Obesity: Pathophysiology and Intervention

{kind=link}
Abstract
:1. Introduction
2. Epidemiological Studies
3. Binge Eating and Food Addiction
3.1. Binge Eating
3.2. Food Addiction
3.3. Prader-Willi Syndrome (PWS)
4. Hormones and Gut Peptides
4.1. Leptin
4.2. Insulin
4.3. Ghrelin
4.4. Peptide YY (PYY)
4.5. Glucagon-Like Peptide 1 (GLP-1)
4.6. Cholecystokinin (CCK)
5. Neuroimaging Studies
5.1. Functional Neuroimaging
5.2. Structural Imaging
6. Brain Circuits Related to Obesity
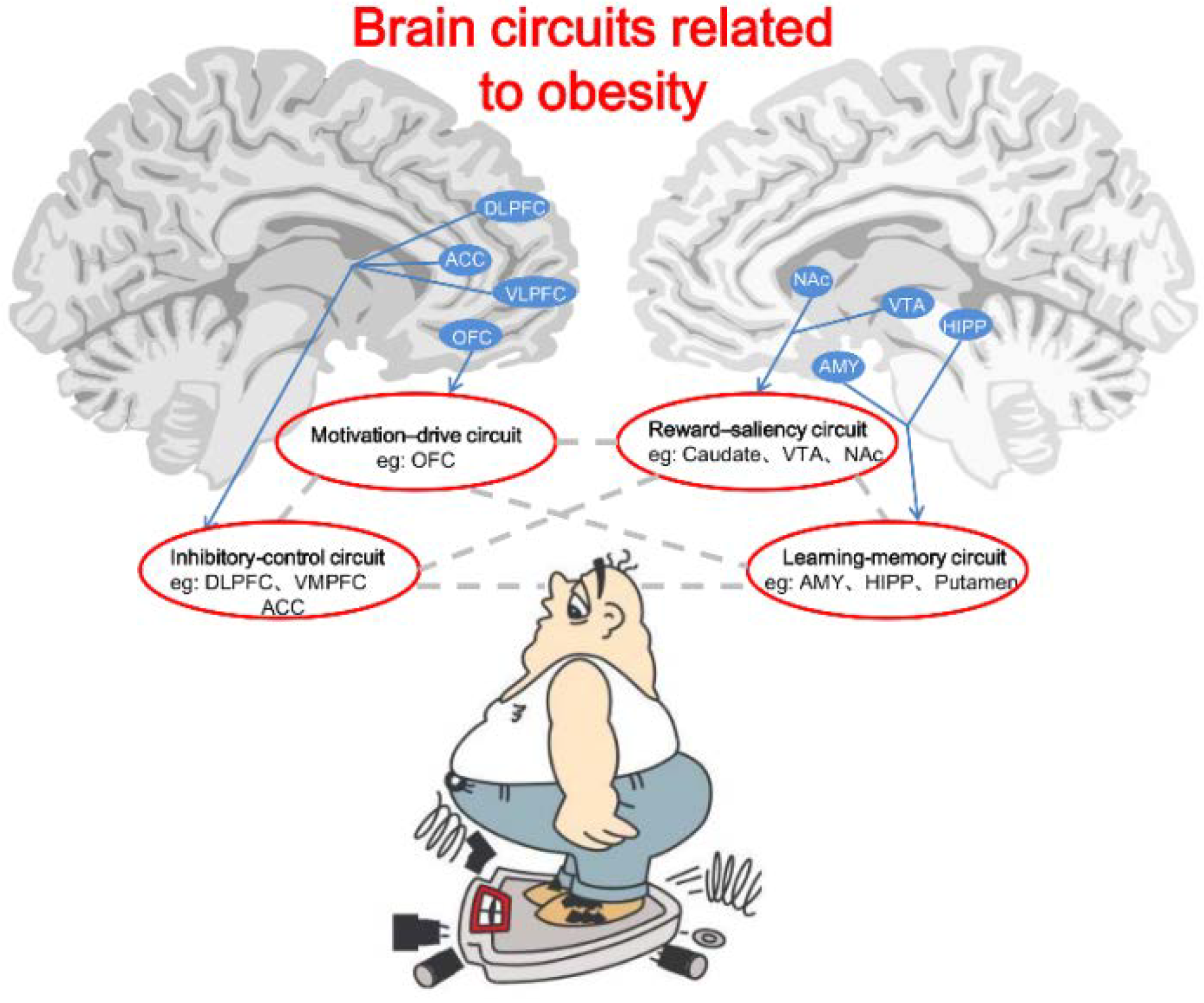
6.1. Reward-Saliency Circuit
6.2. Motivation-Drive Circuit
6.3. Learning-Memory Circuit
6.4. Inhibitory-Control Circuit
7. Therapeutic Interventions
7.1. Dietary and Lifestyle Interventions
7.2. Weight Loss Drugs
7.3. Bariatric Surgery
7.4. Fecal Microbiota Transplantation
8. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Rayner, G.; Lang, T. Obesity: Using the ecologic public health approach to overcome policy cacophony. In Clinical Obesity in Adults and Children; Wiley-Blackwell: Malden, USA, 2009; pp. 452–470. [Google Scholar]
- Pi-Sunyer, X. The medical risks of obesity. Postgrad. Med. 2009, 121, 21–33. [Google Scholar] [CrossRef]
- Campos, P.; Saguy, A.; Ernsberger, P.; Oliver, E.; Gaesser, G. The epidemiology of overweight and obesity: Public health crisis or moral panic? Int. J. Epidemiol. 2006, 35, 55–60. [Google Scholar] [CrossRef] [PubMed]
- Von Deneen, K.M.; Liu, Y. Obesity as an addiction: Why do the obese eat more? Maturitas 2011, 68, 342–345. [Google Scholar] [CrossRef] [PubMed]
- Avena, N.M.; Gold, J.A.; Kroll, C.; Gold, M.S. Further developments in the neurobiology of food and addiction: Update on the state of the science. Nutrition 2012, 28, 341–343. [Google Scholar] [CrossRef] [PubMed]
- Cho, J.; Juon, H.S. Assessing Overweight and Obesity Risk among Korean Americans in California Using World Health Organization Body Mass Index Criteria for Asians. Available online: http://www.cdc.gov/pcd/issues/2006/jul/pdf/05_0198.pdf (accessed on 23 June 2014).
