Neuroprotective Properties of the Marine Carotenoid Astaxanthin and Omega-3 Fatty Acids, and Perspectives for the Natural Combination of Both in Krill Oil



{kind=link}
{kind=link}
Abstract
:1. Oxidative Stress in Brain Tissues
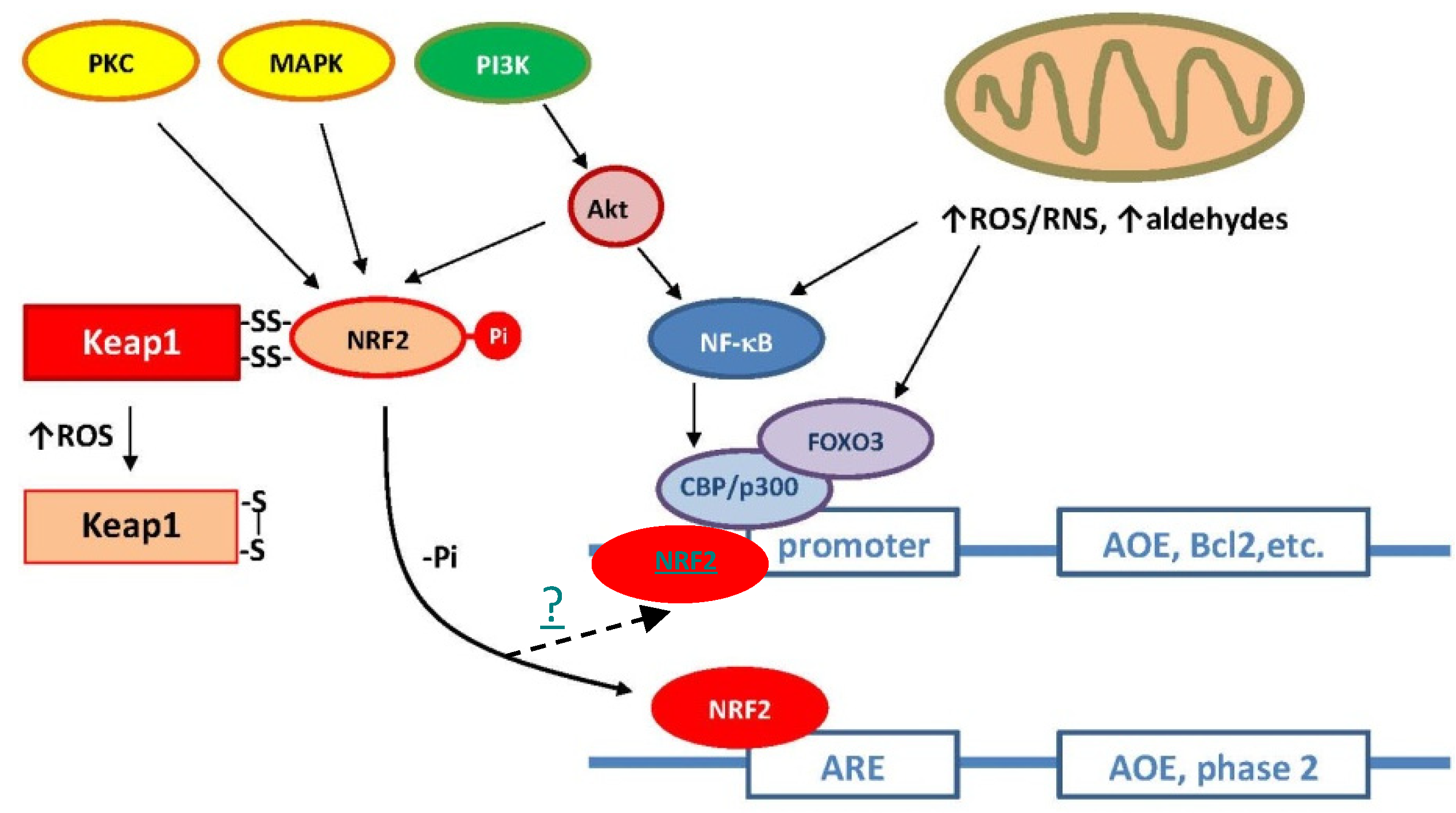
2. Antioxidant Defenses and “Neurohormesis”

3. Redox Imbalances and Neurodegenerative Pathologies
4. Marine Antioxidant/Phytochemical Therapies
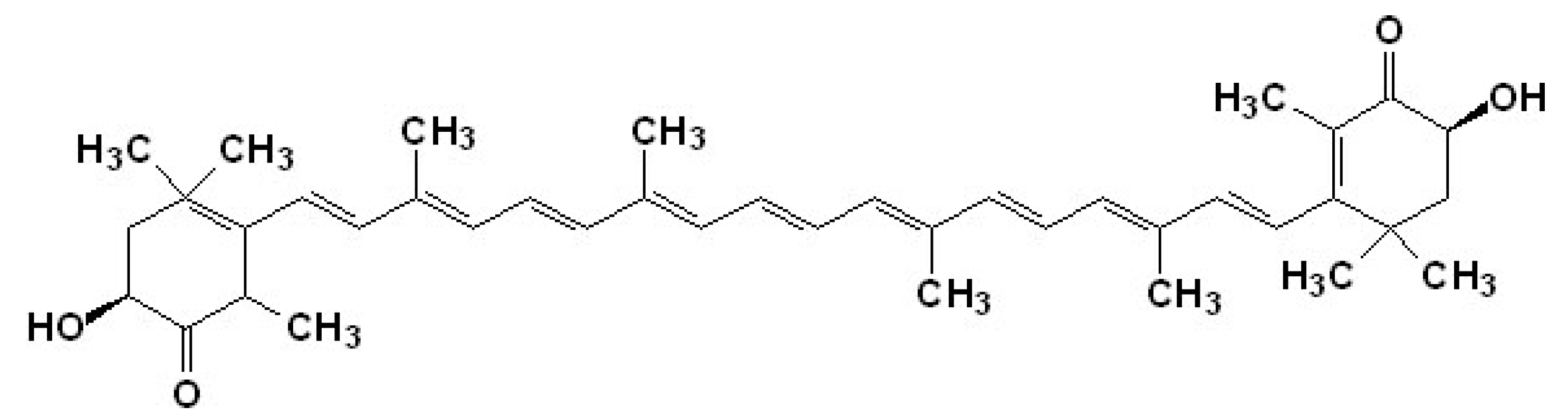
4.1. Astaxanthin

4.2. Polyunsaturated Fatty Acids (n-3/n-6 PUFAs)
4.3. Krill Oil
5. Conclusions
Acknowledgments
Conflicts of Interest
References
- Wang, J.-Y.; Wen, Y.-L.; Huan, Y.-N.; Chen, T.; Ku, M.-C. Dual effects of antioxidants in neurodegeneration: Direct neuroprotection against oxidative stress and indirect protection via suprresion of gliamediated inflammation. Curr. Pharm. Des. 2006, 12, 3521–3533. [Google Scholar] [CrossRef]
- Buonocore, G.; Perrone, S.; Tataranno, M.L. Oxygen toxicity: Chemistry and biology of reactive oxygen species. Semin. Fetal Neonatal Med. 2010, 15, 186–190. [Google Scholar] [CrossRef]
- Schiavone, S.; Jaquet, V.; Trabace, L.; Krause, K.-H. Severe life stress and oxidative stress in the brain: From animal models to human pathology. Antioxid. Redox. Signal. 2013, 18, 1475–1490. [Google Scholar] [CrossRef]
- Emerit, J.; Edeas, M.; Bricaire, F. Neurodegenerative diseases and stress. Biomed. Pharmacother. 2004, 58, 39–46. [Google Scholar] [CrossRef]
- Bedard, K.; Krause, K. The NOX family of ROS-generating NADPH oxidases: Physiology and pathophysiology. Physiol. Rev. 2007, 87, 245–313. [Google Scholar] [CrossRef]
- Drechsel, D.A.; Estévez, A.G.; Barbeito, L.; Beckman, J.S. Nitric oxide-mediated oxidative damage and the progressive demise of motor neurons in ALS. Neurotox. Res. 2012, 22, 251–264. [Google Scholar] [CrossRef]
- Kannan, K.; Jain, S.K. Oxidative stress. Pathophysiology 2000, 7, 153–157. [Google Scholar] [CrossRef]
- Schafer, F.Q.; Buettner, G.R. Redox environment of the cell as viewed through the redox state of the glutathione disulfide/glutathione couple. Free Radic. Biol. Med. 2001, 30, 1191–1212. [Google Scholar] [CrossRef]
- Gomez-Cabrera, M.C.; Viña, J.; Ji, L.L. Interplay of oxidants and antioxidants during exercise: Implications for muscle health. Phys. Sportsmed. 2009, 37, 16–23. [Google Scholar]
- Choi, B.H. Oxygen, antioxidants and brain dysfunction. Yonsei Med. J. 1993, 34, 1–10. [Google Scholar]
- Halliwell, B. Oxidative stress and neurodegeneration: Where are we now? J. Neurochem. 2006, 97, 1634–1658. [Google Scholar] [CrossRef]
- Stoll, G.; Jander, S.; Schroeter, M. Inflammation and glial responses in ischemic brain lesions. Prog. Neurobiol. 1998, 56, 149–171. [Google Scholar] [CrossRef]
- Repetto, M.G.; Reides, C.G.; Evelson, P.; Kohan, S.; de Lustig, E.S.; Llesuy, S.F. Peripheral markers of oxidative stress in probable Alzheimer patients. Eur. J. Clin. Investig. 1999, 29, 643–649. [Google Scholar] [CrossRef]
- Migliore, L.; Fontana, I.; Colognato, R.; Coppede, F.; Siciliano, G.; Murri, L. Searching for the role and the most suitable biomarkers of oxidative stress in Alzheimer’s disease and in other neurodegenerative diseases. Neurobiol. Aging 2005, 26, 587–595. [Google Scholar] [CrossRef]
- Ikeda, K.; Negishi, H.; Yamori, Y. Antioxidant nutrients and hypoxia/ischemia brain injury in rodents. Toxicology 2003, 189, 55–61. [Google Scholar] [CrossRef]
- Gutteridge, J.M.C.; Halliwell, B. Antioxidants: Molecules, medicines, and myths. Biochem. Biophys. Res. Commun. 2010, 393, 561–564. [Google Scholar] [CrossRef]
