: Implications for Cardiovascular Disease
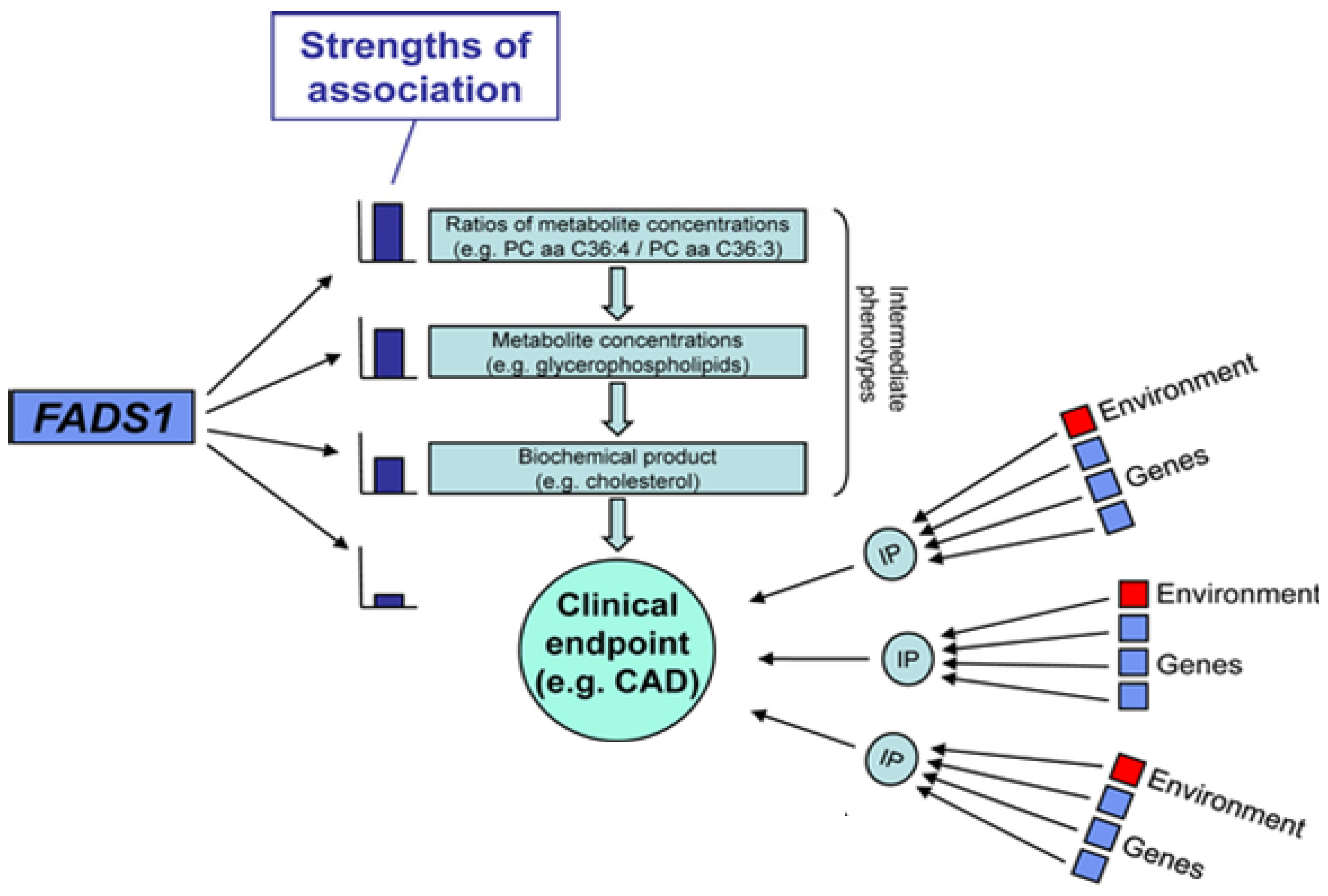
2.7. Interactions between PUFAs and Variants that Code for Enzymes that Mobilize and Metabolize Arachidonic Acid in Cardiovascular Disease
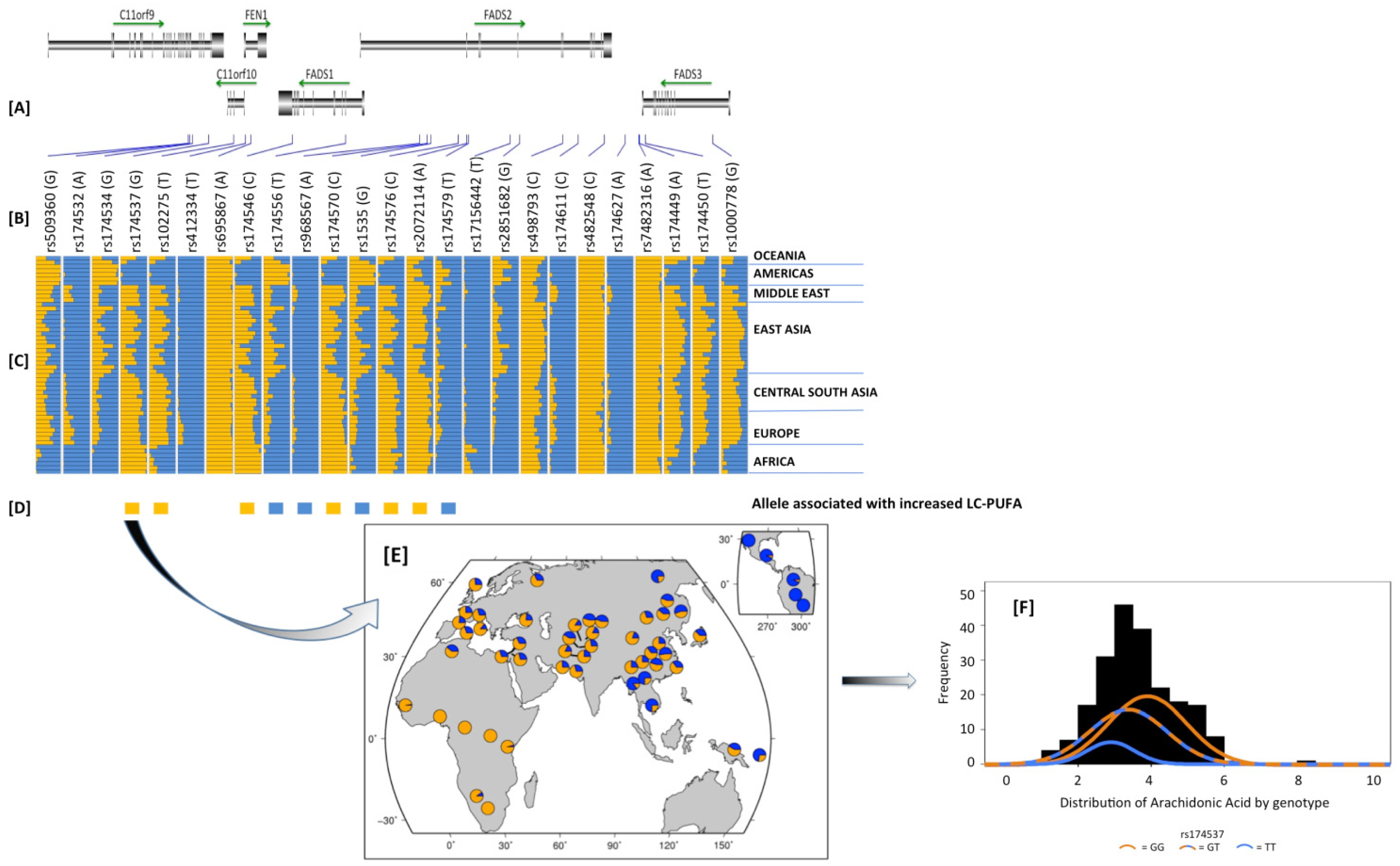
2.8. Potential Interactions between Dietary PUFAs and Genetic Variants in the FADS Cluster and other PUFA Metabolizing Genes; a Potential Contributor to Health Disparities