- Ogden, C.L.; Carroll, M.D.; Curtin, L.R.; McDowell, M.A.; Tabak, C.J.; Flegal, K.M. Prevalence of overweight and obesity in the United States, 1999–2004. JAMA 2006, 295, 1549–1555. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Beydoun, M.A.; Liang, L.; Caballero, B.; Kumanyika, S.K. Will all Americans become overweight or obese? Estimating the progression and cost of the US obesity epidemic. Obesity (Silver Spring) 2008, 16, 2323–2330. [Google Scholar] [CrossRef]
- Fincham, J.E. The expanding public health threat of obesity and overweight. Int. J. Pharm. Pract. 2011, 19, 214–216. [Google Scholar] [CrossRef] [PubMed]
- Flegal, K.M.; Graubard, B.I.; Williamson, D.F.; Gail, M.H. Excess deaths associated with underweight, overweight, and obesity. JAMA 2005, 293, 1861–1867. [Google Scholar] [CrossRef] [PubMed]
- Calle, E.E.; Rodriguez, C.; Walker-Thurmond, K.; Thun, M.J. Overweight, obesity, and mortality from cancer in a prospectively studied cohort of US adults. N. Engl. J. Med. 2003, 348, 1625–1638. [Google Scholar] [CrossRef] [PubMed]
- Adams, K.F.; Schatzkin, A.; Harris, T.B.; Kipnis, V.; Mouw, T.; Ballard-Barbash, R.; Hollenbeck, A.; Leitzmann, M.F. Overweight, obesity, and mortality in a large prospective cohort of persons 50 to 71 years old. N. Engl. J. Med. 2006, 355, 763–778. [Google Scholar] [CrossRef] [PubMed]
- Davis, C.; Carter, J.C. Compulsive overeating as an addiction disorder. A review of theory and evidence. Appetite 2009, 53, 1–8. [Google Scholar] [CrossRef] [PubMed]
- French, S.A.; Story, M.; Fulkerson, J.A.; Gerlach, A.F. Food environment in secondary schools: A la carte, vending machines, and food policies and practices. Am. J. Public Health 2003, 93, 1161–1167. [Google Scholar] [CrossRef] [PubMed]
- Frazao, E.; Allshouse, J. Strategies for intervention: Commentary and debate. J. Nutr. 2003, 133, 844S–847S. [Google Scholar] [PubMed]
- Wadden, T.A.; Clark, V.L. Behavioural treatment of obesity: Achievements and challenges. In Clinical Obesity in Adults and Children; Wiley-Blackwell: Malden, MA, USA, 2005; pp. 350–362. [Google Scholar]
- Stice, E.; Spoor, S.; Ng, J.; Zald, D.H. Relation of obesity to consummatory and anticipatory food reward. Physiol. Behav. 2009, 97, 551–560. [Google Scholar] [CrossRef] [PubMed]
- Swanson, S.A.; Crow, S.J.; le Grange, D.; Swendsen, J.; Merikangas, K.R. Prevalence and correlates of eating disorders in adolescents. Results from the national comorbidity survey replication adolescent supplement. Arch. Gen. Psychiatry 2011, 68, 714–723. [Google Scholar] [CrossRef] [PubMed]
- Lebow, J.; Sim, L.A.; Kransdorf, L.N. Prevalence of a history of overweight and obesity in adolescents with restrictive eating disorders. J. Adolesc. Health 2014, in press. [Google Scholar]
- Baile, J.I. Binge eating disorder: Officially recognized as the new eating disorder. Rev. Med. Chil. 2014, 142, 128–129. [Google Scholar] [CrossRef] [PubMed]
- Iacovino, J.M.; Gredysa, D.M.; Altman, M.; Wilfley, D.E. Psychological treatments for binge eating disorder. Curr. Psychiatry Rep. 2012, 14, 432–446. [Google Scholar] [CrossRef]
- Hudson, J.I.; Hiripi, E.; Pope, H.J.; Kessler, R.C. The prevalence and correlates of eating disorders in the National Comorbidity Survey Replication. Biol. Psychiatry 2007, 61, 348–358. [Google Scholar] [CrossRef] [PubMed]
- Westerburg, D.P.; Waitz, M. Binge-eating disorder. Osteopath. Fam. Phys. 2013, 5, 230–233. [Google Scholar] [CrossRef]
- Gearhardt, A.N.; White, M.A.; Potenza, M.N. Binge eating disorder and food addiction. Curr. Drug Abuse Rev. 2011, 4, 201–207. [Google Scholar] [CrossRef] [PubMed]
- Avena, N.M.; Rada, P.; Hoebel, B.G. Evidence for sugar addiction: Behavioral and neurochemical effects of intermittent, excessive sugar intake. Neurosci. Biobehav. Rev. 2008, 32, 20–39. [Google Scholar] [CrossRef] [PubMed]
- Johnson, P.M.; Kenny, P.J. Dopamine D2 receptors in addiction-like reward dysfunction and compulsive eating in obese rats. Nat. Neurosci. 2010, 13, 635–641. [Google Scholar] [CrossRef] [PubMed]
- Zilberter, T. Food addiction and obesity: Do macronutrients matter? Front. Neuroenergetics 2012, 4, 7. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.J.; Volkow, N.D.; Thanos, P.K.; Fowler, J.S. Similarity between obesity and drug addiction as assessed by neurofunctional imaging: A concept review. J. Addict. Dis. 2004, 23, 39–53. [Google Scholar] [CrossRef] [PubMed]
- Hebebrand, J.; Albayrak, O.; Adan, R.; Antel, J.; Dieguez, C.; de Jong, J.; Leng, G.; Menzies, J.; Mercer, J.G.; Murphy, M.; et al. “Eating addiction”, rather than “food addiciton”, better captures addictive-like eating behavior. Neurosci. Biobehav. Rev. 2014, 47, 295–306. [Google Scholar] [CrossRef]
- Page, R.M.; Brewster, A. Depiction of food as having drug-like properties in televised food advertisements directed at children: Portrayals as pleasure enhancing and addictive. J. Pediatr. Health Care 2009, 23, 150–157. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.J.; Volkow, N.D.; Thanos, P.K.; Fowler, J.S. Imaging of brain dopamine pathways: Implications for understanding obesity. J. Addict. Med. 2009, 3, 8–18. [Google Scholar] [CrossRef]
- Dagher, A. The neurobiology of appetite: Hunger as addiction. Int. J. Obes. (Lond.) 2009, 33, S30–S33. [Google Scholar] [CrossRef]
- Ifland, J.R.; Preuss, H.G.; Marcus, M.T.; Rourke, K.M.; Taylor, W.C.; Burau, K.; Jacobs, W.S.; Kadish, W.; Manso, G. Refined food addiction: A classic substance use disorder. Med. Hypotheses 2009, 72, 518–526. [Google Scholar] [CrossRef] [PubMed]
- Spring, B.; Schneider, K.; Smith, M.; Kendzor, D.; Appelhans, B.; Hedeker, D.; Pagoto, S. Abuse potential of carbohydrates for overweight carbohydrate cravers. Psychopharmacology (Berl.) 2008, 197, 637–647. [Google Scholar] [CrossRef]
- Stice, E.; Spoor, S.; Bohon, C.; Small, D.M. Relation between obesity and blunted striatal response to food is moderated by TaqIA A1 allele. Science 2008, 322, 449–452. [Google Scholar] [CrossRef] [PubMed]
- Noble, E.P.; Blum, K.; Ritchie, T.; Montgomery, A.; Sheridan, P.J. Allelic association of the D2 dopamine receptor gene with receptor-binding characteristics in alcoholism. Arch. Gen. Psychiatry 1991, 48, 648–654. [Google Scholar] [CrossRef] [PubMed]
- Gearhardt, A.N.; Roberto, C.A.; Seamans, M.J.; Corbin, W.R.; Brownell, K.D. Preliminary validation of the Yale Food Addiction Scale for children. Eat. Behav. 2013, 14, 508–512. [Google Scholar] [CrossRef] [PubMed]
- Gearhardt, A.N.; Corbin, W.R.; Brownell, K.D. Preliminary validation of the Yale Food Addiction Scale. Appetite 2009, 52, 430–436. [Google Scholar] [CrossRef] [PubMed]
- Gearhardt, A.N.; Yokum, S.; Orr, P.T.; Stice, E.; Corbin, W.R.; Brownell, K.D. Neural correlates of food addiction. Arch. Gen. Psychiatry 2011, 68, 808–816. [Google Scholar] [CrossRef] [PubMed]