- Koutsilieri, E.; Scheller, C.; Tribl, F.; Riederer, P. Degeneration of neuronal cells due to oxidative stress—Microglial contribution. Parkinsonism Relat. Disord. 2002, 8, 401–406. [Google Scholar] [CrossRef]
- Mattson, M.P.; Cheng, A. Neurohormetic phytochemicals: Low-dose toxins that induce adaptive neuronal stress responses. Trends Neurosci. 2006, 29, 632–639. [Google Scholar] [CrossRef]
- Wang, Q.; Xu, J.; Rottinghaus, G.E.; Simonyi, A.; Lubahn, D.; Sun, G.Y.; Sun, A.Y. Resveratrol protects against global cerebral ischemic injury in gerbils. Brain Res. 2002, 958, 439–447. [Google Scholar] [CrossRef]
- Kaplan, S.; Bisleri, G.; Morgan, J.A.; Cheema, F.H.; Oz, M.C. Resveratrol, a natural red wine polyphenol, reduces ischemia-reperfusion-induced spinal cord injury. Ann. Thorac. Surg. 2005, 80, 2242–2249. [Google Scholar] [CrossRef]
- Ohshima, Y.; Mizuno, T.; Yamada, K.; Matsumoto, S.; Nagakane, Y.; Kondo, M.; Kuriyama, N.; Miyazaki, T.; Takeda, K.; Nishimura, T.; et al. Low vitamin and carotenoid levels are related to cerebral white matter lesions. J. Nutr. Health Aging 2013, 17, 456–460. [Google Scholar] [CrossRef]
- Mattei, R.; Polotow, T.G.; Vardaris, C.V.; Guerra, B.A.; Leite, J.R.; Otton, R.; Barros, M.P. Astaxanthin limits fish oil-related oxidative insult in the anterior forebrain of Wistar rats: Putative anxiolytic effects? Pharmacol. Biochem. Behav. 2011, 99, 349–355. [Google Scholar] [CrossRef]
- Mattson, M.P.; Meffert, M.K. Roles for NF-κB in nerve cell survival, plasticity, and disease. Cell Death Differ. 2006, 13, 852–860. [Google Scholar] [CrossRef]
- West, A.E.; Chen, W.G.; Dalva, M.B.; Dolmetsch, R.E.; Kornhauser, J.M.; Shaywitz, A.J.; Takasu, M.A.; Tao, X.; Greenberg, M.E. Calcium regulation of neuronal gene expression. Proc. Natl. Acad. Sci. USA 2001, 98, 11024–11031. [Google Scholar] [CrossRef]
- Jeong, W.S.; Jun, M.; Kong, A.N. Nrf2: A potential molecular target for cancer chemoprevention by natural compounds. Antioxid. Redox. Signal. 2006, 8, 99–106. [Google Scholar] [CrossRef]
- Liu, J.; Narasimhan, P.; Yu, F.; Chan, P.H. Neuroprotection by hypoxic preconditioning involves oxidative stress-mediated expression of hypoxia-inducible factor and erythropoietin. Stroke 2005, 36, 1264–1269. [Google Scholar]
- Kalyanaraman, B. Teaching the basics of redox biology to medical and graduate students: Oxidants, antioxidants and disease mechanisms. Redox. Biol. 2013, 1, 244–257. [Google Scholar] [CrossRef]
- Zhang, M.; An, C.; Gao, Y.; Leak, R.K.; Chen, J.; Zhang, F. Emerging roles of Nrf2 and phase II antioxidant enzymes in neuroprotection. Prog. Neurobiol. 2013, 100, 30–47. [Google Scholar] [CrossRef]
- De Vries, H.E.; Witte, M.; Hondius, D.; Rozemuller, A.J.M.; Drukarch, B.; Hoozemans, J.; van Horssen, J. Nrf2-induced antioxidant protection: A promising target to counteract ROS-mediated damage in neurodegenerative disease? Free Radic. Biol. Med. 2008, 45, 1375–1383. [Google Scholar] [CrossRef]
- Bains, M.; Hall, E.D. Antioxidant therapies in traumatic brain and spinal cord injury. Biochim. Biophys. Acta. 2012, 1822, 675–684. [Google Scholar]
- Currais, A.; Maher, P. Functional consequences of age-dependent changes in glutathione status in the brain. Antioxid. Redox. Signal. 2013, 19, 813–822. [Google Scholar] [CrossRef]
- Klegeris, A.; Walker, D.G.; McGeer, P.L. Activation of macrophages by Alzheimer beta amyloid peptide. Biochem. Biophys. Res. Commun. 1994, 199, 984–991. [Google Scholar] [CrossRef]
- Meda, L.; Cassatella, M.A.; Szendrei, G.I.; Otvos, L., Jr.; Villalba, M.; Ferrari, D.; Rossi, F. Activation of microglial cells by β-amyloid protein and interferon-gamma. Nature 1995, 374, 647–650. [Google Scholar] [CrossRef]