2.9. How Did These Ancestral Differences Arise?

3. Conclusions
Abbreviations
| ALA | α-linolenic acid |
| ARA | arachidonic acid |
| COX | cyclooxygenase |
| DGLA | dihomo γ-linolenic acid |
| DHA | docosahexaenoic acid |
| EPA | eicosapentaenoic acid |
| ETA | eicosatetraenoic acid |
| CAD | coronary artery disease |
| CVD | cardiovascular disease |
| FADS | fatty acid desaturase |
| GLA | γ-linolenic acid |
| GWAS | genome wide association study |
| IMT | intima-media thickness |
| LA | linoleic acid |
| LC | long chain |
| LD | linkage disequilibrium |
| LTB4 | leukotriene B4 |
| C18 | 18 carbon |
| MWD | modern western diet |
| NSAID | non-steroidal anti-inflammatory drug |
| PC | phosphatidylcholine |
| PE | phosphatidylethanolamine |
| PI | phosphatidylinositol |
| PS | phosphatidylserine |
| PLA2 | phospholipase A2 |
| PUFA | polyunsaturated fatty acid |
| SDA | stearidonic acid |
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Cordain, L.; Eaton, S.B.; Sebastian, A.; Mann, N.; Lindeberg, S.; Watkins, B.A.; O’Keefe, J.H.; Brand-Miller, J. Origins and evolution of the Western diet: Health implications for the 21st century. Am. J. Clin. Nutr. 2005, 81, 341–354. [Google Scholar]
- Ferrante, A.W., Jr. Obesity-induced inflammation: A metabolic dialogue in the language of inflammation. J. Intern. Med. 2007, 262, 408–414. [Google Scholar] [CrossRef]
- Hamminga, E.A.; van der Lely, A.J.; Neumann, H.A.; Thio, H.B. Chronic inflammation in psoriasis and obesity: Implications for therapy. Med. Hypotheses 2006, 67, 768–773. [Google Scholar] [CrossRef]
- Forsythe, L.K.; Wallace, J.M.; Livingstone, M.B. Obesity and inflammation: The effects of weight loss. Nutr. Res. Rev. 2008, 21, 117–133. [Google Scholar] [CrossRef]
- Nguyen, X.-M.T.; Lane, J.; Smith, B.R.; Nguyen, N.T. Changes in inflammatory biomarkers across weight classes in a representative US population: A link between obesity and inflammation. J. Gastrointest. Surg. 2009, 13, 1205–1212. [Google Scholar] [CrossRef]
- Calle, E.E.; Thun, M.J. Obesity and cancer. Oncogene 2004, 23, 6365–6378. [Google Scholar] [CrossRef]
- Beuther, D.A.; Weiss, S.T.; Sutherland, E.R. Obesity and asthma. Am. J. Respir. Crit. Care Med. 2006, 174, 112–119. [Google Scholar] [CrossRef]
- Naderali, E.K.; Ratcliffe, S.H.; Dale, M.C. Obesity and Alzheimer’s disease: A link between body weight and cognitive function in old age. Am. J. Alzheimers Dis. Other. Demen 2009, 24, 445–449. [Google Scholar] [CrossRef]
- Leveille, S.G.; Wee, C.C.; Iezzoni, L.I. Trends in obesity and arthritis among baby boomers and their predecessors, 1971–2002. Am. J. Public Health 2005, 95, 1607–1613. [Google Scholar] [CrossRef]
- Sankar, P.; Cho, M.K.; Condit, C.M.; Hunt, L.M.; Koenig, B.; Marshall, P.; Lee, S.S.; Spicer, P. Genetic research and health disparities. JAMA 2004, 291, 2985–2989. [Google Scholar] [CrossRef]
- Mensah, G.A.; Mokdad, A.H.; Ford, E.S.; Greenlund, K.J.; Croft, J.B. State of disparities in cardiovascular health in the United States. Circulation 2005, 111, 1233–1241. [Google Scholar] [CrossRef]
- Krieger, N.; Chen, J.T.; Waterman, P.D.; Soobader, M.J.; Subramanian, S.V.; Carson, R. Geocoding and monitoring of US socioeconomic inequalities in mortality and cancer incidence: Does the choice of area-based measure and geographic level matter? The Public Health Disparities Geocoding Project. Am. J. Epidemiol. 2002, 156, 471–482. [Google Scholar] [CrossRef]
- Braveman, P. Health disparities and health equity: Concepts and measurement. Annu. Rev. Public Health 2006, 27, 167–194. [Google Scholar] [CrossRef]
- Sankar, P.; Cho, M.K.; Mountain, J. Race and ethnicity in genetic research. Am. J. Med. Genet A 2007, 143A, 961–970. [Google Scholar] [CrossRef]
- Kuzawa, C.W.; Sweet, E. Epigenetics and the embodiment of race: Developmental origins of US racial disparities in cardiovascular health. Am. J. Hum. Biol. 2009, 21, 2–15. [Google Scholar] [CrossRef]
- Hibbeln, J.R.; Nieminen, L.R.; Blasbalg, T.L.; Riggs, J.A.; Lands, W.E. Healthy intakes of n-3 and n-6 fatty acids: Estimations considering worldwide diversity. Am. J. Clin. Nutr. 2006, 83, 1483S–1493S. [Google Scholar]
- Blasbalg, T.L.; Hibbeln, J.R.; Ramsden, C.E.; Majchrzak, S.F.; Rawlings, R.R. Changes in consumption of omega-3 and omega-6 fatty acids in the United States during the 20th century. Am. J. Clin. Nutr. 2011, 93, 950–962. [Google Scholar] [CrossRef]
- Simopoulos, A.P. Importance of the omega-6/omega-3 balance in health and disease: Evolutionary aspects of diet. World Rev. Nutr. Diet 2011, 102, 10–21. [Google Scholar] [CrossRef]
- Mensink, R.P.; Zock, P.L.; Kester, A.D.; Katan, M.B. Effects of dietary fatty acids and carbohydrates on the ratio of serum total to HDL cholesterol and on serum lipids and apolipoproteins: A meta-analysis of 60 controlled trials. Am. J. Clin. Nutr. 2003, 77, 1146–1155. [Google Scholar]
- Siguel, E. A new relationship between total/high density lipoprotein cholesterol and polyunsaturated fatty acids. Lipids 1996, 31, S51–S56. [Google Scholar] [CrossRef]
- Mensink, R.P.; Katan, M.B. Effect of dietary fatty acids on serum lipids and lipoproteins. A meta-analysis of 27 trials. Arterioscler Thromb. 1992, 12, 911–919. [Google Scholar] [CrossRef]
