Out of Balance—Systemic Iron Homeostasis in Iron-Related Disorders

Abstract
:
1. Introduction

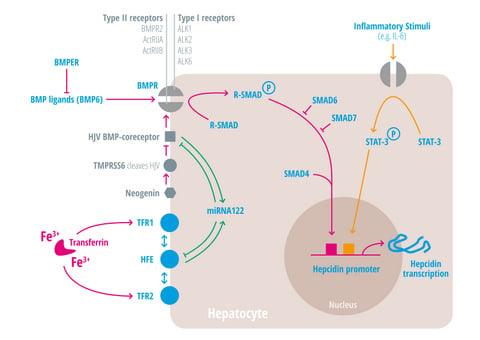
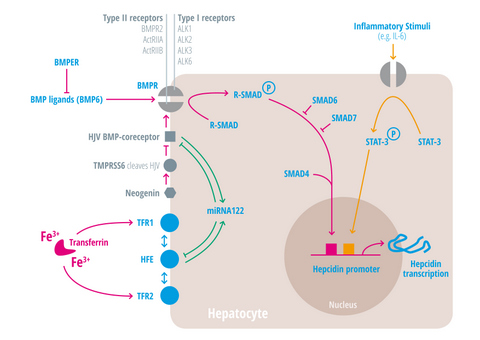{kind=link}
{kind=link}
{kind=link}
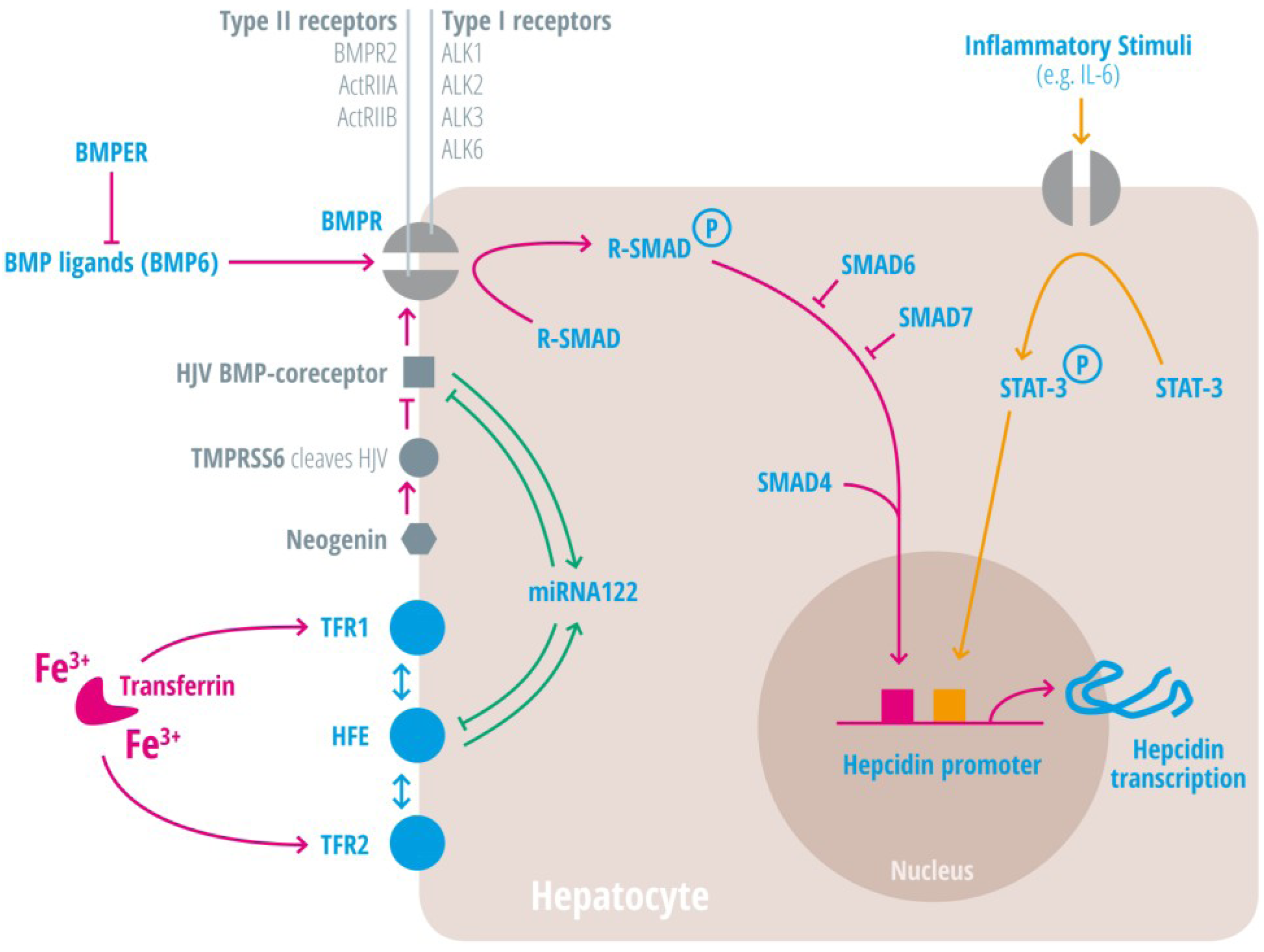{kind=link}
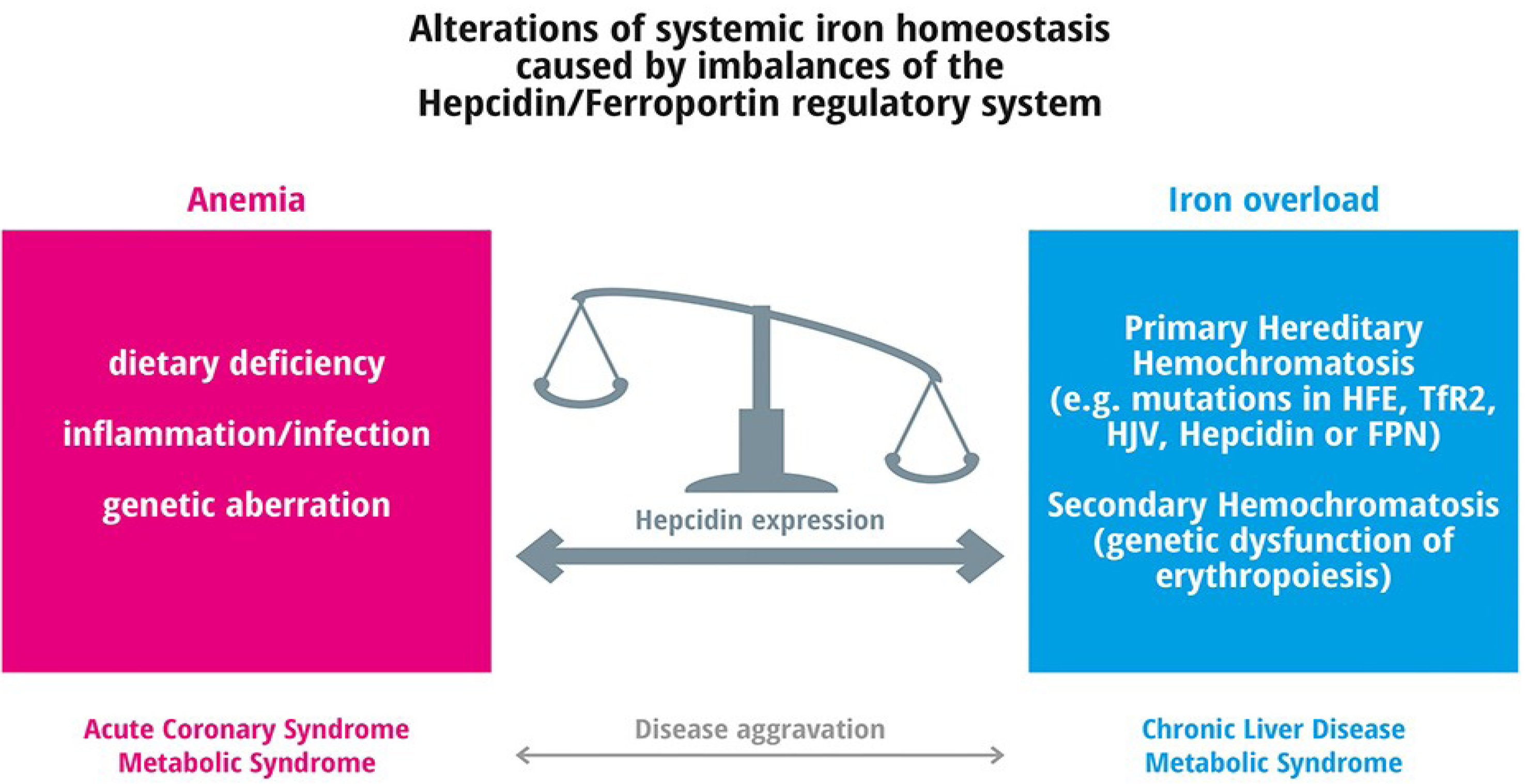{kind=link}
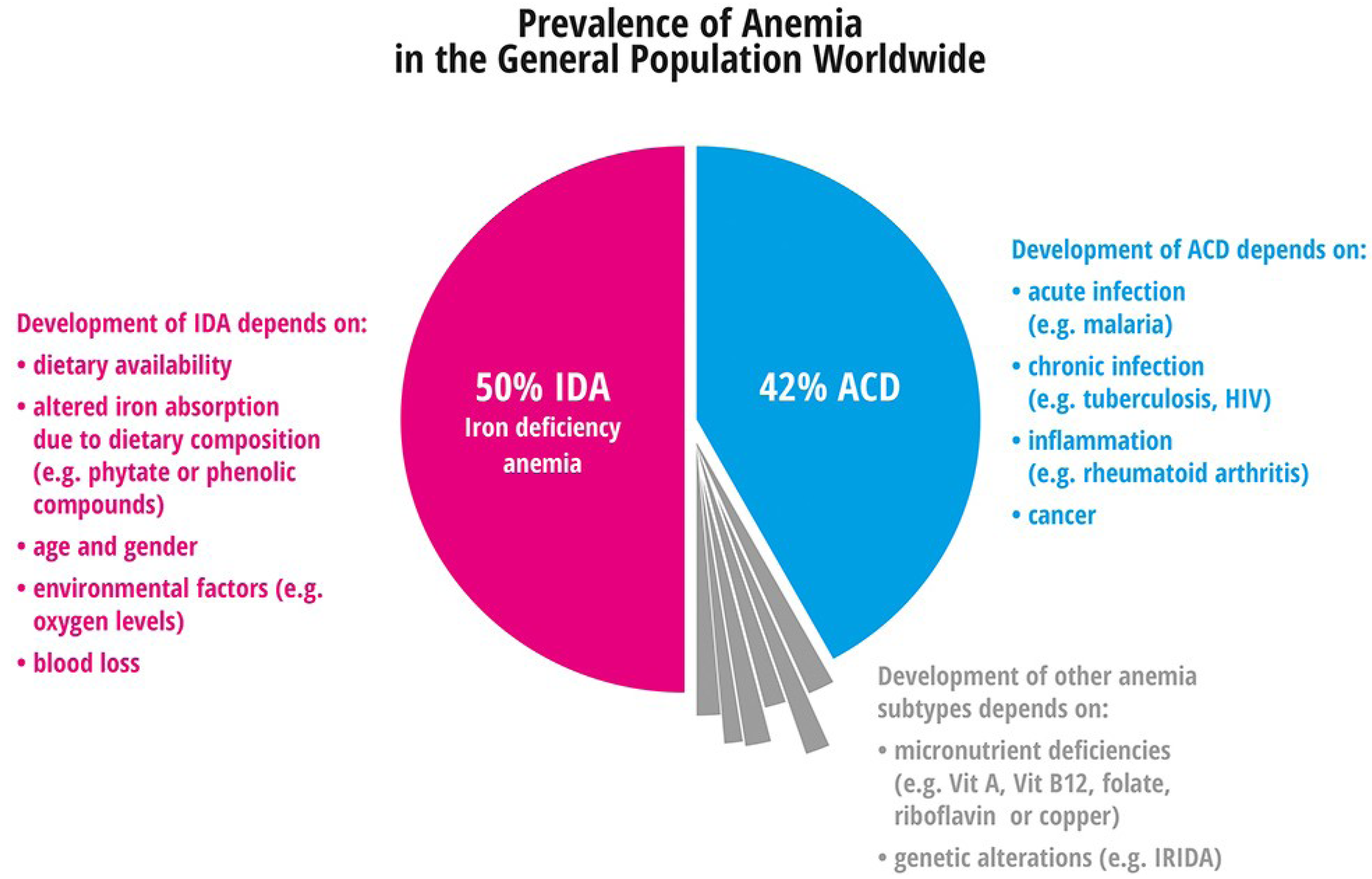{kind=link}
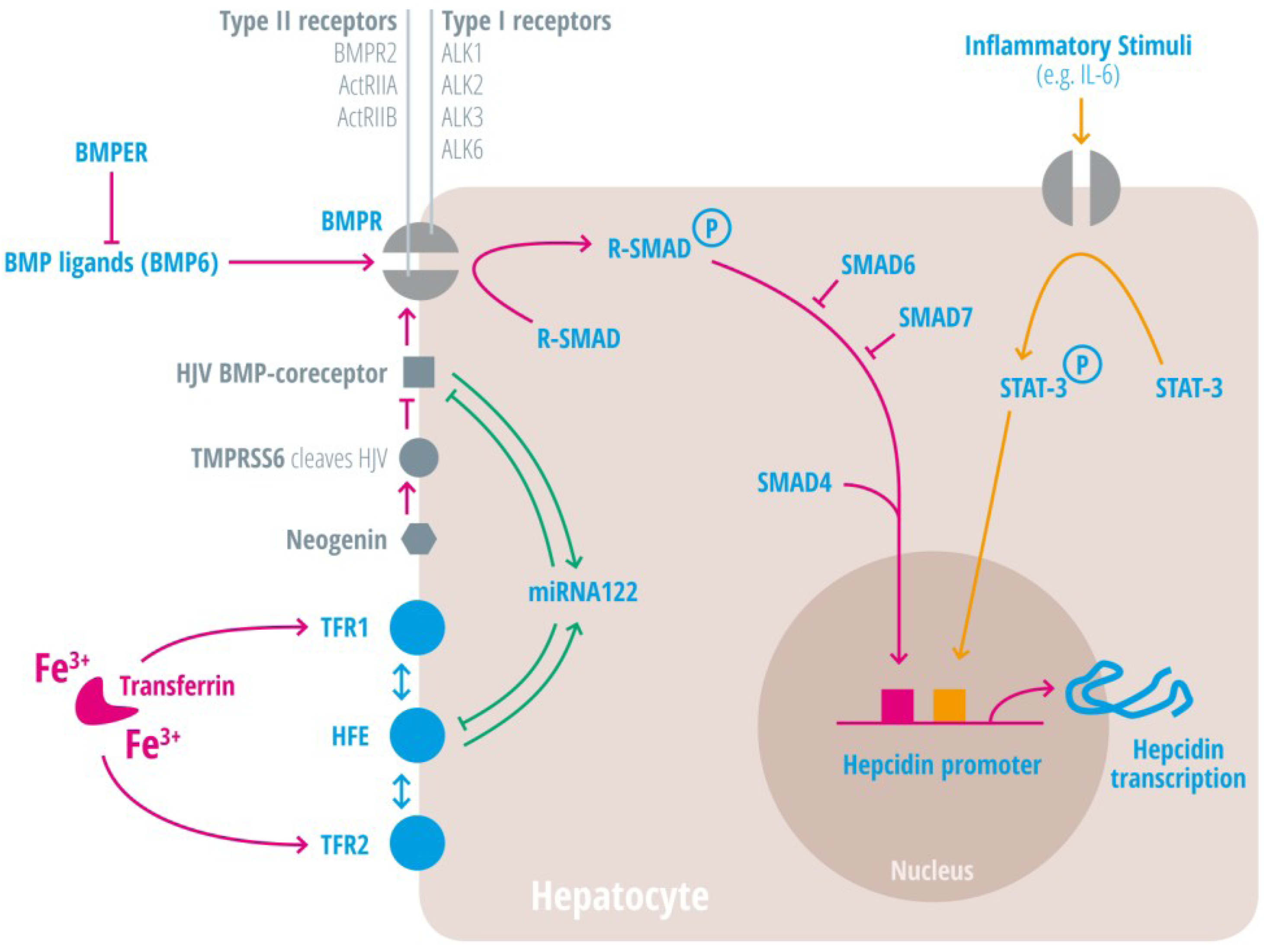
| Regulators of iron homeostasis | Abbreviation | Iron regulatory mechanisms |
|---|---|---|
| Activin-receptor like kinase 2 | Alk2 = ACVRL | BMP Type I receptor, required for hepcidin induction under stimulated conditions. Activation leads to increased hepcidin levels [25,26,27]. |
| Activin-receptor like kinase 3 | Alk3 = BMPR1a | BMP Type I receptor, required for baseline hepcidin expression. Activation leads to hepcidin increase [25,26,28,29,30]. |
| Activin A receptor, type IIA and II B | ActRII a and ActRIIb | BMP Type II receptors. Activation leads to hepcidin induction [31,32,33]. |