- Liu, F.; McCullough, L.D. Inflammatory responses in hypoxic ischemic encephalopathy. Acta Pharmacol. Sin. 2013, 34, 1121–1130. [Google Scholar] [CrossRef]
- Juurlink, B.H. Response of glial cells to ischemia: Roles of reactive oxygen species and glutathione. Neurosci. Biobehav. Rev. 1997, 21, 151–166. [Google Scholar] [CrossRef]
- Thorborne, S.K.; Juurlink, B.H. Low glutathione and high iron govern the susceptibility of oligodendroglial precursors to oxidative stress. J. Neurochem. 1996, 67, 1014–1022. [Google Scholar] [CrossRef]
- Hollensworth, S.B.; Shen, C.; Sim, J.E.; Spitz, D.R.; LeDoux, S.P. Glial cell type-specific responses to menadione-induced oxidative stress. Free Radic. Biol. Med. 2000, 28, 1161–1174. [Google Scholar] [CrossRef]
- Dringen, R. Metabolism and functions of glutathione in brain. Prog. Neurobiol. 2000, 62, 649–671. [Google Scholar] [CrossRef]
- Burgoyne, R.D.; Pearce, I.A.; Cambray-Deakin, M. N-Methyl-d-aspartate raises cytosolic calcium concentration in rat cerebellar granule cells in culture. Neurosci. Lett. 1988, 91, 47–52. [Google Scholar] [CrossRef]
- Kambe, Y.; Nakamichi, N.; Takarada, T.; Fukumori, R.; Nakazato, R.; Hinoi, E.; Yoneda, Y. A possible pivotal role of mitochondrial free calcium in neurotoxicity mediated by N-methyl-d-aspartate receptors in cultured rat hippocampal neurons. Neurochem. Int. 2011, 59, 10–20. [Google Scholar] [CrossRef]
- Le, T.M.; Jiang, H.; Cunningham, G.R.; Magarik, J.A.; Barge, W.S.; Cato, M.C.; Farina, M.; Rocha, J.B.T.; Milatovic, D.; Lee, E.; et al. γ-lutamylcysteine ameliorates oxidative injury in neurons and astrocytes in vitro and increases brain glutathione in vivo. Neurotoxicology 2011, 32, 518–525. [Google Scholar] [CrossRef]
- Butterfield, D.A.; Drake, J.; Pocernich, C.; Castegna, A. Evidence of oxidative damage in Alzheimerʼs disease brain: Central role for amyloid β-peptide. Trends Mol. Med. 2001, 7, 548–554. [Google Scholar] [CrossRef]
- Bandy, B.; Davidson, A.J. Mitochondrial mutations may increase oxidative stress. Free Radic. Biol. Med. 1990, 8, 523–529. [Google Scholar] [CrossRef]
- Reddy, P.H.; Reddy, T.P. Mitochondria as a therapeutic target for aging and neurodegenerative diseases. Curr. Alzheimer Res. 2011, 8, 393–409. [Google Scholar] [CrossRef]
- Federico, A.; Cardaioli, E.; Da Pozzo, P.; Formichi, P.; Gallus, G.N.; Radi, E. Mitochondria, oxidative stress and neurodegeneration. J. Neurol. Sci. 2012, 322, 254–262. [Google Scholar] [CrossRef]
- Cassarino, D.S.; Bennett, J.P., Jr. An evaluation of the role of mitochondria in neurodegenerative diseases: Mitochondrial and oxidative pathology, protective nuclear responses, and cell death in neurodegeneration. Brain Res. Rev. 1999, 29, 1–25. [Google Scholar] [CrossRef]
- Bredt, D.S.; Snyder, S.H. Nitric oxide: A physiologic messenger molecule. Ann. Rev. Biochem. 1994, 63, 175–195. [Google Scholar] [CrossRef]
- Smith, J.A.; Park, S.; Krause, J.S.; Banik, N.L. Oxidative stress, DNA damage, and the telomeric complex as therapeutic targets in acute neurodegeneration. Neurochem. Int. 2013, 62, 764–775. [Google Scholar] [CrossRef]
- Markesbery, W. Oxidative stress hypothesis in Alzheimer’s disease. Free. Radic. Biol. Med. 1997, 23, 134–147. [Google Scholar] [CrossRef]
- Draczynska-Lusiak, B.; Doung, A.; Sun, A.Y. Oxidized lipoproteins may play a role in neuronal cell dath in Alzheimer disease. Mol. Chem. Neuropathol. 1998, 33, 139–148. [Google Scholar] [CrossRef]
- Smith, M.A.; Rottkamp, C.A.; Nunomura, A.; Raina, A.K.; Perry, G. Oxidative stress in Alzheimerʼs disease. Biochim. Biophys. Acta 139–144.