- Warren, M.W.; Gold, M.S. The relationship between obesity and drug use. Am. J. Psychiatry 2007, 164, 1268–1269. [Google Scholar] [CrossRef] [PubMed]
- Gold, M.S.; Frost-Pineda, K.; Jacobs, W.S. Overeating, binge eating, and eating disorders as addiction. Psychiatr. Ann. 2003, 33, 1549–1555. [Google Scholar]
- Zhang, Y.; von Deneen, K.M.; Tian, J.; Gold, M.S.; Liu, Y. Food addiction and neuroimaging. Curr. Pharm. Des. 2011, 17, 1149–1157. [Google Scholar] [CrossRef] [PubMed]
- Von Deneen, K.M.; Gold, M.S.; Liu, Y. Food addiction and cues in Prader-Willi syndrome. J. Addict. Med. 2009, 3, 19–25. [Google Scholar] [CrossRef] [PubMed]
- Shapira, N.A.; Lessig, M.C.; He, A.G.; James, G.A.; Driscoll, D.J.; Liu, Y. Satiety dysfunction in Prader-Willi syndrome demonstrated by fMRI. J. Neurol. Neurosurg. Psychiatry 2005, 76, 260–262. [Google Scholar] [CrossRef] [PubMed]
- Dimitropoulos, A.; Blackford, J.; Walden, T.; Thompson, T. Compulsive behavior in Prader-Willi syndrome: Examining severity in early childhood. Res. Dev. Disabil. 2006, 27, 190–202. [Google Scholar] [CrossRef] [PubMed]
- Dimitropoulos, A.; Schultz, R.T. Food-related neural circuitry in Prader-Willi syndrome: Response to high- versus low-calorie foods. J. Autism Dev. Disord. 2008, 38, 1642–1653. [Google Scholar] [CrossRef] [PubMed]
- Holsen, L.M.; Zarcone, J.R.; Chambers, R.; Butler, M.G.; Bittel, D.C.; Brooks, W.M.; Thompson, T.I.; Savage, C.R. Genetic subtype differences in neural circuitry of food motivation in Prader-Willi syndrome. Int. J. Obes. (Lond.) 2009, 33, 273–283. [Google Scholar] [CrossRef]
- Mantoulan, C.; Payoux, P.; Diene, G.; Glattard, M.; Roge, B.; Molinas, C.; Sevely, A.; Zilbovicius, M.; Celsis, P.; Tauber, M. PET scan perfusion imaging in the Prader-Willi syndrome: New insights into the psychiatric and social disturbances. J. Cereb. Blood Flow Metab. 2011, 31, 275–282. [Google Scholar] [CrossRef] [PubMed]
- Miller, J.L.; James, G.A.; Goldstone, A.P.; Couch, J.A.; He, G.; Driscoll, D.J.; Liu, Y. Enhanced activation of reward mediating prefrontal regions in response to food stimuli in Prader-Willi syndrome. J. Neurol. Neurosurg. Psychiatry 2007, 78, 615–619. [Google Scholar] [CrossRef] [PubMed]
- Ogura, K.; Shinohara, M.; Ohno, K.; Mori, E. Frontal behavioral syndromes in Prader-Willi syndrome. Brain Dev. 2008, 30, 469–476. [Google Scholar] [CrossRef] [PubMed]
- Holsen, L.M.; Zarcone, J.R.; Brooks, W.M.; Butler, M.G.; Thompson, T.I.; Ahluwalia, J.S.; Nollen, N.L.; Savage, C.R. Neural mechanisms underlying hyperphagia in Prader-Willi syndrome. Obesity (Silver Spring) 2006, 14, 1028–1037. [Google Scholar] [CrossRef]
- Kim, S.E.; Jin, D.K.; Cho, S.S.; Kim, J.H.; Hong, S.D.; Paik, K.H.; Oh, Y.J.; Kim, A.H.; Kwon, E.K.; Choe, Y.H. Regional cerebral glucose metabolic abnormality in Prader-Willi syndrome: A 18F-FDG PET study under sedation. J. Nucl. Med. 2006, 47, 1088–1092. [Google Scholar] [PubMed]
- Zhang, Y.; Zhao, H.; Qiu, S.; Tian, J.; Wen, X.; Miller, J.L.; von Deneen, K.M.; Zhou, Z.; Gold, M.S.; Liu, Y. Altered functional brain networks in Prader-Willi syndrome. NMR Biomed. 2013, 26, 622–629. [Google Scholar] [PubMed]
- Liu, Y.; von Deneen, K.M.; Kobeissy, F.H.; Gold, M.S. Food addiction and obesity: Evidence from bench to bedside. J. Psychoact. Drugs 2010, 42, 133–145. [Google Scholar] [CrossRef]
- Avena, N.M.; Rada, P.; Hoebel, B.G. Sugar and fat bingeing have notable differences in addictive-like behavior. J. Nutr. 2009, 139, 623–628. [Google Scholar] [CrossRef] [PubMed]
- Lutter, M.; Nestler, E.J. Homeostatic and hedonic signals interact in the regulation of food intake. J. Nutr. 2009, 139, 629–632. [Google Scholar] [CrossRef] [PubMed]
- Small, D.M.; Jones-Gotman, M.; Dagher, A. Feeding-induced dopamine release in dorsal striatum correlates with meal pleasantness ratings in healthy human volunteers. Neuroimage 2003, 19, 1709–1715. [Google Scholar] [CrossRef] [PubMed]
- Lenard, N.R.; Berthoud, H.R. Central and peripheral regulation of food intake and physical activity: Pathways and genes. Obesity (Silver Spring) 2008, 16, S11–S22. [Google Scholar] [CrossRef]
- Myers, M.G.; Cowley, M.A.; Munzberg, H. Mechanisms of leptin action and leptin resistance. Annu. Rev. Physiol. 2008, 70, 537–556. [Google Scholar] [CrossRef] [PubMed]
- Palmiter, R.D. Is dopamine a physiologically relevant mediator of feeding behavior? Trends Neurosci. 2007, 30, 375–381. [Google Scholar] [CrossRef] [PubMed]
- Abizaid, A.; Liu, Z.W.; Andrews, Z.B.; Shanabrough, M.; Borok, E.; Elsworth, J.D.; Roth, R.H.; Sleeman, M.W.; Picciotto, M.R.; Tschop, M.H.; et al. Ghrelin modulates the activity and synaptic input organization of midbrain dopamine neurons while promoting appetite. J. Clin. Investig. 2006, 116, 3229–3239. [Google Scholar] [CrossRef] [PubMed]
- Fried, S.K.; Ricci, M.R.; Russell, C.D.; Laferrere, B. Regulation of leptin production in humans. J. Nutr. 2000, 130, 3127S–3131S. [Google Scholar] [PubMed]
- Arora, S.; Anubhut. Role of neuropeptides in appetite regulation and obesity—A review. Neuropeptides 2006, 40, 375–401. [Google Scholar] [CrossRef] [PubMed]
- Farooqi, I.S.; O’Rahilly, S. Recent advances in the genetics of severe childhood obesity. Arch. Dis. Child 2000, 83, 31–34. [Google Scholar] [CrossRef] [PubMed]
- Benoit, S.C.; Clegg, D.J.; Seeley, R.J.; Woods, S.C. Insulin and leptin as adiposity signals. Recent Prog. Horm. Res. 2004, 59, 267–285. [Google Scholar] [CrossRef] [PubMed]
- Farooqi, I.S.; Bullmore, E.; Keogh, J.; Gillard, J.; O’Rahilly, S.; Fletcher, P.C. Leptin regulates striatal regions and human eating behavior. Science 2007, 317, 1355. [Google Scholar] [CrossRef] [PubMed]
- Hukshorn, C.J.; van Dielen, F.M.; Buurman, W.A.; Westerterp-Plantenga, M.S.; Campfield, L.A.; Saris, W.H. The effect of pegylated recombinant human leptin (PEG-OB) on weight loss and inflammatory status in obese subjects. Int. J. Obes. Relat. Metab. Disord. 2002, 26, 504–509. [Google Scholar] [CrossRef] [PubMed]
- Figlewicz, D.P.; Bennett, J.; Evans, S.B.; Kaiyala, K.; Sipols, A.J.; Benoit, S.C. Intraventricular insulin and leptin reverse place preference conditioned with high-fat diet in rats. Behav. Neurosci. 2004, 118, 479–487. [Google Scholar] [CrossRef] [PubMed]
- Maffeis, C.; Manfredi, R.; Trombetta, M.; Sordelli, S.; Storti, M.; Benuzzi, T.; Bonadonna, R.C. Insulin sensitivity is correlated with subcutaneous but not visceral body fat in overweight and obese prepubertal children. J. Clin. Endocrinol. Metab. 2008, 93, 2122–2128. [Google Scholar] [CrossRef] [PubMed]
- Bjorntorp, P. Obesity, atherosclerosis and diabetes mellitus. Verh. Dtsch. Ges. Inn. Med. 1987, 93, 443–448. [Google Scholar] [PubMed]
- Rushing, P.A.; Lutz, T.A.; Seeley, R.J.; Woods, S.C. Amylin and insulin interact to reduce food intake in rats. Horm. Metab. Res. 2000, 32, 62–65. [Google Scholar] [CrossRef] [PubMed]