| Bone morphogenic protein receptor 2 | BMPRII | BMP Type II receptor. Activation leads to hepcidin induction [31,32,33]. |
| Bone morphogenetic protein 6 | BMP6 | Agonist of the BMP receptor, ligand for the BMP-SMAD signaling pathway in cells and mice; levels increased by hepatic iron; induces hepcidin expression [34,35,36]. |
| Bone morphogenetic protein receptor
BMPER | BMPR
BMPER | Receptor for BMP ligands. Induces SMAD phosphorylation, which activates a signaling cascade to stimulate hepcidin expression [25,26,28,29,30]. BMP endothelial cell precursor-derived regulator inhibits BMP signaling and decreases hepatic hepcidin expression [37,38]. |
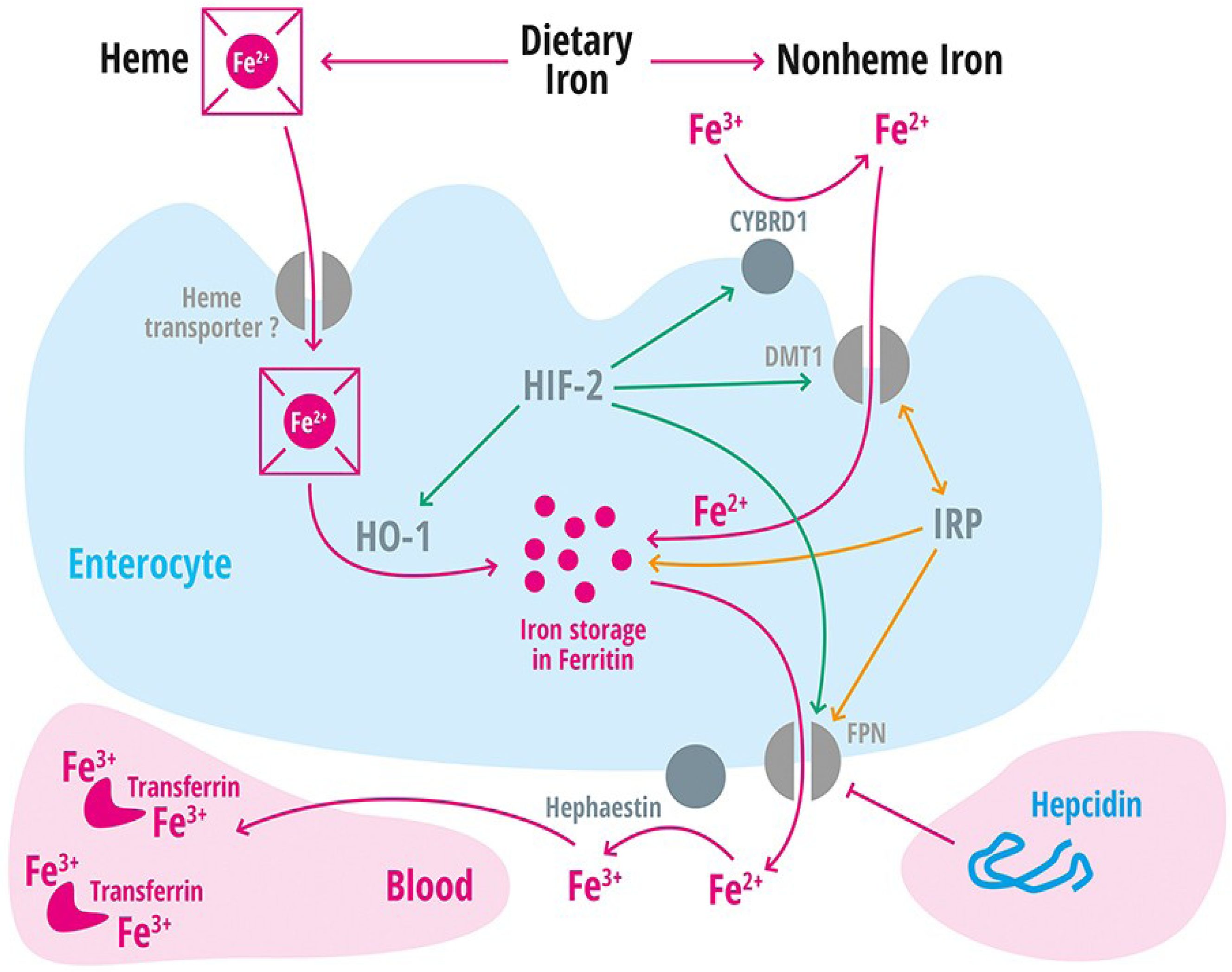
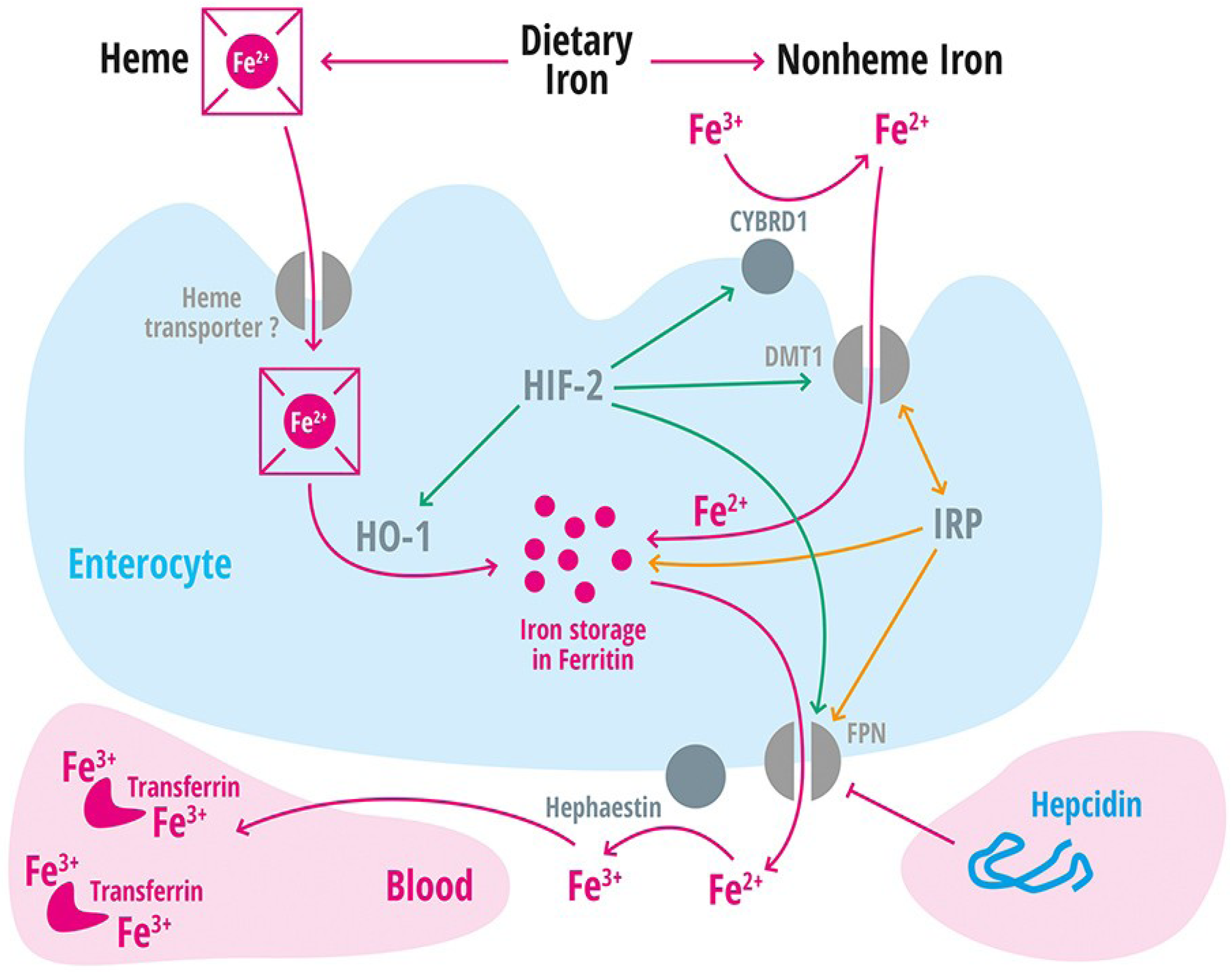
| Divalent metal transporter 1 | DMT1 | Iron transporter (Fe2+) in duodenal enterocytes and endosomes of most cell types [39]. |
| Membrane-associated ferrireductase Cybrd1 (DcytB) | Cybrd1 | Ferrireductase located at the apical membrane of enterocytes, reduces Fe3+ to Fe2+ [40]. |