- Varadarajan, S.; Yatin, S.; Aksenova, M.; Butterfield, D.A. Alzheimer’s amyloid β-peptide-associated free radical oxidative stress. J. Struct. Biol. 2000, 130, 184–208. [Google Scholar] [CrossRef]
- Butterfield, D.A. Amyloid beta-peptide (1,42)-induced oxidative stress and neurotoxicity: Implications form neurodegeneration in Alzheimerʼs disease brain. A review. Free Radic. Res. 2002, 12, 1307–1313. [Google Scholar] [CrossRef]
- Butterfield, D.A.; Castegna, A.; Lauderback, C.M.; Drake, J. Evidence that amyloid beta-peptide-induced peroxidation and its sequelae in Alzheimerʼs disease brain contribute to neuronal death. Neurobiol. Aging 2002, 23, 655–664. [Google Scholar] [CrossRef]
- Butterfield, D.A.; Lauderback, C.M. Lipid peroxidation and protein oxidation in Alzheimer’s disease brain: Potential causes and consequences involving amyloid β-peptide-associated free radical oxidative stress. Free. Radic. Biol. Med. 2002, 32, 1050–1060. [Google Scholar] [CrossRef]
- Montine, T.J.; Neely, M.D.; Quinn, J.F.; Beal, M.F.; Markesbery, W.R.; Roberts, L.J., II; Morrow, J.D. Lipid peroxidation in aging brain and Alzheimer’s disease. Free. Radic. Biol. Med. 2002, 33, 620–626. [Google Scholar] [CrossRef]
- Perry, G.; Taddeo, M.A.; Nunomura, A.; Zhu, X.; Zenteno-Svin, T.; Drew, K.L.; Shimohamam, S.; Avila, J.; Castellani, R.J.; Smith, M.A. Comparative biology and pathology of oxidative stress in Alzheimer and other neurodegenerative diseases: Beyond damage and response. Comp. Biochem. Physiol. 2002, 133, 507–513. [Google Scholar]
- Gilkun-Sherki, Y.; Melamed, E.; Offen, D. Antioxidant treatment in Alzheimer’s disease: Current state. J. Mol. Neurosci. 2003, 21, 1–11. [Google Scholar] [CrossRef]
- Meraz-Ríos, M.A.; Franco-Bocanegra, D.; Toral-Rios, D.; Campos-Peña, V. Early onset Alzheimer’s disease and oxidative stress. Oxid. Med. Cell. Longev. 2014, 2014, 375968. [Google Scholar]
- Chauhan, V.; Chauhan, A. Oxidative stress in Alzheimer’s disease. Pathophysiology 2006, 13, 195–208. [Google Scholar] [CrossRef]
- Nunomura, A.; Moreira, P.I.; Lee, H.G.; Zhu, X.; Castellani, R.J.; Smith, M.A.; Perry, G. Neuronal cell death and survival under oxidative stress in Alzheimer and Parkinson disease. CNS Neurol. Disord. Drug Targets 2007, 6, 411–423. [Google Scholar] [CrossRef]
- Marlatt, M.W.; Lucassen, P.J.; Perry, G.; Smith, M.A.; Zhu, X. Alzheimer’s disease: Cerebrovascular dysfunction, oxidative stress, and advanced clinical therapies. J. Alzheimer Dis. 2008, 15, 199–210. [Google Scholar]
- Koudinov, A.; Koudinova, K.E.; Berezov, T. Amyloid-β, tau protein, and oxidative changes as a physiological compensatory mechanism to maintain CNS plasticity under Alzheimer’s disease and other degenerative conditions. J. Alzheimer Dis. 2009, 18, 381–400. [Google Scholar]
- Butterfield, D.A.; Langue, M.L.B.; Sultana, R. Involvements of the lipid peroxidation production, HNE, in the pathogenesis and progression of Alzheimerʼs disease. Biochim. Biophys. Acta 2010, 180, 924–929. [Google Scholar] [CrossRef]
- Whitton, P.S. Inflammation as a causative factor in the aethiology of Parkinson’s disease. Br. J. Pharmacol. 2007, 150, 963–976. [Google Scholar] [CrossRef]
- Qian, L.; Flood, P.M.; Hong, J.-S. Neuroinflammation is a key player in Parkinson’s disease and a prime target for therapy. J. Neural Transm. 2010, 117, 971–979. [Google Scholar] [CrossRef]
- Sierra, A.; Abiega, O.; Shahraz, A.; Neumann, H. Janus-faced microglia: Beneficial and detrimental consequences of microglial phagocytosis. Front. Cell. Neurosci. 2013, 7, 6. [Google Scholar]
- Koppula, C.; Kumar, H.; More, S.V.; Kim, B.W.; Kim, I.S.; Choi, D.K. Recent advances on the neuroprotective potential of antioxidants in experimental models of Parkinson’s disease. Int. J. Mol. Sci. 2012, 13, 10608–10629. [Google Scholar] [CrossRef]
- Barber, S.C.; Shaw, P.J. Oxidative stress in ALS: A key role in motor neuron injury and therapeutic target. Free Radic. Biol. Med. 2010, 48, 629–641. [Google Scholar] [CrossRef]
- Galea, E.; Launay, N.; Portero-Otin, M.; Ruiz, M.; Pamplona, R.; Aubourg, P.; Ferrer, I.; Pujol, A. Oxidative stress underlying axonal degeneration in adrenoleukodystrophy: A paradigm for multifactorial neurodegenerative disease? Biochim. Biophys. Acta 1475–1488.
- Reddy, P.H.; Mao, P.; Manczak, M. Mitochondrial structural and functional dynamics in Huntington’s disease. Brain Res. Rev. 2009, 61, 33–48. [Google Scholar] [CrossRef]
- Shin, E.-J.; Jeong, J.H.; Chung, Y.H.; Kim, W.-K.; KO, K.-H.; Bach, J.-H.; Hong, J.-S.; Yoneda, Y.; Kim, H.-C. Role of oxidative stress in epileptic seizures. Neurochem. Int. 2011, 59, 122–137. [Google Scholar] [CrossRef]
- Mariani, E.; Polidori, M.C.; Cherubini, A.; Mecocci, P. Oxidative stress in brain aging, neurodegenerative and vascular diseases: An overview. J. Chromatogr. 2005, 827, 65–75. [Google Scholar]
- Valcour, V.; Shiramizu, B. HIV-associated dementia, mitochondrial dysfunction, and oxidative stress. Mitochondrion 2004, 4, 119–129. [Google Scholar] [CrossRef]
- Rawdin, B.J.; Mellon, S.H.; Dhabhar, F.S.; Epel, E.S.; Puterman, E.; Burke, H.M.; Reus, V.I.; Rosser, R.; Hamilton, S.P.; Nelson, J.C.; et al. Dysregulated relationship of inflammation and oxidative stress in major depression. Brain Behav. Immun. 2013, 31, 143–152. [Google Scholar] [CrossRef]
- Anderson, G.; Maes, M.; Berk, M. Schizophrenia is primed for an increased expression of depression through activation of immune-inflammatory, oxidative and nitrosative stress, and tryptophan catabolite pathways. Prog. Neuro-Psycopharmacol. Biol. Psichyatry 2013, 42, 101–114. [Google Scholar] [CrossRef]
- Frustaci, A.; Neri, M.; Cesario, A.; Adams, J.B.; Domeneci, E.; Dalla Bernardina, B.; Bonassi, S. Oxidative stress-related biomarkers in autism: Systematic review and meta-analysis. Free Radic. Biol. Med. 2012, 52, 2128–2141. [Google Scholar] [CrossRef]