- Qatanani, M.; Lazar, M.A. Mechanisms of obesity-associated insulin resistance: Many choices on the menu. Genes Dev. 2007, 21, 1443–1455. [Google Scholar] [CrossRef] [PubMed]
- Yang, R.; Barouch, L.A. Leptin signaling and obesity: Cardiovascular consequences. Circ. Res. 2007, 101, 545–559. [Google Scholar] [CrossRef] [PubMed]
- Anthony, K.; Reed, L.J.; Dunn, J.T.; Bingham, E.; Hopkins, D.; Marsden, P.K.; Amiel, S.A. Attenuation of insulin-evoked responses in brain networks controlling appetite and reward in insulin resistance: The cerebral basis for impaired control of food intake in metabolic syndrome? Diabetes 2006, 55, 2986–2992. [Google Scholar] [CrossRef] [PubMed]
- Figlewicz, D.P.; Bennett, J.L.; Naleid, A.M.; Davis, C.; Grimm, J.W. Intraventricular insulin and leptin decrease sucrose self-administration in rats. Physiol. Behav. 2006, 89, 611–616. [Google Scholar] [CrossRef] [PubMed]
- Korbonits, M.; Goldstone, A.P.; Gueorguiev, M.; Grossman, A.B. Ghrelin—A hormone with multiple functions. Front. Neuroendocrinol. 2004, 25, 27–68. [Google Scholar] [CrossRef] [PubMed]
- Wren, A.M.; Small, C.J.; Abbott, C.R.; Dhillo, W.S.; Seal, L.J.; Cohen, M.A.; Batterham, R.L.; Taheri, S.; Stanley, S.A.; Ghatei, M.A.; et al. Ghrelin causes hyperphagia and obesity in rats. Diabetes 2001, 50, 2540–2547. [Google Scholar] [CrossRef] [PubMed]
- Wren, A.M.; Seal, L.J.; Cohen, M.A.; Brynes, A.E.; Frost, G.S.; Murphy, K.G.; Dhillo, W.S.; Ghatei, M.A.; Bloom, S.R. Ghrelin enhances appetite and increases food intake in humans. J. Clin. Endocrinol. Metab. 2001, 86, 5992. [Google Scholar] [CrossRef] [PubMed]
- Cummings, D.E.; Weigle, D.S.; Frayo, R.S.; Breen, P.A.; Ma, M.K.; Dellinger, E.P.; Purnell, J.Q. Plasma ghrelin levels after diet-induced weight loss or gastric bypass surgery. N. Engl. J. Med. 2002, 346, 1623–1630. [Google Scholar] [CrossRef] [PubMed]
- Tschop, M.; Smiley, D.L.; Heiman, M.L. Ghrelin induces adiposity in rodents. Nature 2000, 407, 908–913. [Google Scholar] [CrossRef] [PubMed]
- Tschop, M.; Weyer, C.; Tataranni, P.A.; Devanarayan, V.; Ravussin, E.; Heiman, M.L. Circulating ghrelin levels are decreased in human obesity. Diabetes 2001, 50, 707–709. [Google Scholar] [CrossRef] [PubMed]
- Shiiya, T.; Nakazato, M.; Mizuta, M.; Date, Y.; Mondal, M.S.; Tanaka, M.; Nozoe, S.; Hosoda, H.; Kangawa, K.; Matsukura, S. Plasma ghrelin levels in lean and obese humans and the effect of glucose on ghrelin secretion. J. Clin. Endocrinol. Metab. 2002, 87, 240–244. [Google Scholar] [CrossRef] [PubMed]
- Malik, S.; McGlone, F.; Bedrossian, D.; Dagher, A. Ghrelin modulates brain activity in areas that control appetitive behavior. Cell Metab. 2008, 7, 400–409. [Google Scholar] [CrossRef] [PubMed]
- Jerlhag, E.; Egecioglu, E.; Dickson, S.L.; Douhan, A.; Svensson, L.; Engel, J.A. Ghrelin administration into tegmental areas stimulates locomotor activity and increases extracellular concentration of dopamine in the nucleus accumbens. Addict. Biol. 2007, 12, 6–16. [Google Scholar] [CrossRef] [PubMed]
- Valassi, E.; Scacchi, M.; Cavagnini, F. Neuroendocrine control of food intake. Nutr. Metab. Cardiovasc. Dis. 2008, 18, 158–168. [Google Scholar] [CrossRef] [PubMed]
- Naslund, E.; Hellstrom, P.M. Appetite signaling: From gut peptides and enteric nerves to brain. Physiol. Behav. 2007, 92, 256–262. [Google Scholar] [CrossRef] [PubMed]
- Woods, S.C. Gastrointestinal satiety signals I. An overview of gastrointestinal signals that influence food intake. Am. J. Physiol. Gastrointest. Liver Physiol. 2004, 286, G7–G13. [Google Scholar] [CrossRef] [PubMed]
- Alvarez, B.M.; Borque, M.; Martinez-Sarmiento, J.; Aparicio, E.; Hernandez, C.; Cabrerizo, L.; Fernandez-Represa, J.A.; Peptide, Y.Y. Secretion in morbidly obese patients before and after vertical banded gastroplasty. Obes. Surg. 2002, 12, 324–327. [Google Scholar] [CrossRef] [PubMed]
- Batterham, R.L.; Cohen, M.A.; Ellis, S.M.; le Roux, C.W.; Withers, D.J.; Frost, G.S.; Ghatei, M.A.; Bloom, S.R. Inhibition of food intake in obese subjects by peptide YY3–36. N. Engl. J. Med. 2003, 349, 941–948. [Google Scholar] [CrossRef] [PubMed]
- Murphy, K.G.; Bloom, S.R. Gut hormones and the regulation of energy homeostasis. Nature 2006, 444, 854–859. [Google Scholar] [CrossRef] [PubMed]
- Holst, J.J. The physiology of glucagon-like peptide 1. Physiol. Rev. 2007, 87, 1409–1439. [Google Scholar] [CrossRef] [PubMed]
- Tang-Christensen, M.; Vrang, N.; Larsen, P.J. Glucagon-like peptide containing pathways in the regulation of feeding behaviour. Int. J. Obes. Relat. Metab. Disord. 2001, 25, S42–S47. [Google Scholar] [CrossRef] [PubMed]
- Naslund, E.; King, N.; Mansten, S.; Adner, N.; Holst, J.J.; Gutniak, M.; Hellstrom, P.M. Prandial subcutaneous injections of glucagon-like peptide-1 cause weight loss in obese human subjects. Br. J. Nutr. 2004, 91, 439–446. [Google Scholar] [CrossRef] [PubMed]
- Verdich, C.; Toubro, S.; Buemann, B.; Lysgard, M.J.; Juul, H.J.; Astrup, A. The role of postprandial releases of insulin and incretin hormones in meal-induced satiety—Effect of obesity and weight reduction. Int. J. Obes. Relat. Metab. Disord. 2001, 25, 1206–1214. [Google Scholar] [CrossRef] [PubMed]
- Ochner, C.N.; Gibson, C.; Shanik, M.; Goel, V.; Geliebter, A. Changes in neurohormonal gut peptides following bariatric surgery. Int. J. Obes. (Lond.) 2011, 35, 153–166. [Google Scholar] [CrossRef]
- Liddle, R.A.; Goldfine, I.D.; Rosen, M.S.; Taplitz, R.A.; Williams, J.A. Cholecystokinin bioactivity in human plasma. Molecular forms, responses to feeding, and relationship to gallbladder contraction. J. Clin. Investig. 1985, 75, 1144–1152. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, S.; Ramos, E.J.; Goncalves, C.G.; Chen, C.; Meguid, M.M. Changes in GI hormones and their effect on gastric emptying and transit times after Roux-en-Y gastric bypass in rat model. Surgery 2005, 138, 283–290. [Google Scholar] [CrossRef] [PubMed]
- Carnell, S.; Gibson, C.; Benson, L.; Ochner, C.N.; Geliebter, A. Neuroimaging and obesity: Current knowledge and future directions. Obes. Rev. 2012, 13, 43–56. [Google Scholar] [CrossRef] [PubMed]
- Rothemund, Y.; Preuschhof, C.; Bohner, G.; Bauknecht, H.C.; Klingebiel, R.; Flor, H.; Klapp, B.F. Differential activation of the dorsal striatum by high-calorie visual food stimuli in obese individuals. Neuroimage 2007, 37, 410–421. [Google Scholar] [CrossRef] [PubMed]
- Bragulat, V.; Dzemidzic, M.; Bruno, C.; Cox, C.A.; Talavage, T.; Considine, R.V.; Kareken, D.A. Food-related odor probes of brain reward circuits during hunger: A pilot FMRI study. Obesity (Silver Spring) 2010, 18, 1566–1571. [Google Scholar] [CrossRef]