| Ferroportin | FPN | Iron export protein, internalized and degraded by hepcidin [20,21,22,41,42,43]. |
| Growth and differentiation factor 15 | GDF15 | Possible erythropoietic-derived suppressor of hepcidin levels [44,45,46]. |
| Hepcidin | HAMP1, Leap1 | Iron regulatory hormone, synthesized mainly by the liver [12,23,47,48,49,50,51,52] (only some articles are cited here, please consider the citation index at the end of the manuscript). |
| HFE | HFE | Name of a gene mutated in the most frequent HH subtype. MHC class1-like protein involved in iron sensing; sensitizes cells to BMP stimuli; activator of hepcidin transcription [53,54,55,56,57,58,59]. |
| Heme oxygenase-1 | HO-1 | Releases intracellular iron from heme [60]. |
| Hemojuvelin | HJV | Mutation in HJV gene cause a juvenile hemochromatosis subtype, BMP co-receptor that sensitizes hepatocytes to low endogenous BMP levels and activator of hepcidin transcription [61]. |
| Hephaestin | A multicopper oxidase homologous to ceruloplasmin, which oxidases Fe2+ to Fe3+ [11]. | |
| Interleukin-6 | IL-6 | Cytokine, induced by inflammation. Binds to the IL-6 receptor. Activates hepcidin expression via STAT-3 phosphorylation [62,63,64,65]. |
| Iron regulatory protein 1 and 2 | IRP-1 and IRP2 | Cellular regulators of iron homeostasis that control expression of iron-regulated mRNA on a post-transcriptional level [2]. |
| Neogenin | Interacts with HJV and BMPs, may regulate secretion of HJV and iron uptake [31,66,67]. | |
| Solute Carrier Family 11, member 2 | SLC11A2 | Gene encoding the divalent metal transporter 1 (DMT1 = NRAMP2 = DCT1). Iron absorption channel expressed at the brush border side of duodenal enterocytes [12,68,69,70]. |
| Smad 1/5/8 | Signaling molecules phosphorylated by BMP receptors [71]. | |
| Smad 4 | Transcription factor that controls BMP-mediated signalling and activator of hepcidin expression [71]. | |
| Smad 6/7 | Inhibitory SMAD proteins that regulate BMP and/or TGFbeta signaling in a negative feedback manner [72]. | |