- Corsinovi, L.; Biasi, F.; Poli, G.; Leonarduzzi, G.; Isaia, G. Dietary lipids and their oxidized products in Alzheimerʼs disease. Mol. Nutr. Food Res. 2011, 55, S161–S172. [Google Scholar] [CrossRef]
- Rodriguez-Amaya, D.B. Quantitative analysis, in vitro assessment of bioavailability and antioxidant activity of food carotenoids—A review. J. Food Comp. Anal. 2010, 23, 726–740. [Google Scholar] [CrossRef]
- Britton, G. Carotenoids: Spectroscopy, 1st ed.; Britton, G., Liaaen-Jensen, S., Pfander, H., Eds.; Birkhäuser: Basel, Switzerland, 1995; Volume 1B, pp. 13–62. [Google Scholar]
- Clydesdale, F.M. Color as a factor in food choice. Crit. Rev. Food Sci. Nutr. 1993, 33, 83–101. [Google Scholar] [CrossRef]
- Meléndez-Martínez, A.J.; Britton, G.; Vicario, I.M.; Heredia, F.J. Relationship between the colour and the chemical structure of carotenoid pigments. Food Chem. 2007, 101, 1145–1150. [Google Scholar] [CrossRef]
- Young, A.J.; Lowe, G.M. Antioxidant and prooxidant properties of carotenoids. Arch. Biochem. Biophys. 2001, 385, 20–27. [Google Scholar] [CrossRef]
- Palozza, P. Prooxidant actions of carotenoids in biologic systems. Nutr. Rev. 1998, 56, 257–265. [Google Scholar] [CrossRef]
- Barros, M.P.; Colepicolo, P.; Guaratini, T. Catalysis and Photochemistry in Heterogeneous Media; Nantes, I.L., Brochsztain, S., Eds.; Research Signpost: Kerala, India, 2007; pp. 161–186. [Google Scholar]
- Barros, M.P.; Pinto, E.; Colepicolo, P.; Pedersén, M. Astaxanthin and peridinin inhibit oxidative damage in Fe2+-loaded liposomes: Scavenging oxyradicals or changing membrane permeability? Biochem. Biophys. Res. Commun. 2001, 288, 225–232. [Google Scholar] [CrossRef]
- Otton, R.; Marin, D.P.; Bolin, A.P.; Santos, R.C.; Polotow, T.G.; Sampaio, S.C.; Barros, M.P. Astaxanthin ameliorates the redox imbalance in lymphocytes of experimental diabetic rats. Chem. Biol. Interact. 2010, 186, 306–315. [Google Scholar] [CrossRef]
- Bolin, A.P.; Macedo, R.C.; Marin, D.P.; Barros, M.P.; Otton, R. Astaxanthin prevents in vitro auto-oxidative injury in human lymphocytes. Cell Biol. Toxicol. 2010, 26, 457–467. [Google Scholar] [CrossRef]
- Guerin, M.; Huntley, M.E.; Olaizola, M. Haematococcus astaxanthin: Applications for human health and nutrition. Trends Biotechnol. 2003, 21, 210–216. [Google Scholar] [CrossRef]
- Barros, M.P.; Poppe, S.C.; Souza-Junior, T.P. Putative benefits of microalgal astaxanthin on exercise and human health. Braz. J. Pharmacognos. 2011, 21, 283–289. [Google Scholar]
- Ghosh, S.; Hayden, M.S. New regulators of NF-κB in inflammation. Nat. Rev.Immunol. 2008, 8, 837–848. [Google Scholar] [CrossRef]
- Karin, M. Nuclear factor-κB in cancer development and progression. Nature 2006, 441, 431–436. [Google Scholar] [CrossRef]
- O’Neill, L.A.J.; Kaltschmidt, C. NF-κB: A crucial transcription factor for glial and neuronal cell function. Trends Neurosci. 1997, 20, 252–258. [Google Scholar] [CrossRef]
- Folmer, F.; Jaspars, M.; Solano, G.; Cristofanon, S.; Henry, E.; Tabudravu, J.; Black, K.; Green, D.H.; Küpper, F.C.; Aalbersberg, W.; et al. The inhibition of TNF-α-induced NF-κB activation by marine natural products. Biochem. Pharmacol. 2009, 78, 592–606. [Google Scholar] [CrossRef]
- Kim, Y.H.; Koh, H.-K.; Kim, D.-S. Down-regulation of IL-6 production by astaxanthin via ERK-, MSK-, and NF-κB-mediated signals in activated microglia. Int. Immunopharmacol. 2010, 10, 1560–1572. [Google Scholar] [CrossRef]
- Lee, S.J.; Bai, S.K.; Lee, K.S.; Namkoong, S.; Na, H.J.; Ha, K.S.; Han, J.A.; Yim, S.V.; Chang, K.; Kwon, Y.G.; et al. Astaxanthin inhibits nitric oxide production and inflammatory gene expression by suppressing I(kappa)B kinase-dependent NF-kappaB activation. Mol. Cells 2003, 16, 97–105. [Google Scholar]
- Nagendraprabhu, P.; Sudhandiran, G. Astaxanthin inhibits tumor invasion by decreasing extracellular matrix production and induces apoptosis in experimental rat colon carcinogenesis by modulating the expressions of ERK-2, NFκB and COX-2. Investig. New Drugs 2011, 29, 207–224. [Google Scholar] [CrossRef]
- Chew, B.P.; Park, J.S. Carotenoid action on the immune response. J. Nutr. 2004, 134, 257S–261S. [Google Scholar]
- Ohgami, K.; Shiratori, K.; Kotake, S.; Nishida, T.; Mizuki, N.; Yazawa, K.; Ohno, S. Effects of astaxanthin on lipopolysaccharide-induced inflammation in vitro and in vivo. Investig. Ophthalmol. Vis. Sci. 2003, 44, 2694–2701. [Google Scholar] [CrossRef]
- Chew, B.P.; Wong, M.W.; Park, J.S.; Wong, T.S. Dietary β-carotene and astaxanthin but not canthaxanthin stimulate splenocyte function in mice. Anticancer Res. 1999, 19, 5223–5227. [Google Scholar]
- Jyonouchi, H.; Zhang, L.; Gross, M.; Tomita, Y. Immunomodulating actions of carotenoids: Enhancement of in vivo and in vitro antibody production to T-dependent antigens. Nutr. Cancer 1994, 21, 47–58. [Google Scholar] [CrossRef]
- Park, J.S.; Chyun, J.H.; Kim, Y.K.; Line, L.L.; Chew, B.P. Astaxanthin decreased oxidative stress and inflammation and enhanced immune response in humans. Nutr. Metab. 2010, 7, 18. [Google Scholar] [CrossRef]
- Kurihara, H.; Koda, H.; Asami, S.; Kiso, Y.; Tanaka, T. Contribution of the antioxidative property of astaxanthin to its protective effect on the promotion of cancer metastasis in mice treated with restraint stress. Life Sci. 2002, 70, 2509–2520. [Google Scholar] [CrossRef]
- Marin, D.P.; Bolin, A.P.; Macedo, R.C.; Sampaio, S.C.; Otton, R. ROS production in neutrophils from alloxan-induced diabetic rats treated in vivo with astaxanthin. Int. Immunopharmacol. 2011, 11, 103–109. [Google Scholar] [CrossRef]
- Ye, Q.; Huang, B.; Zhang, X.; Zhu, Y.; Chen, X. Astaxanthin protects against MPP+-induced oxidative stress in PC12 cells via the HO-1/NOX2 axis. BMC Neurosci. 2012, 13, 156. [Google Scholar] [CrossRef]