- Gautier, J.F.; Chen, K.; Salbe, A.D.; Bandy, D.; Pratley, R.E.; Heiman, M.; Ravussin, E.; Reiman, E.M.; Tataranni, P.A. Differential brain responses to satiation in obese and lean men. Diabetes 2000, 49, 838–846. [Google Scholar] [CrossRef] [PubMed]
- Soto-Montenegro, M.L.; Pascau, J.; Desco, M. Response to deep brain stimulation in the lateral hypothalamic area in a rat model of obesity: In vivo assessment of brain glucose metabolism. Mol. Imaging Biol. 2014, in press. [Google Scholar]
- Melega, W.P.; Lacan, G.; Gorgulho, A.A.; Behnke, E.J.; de Salles, A.A. Hypothalamic deep brain stimulation reduces weight gain in an obesity-animal model. PLoS One 2012, 7, e30672. [Google Scholar] [CrossRef] [PubMed]
- Whiting, D.M.; Tomycz, N.D.; Bailes, J.; de Jonge, L.; Lecoultr, V.; Wilent, B.; Alcindor, D.; Prostko, E.R.; Cheng, B.C.; Angle, C.; et al. Lateral hypothalamic area deep brain stimulation for refractory obesity: A pilot study with preliminary data on safety, body weight, and energy metabolism. J. Neurosurg. 2013, 119, 56–63. [Google Scholar] [CrossRef] [PubMed]
- Orava, J.; Nummenmaa, L.; Noponen, T.; Viljanen, T.; Parkkola, R.; Nuutila, P.; Virtanen, K.A. Brown adipose tissue function is accompanied by cerebral activation in lean but not in obese humans. J. Cereb. Blood Flow Metab. 2014, 34, 1018–1023. [Google Scholar] [CrossRef] [PubMed]
- Lavie, C.J.; de Schutter, A.; Patel, D.A.; Milani, R.V. Does fitness completely explain the obesity paradox? Am. Heart J. 2013, 166, 1–3. [Google Scholar] [CrossRef] [PubMed]
- Van de Giessen, E.; Celik, F.; Schweitzer, D.H.; van den Brink, W.; Booij, J. Dopamine D2/3 receptor availability and amphetamine-induced dopamine release in obesity. J. Psychopharmacol. 2014, 28, 866–873. [Google Scholar] [CrossRef] [PubMed]
- Hung, C.S.; Wu, Y.W.; Huang, J.Y.; Hsu, P.Y.; Chen, M.F. Evaluation of circulating adipokines and abdominal obesity as predictors of significant myocardial ischemia using gated single-photon emission computed tomography. PLoS One 2014, 9, e97710. [Google Scholar] [CrossRef] [PubMed]
- Chow, B.J.; Dorbala, S.; di Carli, M.F.; Merhige, M.E.; Williams, B.A.; Veledar, E.; Min, J.K.; Pencina, M.J.; Yam, Y.; Chen, L.; et al. Prognostic value of PET myocardial perfusion imaging in obese patients. JACC Cardiovasc. Imaging 2014, 7, 278–287. [Google Scholar] [CrossRef] [PubMed]
- Ogura, K.; Fujii, T.; Abe, N.; Hosokai, Y.; Shinohara, M.; Fukuda, H.; Mori, E. Regional cerebral blood flow and abnormal eating behavior in Prader-Willi syndrome. Brain Dev. 2013, 35, 427–434. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.; Kyung, C.; Park, J.S.; Kim, S.; Lee, S.P.; Kim, M.K.; Kim, H.K.; Kim, K.R.; Jeon, T.J.; Ahn, C.W. Subclinical vascular inflammation in subjects with normal weight obesity and its association with body fat: An 18 F-FDG-PET/CT study. Cardiovasc. Diabetol. 2014, 13, 70. [Google Scholar] [CrossRef] [PubMed]
- Le, D.S.; Pannacciulli, N.; Chen, K.; Del, P.A.; Salbe, A.D.; Reiman, E.M.; Krakoff, J. Less activation of the left dorsolateral prefrontal cortex in response to a meal: A feature of obesity. Am. J. Clin. Nutr. 2006, 84, 725–731. [Google Scholar] [PubMed]
- Green, E.; Jacobson, A.; Haase, L.; Murphy, C. Reduced nucleus accumbens and caudate nucleus activation to a pleasant taste is associated with obesity in older adults. Brain Res. 2011, 1386, 109–117. [Google Scholar] [CrossRef] [PubMed]
- Walther, K.; Birdsill, A.C.; Glisky, E.L.; Ryan, L. Structural brain differences and cognitive functioning related to body mass index in older females. Hum. Brain Mapp. 2010, 31, 1052–1064. [Google Scholar] [CrossRef] [PubMed]
- Taki, Y.; Kinomura, S.; Sato, K.; Inoue, K.; Goto, R.; Okada, K.; Uchida, S.; Kawashima, R.; Fukuda, H. Relationship between body mass index and gray matter volume in 1428 healthy individuals. Obesity (Silver Spring) 2008, 16, 119–124. [Google Scholar] [CrossRef]
- Pannacciulli, N.; Del, P.A.; Chen, K.; Le, D.S.; Reiman, E.M.; Tataranni, P.A. Brain abnormalities in human obesity: A voxel-based morphometric study. Neuroimage 2006, 31, 1419–1425. [Google Scholar] [CrossRef] [PubMed]
- Ward, M.A.; Carlsson, C.M.; Trivedi, M.A.; Sager, M.A.; Johnson, S.C. The effect of body mass index on global brain volume in middle-aged adults: A cross sectional study. BMC Neurol. 2005, 5, 23. [Google Scholar] [CrossRef] [PubMed]
- Gunstad, J.; Paul, R.H.; Cohen, R.A.; Tate, D.F.; Spitznagel, M.B.; Grieve, S.; Gordon, E. Relationship between body mass index and brain volume in healthy adults. Int. J. Neurosci. 2008, 118, 1582–1593. [Google Scholar] [CrossRef] [PubMed]
- Raji, C.A.; Ho, A.J.; Parikshak, N.N.; Becker, J.T.; Lopez, O.L.; Kuller, L.H.; Hua, X.; Leow, A.D.; Toga, A.W.; Thompson, P.M. Brain structure and obesity. Hum. Brain Mapp. 2010, 31, 353–364. [Google Scholar] [PubMed]
- Kivipelto, M.; Ngandu, T.; Fratiglioni, L.; Viitanen, M.; Kareholt, I.; Winblad, B.; Helkala, E.L.; Tuomilehto, J.; Soininen, H.; Nissinen, A. Obesity and vascular risk factors at midlife and the risk of dementia and Alzheimer disease. Arch. Neurol. 2005, 62, 1556–1560. [Google Scholar] [PubMed]
- Whitmer, R.A.; Gustafson, D.R.; Barrett-Connor, E.; Haan, M.N.; Gunderson, E.P.; Yaffe, K. Central obesity and increased risk of dementia more than three decades later. Neurology 2008, 71, 1057–1064. [Google Scholar] [CrossRef] [PubMed]
- Dahl, A.; Hassing, L.B.; Fransson, E.; Berg, S.; Gatz, M.; Reynolds, C.A.; Pedersen, N.L. Being overweight in midlife is associated with lower cognitive ability and steeper cognitive decline in late life. J. Gerontol. A Biol. Sci. Med. Sci. 2010, 65, 57–62. [Google Scholar] [CrossRef] [PubMed]
- Lim, D.C.; Veasey, S.C. Neural injury in sleep apnea. Curr. Neurol. Neurosci. Rep. 2010, 10, 47–52. [Google Scholar] [CrossRef] [PubMed]
- Bruce-Keller, A.J.; Keller, J.N.; Morrison, C.D. Obesity and vulnerability of the CNS. Biochim. Biophys. Acta 2009, 1792, 395–400. [Google Scholar] [CrossRef] [PubMed]
- Pistell, P.J.; Morrison, C.D.; Gupta, S.; Knight, A.G.; Keller, J.N.; Ingram, D.K.; Bruce-Keller, A.J. Cognitive impairment following high fat diet consumption is associated with brain inflammation. J. Neuroimmunol. 2010, 219, 25–32. [Google Scholar] [CrossRef] [PubMed]
- Widya, R.L.; de Roos, A.; Trompet, S.; de Craen, A.J.; Westendorp, R.G.; Smit, J.W.; van Buchem, M.A.; van der Grond, J. Increased amygdalar and hippocampal volumes in elderly obese individuals with or at risk of cardiovascular disease. Am. J. Clin. Nutr. 2011, 93, 1190–1195. [Google Scholar] [CrossRef] [PubMed]
- Purnell, J.Q.; Lahna, D.L.; Samuels, M.H.; Rooney, W.D.; Hoffman, W.F. Loss of pons-to-hypothalamic white matter tracks in brainstem obesity. Int. J. Obes. (Lond.) 2014, in press. [Google Scholar]