| STAT3 | Intracellular signaling molecule of the IL-6 pathway, its phosphorylation causes hepcidin induction [62,63,65,73]. | |
| Transferrin receptor 1 | TFR1 | Receptor for iron-bound transferrin, possibly involved in iron sensing by interacting with HFE [57,74,75]. |
| Transferrin receptor 2 | TFR2 | Receptor for iron-bound transferring, possibly involved in iron sensing by interacting with HFE [76,77]. |
| Transmembrane protease serine 6 | TMPRSS6 | Inhibits hepcidin expression by cleaving HJV, iron-deficiency sensor; phosphorylates Smad5 [78,79,80]. |
| Twisted gastrulation homolog 1 | TWSG1 | Possible suppressor secreted from erythropoietic precursor cells to repress hepcidin levels [81]. |
1.1. Iron Availability
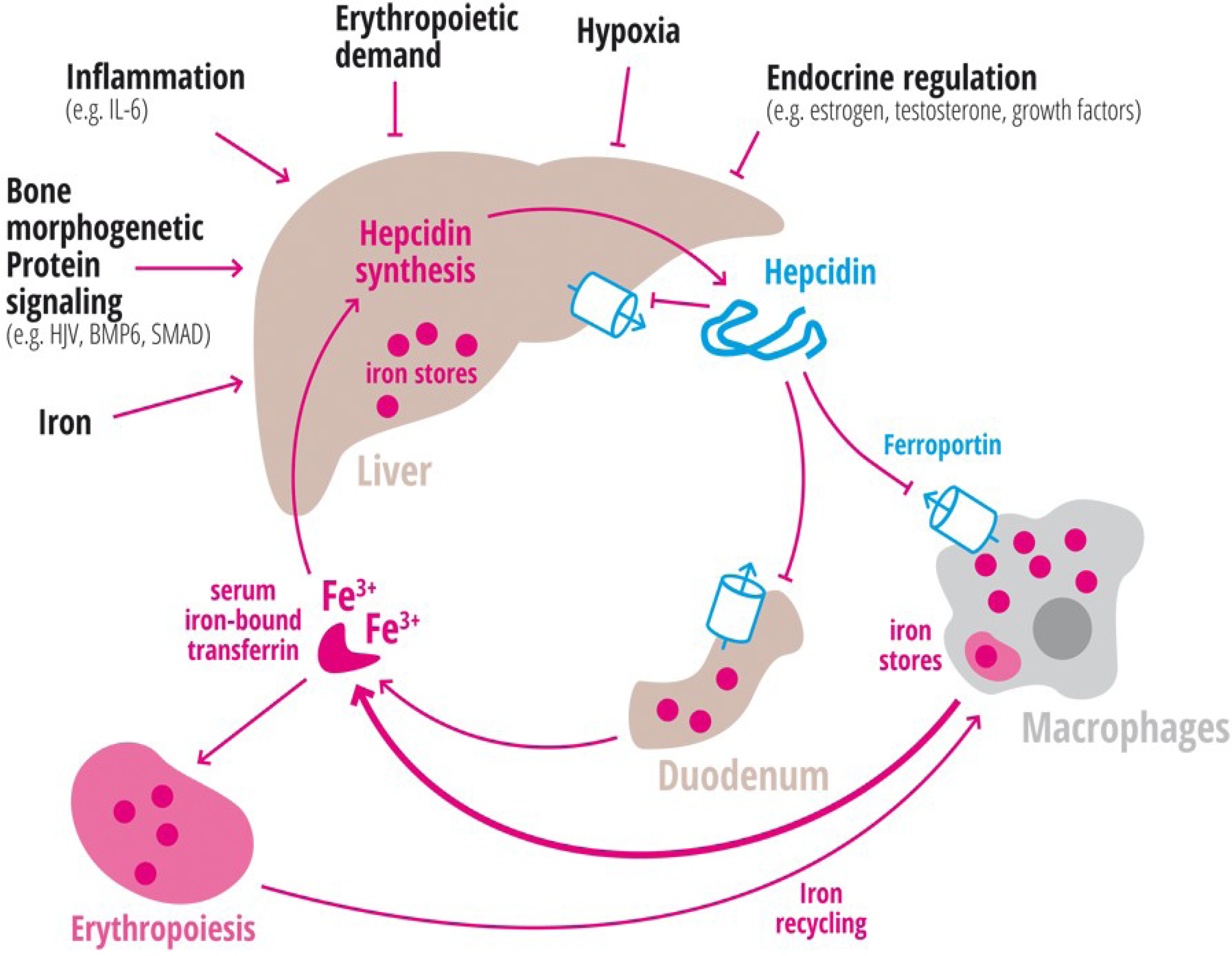
1.2. Inflammation
1.3. Erythropoiesis
1.4. Hypoxia
1.5. Endocrine Regulation
2. Iron Is a Critical Nutrient
3. Iron Supplementation
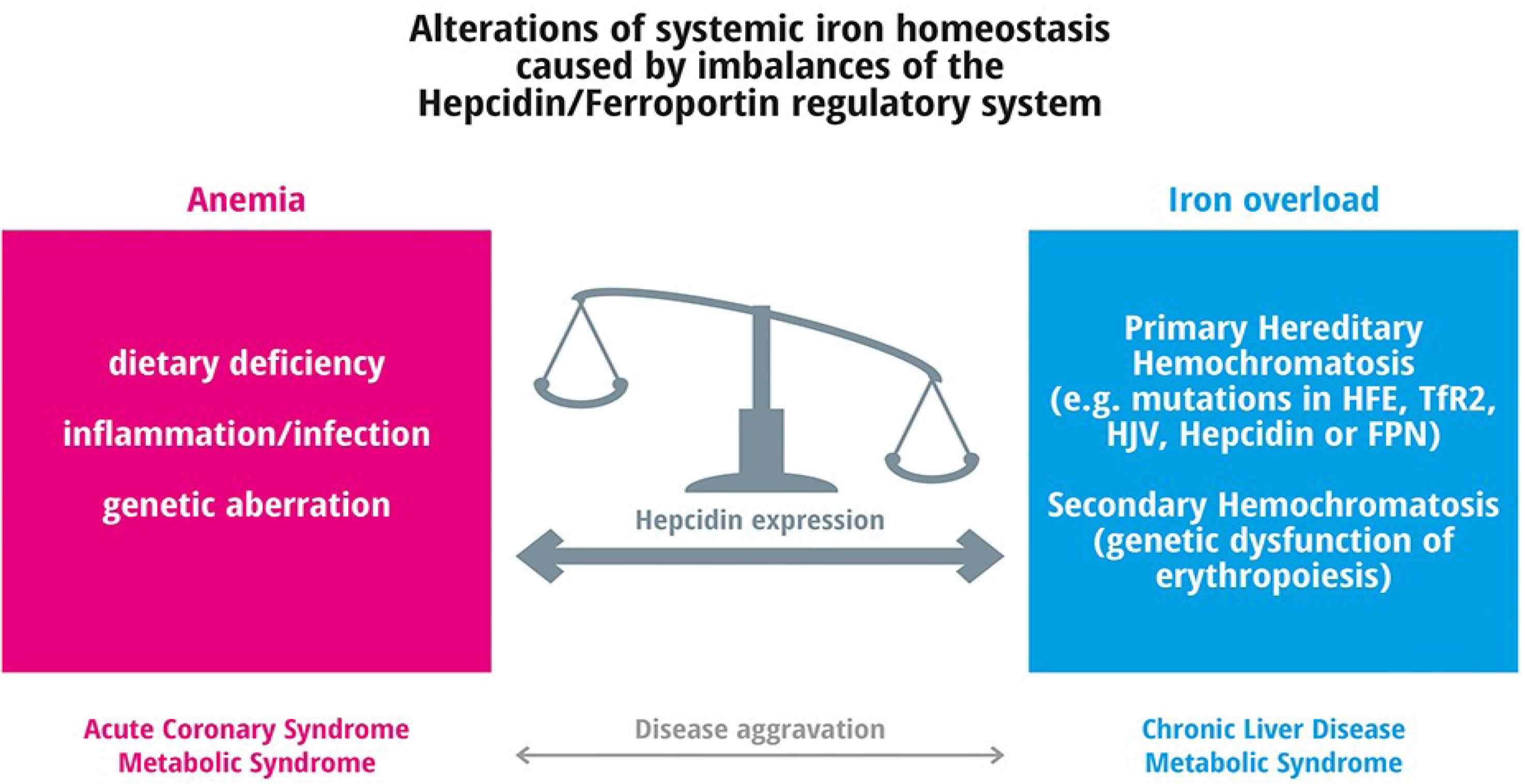
4. Frequent Iron Related Disorders
4.1. Anemia
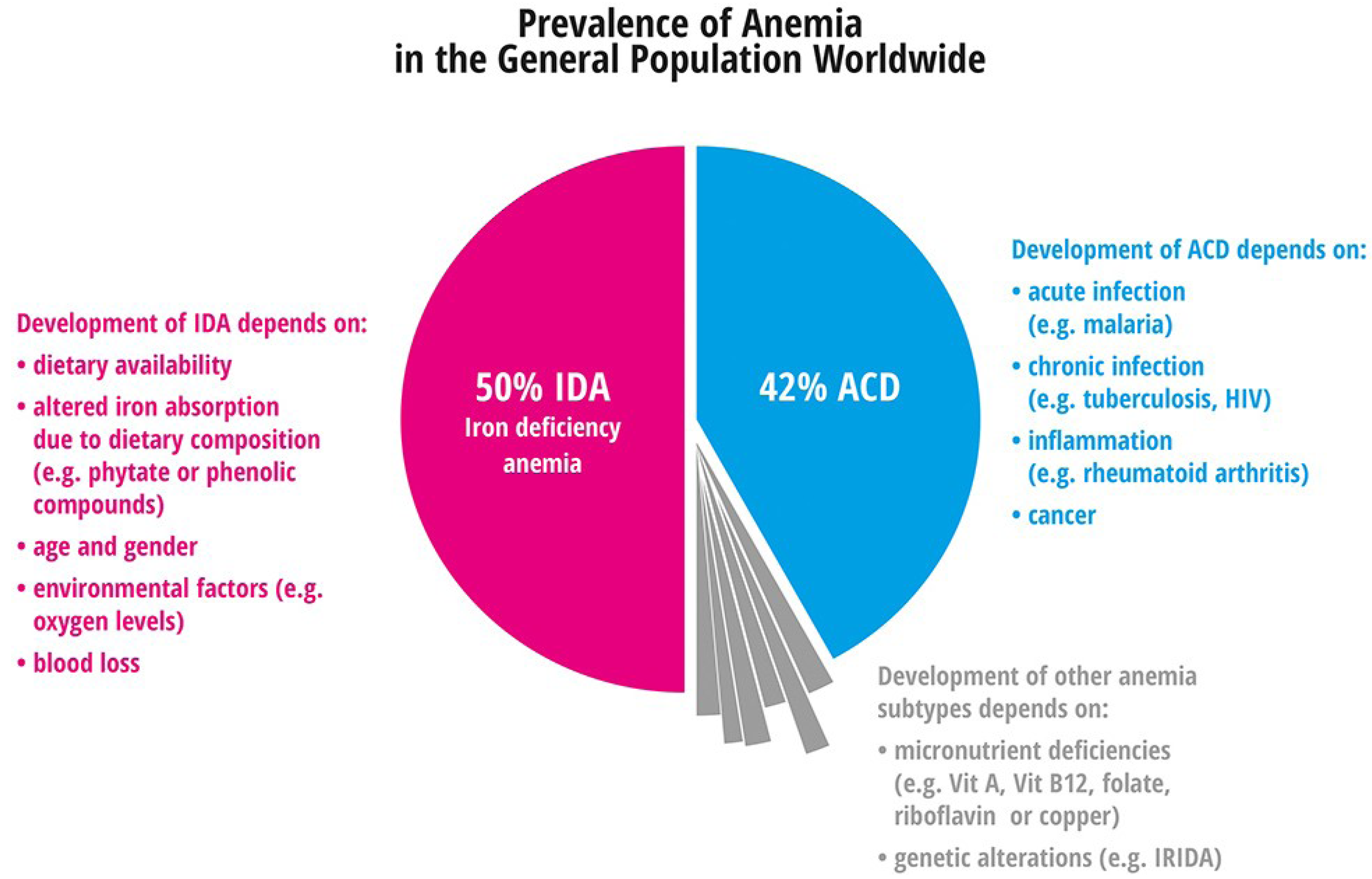