- Harris, W.S.; Mozaffarian, D.; Rimm, E.; Kris-Etherton, P.; Rudel, L.L.; Appel, L.J.; Engler, M.M.; Engler, M.B.; Sacks, F. Omega-6 fatty acids and risk for cardiovascular disease: A science advisory from the American Heart Association Nutrition Subcommittee of the Council on Nutrition, Physical Activity, and Metabolism; Council on Cardiovascular Nursing; and Council on Epidemiology and Prevention. Circulation 2009, 119, 902–907. [Google Scholar] [CrossRef]
- Ramsden, C.E.; Hibbeln, J.R.; Majchrzak-Hong, S.F. All PUFAs are not created equal: Absence of CHD benefit specific to linoleic acid in randomized controlled trials and prospective observational cohorts. World Rev. Nutr. Diet. 2011, 102, 30–43. [Google Scholar] [CrossRef]
- Ramsden, C.E.; Hibbeln, J.R.; Majchrzak, S.F.; Davis, J.M. n-6 Fatty acid-specific and mixed polyunsaturate dietary interventions have different effects on CHD risk: A meta-analysis of randomised controlled trials. Br. J. Nutr. 2010, 104, 1586–1600. [Google Scholar] [CrossRef]
- Ramsden, C.E.; Zamora, D.; Leelarthaepin, B.; Majchrzak-Hong, S.F.; Faurot, K.R.; Suchindran, C.M.; Ringel, A.; Davis, J.M.; Hibbeln, J.R. Use of dietary linoleic acid for secondary prevention of coronary heart disease and death: Evaluation of recovered data from the Sydney Diet Heart Study and updated meta-analysis. BMJ 2013, 346, e8707. [Google Scholar] [CrossRef]
- Smith, W.L. The eicosanoids and their biochemical mechanisms of action. Biochem. J. 1989, 259, 315–324. [Google Scholar]
- Haeggstrom, J.Z.; Funk, C.D. Lipoxygenase and leukotriene pathways: Biochemistry, biology, and roles in disease. Chem. Rev. 2011, 111, 5866–5898. [Google Scholar] [CrossRef]
- Wang, D.; Dubois, R.N. Eicosanoids and cancer. Nat. Rev. Cancer 2010, 10, 181–193. [Google Scholar] [CrossRef]
- Panigrahy, D.; Kalish, B.T.; Huang, S.; Bielenberg, D.R.; Le, H.D.; Yang, J.; Edin, M.L.; Lee, C.R.; Benny, O.; Mudge, D.K.; Butterfield, C.E.; et al. Epoxyeicosanoids promote organ and tissue regeneration. Proc. Natl. Acad. Sci. USA 2013, 110, 13528–13533. [Google Scholar] [CrossRef]
- Harris, T.R.; Hammock, B.D. Soluble epoxide hydrolase: Gene structure, expression and deletion. Gene 2013, 526, 61–74. [Google Scholar] [CrossRef]
- Serhan, C.N.; Chiang, N. Resolution phase lipid mediators of inflammation: Agonists of resolution. Curr. Opin. Pharmacol. 2013, 13, 632–640. [Google Scholar] [CrossRef]
- Innis, S. Maternal nutrition, genetics, and human milk lipids. Curr. Nutr. Rep. 2013, 2, 151–158. [Google Scholar] [CrossRef]
- Park, W.J.; Kothapalli, K.S.; Lawrence, P.; Tyburczy, C.; Brenna, J.T. An alternate pathway to long-chain polyunsaturates: The FADS2 gene product Delta8-desaturates 20:2n-6 and 20:3n-3. J. Lipid. Res. 2009, 50, 1195–1202. [Google Scholar] [CrossRef]
- Koletzko, B.; Lattka, E.; Zeilinger, S.; Illig, T.; Steer, C. Genetic variants of the fatty acid desaturase gene cluster predict amounts of red blood cell docosahexaenoic and other polyunsaturated fatty acids in pregnant women: Findings from the Avon Longitudinal Study of Parents and Children. Am. J. Clin. Nutr. 2011, 93, 211–219. [Google Scholar] [CrossRef]
- Meyer, B.J.; Mann, N.J.; Lewis, J.L.; Milligan, G.C.; Sinclair, A.J.; Howe, P.R. Dietary intakes and food sources of omega-6 and omega-3 polyunsaturated fatty acids. Lipids 2003, 38, 391–398. [Google Scholar]
- Calder, P.C. Dietary arachidonic acid: Harmful, harmless or helpful? Br. J. Nutr. 2007, 98, 451–453. [Google Scholar] [CrossRef]
- Weaver, K.L.; Ivester, P.; Chilton, J.A.; Wilson, M.D.; Pandey, P.; Chilton, F.H. The content of favorable and unfavorable polyunsaturated fatty acids found in commonly eaten fish. J. Am. Diet. Assoc. 2008, 108, 1178–1185. [Google Scholar] [CrossRef]
- Simopoulos, A.P. The importance of the omega-6/omega-3 fatty acid ratio in cardiovascular disease and other chronic diseases. Exp. Biol. Med. 2008, 233, 674–688. [Google Scholar] [CrossRef]
- Saravanan, P.; Davidson, N.C.; Schmidt, E.B.; Calder, P.C. Cardiovascular effects of marine omega-3 fatty acids. Lancet 2010, 376, 540–550. [Google Scholar] [CrossRef]
- Mozaffarian, D.; Wu, J.H. Omega-3 fatty acids and cardiovascular disease: Effects on risk factors, molecular pathways, and clinical events. J. Am. Coll. Cardiol. 2011, 58, 2047–2067. [Google Scholar] [CrossRef]
- Sprecher, H. Biochemistry of essential fatty acids. Prog. Lipid. Res. 1981, 20, 13–22. [Google Scholar] [CrossRef]
- Christophersen, B.O.; Hagve, T.A.; Christensen, E.; Johansen, Y.; Tverdal, S. Eicosapentaenoic and arachidonic acid metabolism in isolated liver cells. Scand. J. Clin. Lab. Investing. Suppl. 1986, 184, 55–60. [Google Scholar]
- Poisson, J.P.; Dupuy, R.P.; Sarda, P.; Descomps, B.; Narce, M.; Rieu, D.; Crastes de, P.A. Evidence that liver microsomes of human neonates desaturate essential fatty acids. Biochim. Biophys. Acta 1993, 1167, 109–113. [Google Scholar] [CrossRef]
- Horrobin, D.F. Fatty acid metabolism in health and disease: The role of delta-6-desaturase. Am. J. Clin. Nutr. 1993, 57, 732S–736S. [Google Scholar]
- El Boustani, S.; Causse, J.E.; Descomps, B.; Monnier, L.; Mendy, F.; Crastes de, P.A. Direct in vivo characterization of delta 5 desaturase activity in humans by deuterium labeling: Effect of insulin. Metabolism 1989, 38, 315–321. [Google Scholar] [CrossRef]
- Glaser, C.; Lattka, E.; Rzehak, P.; Steer, C.; Koletzko, B. Genetic variation in polyunsaturated fatty acid metabolism and its potential relevance for human development and health. Matern. Child Nutr. 2011, 7, 27–40. [Google Scholar]