- Karlsson, H.K.; Tuulari, J.J.; Hirvonen, J.; Lepomaki, V.; Parkkola, R.; Hiltunen, J.; Hannukainen, J.C.; Soinio, M.; Pham, T.; Salminen, P.; et al. Obesity is associated with white matter atrophy: A combined diffusion tensor imaging and voxel-based morphometric study. Obesity (Silver Spring) 2013, 21, 2530–2537. [Google Scholar] [CrossRef]
- Volkow, N.D.; Wang, G.J.; Fowler, J.S.; Telang, F. Overlapping neuronal circuits in addiction and obesity: Evidence of systems pathology. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2008, 363, 3191–3200. [Google Scholar] [CrossRef] [PubMed]
- Volkow, N.D.; Wang, G.J.; Baler, R.D. Reward, dopamine and the control of food intake: Implications for obesity. Trends Cogn. Sci. 2011, 15, 37–46. [Google Scholar] [CrossRef] [PubMed]
- Steele, K.E.; Prokopowicz, G.P.; Schweitzer, M.A.; Magunsuon, T.H.; Lidor, A.O.; Kuwabawa, H.; Kumar, A.; Brasic, J.; Wong, D.F. Alterations of central dopamine receptors before and after gastric bypass surgery. Obes. Surg. 2010, 20, 369–374. [Google Scholar] [CrossRef] [PubMed]
- Salamone, J.D.; Cousins, M.S.; Snyder, B.J. Behavioral functions of nucleus accumbens dopamine: Empirical and conceptual problems with the anhedonia hypothesis. Neurosci. Biobehav. Rev. 1997, 21, 341–359. [Google Scholar] [CrossRef] [PubMed]
- Wise, R.A.; Bozarth, M.A. Brain reward circuitry: Four circuit elements “wired” in apparent series. Brain Res. Bull. 1984, 12, 203–208. [Google Scholar] [CrossRef] [PubMed]
- Bassareo, V.; di Chiara, G. Modulation of feeding-induced activation of mesolimbic dopamine transmission by appetitive stimuli and its relation to motivational state. Eur. J. Neurosci. 1999, 11, 4389–4397. [Google Scholar] [CrossRef] [PubMed]
- Volkow, N.D.; Wang, G.J.; Maynard, L.; Jayne, M.; Fowler, J.S.; Zhu, W.; Logan, J.; Gatley, S.J.; Ding, Y.S.; Wong, C.; et al. Brain dopamine is associated with eating behaviors in humans. Int. J. Eat. Disord. 2003, 33, 136–142. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, M.W.; Woods, S.C.; Porte, D.J.; Seeley, R.J.; Baskin, D.G. Central nervous system control of food intake. Nature 2000, 404, 661–671. [Google Scholar] [PubMed]
- Wang, G.J.; Volkow, N.D.; Felder, C.; Fowler, J.S.; Levy, A.V.; Pappas, N.R.; Wong, C.T.; Zhu, W.; Netusil, N. Enhanced resting activity of the oral somatosensory cortex in obese subjects. Neuroreport 2002, 13, 1151–1155. [Google Scholar] [CrossRef] [PubMed]
- Huttunen, J.; Kahkonen, S.; Kaakkola, S.; Ahveninen, J.; Pekkonen, E. Effects of an acute D2-dopaminergic blockade on the somatosensory cortical responses in healthy humans: Evidence from evoked magnetic fields. Neuroreport 2003, 14, 1609–1612. [Google Scholar] [CrossRef] [PubMed]
- Rossini, P.M.; Bassetti, M.A.; Pasqualetti, P. Median nerve somatosensory evoked potentials. Apomorphine-induced transient potentiation of frontal components in Parkinson’s disease and in parkinsonism. Electroencephalogr. Clin. Neurophysiol. 1995, 96, 236–247. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.I.; Ren, J.; Wang, F.N.; Xu, H.; Mandeville, J.B.; Kim, Y.; Rosen, B.R.; Jenkins, B.G.; Hui, K.K.; Kwong, K.K. Inhibition of stimulated dopamine release and hemodynamic response in the brain through electrical stimulation of rat forepaw. Neurosci. Lett. 2008, 431, 231–235. [Google Scholar] [CrossRef] [PubMed]
- Wise, R.A. Role of brain dopamine in food reward and reinforcement. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2006, 361, 1149–1158. [Google Scholar] [CrossRef] [PubMed]
- McFarland, K.; Ettenberg, A. Haloperidol does not affect motivational processes in an operant runway model of food-seeking behavior. Behav. Neurosci. 1998, 112, 630–635. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.J.; Volkow, N.D.; Logan, J.; Pappas, N.R.; Wong, C.T.; Zhu, W.; Netusil, N.; Fowler, J.S. Brain dopamine and obesity. Lancet 2001, 357, 354–357. [Google Scholar] [CrossRef] [PubMed]
- Haltia, L.T.; Rinne, J.O.; Merisaari, H.; Maguire, R.P.; Savontaus, E.; Helin, S.; Nagren, K.; Kaasinen, V. Effects of intravenous glucose on dopaminergic function in the human brain in vivo. Synapse 2007, 61, 748–756. [Google Scholar] [CrossRef] [PubMed]
- Restaino, L.; Frampton, E.W.; Turner, K.M.; Allison, D.R. A chromogenic plating medium for isolating Escherichia coli O157:H7 from beef. Lett. Appl. Microbiol. 1999, 29, 26–30. [Google Scholar] [CrossRef] [PubMed]
- Rolls, E.T. The functions of the orbitofrontal cortex. Brain Cogn. 2004, 55, 11–29. [Google Scholar] [CrossRef] [PubMed]
- Szalay, C.; Aradi, M.; Schwarcz, A.; Orsi, G.; Perlaki, G.; Nemeth, L.; Hanna, S.; Takacs, G.; Szabo, I.; Bajnok, L.; et al. Gustatory perception alterations in obesity: An fMRI study. Brain Res. 2012, 1473, 131–140. [Google Scholar] [CrossRef] [PubMed]
- Volkow, N.D.; Fowler, J.S. Addiction, a disease of compulsion and drive: Involvement of the orbitofrontal cortex. Cereb Cortex 2000, 10, 318–325. [Google Scholar] [CrossRef] [PubMed]
- Volkow, N.D.; Fowler, J.S.; Wang, G.J. The addicted human brain: Insights from imaging studies. J. Clin. Investig. 2003, 111, 1444–1451. [Google Scholar] [CrossRef] [PubMed]
- White, N.M. Addictive drugs as reinforcers: Multiple partial actions on memory systems. Addiction 1996, 91, 921–949. [Google Scholar] [CrossRef] [PubMed]
- Healy, S.D.; de Kort, S.R.; Clayton, N.S. The hippocampus, spatial memory and food hoarding: A puzzle revisited. Trends Ecol. Evol. 2005, 20, 17–22. [Google Scholar] [CrossRef] [PubMed]
- Breiter, H.C.; Gollub, R.L.; Weisskoff, R.M.; Kennedy, D.N.; Makris, N.; Berke, J.D.; Goodman, J.M.; Kantor, H.L.; Gastfriend, D.R.; Riorden, J.P.; et al. Acute effects of cocaine on human brain activity and emotion. Neuron 1997, 19, 591–611. [Google Scholar] [CrossRef] [PubMed]
- Stein, E.A.; Pankiewicz, J.; Harsch, H.H.; Cho, J.K.; Fuller, S.A.; Hoffmann, R.G.; Hawkins, M.; Rao, S.M.; Bandettini, P.A.; Bloom, A.S. Nicotine-induced limbic cortical activation in the human brain: A functional MRI study. Am. J. Psychiatry 1998, 155, 1009–1015. [Google Scholar] [PubMed]
- Grant, S.; London, E.D.; Newlin, D.B.; Villemagne, V.L.; Liu, X.; Contoreggi, C.; Phillips, R.L.; Kimes, A.S.; Margolin, A. Activation of memory circuits during cue-elicited cocaine craving. Proc. Natl. Acad. Sci. USA 1996, 93, 12040–12045. [Google Scholar] [CrossRef] [PubMed]
- Childress, A.R.; Mozley, P.D.; McElgin, W.; Fitzgerald, J.; Reivich, M.; O’Brien, C.P. Limbic activation during cue-induced cocaine craving. Am. J. Psychiatry 1999, 156, 11–18. [Google Scholar] [PubMed]
- Kilts, C.D.; Schweitzer, J.B.; Quinn, C.K.; Gross, R.E.; Faber, T.L.; Muhammad, F.; Ely, T.D.; Hoffman, J.M.; Drexler, K.P. Neural activity related to drug craving in cocaine addiction. Arch. Gen. Psychiatry 2001, 58, 334–341. [Google Scholar] [CrossRef] [PubMed]
- Ito, R.; Dalley, J.W.; Robbins, T.W.; Everitt, B.J. Dopamine release in the dorsal striatum during cocaine-seeking behavior under the control of a drug-associated cue. J. Neurosci. 2002, 22, 6247–6253. [Google Scholar] [PubMed]