4.1.1. Iron Deficiency Anemia
4.1.2. Anemia of Chronic Disease
- -
- Anemia of heart failure;
- -
- Anemia of chronic kidney disease;
- -
- Anemia in inflammatory rheumatic diseases;
- -
- Anemia of the elderly.
4.1.3. Myelodysplastic Syndrome
4.1.4. Anemias Caused by Genetic Defects
5. Iron Deficiency and Frequent Diseases
Iron Overload
6. Iron Overload and Frequent Diseases
7. Conclusions
8. Perspective
Acknowledgements
Conflict of Interests
References
- McLean, E.; Cogswell, M.; Egli, I.; Wojdyla, D.; de Benoist, B. Worldwide prevalence of anaemia, who vitamin and mineral nutrition information system, 1993–2005. Public Health Nutr. 2009, 12, 444–454. [Google Scholar] [CrossRef]
- Muckenthaler, M.U.; Galy, B.; Hentze, M.W. Systemic iron homeostasis and the iron-responsive element/iron-regulatory protein (IRE/IRP) regulatory network. Annu. Rev. Nutr. 2008, 28, 197–213. [Google Scholar] [CrossRef]
- Haase, V.H. Regulation of erythropoiesis by hypoxia-inducible factors. Blood Rev. 2013, 27, 41–53. [Google Scholar] [CrossRef]