- Reardon, H.T.; Zhang, J.; Kothapalli, K.S.; Kim, A.J.; Park, W.J.; Brenna, J.T. Insertion-deletions in a FADS2 intron 1 conserved regulatory locus control expression of fatty acid desaturases 1 and 2 and modulate response to simvastatin. Prostaglandins Leukot. Essent. Fatty Acids 2012, 87, 25–33. [Google Scholar] [CrossRef]
- Gregory, M.K.; Cleland, L.G.; James, M.J. Molecular basis for differential elongation of omega-3 docosapentaenoic acid by the rat Elovl5 and Elovl2. J. Lipid. Res. 2013, 54, 2851–2857. [Google Scholar] [CrossRef]
- Sprecher, H.; Chen, Q. Polyunsaturated fatty acid biosynthesis: A microsomal-peroxisomal process. Prostaglandins Leukot. Essent. Fatty Acids 1999, 60, 317–321. [Google Scholar] [CrossRef]
- Sprecher, H. Metabolism of highly unsaturated n-3 and n-6 fatty acids. Biochim. Biophys. Acta 2000, 1486, 219–231. [Google Scholar] [CrossRef]
- Sprecher, H. The roles of anabolic and catabolic reactions in the synthesis and recycling of polyunsaturated fatty acids. Prostaglandins Leukot. Essent. Fatty Acids 2002, 67, 79–83. [Google Scholar] [CrossRef]
- Hodson, L.; Skeaff, C.M.; Fielding, B.A. Fatty acid composition of adipose tissue and blood in humans and its use as a biomarker of dietary intake. Prog. Lipid. Res. 2008, 47, 348–380. [Google Scholar] [CrossRef]
- Gregory, M.K.; Gibson, R.A.; Cook-Johnson, R.J.; Cleland, L.G.; James, M.J. Elongase reactions as control points in long-chain polyunsaturated fatty acid synthesis. PLoS One 2011, 6, e29662. [Google Scholar]
- Blank, C.; Neumann, M.A.; Makrides, M.; Gibson, R.A. Optimizing DHA levels in piglets by lowering the linoleic acid to alpha-linolenic acid ratio. J. Lipid Res. 2002, 43, 1537–1543. [Google Scholar] [CrossRef]
- Gibson, R.A.; Neumann, M.A.; Lien, E.L.; Boyd, K.A.; Tu, W.C. Docosahexaenoic acid synthesis from alpha-linolenic acid is inhibited by diets high in polyunsaturated fatty acids. Prostaglandins Leukot. Essent. Fatty Acids 2013, 88, 139–146. [Google Scholar] [CrossRef]
- Mohrhauer, H.; Holman, R.T. The effect of dose level of essential fatty acids upon fatty acid composition of the rat liver. J. Lipid Res. 1963, 4, 151–159. [Google Scholar]
- Sarkkinen, E.S.; Agren, J.J.; Ahola, I.; Ovaskainen, M.L.; Uusitupa, M.I. Fatty acid composition of serum cholesterol esters, and erythrocyte and platelet membranes as indicators of long-term adherence to fat-modified diets. Am. J. Clin. Nutr. 1994, 59, 364–370. [Google Scholar]
- Hussein, N.; Ah-Sing, E.; Wilkinson, P.; Leach, C.; Griffin, B.A.; Millward, D.J. Long-chain conversion of [13C] linoleic acid and alpha-linolenic acid in response to marked changes in their dietary intake in men. J. Lipid Res. 2005, 46, 269–280. [Google Scholar]
- Spector, A.A. Plasma free fatty acid and lipoproteins as sources of polyunsaturated fatty acid for the brain. J. Mol. Neurosci. 2001, 16, 159–165. [Google Scholar]
- Lands, W.E.; Hart, P. Metabolism of glycerolipids.VI. Specificities of acyl coenzyme A: Phospolipid acyltransferases. J. Biol. Chem. 1965, 240, 1905–1911. [Google Scholar]
- Chilton, F.H.; Murphy, R.C. Remodeling of arachidonate-containing phosphoglycerides within the human neutrophil. J. Biol. Chem. 1986, 261, 7771–7777. [Google Scholar]
- Kang, M.J.; Fujino, T.; Sasano, H.; Minekura, H.; Yabuki, N.; Nagura, H.; Iijima, H.; Yamamoto, T.T. A novel arachidonate-preferring acyl-CoA synthetase is present in steroidogenic cells of the rat adrenal, ovary, and testis. Proc. Natl. Acad. Sci. USA 1997, 94, 2880–2884. [Google Scholar]
- Matsuda, S.; Inoue, T.; Lee, H.C.; Kono, N.; Tanaka, F.; Gengyo-Ando, K.; Mitani, S.; Arai, H. Member of the membrane-bound O-acyltransferase (MBOAT) family encodes a lysophospholipid acyltransferase with broad substrate specificity. Genes Cells 2008, 13, 879–888. [Google Scholar] [CrossRef]
- Chilton, F.H.; Fonteh, A.N.; Surette, M.E.; Triggiani, M.; Winkler, J.D. Control of arachidonate levels within inflammatory cells. Biochim. Biophys. Acta 1996, 1299, 1–15. [Google Scholar]
- Astudillo, A.M.; Balgoma, D.; Balboa, M.A.; Balsinde, J. Dynamics of arachidonic acid mobilization by inflammatory cells. Biochim. Biophys. Acta 2012, 1821, 249–256. [Google Scholar] [Green Version]
- Clark, J.D.; Lin, L.L.; Kriz, R.W.; Ramesha, C.S.; Sultzman, L.A.; Lin, A.Y.; Milona, N.; Knopf, J.L. A novel arachidonic acid-selective cytosolic PLA2 contains a Ca2+-dependent translocation domain with homology to PKC and GAP. Cell 1991, 65, 1043–1051. [Google Scholar] [CrossRef]
- Murakami, M.; Lambeau, G. Emerging roles of secreted phospholipase A2 enzymes: An update. Biochimie 2013, 95, 43–50. [Google Scholar] [CrossRef]
- Oskolkova, O.V.; Afonyushkin, T.; Preinerstorfer, B.; Bicker, W.; von, S.E.; Hainzl, E.; Demyanets, S.; Schabbauer, G.; Lindner, W.; Tselepis, A.D.; et al. Oxidized phospholipids are more potent antagonists of lipopolysaccharide than inducers of inflammation. J. Immunol. 2010, 185, 7706–7712. [Google Scholar] [CrossRef]
- Schmitz, G.; Ecker, J. The opposing effects of n-3 and n-6 fatty acids. Prog. Lipid. Res. 2008, 47, 147–155. [Google Scholar] [CrossRef]
- Boyce, J.A. Eicosanoids in asthma, allergic inflammation, and host defense. Curr. Mol. Med. 2008, 8, 335–349. [Google Scholar] [CrossRef]
- Calder, P.C. Long chain fatty acids and gene expression in inflammation and immunity. Curr. Opin. Clin. Nutr. Metab. Care 2013, 16, 425–433. [Google Scholar] [CrossRef]
- Soberman, R.J.; Christmas, P. The organization and consequences of eicosanoid signaling. J. Clin. Investig. 2003, 111, 1107–1113. [Google Scholar] [CrossRef]