- Letchworth, S.R.; Nader, M.A.; Smith, H.R.; Friedman, D.P.; Porrino, L.J. Progression of changes in dopamine transporter binding site density as a result of cocaine self-administration in rhesus monkeys. J. Neurosci. 2001, 21, 2799–2807. [Google Scholar] [PubMed]
- Knight, R.T.; Staines, W.R.; Swick, D.; Chao, L.L. Prefrontal cortex regulates inhibition and excitation in distributed neural networks. Acta Psychol. (Amst.) 1999, 101, 159–178. [Google Scholar] [CrossRef]
- Hollmann, M.; Hellrung, L.; Pleger, B.; Schlogl, H.; Kabisch, S.; Stumvoll, M.; Villringer, A.; Horstmann, A. Neural correlates of the volitional regulation of the desire for food. Int. J. Obes. (Lond.) 2012, 36, 648–655. [Google Scholar] [CrossRef]
- Hare, T.A.; Camerer, C.F.; Rangel, A. Self-control in decision-making involves modulation of the vmPFC valuation system. Science 2009, 324, 646–648. [Google Scholar] [CrossRef] [PubMed]
- Holsen, L.M.; Savage, C.R.; Martin, L.E.; Bruce, A.S.; Lepping, R.J.; Ko, E.; Brooks, W.M.; Butler, M.G.; Zarcone, J.R.; Goldstein, J.M. Importance of reward and prefrontal circuitry in hunger and satiety: Prader-Willi syndrome vs. simple obesity. Int. J. Obes. (Lond.) 2012, 36, 638–647. [Google Scholar] [CrossRef]
- Goldstein, R.Z.; Volkow, N.D. Drug addiction and its underlying neurobiological basis: Neuroimaging evidence for the involvement of the frontal cortex. Am. J. Psychiatry 2002, 159, 1642–1652. [Google Scholar] [CrossRef] [PubMed]
- Royall, D.R.; Lauterbach, E.C.; Cummings, J.L.; Reeve, A.; Rummans, T.A.; Kaufer, D.I.; LaFrance, W.J.; Coffey, C.E. Executive control function: A review of its promise and challenges for clinical research. A report from the Committee on Research of the American Neuropsychiatric Association. J. Neuropsychiatry Clin. Neurosci. 2002, 14, 377–405. [Google Scholar] [CrossRef] [PubMed]
- Bechara, A.; Damasio, H. Decision-making and addiction (part I): Impaired activation of somatic states in substance dependent individuals when pondering decisions with negative future consequences. Neuropsychologia 2002, 40, 1675–1689. [Google Scholar] [CrossRef] [PubMed]
- Ernst, M.; Grant, S.J.; London, E.D.; Contoreggi, C.S.; Kimes, A.S.; Spurgeon, L. Decision making in adolescents with behavior disorders and adults with substance abuse. Am. J. Psychiatry 2003, 160, 33–40. [Google Scholar] [CrossRef] [PubMed]
- Robinson, T.E.; Gorny, G.; Mitton, E.; Kolb, B. Cocaine self-administration alters the morphology of dendrites and dendritic spines in the nucleus accumbens and neocortex. Synapse 2001, 39, 257–266. [Google Scholar] [CrossRef] [PubMed]
- Ernst, M.; Matochik, J.A.; Heishman, S.J.; van Horn, J.D.; Jons, P.H.; Henningfield, J.E.; London, E.D. Effect of nicotine on brain activation during performance of a working memory task. Proc. Natl. Acad. Sci. USA 2001, 98, 4728–4733. [Google Scholar] [CrossRef] [PubMed]
- Rosenkranz, J.A.; Grace, A.A. Dopamine attenuates prefrontal cortical suppression of sensory inputs to the basolateral amygdala of rats. J. Neurosci. 2001, 21, 4090–4103. [Google Scholar] [PubMed]
- Lau, D.C.; Douketis, J.D.; Morrison, K.M.; Hramiak, I.M.; Sharma, A.M.; Ur, E. 2006 Canadian clinical practice guidelines on the management and prevention of obesity in adults and children (summary). CMAJ 2007, 176, S1–S13. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Hong, K.; Yip, I.; Huerta, S.; Bowerman, S.; Walker, J.; Wang, H.; Elashoff, R.; Go, V.L.; Heber, D. Body weight loss with phentermine alone versus phentermine and fenfluramine with very-low-calorie diet in an outpatient obesity management program: A retrospective study. Curr. Ther. Res. Clin. Exp. 2003, 64, 447–460. [Google Scholar] [CrossRef] [PubMed]
- Munro, I.A.; Bore, M.R.; Munro, D.; Garg, M.L. Using personality as a predictor of diet induced weight loss and weight management. Int. J. Behav. Nutr. Phys. Act 2011, 8, 129. [Google Scholar] [CrossRef] [PubMed]
- Tate, D.F.; Jeffery, R.W.; Sherwood, N.E.; Wing, R.R. Long-term weight losses associated with prescription of higher physical activity goals. Are higher levels of physical activity protective against weight regain? Am. J. Clin. Nutr. 2007, 85, 954–959. [Google Scholar] [PubMed]
- Hansen, D.; Dendale, P.; Berger, J.; van Loon, L.J.; Meeusen, R. The effects of exercise training on fat-mass loss in obese patients during energy intake restriction. Sports Med. 2007, 37, 31–46. [Google Scholar] [CrossRef] [PubMed]
- Sahlin, K.; Sallstedt, E.K.; Bishop, D.; Tonkonogi, M. Turning down lipid oxidation during heavy exercise—What is the mechanism? J. Physiol. Pharmacol. 2008, 59, 19–30. [Google Scholar] [PubMed]
- Huang, S.C.; Freitas, T.C.; Amiel, E.; Everts, B.; Pearce, E.L.; Lok, J.B.; Pearce, E.J. Fatty acid oxidation is essential for egg production by the parasitic flatworm Schistosoma mansoni. PLoS Pathog. 2012, 8, e1002996. [Google Scholar] [CrossRef] [PubMed]
- Haskell, W.L.; Lee, I.M.; Pate, R.R.; Powell, K.E.; Blair, S.N.; Franklin, B.A.; Macera, C.A.; Heath, G.W.; Thompson, P.D.; Bauman, A. Physical activity and public health: Updated recommendation for adults from the American College of Sports Medicine and the American Heart Association. Med. Sci. Sports Exerc. 2007, 39, 1423–1434. [Google Scholar] [CrossRef] [PubMed]
- Tuah, N.A.; Amiel, C.; Qureshi, S.; Car, J.; Kaur, B.; Majeed, A. Transtheoretical model for dietary and physical exercise modification in weight loss management for overweight and obese adults. Cochrane Database Syst. Rev. 2011, 10, CD008066. [Google Scholar] [CrossRef] [PubMed]
- Mastellos, N.; Gunn, L.H.; Felix, L.M.; Car, J.; Majeed, A. Transtheoretical model stages of change for dietary and physical exercise modification in weight loss management for overweight and obese adults. Cochrane Database Syst. Rev. 2014, 2, CD008066. [Google Scholar] [CrossRef] [PubMed]
- Blackburn, G.L.; Walker, W.A. Science-based solutions to obesity: What are the roles of academia, government, industry, and health care? Am. J. Clin. Nutr. 2005, 82, 207S–210S. [Google Scholar] [PubMed]
- Thangaratinam, S.; Rogozinska, E.; Jolly, K.; Glinkowski, S.; Roseboom, T.; Tomlinson, J.W.; Kunz, R.; Mol, B.W.; Coomarasamy, A.; Khan, K.S. Effects of interventions in pregnancy on maternal weight and obstetric outcomes: Meta-analysis of randomised evidence. BMJ 2012, 344, e2088. [Google Scholar] [CrossRef] [PubMed]
- Siebenhofer, A.; Jeitler, K.; Horvath, K.; Berghold, A.; Siering, U.; Semlitsch, T. Long-term effects of weight-reducing drugs in hypertensive patients. Cochrane Database Syst. Rev. 2013, 3, CD007654. [Google Scholar] [CrossRef] [PubMed]