- Domke, A.; Großklaus, R.; Niemann, B.; Przyrembel, H.; Richter, K.; Schmidt, E.; Weißenborn, A.; Wörner, B.; Ziegenhagen, R. Utilisation of Minerals in Nutrients—Toxicologic and Nutrition-Physiologic Aspects; Wissenschaft, B., Ed.; BfR: Halem, Germany, 2004; p. 323. [Google Scholar]
- Gunshin, H.; Starr, C.N.; Direnzo, C.; Fleming, M.D.; Jin, J.; Greer, E.L.; Sellers, V.M.; Galica, S.M.; Andrews, N.C. Cybrd1 (duodenal cytochrome b) is not necessary for dietary iron absorption in mice. Blood 2005, 106, 2879–2883. [Google Scholar] [CrossRef]
- Choi, J.; Masaratana, P.; Latunde-Dada, G.O.; Arno, M.; Simpson, R.J.; McKie, A.T. Duodenal reductase activity and spleen iron stores are reduced and erythropoiesis is abnormal in Dcytb knockout mice exposed to hypoxic conditions. J. Nutr. 2012, 142, 1929–1934. [Google Scholar] [CrossRef]
- Vanoaica, L.; Darshan, D.; Richman, L.; Schumann, K.; Kuhn, L.C. Intestinal ferritin H is required for an accurate control of iron absorption. Cell Metab. 2010, 12, 273–282. [Google Scholar] [CrossRef]