- Santos, S.D.; Cahú, T.B.; Firmino, G.O.; de Castro, C.C.; Carvalho, L.B., Jr.; Bezerra, R.S.; Filho, J.L. Shrimp waste extract and astaxanthin: Rat alveolar macrophage, oxidative stress and inflammation. J. Food Sci. 2012, 77, H141–H146. [Google Scholar] [CrossRef]
- Souza-Junior, T.P.; Yamada, A.K.; Simão, R.; Polotow, T.G.; Curi, R.; Pope, Z.; Willardson, J.M.; Barros, M.P. Supra-physiological doses of testosterone affect membrane oxidation of human neutrophils monitored by the fluorescent probe C11-BODIPY581/591. Eur. J. Appl. Physiol. 2013, 113, 1241–1248. [Google Scholar] [CrossRef]
- Li, Z.R.; Dong, X.; Liu, H.L.; Chen, X.; Shi, H.Q.; Fan, Y.S.; Hou, D.S.; Zhang, X.M. Astaxanthin protects ARPE-19 cells from oxidative stress via upregulation of Nrf2-regulated phase II enzymes through activation of PI3K/Akt. Mol. Vis. 2013, 19, 1656–1666. [Google Scholar]
- Kuroki, T.; Ikeda, S.; Okada, T.; Maoka, T.; Kitamura, A.; Sugimoto, M.; Kume, S. Astaxanthin ameliorates heat stress-induced impairment of blastocyst development in vitro: Astaxanthin colocalization with and action on mitochondria. J. Assist. Reprod. Genet. 2013, 30, 623–631. [Google Scholar] [CrossRef]
- Park, J.S.; Mathison, B.D.; Hayek, M.G.; Zhang, J.; Reinhart, G.A.; Chew, B.P. Astaxanthin modulates age-associated mitochondrial dysfunction in healthy dogs. J. Anim. Sci. 2013, 91, 268–275. [Google Scholar] [CrossRef]
- Lee, D.H.; Kim, C.S.; Lee, Y.J. Astaxanthin protects against MPTP/MPP+-induced mitochondrial dysfunction and ROS production in vivo and in vitro. Food Chem. Toxicol. 2011, 49, 271–280. [Google Scholar] [CrossRef]
- Wolf, A.M.; Asoh, S.; Hiranuma, H.; Ohsawa, I.; Iio, K.; Satou, A.; Ishikura, M.; Ohta, S. Astaxanthin protects mitochondrial redox state and functional integrity against oxidative stress. J. Nutr. Biochem. 2010, 21, 381–389. [Google Scholar] [CrossRef]
- Song, X.D.; Zhang, J.J.; Wang, M.R.; Liu, W.B.; Gu, X.B.; Ly, C.J. Astaxanthin induces mitochondria-mediated apoptosis in rat hepatocellular carcinoma CBRH-7919 cells. Biol. Pharm. Bull. 2011, 34, 839–844. [Google Scholar] [CrossRef]
- Kavitha, K.; Kowshik, J.; Kishore, T.K.; Baba, A.B.; Nagini, S. Astaxanthin inhibits NF-κB and Wnt/β-catenin signaling pathways via inactivation of Erk/MAPK and PI3K/Akt to induce intrinsic apoptosis in a hamster model of oral cancer. Biochim. Biophys. Acta 2013, 1830, 4433–4444. [Google Scholar]
- Dong, L.Y.; Jin, J.; Lu, G.; Kang, X.L. Astaxanthin attenuates the apoptosis of retinal ganglion cells in db/db mice by inhibition of oxidative stress. Mar. Drugs 2013, 11, 960–974. [Google Scholar] [CrossRef]
- Otton, R.; Marin, D.P.; Bolin, A.P.; Macedo, R.C.S.; Campoio, T.R.; Fineto, C., Jr.; Guerra, B.A.; Leite, J.R.; Barros, M.P.; Mattei, R. Combined fish oil and astaxanthin supplementation modulates rat lymphocyte function. Eur. J. Nutr. 2012, 51, 707–718. [Google Scholar] [CrossRef]
- Dhillon, V.S.; Fenech, M. Mutations that affect mitochondrial functions and their association with neurodegenerative diseases. Mutat. Res. 2014, 759, 1–13. [Google Scholar] [CrossRef]
- Mao, P.; Manczak, M.; Shirendeb, U.P.; Reddy, P.H. MitoQ, a mitochondria-targeted antioxidant, delays disease progression and alleviates pathogenesis in an experimental autoimmune encephalomyelitis mouse model of multiple sclerosis. Biochim. Biophys. Acta 2013, 1832, 2322–2331. [Google Scholar]
- Liu, X.; Osawa, T. Astaxanthin protects neuronal cells against oxidative damage and is a potent candidate for brain food. Forum Nutr. 2009, 61, 129–135. [Google Scholar] [CrossRef]
- Wang, H.-Q.; Sun, X.-B.; Xu, Y.-X.; Zhao, H.; Zhu, Q.-Y.; Zhu, C.-Q. Astaxanthin upregulates heme oxygenase-1 expression through ERK1/2 pathway and its protective effect against beta-amyloid-induced cytotoxicity in SH-SY5Y cells. Brain Res. 2010, 1360, 159–167. [Google Scholar]
- Lu, Y.-P.; Liu, S.-Y.; Sun, H.; Wu, X.-M.; Li, J.-J.; Zhu, L. Neuroprotective effect of astaxanthin on H2O2-induced neurotoxicity in vitro and on focal cerebral ischemia in vivo. Brain Res. 2010, 1360, 40–48. [Google Scholar]
- Chan, K.-C.; Mong, M.-C.; Yin, A.-C. Antioxidative and anti-inflammatory, neuroprotective effects of astaxanthin and canthaxanthin in nerve growth factor differentiated PC12 cells. J. Food Sci. 2009, 74, H225–H231. [Google Scholar] [CrossRef]
- Stillwell, W.; Wassall, S.R. Docosahexaenoic acid: Membrane properties of a unique fatty acid. Chem. Phys. Lipids 2003, 126, 1–27. [Google Scholar] [CrossRef]
- Weintraub, H. Update on marine omega-3 fatty acids: Management of dyslipidemia and current omega-3 treatment options. Atherosclerosis 2013, 230, 381–389. [Google Scholar] [CrossRef]
- Freemantle, E.; Vandal, M.; Tremblay-Mercier, J.; Tremblay, S.; Blachère, J.C.; Bégin, M.E.; Brenna, J.T.; Windust, A.; Cunnane, S.C. Omega-3 fatty acids, energy substrates, and brain function during aging. Prostagl. Leukot. Essent. Fatty Acids 2006, 75, 213–220. [Google Scholar] [CrossRef]
- Yehuda, S.; Rabinovitz, S.; Mostofsky, D.I. Essential fatty acids and the brain: From infancy to aging. Neurobiol. Aging 2005, 26, 98–102. [Google Scholar] [CrossRef]
- Whelan, J.; Rust, C. Innovative dietary sources of n-3 fatty acids. Annu. Rev. Nutr. 2006, 26, 75–103. [Google Scholar] [CrossRef]
- Lukiw, W.J.; Bazan, N.G. Docosahexaenoic acid and the aging brain. J. Nutr. 2008, 138, 2510–2514. [Google Scholar] [CrossRef]
- Connor, W.E.; Neuringer, M.; Lin, D.S. Dietary effects on brain fatty acid composition: The reversibility of n-3 fatty acid deficiency and turnover of docosahexaenoic acid in the brain, erythrocytes, and plasma of rhesus monkeys. J. Lipid Res. 2009, 31, 237–247. [Google Scholar]
- Hichami, A.; Datiche, F.; Ullah, S.; Liénard, F.; Chardigny, J.M.; Cattarelli, M.; Khan, N.A. Olfactory discrimination ability and brain expression of c-fos, Gir and Glut1 mRNA are altered in n-3 fatty acid-depleted rats. Behav. Brain Res. 2007, 184, 1–10. [Google Scholar] [CrossRef]