- Chinetti, G.; Fruchart, J.C.; Staels, B. Peroxisome proliferator-activated receptors (PPARs): Nuclear receptors at the crossroads between lipid metabolism and inflammation. Inflamm. Res. 2000, 49, 497–505. [Google Scholar] [CrossRef]
- Jump, D.B.; Clarke, S.D. Regulation of gene expression by dietary fat. Annu. Rev. Nutr. 1999, 19, 63–90. [Google Scholar] [CrossRef]
- Deckelbaum, R.J.; Worgall, T.S.; Seo, T. n-3 Fatty acids and gene expression. Am. J. Clin. Nutr. 2006, 83, 1520S–1525S. [Google Scholar]
- Jung, U.J.; Torrejon, C.; Chang, C.L.; Hamai, H.; Worgall, T.S.; Deckelbaum, R.J. Fatty acids regulate endothelial lipase and inflammatory markers in macrophages and in mouse aorta: A role for PPARgamma. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 2929–2937. [Google Scholar] [CrossRef]
- Berger, A.; Roberts, M.A.; Hoff, B. How dietary arachidonic- and docosahexaenoic-acid rich oils differentially affect the murine hepatic transcriptome. Lipids Health Dis. 2006, 5, 10. [Google Scholar] [CrossRef]
- Caputo, M.; Zirpoli, H.; Torino, G.; Tecce, M.F. Selective regulation of UGT1A1 and SREBP-1c mRNA expression by docosahexaenoic, eicosapentaenoic, and arachidonic acids. J. Cell Physiol. 2011, 226, 187–193. [Google Scholar] [CrossRef]
- Vanden Heuvel, J.P. Nutrigenomics and nutrigenetics of omega-3 polyunsaturated fatty acids. Prog. Mol. Biol. Transl. Sci. 2012, 108, 75–112. [Google Scholar] [CrossRef]
- Alvheim, A.R.; Malde, M.K.; Osei-Hyiaman, D.; Lin, Y.H.; Pawlosky, R.J.; Madsen, L.; Kristiansen, K.; Froyland, L.; Hibbeln, J.R. Dietary linoleic acid elevates endogenous 2-AG and anandamide and induces obesity. Obesity 2012, 20, 1984–1994. [Google Scholar] [CrossRef]
- Innis, S.M. Dietary omega 3 fatty acids and the developing brain. Brain Res. 2008, 1237, 35–43. [Google Scholar] [CrossRef]
- Bradbury, J. Docosahexaenoic acid (DHA): An ancient nutrient for the modern human brain. Nutrients 2011, 3, 529–554. [Google Scholar] [CrossRef]
- Salem, N., Jr.; Litman, B.; Kim, H.Y.; Gawrisch, K. Mechanisms of action of docosahexaenoic acid in the nervous system. Lipids 2001, 36, 945–959. [Google Scholar] [CrossRef]
- Komatsu, W.; Ishihara, K.; Murata, M.; Saito, H.; Shinohara, K. Docosahexaenoic acid suppresses nitric oxide production and inducible nitric oxide synthase expression in interferon-gamma plus lipopolysaccharide-stimulated murine macrophages by inhibiting the oxidative stress. Free Radic. Biol. Med. 2003, 34, 1006–1016. [Google Scholar] [CrossRef]
- Spencer, L.; Mann, C.; Metcalfe, M.; Webb, M.; Pollard, C.; Spencer, D.; Berry, D.; Steward, W.; Dennison, A. The effect of omega-3 FAs on tumour angiogenesis and their therapeutic potential. Eur. J. Cancer 2009, 45, 2077–2086. [Google Scholar] [CrossRef]
- Serhan, C.N.; Hong, S.; Gronert, K.; Colgan, S.P.; Devchand, P.R.; Mirick, G.; Moussignac, R.L. Resolvins: A family of bioactive products of omega-3 fatty acid transformation circuits initiated by aspirin treatment that counter proinflammation signals. J. Exp. Med. 2002, 196, 1025–1037. [Google Scholar] [CrossRef]
- Serhan, C.N.; Arita, M.; Hong, S.; Gotlinger, K. Resolvins, docosatrienes, and neuroprotectins, novel omega-3-derived mediators, and their endogenous aspirin-triggered epimers. Lipids 2004, 39, 1125–1132. [Google Scholar] [CrossRef]
- Ariel, A.; Serhan, C.N. Resolvins and protectins in the termination program of acute inflammation. Trends Immunol. 2007, 28, 176–183. [Google Scholar] [CrossRef]
- Spector, A.A. Essentiality of fatty acids. Lipids 1999, 34, S1–S3. [Google Scholar] [CrossRef]
- Calder, P.C. The role of marine omega-3 (n-3) fatty acids in inflammatory processes, atherosclerosis and plaque stability. Mol. Nutr. Food Res. 2012, 56, 1073–1080. [Google Scholar] [CrossRef]
- Smith, W.L. Cyclooxygenases, peroxide tone and the allure of fish oil. Curr. Opin. Cell Biol. 2005, 17, 174–182. [Google Scholar] [CrossRef]
- Smith, W.L. Nutritionally essential fatty acids and biologically indispensable cyclooxygenases. Trends Biochem. Sci. 2008, 33, 27–37. [Google Scholar] [CrossRef]
- Terano, T.; Salmon, J.A.; Moncada, S. Biosynthesis and biological activity of leukotriene B5. Prostaglandins 1984, 27, 217–232. [Google Scholar] [CrossRef]
- Balvers, M.G.; Verhoeckx, K.C.; Bijlsma, S.; Rubingh, C.M.; Meijerink, J.; Wortelboer, H.M.; Witkamp, R.F. Fish oil and inflammatory status alter the n-3 to n-6 balance of the endocannabinoid and oxylipin metabolomes in mouse plasma and tissues. Metabolomics 2012, 8, 1130–1147. [Google Scholar] [CrossRef]
- Marquardt, A.; Stohr, H.; White, K.; Weber, B.H.F. CDNA cloning, genomic structure, and chromosomal localization of three members of the human fatty acid desaturase family. Genomics 2000, 66, 175–183. [Google Scholar] [CrossRef]
- Nakamura, M.T.; Nara, T.Y. Structure, function, and dietary regulation of delta6, delta5, and delta9 desaturases. Annu. Rev. Nutr. 2004, 24, 345–376. [Google Scholar] [CrossRef]
- Schadt, E.E. Molecular networks as sensors and drivers of common human diseases. Nature 2009, 461, 218–223. [Google Scholar] [CrossRef]
- Gieger, C.; Geistlinger, L.; Altmaier, E.; Hrabe de, A.M.; Kronenberg, F.; Meitinger, T.; Mewes, H.W.; Wichmann, H.E.; Weinberger, K.M.; Adamski, J.; et al. Genetics meets metabolomics: A genome-wide association study of metabolite profiles in human serum. PLoS Genet 2008, 4, e1000282. [Google Scholar] [CrossRef] [Green Version]