- O’Neil, P.M.; Smith, S.R.; Weissman, N.J.; Fidler, M.C.; Sanchez, M.; Zhang, J.; Raether, B.; Anderson, C.M.; Shanahan, W.R. Randomized placebo-controlled clinical trial of lorcaserin for weight loss in type 2 diabetes mellitus: The BLOOM-DM study. Obesity (Silver Spring) 2012, 20, 1426–1436. [Google Scholar] [CrossRef]
- Sinnayah, P.; Jobst, E.E.; Rathner, J.A.; Caldera-Siu, A.D.; Tonelli-Lemos, L.; Eusterbrock, A.J.; Enriori, P.J.; Pothos, E.N.; Grove, K.L.; Cowley, M.A. Feeding induced by cannabinoids is mediated independently of the melanocortin system. PLoS One 2008, 3, e2202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ochner, C.N.; Gibson, C.; Carnell, S.; Dambkowski, C.; Geliebter, A. The neurohormonal regulation of energy intake in relation to bariatric surgery for obesity. Physiol. Behav. 2010, 100, 549–559. [Google Scholar] [CrossRef] [PubMed]
- Samuel, I.; Mason, E.E.; Renquist, K.E.; Huang, Y.H.; Zimmerman, M.B.; Jamal, M. Bariatric surgery trends: An 18-year report from the International Bariatric Surgery Registry. Am. J. Surg. 2006, 192, 657–662. [Google Scholar] [CrossRef] [PubMed]
- Paluszkiewicz, R.; Kalinowski, P.; Wroblewski, T.; Bartoszewicz, Z.; Bialobrzeska-Paluszkiewicz, J.; Ziarkiewicz-Wroblewska, B.; Remiszewski, P.; Grodzicki, M.; Krawczyk, M. Prospective randomized clinical trial of laparoscopic sleeve gastrectomy versus open Roux-en-Y gastric bypass for the management of patients with morbid obesity. Wideochir. Inne Tech. Malo Inwazyjne 2012, 7, 225–232. [Google Scholar] [PubMed]
- Ochner, C.N.; Kwok, Y.; Conceicao, E.; Pantazatos, S.P.; Puma, L.M.; Carnell, S.; Teixeira, J.; Hirsch, J.; Geliebter, A. Selective reduction in neural responses to high calorie foods following gastric bypass surgery. Ann. Surg. 2011, 253, 502–507. [Google Scholar] [CrossRef] [PubMed]
- Doucet, E.; Cameron, J. Appetite control after weight loss: What is the role of bloodborne peptides? Appl. Physiol. Nutr. Metab. 2007, 32, 523–532. [Google Scholar] [CrossRef] [PubMed]
- Cohen, M.A.; Ellis, S.M.; le Roux, C.W.; Batterham, R.L.; Park, A.; Patterson, M.; Frost, G.S.; Ghatei, M.A.; Bloom, S.R. Oxyntomodulin suppresses appetite and reduces food intake in humans. J. Clin. Endocrinol. Metab. 2003, 88, 4696–4701. [Google Scholar] [CrossRef] [PubMed]
- Bose, M.; Teixeira, J.; Olivan, B.; Bawa, B.; Arias, S.; Machineni, S.; Pi-Sunyer, F.X.; Scherer, P.E.; Laferrere, B. Weight loss and incretin responsiveness improve glucose control independently after gastric bypass surgery. J. Diabetes 2010, 2, 47–55. [Google Scholar] [CrossRef] [PubMed]
- Rao, R.S. Bariatric surgery and the central nervous system. Obes. Surg. 2012, 22, 967–978. [Google Scholar] [CrossRef] [PubMed]
- Halmi, K.A.; Mason, E.; Falk, J.R.; Stunkard, A. Appetitive behavior after gastric bypass for obesity. Int. J. Obes. 1981, 5, 457–464. [Google Scholar] [PubMed]
- Thomas, J.R.; Marcus, E. High and low fat food selection with reported frequency intolerance following Roux-en-Y gastric bypass. Obes. Surg. 2008, 18, 282–287. [Google Scholar] [CrossRef] [PubMed]
- Olbers, T.; Bjorkman, S.; Lindroos, A.; Maleckas, A.; Lonn, L.; Sjostrom, L.; Lonroth, H. Body composition, dietary intake, and energy expenditure after laparoscopic Roux-en-Y gastric bypass and laparoscopic vertical banded gastroplasty: A randomized clinical trial. Ann. Surg. 2006, 244, 715–722. [Google Scholar] [CrossRef] [PubMed]
- Kenler, H.A.; Brolin, R.E.; Cody, R.P. Changes in eating behavior after horizontal gastroplasty and Roux-en-Y gastric bypass. Am. J. Clin. Nutr. 1990, 52, 87–92. [Google Scholar] [PubMed]
- Thirlby, R.C.; Bahiraei, F.; Randall, J.; Drewnoski, A. Effect of Roux-en-Y gastric bypass on satiety and food likes: The role of genetics. J. Gastrointest. Surg. 2006, 10, 270–277. [Google Scholar] [CrossRef] [PubMed]
- Brown, E.K.; Settle, E.A.; van Rij, A.M. Food intake patterns of gastric bypass patients. J. Am. Diet. Assoc. 1982, 80, 437–443. [Google Scholar] [PubMed]
- Bueter, M.; Miras, A.D.; Chichger, H.; Fenske, W.; Ghatei, M.A.; Bloom, S.R.; Unwin, R.J.; Lutz, T.A.; Spector, A.C.; le Roux, C.W. Alterations of sucrose preference after Roux-en-Y gastric bypass. Physiol. Behav. 2011, 104, 709–721. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sjostrom, L.; Peltonen, M.; Jacobson, P.; Sjostrom, C.D.; Karason, K.; Wedel, H.; Ahlin, S.; Anveden, A.; Bengtsson, C.; Bergmark, G.; et al. Bariatric surgery and long-term cardiovascular events. JAMA 2012, 307, 56–65. [Google Scholar] [CrossRef] [PubMed]
- Dunn, J.P.; Cowan, R.L.; Volkow, N.D.; Feurer, I.D.; Li, R.; Williams, D.B.; Kessler, R.M.; Abumrad, N.N. Decreased dopamine type 2 receptor availability after bariatric surgery: Preliminary findings. Brain Res. 2010, 1350, 123–130. [Google Scholar] [CrossRef] [PubMed]
- Scholtz, S.; Miras, A.D.; Chhina, N.; Prechtl, C.G.; Sleeth, M.L.; Daud, N.M.; Ismail, N.A.; Durighel, G.; Ahmed, A.R.; Olbers, T.; et al. Obese patients after gastric bypass surgery have lower brain-hedonic responses to food than after gastric banding. Gut 2014, 63, 891–902. [Google Scholar] [CrossRef] [PubMed]
- DiBaise, J.K.; Frank, D.N.; Mathur, R. Impact of the gut microbiota on the development of obesity: Current concepts. Am. J. Gastroenterol. 2012, 5, 22–27. [Google Scholar] [CrossRef]
- Aroniadis, O.C.; Brandt, L.J. Fecal microbiota transplantation: Past, present and future. Curr. Opin. Gastroenterol. 2013, 29, 79–84. [Google Scholar] [CrossRef] [PubMed]
- Turnbaugh, P.J.; Ley, R.E.; Mahowald, M.A.; Magrini, V.; Mardis, E.R.; Gordon, J.I. An obesity-associated gut microbiome with increased capacity for energy harvest. Nature 2006, 444, 1027–1031. [Google Scholar] [CrossRef] [PubMed]
- Backhed, F.; Ding, H.; Wang, T.; Hooper, L.V.; Koh, G.Y.; Nagy, A.; Semenkovich, C.F.; Gordon, J.I. The gut microbiota as an environmental factor that regulates fat storage. Proc. Natl. Acad. Sci. USA 2004, 101, 15718–15723. [Google Scholar] [CrossRef] [PubMed]
- Van Reenen, C.A.; Dicks, L.M. Horizontal gene transfer amongst probiotic lactic acid bacteria and other intestinal microbiota: What are the possibilities? A review. Arch. Microbiol. 2011, 193, 157–168. [Google Scholar] [CrossRef] [PubMed]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, Y.; Liu, J.; Yao, J.; Ji, G.; Qian, L.; Wang, J.; Zhang, G.; Tian, J.; Nie, Y.; Zhang, Y.E.; et al. Obesity: Pathophysiology and Intervention. Nutrients 2014, 6, 5153-5183. https://doi.org/10.3390/nu6115153
Zhang Y, Liu J, Yao J, Ji G, Qian L, Wang J, Zhang G, Tian J, Nie Y, Zhang YE, et al. Obesity: Pathophysiology and Intervention. Nutrients. 2014; 6(11):5153-5183. https://doi.org/10.3390/nu6115153
Chicago/Turabian StyleZhang, Yi, Ju Liu, Jianliang Yao, Gang Ji, Long Qian, Jing Wang, Guansheng Zhang, Jie Tian, Yongzhan Nie, Yi Edi. Zhang, and et al. 2014. "Obesity: Pathophysiology and Intervention" Nutrients 6, no. 11: 5153-5183. https://doi.org/10.3390/nu6115153