- Shirai, N.; Suzuki, H. Effect of dietary docosahexaenoic acid and catechins on maze behavior in mice. Ann. Nutr. Metab. 2004, 48, 51–58. [Google Scholar] [CrossRef]
- Uauy, R.; Dangour, A.D. Nutrition in brain development and aging: Role of essential fatty acids. Nutr. Rev. 2006, 64, S24–S33. [Google Scholar] [CrossRef]
- Giusto, N.M.; Salvador, G.A.; Castagnet, P.I.; Pasquaré, S.J.; Ilincheta de Boschero, M.G. Age-associated changes in central nervous system glycerolipid composition and metabolism. Neurochem. Res. 2002, 27, 1513–1523. [Google Scholar] [CrossRef]
- Hardy, J.A.; Higgins, G.A. Alzheimer’s disease: The amyloid cascade hypothesis. Science 1992, 256, 184–185. [Google Scholar]
- Moore, S.A.; Yoder, E.; Murphy, S.; Dutton, G.R.; Spector, A.A. Astrocytes, not neurons, produce docosahexaenoic acid (22:6 omega-3) and arachidonic acid (20:4 omega-6). J. Neurochem. 1991, 56, 518–524. [Google Scholar] [CrossRef]
- Hashimoto, M.; Tanabe, Y.; Fujii, Y.; Kikuta, T.; Shibata, H.; Shido, O. Chronic administration of docosahexaenoic acid ameliorates the impairment of spatial cognition learning ability in amyloid beta-infused rats. J. Nutr. 2005, 13, 549–555. [Google Scholar]
- Hashimoto, M.; Hossain, S.; Shimada, T.; Shido, O. Docosahexaenoic acid-induced protective effect against impaired learning in amyloid beta-infused rats is associated with increased synaptosomal membrane fluidity. Clin. Exp. Pharmacol. Physiol. 2006, 33, 934–939. [Google Scholar] [CrossRef]
- Calon, F.; Lim, G.P.; Morihara, T.; Yang, F.; Ubeda, O.; Salem, N., Jr.; Frautschy, S.A.; Cole, G.M. Dietary n-3 polyunsaturated fatty acid depletion activates caspases and decreases NMDA receptors in the brain of a transgenic mouse model of Alzheimer’s disease. Eur. J. Neurosci. 2005, 22, 617–626. [Google Scholar] [CrossRef]
- Lim, G.P.; Calon, F.; Morihara, T.; Yang, F.; Teter, B.; Ubeda, O.; Salem, N., Jr.; Frautschy, S.A.; Cole, G.M. A diet enriched with the omega-3 fatty acid docosahexaenoic acid reduces amyloid burden in an aged Alzheimer mouse model. J. Neurosci. 2005, 25, 3032–3040. [Google Scholar] [CrossRef]
- Ma, Q.L.; Teter, B.; Ubeda, O.J.; Morihara, T.; Dhoot, D.; Nyby, M.D.; Tuck, M.L.; Frautschy, S.A.; Cole, G.M. Omega-3 fatty acid docosahexaenoic acid increases SorLA/LR11, a sorting protein with reduced expression in sporadic Alzheimer’s disease (AD): Relevance to AD prevention. J. Neurosci. 2007, 27, 14299–14307. [Google Scholar] [CrossRef]
- Arendash, G.W.; Jensen, M.T.; Salem, N., Jr.; Hussein, N.; Cracchiolo, J.; Dickson, A.; Leighty, R.; Potter, H. A diet high in omega-3 fatty acids does not improve or protect cognitive performance in Alzheimer’s transgenic mice. Neuroscience 2007, 149, 286–302. [Google Scholar] [CrossRef]
- Gillette-Guyonnet, S.; Abellan Van Kan, G.; Andrieu, S.; Barberger-Gateau, P.; Berr, C.; Bonnefoy, M.; Dartigues, J.F.; de Groot, L.; Ferry, M.; Galan, P.; et al. IANA task force on nutrition and cognitive decline with aging. J. Nutr. Health Aging 2007, 11, 132–152. [Google Scholar]
- Barberger-Gateau, P.; Raffaitin, C.; Letenneur, L.; Berr, C.; Tzourio, C.; Dartigues, J.F.; Alpérovitch, A. Dietary patterns and risk of dementia: The Three-City cohort study. Neurology 2007, 69, 1921–1930. [Google Scholar] [CrossRef]
- Calon, F. Omega-3 polyunsaturated fatty acids in Alzheimer’s disease: Key questions and partial answers. Curr. Alzheimer Res. 2011, 8, 470–478. [Google Scholar] [CrossRef]
- Calon, F.; Cole, G. Neuroprotective action of omega-3 polyunsaturated fatty acids against neurodegenerative diseases: Evidence from animal studies. Prostaglandins Leukot. Essent. Fatty Acids 2007, 77, 287–293. [Google Scholar]
- Bousquet, M.; Saint-Pierre, M.; Julien, C.; Salem, N., Jr.; Cicchetti, F.; Calon, F. Beneficial effects of dietary omega-3 polyunsaturated fatty acid on toxin-induced neuronal degeneration in an animal model of Parkinson’s disease. FASEB J. 2008, 22, 1213–1325. [Google Scholar]
- De Lau, L.M.; Schipper, C.M.; Hofman, A.; Koudstaal, P.J.; Breteler, M.M. Prognosis of Parkinson disease: Risk of dementia and mortality: The Rotterdam Study. Arch. Neurol. 2005, 62, 1265–1269. [Google Scholar] [CrossRef]
- Da Silva, T.M.; Munhoz, R.P.; Alvarez, C.; Naliwaiko, K.; Kiss, A.; Andreatini, R.; Ferraz, A.C. Depression in Parkinson’s disease: A double-blind, randomized, placebo-controlled pilot study of omega-3 fatty-acid supplementation. J. Affect. Disord. 2008, 111, 351–359. [Google Scholar] [CrossRef]
- Gorjão, R.; Azevedo-Martins, A.K.; Rodrigues, H.G.; Abdulkader, F.; Arcisio-Miranda, M.; Procopio, J.; Curi, R. Comparative effects of DHA and EPA on cell function. Pharmacol. Ther. 2009, 122, 56–64. [Google Scholar] [CrossRef]
- Choi, H.D.; Kang, H.E.; Yang, S.H.; Lee, M.G.; Shin, W.G. Pharmacokinetics and first-pass metabolism of astaxanthin in rats. Br. J. Nutr. 2011, 105, 220–227. [Google Scholar] [CrossRef]
- Cunnane, S.C.; Plourde, M.; Pifferi, F.; Bégin, M.; Féart, C.; Barberger-Gateau, P. Fish, docosahexaenoic acid and Alzheimerʼs disease. Prog. Lipid Res. 2009, 48, 239–256. [Google Scholar] [CrossRef]
- Appleton, K.M.; Gunnell, D.; Peters, T.J.; Ness, A.R.; Kessler, D.; Rogers, P.J. No clear evidence of an association between plasma concentrations of n-3 long-chain polyunsaturated fatty acids and depressed mood in a non-clinical population. Prostaglandins Leukot. Essent. Fatty Acids 2008, 78, 337–342. [Google Scholar] [CrossRef]
- Bas, O.; Songur, A.; Sahin, O.; Mollaoglu, H.; Ozen, O.A.; Yaman, M.; Eser, O.; Fidan, H.; Yagmurca, M. The protective effect of fish n-3 fatty acids on cerebral ischemia in rat hippocampus. Neurochem. Int. 2007, 50, 548–554. [Google Scholar] [CrossRef]