- Illig, T.; Gieger, C.; Zhai, G.; Romisch-Margl, W.; Wang-Sattler, R.; Prehn, C.; Altmaier, E.; Kastenmuller, G.; Kato, B.S.; Mewes, H.W.; et al. A genome-wide perspective of genetic variation in human metabolism. Nat. Genet 2010, 42, 137–141. [Google Scholar] [CrossRef]
- Kathiresan, S.; Melander, O.; Guiducci, C.; Surti, A.; Burtt, N.P.; Rieder, M.J.; Cooper, G.M.; Roos, C.; Voight, B.F.; Havulinna, A.S.; et al. Six new loci associated with blood low-density lipoprotein cholesterol, high-density lipoprotein cholesterol or triglycerides in humans. Nat. Genet 2008, 40, 189–197. [Google Scholar] [CrossRef]
- Willer, C.J.; Sanna, S.; Jackson, A.U.; Scuteri, A.; Bonnycastle, L.L.; Clarke, R.; Heath, S.C.; Timpson, N.J.; Najjar, S.S.; Stringham, H.M.; et al. Newly identified loci that influence lipid concentrations and risk of coronary artery disease. Nat. Genet 2008, 40, 161–169. [Google Scholar] [CrossRef]
- Kathiresan, S.; Willer, C.J.; Peloso, G.M.; Demissie, S.; Musunuru, K.; Schadt, E.E.; Kaplan, L.; Bennett, D.; Li, Y.; Tanaka, T.; et al. Common variants at 30 loci contribute to polygenic dyslipidemia. Nat. Genet 2009, 41, 56–65. [Google Scholar] [CrossRef]
- Aulchenko, Y.S.; Ripatti, S.; Lindqvist, I.; Boomsma, D.; Heid, I.M.; Pramstaller, P.P.; Penninx, B.W.; Janssens, A.C.; Wilson, J.F.; Spector, T.; et al. Loci influencing lipid levels and coronary heart disease risk in 16 European population cohorts. Nat. Genet 2009, 41, 47–55. [Google Scholar] [CrossRef]
- Teslovich, T.M.; Musunuru, K.; Smith, A.V.; Edmondson, A.C.; Stylianou, I.M.; Koseki, M.; Pirruccello, J.P.; Ripatti, S.; Chasman, D.I.; Willer, C.J.; et al. Biological, clinical and population relevance of 95 loci for blood lipids. Nature 2010, 466, 707–713. [Google Scholar] [CrossRef]
- Malerba, G.; Schaeffer, L.; Xumerle, L.; Klopp, N.; Trabetti, E.; Biscuola, M.; Cavallari, U.; Galavotti, R.; Martinelli, N.; Guarini, P.; et al. SNPs of the FADS gene cluster are associated with polyunsaturated fatty acids in a cohort of patients with cardiovascular disease. Lipids 2008, 43, 289–299. [Google Scholar] [CrossRef]
- Xie, L.; Innis, S.M. Genetic variants of the FADS1 FADS2 gene cluster are associated with altered (n-6) and (n-3) essential fatty acids in plasma and erythrocyte phospholipids in women during pregnancy and in breast milk during lactation. J. Nutr. 2008, 138, 2222–2228. [Google Scholar] [CrossRef]
- Schaeffer, L.; Gohlke, H.; Muller, M.; Heid, I.M.; Palmer, L.J.; Kompauer, I.; Demmelmair, H.; Illig, T.; Koletzko, B.; Heinrich, J. Common genetic variants of the FADS1 FADS2 gene cluster and their reconstructed haplotypes are associated with the fatty acid composition in phospholipids. Hum. Mol. Genet 2006, 15, 1745–1756. [Google Scholar] [CrossRef]
- Rzehak, P.; Heinrich, J.; Klopp, N.; Schaeffer, L.; Hoff, S.; Wolfram, G.; Illig, T.; Linseisen, J. Evidence for an association between genetic variants of the fatty acid desaturase 1 fatty acid desaturase 2 (FADS1 FADS2) gene cluster and the fatty acid composition of erythrocyte membranes. Br. J. Nutr. 2009, 101, 20–26. [Google Scholar] [CrossRef]
- Bokor, S.; Dumont, J.; Spinneker, A.; Gonzalez-Gross, M.; Nova, E.; Widhalm, K.; Moschonis, G.; Stehle, P.; Amouyel, P.; De, H.S.; et al. Single nucleotide polymorphisms in the FADS gene cluster are associated with delta-5 and delta-6 desaturase activities estimated by serum fatty acid ratios. J. Lipid Res. 2010, 51, 2325–2333. [Google Scholar]
- De Antueno, R.J.; Knickle, L.C.; Smith, H.; Elliot, M.L.; Allen, S.J.; Nwaka, S.; Winther, M.D. Activity of human Delta5 and Delta6 desaturases on multiple n-3 and n-6 polyunsaturated fatty acids. FEBS Lett. 2001, 509, 77–80. [Google Scholar] [CrossRef]
- Mathias, R.A.; Vergara, C.; Gao, L.; Rafaels, N.; Hand, T.; Campbell, M.; Bickel, C.; Ivester, P.; Sergeant, S.; Barnes, K.C.; et al. FADS genetic variants and omega-6 polyunsaturated fatty acid metabolism in a homogeneous island population. J. Lipid Res. 2010, 51, 2766–2774. [Google Scholar] [CrossRef]
- Mathias, R.A.; Sergeant, S.; Ruczinski, I.; Torgerson, D.G.; Hugenschmidt, C.E.; Kubala, M.; Vaidya, D.; Suktitipat, B.; Ziegler, J.T.; Ivester, P.; et al. The impact of FADS genetic variants on omega6 polyunsaturated fatty acid metabolism in African Americans. BMC Genet 2011, 12, 50. [Google Scholar]
- Sergeant, S.; Hugenschmidt, C.E.; Rudock, M.E.; Ziegler, J.T.; Ivester, P.; Ainsworth, H.C.; Vaidya, D.; Case, L.D.; Langefeld, C.D.; Freedman, B.I.; et al. Differences in arachidonic acid levels and fatty acid desaturase (FADS) gene variants in African Americans and European Americans with diabetes or the metabolic syndrome. Br. J. Nutr. 2012, 107, 547–555. [Google Scholar] [CrossRef]
- Tanaka, T.; Shen, J.; Abecasis, G.R.; Kisialiou, A.; Ordovas, J.M.; Guralnik, J.M.; Singleton, A.; Bandinelli, S.; Cherubini, A.; Arnett, D.; et al. Genome-wide association study of plasma polyunsaturated fatty acids in the InCHIANTI Study. PLoS Genet 2009, 5, e1000338. [Google Scholar] [CrossRef]
- Lemaitre, R.N.; Tanaka, T.; Tang, W.; Manichaikul, A.; Foy, M.; Kabagambe, E.K.; Nettleton, J.A.; King, I.B.; Weng, L.C.; Bhattacharya, S.; et al. Genetic loci associated with plasma phospholipid n-3 fatty acids: A meta-analysis of genome-wide association studies from the CHARGE Consortium. PLoS Genet 2011, 7, e1002193. [Google Scholar] [CrossRef]
- Lattka, E.; Illig, T.; Heinrich, J.; Koletzko, B. Do FADS genotypes enhance our knowledge about fatty acid related phenotypes? Clin. Nutr. 2010, 29, 277–287. [Google Scholar] [CrossRef]
- Baylin, A.; Ruiz-Narvaez, E.; Kraft, P.; Campos, H. Alpha-Linolenic acid, Delta6-desaturase gene polymorphism, and the risk of nonfatal myocardial infarction. Am. J. Clin. Nutr. 2007, 85, 554–560. [Google Scholar]
- Kwak, J.H.; Paik, J.K.; Kim, O.Y.; Jang, Y.; Lee, S.H.; Ordovas, J.M.; Lee, J.H. FADS gene polymorphisms in Koreans: Association with omega6 polyunsaturated fatty acids in serum phospholipids, lipid peroxides, and coronary artery disease. Atherosclerosis 2011, 214, 94–100. [Google Scholar] [CrossRef]
- Martinelli, N.; Girelli, D.; Malerba, G.; Guarini, P.; Illig, T.; Trabetti, E.; Sandri, M.; Friso, S.; Pizzolo, F.; Schaeffer, L.; et al. FADS genotypes and desaturase activity estimated by the ratio of arachidonic acid to linoleic acid are associated with inflammation and coronary artery disease. Am. J. Clin. Nutr. 2008, 88, 941–949. [Google Scholar]
- Li, S.W.; Lin, K.; Ma, P.; Zhang, Z.L.; Zhou, Y.D.; Lu, S.Y.; Zhou, X.; Liu, S.M. FADS gene polymorphisms confer the risk of coronary artery disease in a Chinese Han population through the altered desaturase activities: Based on high-resolution melting analysis. PLoS One 2013, 8, e55869. [Google Scholar]
- Lu, Y.; Vaarhorst, A.; Merry, A.H.; Dolle, M.E.; Hovenier, R.; Imholz, S.; Schouten, L.J.; Heijmans, B.T.; Muller, M.; Slagboom, P.E.; et al. Markers of endogenous desaturase activity and risk of coronary heart disease in the CAREMA cohort study. PLoS One 2012, 7, e41681. [Google Scholar]
- Schwedhelm, E.; Bartling, A.; Lenzen, H.; Tsikas, D.; Maas, R.; Brummer, J.; Gutzki, F.M.; Berger, J.; Frolich, J.C.; Boger, R.H. Urinary 8-iso-prostaglandin F2alpha as a risk marker in patients with coronary heart disease: A matched case-control study. Circulation 2004, 109, 843–848. [Google Scholar] [CrossRef]
- Wolfram, R.; Oguogho, A.; Palumbo, B.; Sinzinger, H. Enhanced oxidative stress in coronary heart disease and chronic heart failure as indicated by an increased 8-epi-PGF(2 alpha). Eur. J. Heart Fail 2005, 7, 167–172. [Google Scholar] [CrossRef]
- Park, J.Y.; Paik, J.K.; Kim, O.Y.; Chae, J.S.; Jang, Y.; Lee, J.H. Interactions between the APOA5 -1131 T > C and the FEN1 10154 G > T polymorphisms on omega6 polyunsaturated fatty acids in serum phospholipids and coronary artery disease. J. Lipid. Res. 2010, 51, 3281–3288. [Google Scholar] [CrossRef]
- Hong, S.H.; Kwak, J.H.; Paik, J.K.; Chae, J.S.; Lee, J.H. Association of polymorphisms in FADS gene with age-related changes in serum phospholipid polyunsaturated fatty acids and oxidative stress markers in middle-aged nonobese men. Clin. Interv. Aging 2013, 8, 585–596. [Google Scholar]
- Reardon, H.T.; Hsieh, A.T.; Park, W.J.; Kothapalli, K.S.; Anthony, J.C.; Nathanielsz, P.W.; Brenna, J.T. Dietary long-chain polyunsaturated fatty acids upregulate expression of FADS3 transcripts. Prostaglandins Leukot. Essent. Fatty Acids 2013, 88, 15–19. [Google Scholar] [CrossRef]
- Lattka, E.; Eggers, S.; Moeller, G.; Heim, K.; Weber, M.; Mehta, D.; Prokisch, H.; Illig, T.; Adamski, J. A common FADS2 promoter polymorphism increases promoter activity and facilitates binding of transcription factor ELK1. J. Lipid Res. 2010, 51, 182–191. [Google Scholar] [CrossRef] [Green Version]
- Von, S.C.; Fischer, S.; Weber, P.C. Long-term effects of dietary marine omega-3 fatty acids upon plasma and cellular lipids, platelet function, and eicosanoid formation in humans. J. Clin. Investig. 1985, 76, 1626–1631. [Google Scholar] [CrossRef]
- Wijendran, V.; Downs, I.; Srigley, C.T.; Kothapalli, K.S.; Park, W.J.; Blank, B.S.; Zimmer, J.P.; Butt, C.M.; Salem, N., Jr.; Brenna, J.T. Dietary arachidonic acid and docosahexaenoic acid regulate liver fatty acid desaturase (FADS) alternative transcript expression in suckling piglets. Prostaglandins Leukot. Essent. Fatty Acids 2013, 89, 345–350. [Google Scholar] [CrossRef]
- Garagnani, P.; Bacalini, M.G.; Pirazzini, C.; Gori, D.; Giuliani, C.; Mari, D.; Di Blasio, A.M.; Gentilini, D.; Vitale, G.; Collino, S.; et al. Methylation of ELOVL2 gene as a new epigenetic marker of age. Aging Cell 2012, 11, 1132–1134. [Google Scholar] [CrossRef]
- Helgadottir, A.; Gretarsdottir, S.; St, C.D.; Manolescu, A.; Cheung, J.; Thorleifsson, G.; Pasdar, A.; Grant, S.F.; Whalley, L.J.; Hakonarson, H.; Thorsteinsdottir, U.; et al. Association between the gene encoding 5-lipoxygenase-activating protein and stroke replicated in a Scottish population. Am. J. Hum. Genet 2005, 76, 505–509. [Google Scholar] [CrossRef]
- Helgadottir, A.; Manolescu, A.; Thorleifsson, G.; Gretarsdottir, S.; Jonsdottir, H.; Thorsteinsdottir, U.; Samani, N.J.; Gudmundsson, G.; Grant, S.F.; Thorgeirsson, G.; et al. The gene encoding 5-lipoxygenase activating protein confers risk of myocardial infarction and stroke. Nat. Genet 2004, 36, 233–239. [Google Scholar] [CrossRef] [Green Version]
- Lohmussaar, E.; Gschwendtner, A.; Mueller, J.C.; Org, T.; Wichmann, E.; Hamann, G.; Meitinger, T.; Dichgans, M. ALOX5AP gene and the PDE4D gene in a central European population of stroke patients. Stroke 2005, 36, 731–736. [Google Scholar] [CrossRef]
- Funk, C.D. Leukotriene modifiers as potential therapeutics for cardiovascular disease. Nat. Rev. Drug Discov. 2005, 4, 664–672. [Google Scholar] [CrossRef]