- King, V.R.; Huang, W.L.; Dyall, S.C.; Curran, O.E.; Priestley, J.V.; Michael-Titus, A.T. Omega-3 fatty acids improve recovery, whereas omega-6 fatty acids worsen outcome, after spinal cord injury in the adult rat. J. Neurosci. 2006, 26, 4672–4680. [Google Scholar] [CrossRef]
- Calabrese, V.; Cornelius, C.; Dinkova-Kostova, A.T.; Calabrese, E.J.; Mattson, M.P. Cellular stress responses, the hormesis paradigm, and vitagenes: Novel targets for therapeutic intervention in neurodegenerative disorders. Antioxid. Redox Signal. 2010, 13, 1763–1811. [Google Scholar] [CrossRef]
- Jicha, G.A.; Markesbery, W.R. Omega-3 fatty acids: Potential role in the management of early Alzheimer’s disease. Clin. Interv. Aging 2010, 5, 45–61. [Google Scholar] [CrossRef]
- Tarling, G.A. Population dynamics of Northern krill (Meganyctiphanes norvegica Sars). Adv. Mar. Biol. 2010, 57, 59–90. [Google Scholar] [CrossRef]
- Tou, J.C.; Jaczynski, J.; Chen, Y. Krill for human consumption: Nutritional value and potential health benefits. Nutr. Rev. 2007, 65, 63–77. [Google Scholar]
- Ulven, S.M.; Kirkhus, B.; Lamglait, A.; Basu, S.; Elind, E.; Haider, T.; Berge, K.; Vik, H.; Pedersen, J.I. Metabolic effects of krill oil are essentially similar to those of fish oil but at lower dose of EPA and DHA, in healthy volunteers. Lipids 2011, 46, 37–46. [Google Scholar] [CrossRef]
- Grantham, G.J. The Southern Ocean: The Utilization of Krill; Southern Ocean Fisheries Survey Programme, GLO/SO/7/3; Food and Agriculture Organization: Rome, Italy, 1977; pp. 1–61. [Google Scholar]
- Grynbaum, M.D.; Hentschel, P.; Putzbach, K.; Rehbein, J.; Krucker, M.; Nicholson, G.; Albert, K. Unambiguous detection of astaxanthin and astaxanthin fatty acid esters in krill (Euphausia superba Dana). J. Sep. Sci. 2005, 28, 1685–1693. [Google Scholar] [CrossRef]
- Miyashita, K. Function of marine carotenoids. Forum Nutr. 2009, 61, 136–146. [Google Scholar] [CrossRef]
- Gigliotti, J.C.; Davenport, M.P.; Beamer, S.K.; Tou, J.C.; Jaczynski, J. Extraction and characterisation of lipids from Antarctic krill (Euphausia superba). Food Chem. 2011, 125, 1028–1036. [Google Scholar]
- Suzuki, T.; Shibata, N. The utilization of Antarctic krill for human food. Food Rev. Int. 1990, 6, 119–147. [Google Scholar] [CrossRef]
- Maki, K.C.; Reeves, M.S.; Farmer, M.; Griinari, M.; Berge, K.; Vik, H.; Hubacher, R.; Rains, T.M. Krill oil supplementation increases plasma concentrations of eicosapentaenoic and docosahexaenoic acids in overweight and obese men and women. Nutr. Res. 2009, 29, 609–615. [Google Scholar] [CrossRef]
- Mancuso, J.R.; McClements, D.J.; Decker, E.A. The effects of surfactant type, pH, and chelators on the oxidation of salmon oil-in-water emulsions. J. Agric. Food Chem. 1999, 47, 4112–4116. [Google Scholar] [CrossRef]
- Tou, J.C.; Altman, S.N.; Gigliotti, J.C.; Benedito, V.A.; Cordonier, E.L. Different sources of omega-3 polyunsaturated fatty acids affects apparent digestibility, tissue deposition, and tissue oxidative stability in growing female rats. Lipids Health Dis. 2011, 10, 179. [Google Scholar] [CrossRef]
- Venkatraman, J.T.; Chandrasekar, B.; Kim, J.D.; Fernandes, G. Effects of n-3 and n-6 fatty acids on the activities and expression of hepatic antioxidant enzymes in autoimmune-prone NZB × NZW F1 mice. Lipids 1994, 29, 561–568. [Google Scholar] [CrossRef]
- Di Marzo, V.; Griinari, M.; Carta, G.; Murru, E.; Ligresti, A.; Cordeddu, L.; Giordano, E.; Bisogno, T.; Collu, M.; Batetta, B.; et al. Dietary krill oil increases docosahexaenoic acid and reduces 2-arachidonoylglycerol but not N-acylethanolamine levels in the brain of obese Zucker rats. Int. Dairy J. 2010, 20, 231–235. [Google Scholar] [CrossRef]
- Tillander, V.; Burri, L.; Alexson, S. Fish oil and Krill oil differentially regulate gene expression. Chem. Phys. Lipids 2010, 163S, S22–S33. [Google Scholar]
- Vigerust, N.F.; Bjørndal, B.; Bohov, P.; Brattelid, T.; Svardal, A.; Berge, R.K. Krill oil versus fish oil in modulation of inflammation and lipid metabolism in mice transgenic for TNF-α. Eur. J. Nutr. 2013, 52, 1315–1325. [Google Scholar] [CrossRef]
- Deutsch, L. Evaluation of the effect of neptune krill oil on chronic inflammation and arthritic symptoms. J. Am. Coll. Nutr. 2007, 26, 39–48. [Google Scholar] [CrossRef]
- Wibrand, K.; Berge, K.; Messaoudi, M.; Duffaud, A.; Panja, D.; Bramham, C.R.; Burri, L. Enhanced cognitive function and antidepressant-like effects after krill oil supplementation in rats. Lipids Health Dis. 2013, 12, 6. [Google Scholar] [CrossRef]
- Lee, B.; Sur, B.-J.; Han, J.-J.; Shim, I.; Her, S.; Lee, H.-J.; Hahm, D.-H. Krill phosphatidylserine improves learning and memory in Morris water maze in aged rats. Progr. Neuro-Psychopharmacol. Biol. Psych. 2010, 34, 1085–1093. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Barros, M.P.; Poppe, S.C.; Bondan, E.F. Neuroprotective Properties of the Marine Carotenoid Astaxanthin and Omega-3 Fatty Acids, and Perspectives for the Natural Combination of Both in Krill Oil. Nutrients 2014, 6, 1293-1317. https://doi.org/10.3390/nu6031293
Barros MP, Poppe SC, Bondan EF. Neuroprotective Properties of the Marine Carotenoid Astaxanthin and Omega-3 Fatty Acids, and Perspectives for the Natural Combination of Both in Krill Oil. Nutrients. 2014; 6(3):1293-1317. https://doi.org/10.3390/nu6031293
Chicago/Turabian StyleBarros, Marcelo P., Sandra C. Poppe, and Eduardo F. Bondan. 2014. "Neuroprotective Properties of the Marine Carotenoid Astaxanthin and Omega-3 Fatty Acids, and Perspectives for the Natural Combination of Both in Krill Oil" Nutrients 6, no. 3: 1293-1317. https://doi.org/10.3390/nu6031293
APA StyleBarros, M. P., Poppe, S. C., & Bondan, E. F. (2014). Neuroprotective Properties of the Marine Carotenoid Astaxanthin and Omega-3 Fatty Acids, and Perspectives for the Natural Combination of Both in Krill Oil. Nutrients, 6(3), 1293-1317. https://doi.org/10.3390/nu6031293