- Riccioni, G.; Back, M.; Capra, V. Leukotrienes and atherosclerosis. Curr. Drug Targets 2010, 11, 882–887. [Google Scholar] [CrossRef]
- Mehrabian, M.; Allayee, H.; Wong, J.; Shi, W.; Wang, X.P.; Shaposhnik, Z.; Funk, C.D.; Lusis, A.J. Identification of 5-lipoxygenase as a major gene contributing to atherosclerosis susceptibility in mice. Circ. Res. 2002, 91, 120–126. [Google Scholar] [CrossRef]
- Drazen, J.M.; Yandava, C.N.; Dube, L.; Szczerback, N.; Hippensteel, R.; Pillari, A.; Israel, E.; Schork, N.; Silverman, E.S.; Katz, D.A.; et al. Pharmacogenetic association between ALOX5 promoter genotype and the response to anti-asthma treatment. Nat. Genet 1999, 22, 168–170. [Google Scholar] [CrossRef]
- Dwyer, J.H.; Allayee, H.; Dwyer, K.M.; Fan, J.; Wu, H.; Mar, R.; Lusis, A.J.; Mehrabian, M. Arachidonate 5-lipoxygenase promoter genotype, dietary arachidonic acid, and atherosclerosis. N. Engl. J. Med. 2004, 350, 29–37. [Google Scholar] [CrossRef]
- Allayee, H.; Baylin, A.; Hartiala, J.; Wijesuriya, H.; Mehrabian, M.; Lusis, A.J.; Campos, H. Nutrigenetic association of the 5-lipoxygenase gene with myocardial infarction. Am. J. Clin. Nutr. 2008, 88, 934–940. [Google Scholar]
- Hartiala, J.; Li, D.; Conti, D.V.; Vikman, S.; Patel, Y.; Tang, W.H.; Brennan, M.L.; Newman, J.W.; Stephensen, C.B.; Armstrong, P.; et al. Genetic contribution of the leukotriene pathway to coronary artery disease. Hum. Genet 2011, 129, 617–627. [Google Scholar] [CrossRef]
- Hartiala, J.; Gilliam, E.; Vikman, S.; Campos, H.; Allayee, H. Association of PLA2G4A with myocardial infarction is modulated by dietary PUFAs. Am. J. Clin. Nutr. 2012, 95, 959–965. [Google Scholar] [CrossRef]
- Johnson, G.H.; Fritsche, K. Effect of dietary linoleic acid on markers of inflammation in healthy persons: A systematic review of randomized controlled trials. J. Acad. Nutr. Diet 2012, 112, 1029–1041. [Google Scholar] [CrossRef]
- Ferrucci, L.; Cherubini, A.; Bandinelli, S.; Bartali, B.; Corsi, A.; Lauretani, F.; Martin, A.; Andres-Lacueva, C.; Senin, U.; Guralnik, J.M. Relationship of plasma polyunsaturated fatty acids to circulating inflammatory markers. J. Clin. Endocrinol. Metab. 2006, 91, 439–446. [Google Scholar] [CrossRef]
- Montoya, M.T.; Porres, A.; Serrano, S.; Fruchart, J.C.; Mata, P.; Gerique, J.A.; Castro, G.R. Fatty acid saturation of the diet and plasma lipid concentrations, lipoprotein particle concentrations, and cholesterol efflux capacity. Am. J. Clin. Nutr. 2002, 75, 484–491. [Google Scholar]
- Chan, J.K.; McDonald, B.E.; Gerrard, J.M.; Bruce, V.M.; Weaver, B.J.; Holub, B.J. Effect of dietary alpha-linolenic acid and its ratio to linoleic acid on platelet and plasma fatty acids and thrombogenesis. Lipids 1993, 28, 811–817. [Google Scholar] [CrossRef]
- Thijssen, M.A.; Mensink, R.P. Small differences in the effects of stearic acid, oleic acid, and linoleic acid on the serum lipoprotein profile of humans. Am. J. Clin. Nutr. 2005, 82, 510–516. [Google Scholar]
- Liou, Y.A.; King, D.J.; Zibrik, D.; Innis, S.M. Decreasing linoleic acid with constant alpha-linolenic acid in dietary fats increases (n-3) eicosapentaenoic acid in plasma phospholipids in healthy men. J. Nutr. 2007, 137, 945–952. [Google Scholar]
- Lands, W.E.; Morris, A.; Libelt, B. Quantitative effects of dietary polyunsaturated fats on the composition of fatty acids in rat tissues. Lipids 1990, 25, 505–516. [Google Scholar] [CrossRef]
- Mathias, R.A.; Fu, W.Q.; Akey, J.M.; Ainsworth, H.C.; Torgerson, D.G.; Ruczinski, I.; Sergeant, S.; Barnes, K.C.; Chilton, F.H. Adaptive Evolution of the FADS Gene Cluster within Africa. PLoS One 2012, 7, e44926. [Google Scholar]
- Armstrong, P.; Kelley, D.S.; Newman, J.W.; Staggers, F.E., Sr.; Hartiala, J.; Allayee, H.; Stephensen, C.B. Arachidonate 5-lipoxygenase gene variants affect response to fish oil supplementation by healthy African Americans. J. Nutr. 2012, 142, 1417–1428. [Google Scholar] [CrossRef]
- Ameur, A.; Enroth, S.; Johansson, A.; Zaboli, G.; Igl, W.; Johansson, A.C.; Rivas, M.A.; Daly, M.J.; Schmitz, G.; Hicks, A.A.; et al. Genetic adaptation of fatty-acid metabolism: A human-specific haplotype increasing the biosynthesis of long-chain omega-3 and omega-6 fatty acids. Am. J. Hum. Genet 2012, 90, 809–820. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Chilton, F.H.; Murphy, R.C.; Wilson, B.A.; Sergeant, S.; Ainsworth, H.; Seeds, M.C.; Mathias, R.A. Diet-Gene Interactions and PUFA Metabolism: A Potential Contributor to Health Disparities and Human Diseases. Nutrients 2014, 6, 1993-2022. https://doi.org/10.3390/nu6051993
Chilton FH, Murphy RC, Wilson BA, Sergeant S, Ainsworth H, Seeds MC, Mathias RA. Diet-Gene Interactions and PUFA Metabolism: A Potential Contributor to Health Disparities and Human Diseases. Nutrients. 2014; 6(5):1993-2022. https://doi.org/10.3390/nu6051993
Chicago/Turabian StyleChilton, Floyd H., Robert C. Murphy, Bryan A. Wilson, Susan Sergeant, Hannah Ainsworth, Michael C. Seeds, and Rasika A. Mathias. 2014. "Diet-Gene Interactions and PUFA Metabolism: A Potential Contributor to Health Disparities and Human Diseases" Nutrients 6, no. 5: 1993-2022. https://doi.org/10.3390/nu6051993
APA StyleChilton, F. H., Murphy, R. C., Wilson, B. A., Sergeant, S., Ainsworth, H., Seeds, M. C., & Mathias, R. A. (2014). Diet-Gene Interactions and PUFA Metabolism: A Potential Contributor to Health Disparities and Human Diseases. Nutrients, 6(5), 1993-2022. https://doi.org/10.3390/nu6051993