- Peyssonnaux, C.; Zinkernagel, A.S.; Schuepbach, R.A.; Rankin, E.; Vaulont, S.; Haase, V.H.; Nizet, V.; Johnson, R.S. Regulation of iron homeostasis by the hypoxia-inducible transcription factors (HIFs). J. Clin. Investig. 2007, 117, 1926–1932. [Google Scholar] [CrossRef] [Green Version]
- Hentze, M.W. Translational control by iron-responsive elements. Adv. Exp. Med. Biol. 1994, 356, 119–126. [Google Scholar] [CrossRef]
- Hentze, M.W.; Kuhn, L.C. Molecular control of vertebrate iron metabolism: mRNA-Based regulatory circuits operated by iron, nitric oxide, and oxidative stress. Proc. Natl. Acad. Sci. USA 1996, 93, 8175–8182. [Google Scholar] [CrossRef]
- Vulpe, C.D.; Kuo, Y.M.; Murphy, T.L.; Cowley, L.; Askwith, C.; Libina, N.; Gitschier, J.; Anderson, G.J. Hephaestin, a ceruloplasmin homologue implicated in intestinal iron transport, is defective in the sla mouse. Nat. Genet. 1999, 21, 195–199. [Google Scholar] [CrossRef]
- Hentze, M.W.; Muckenthaler, M.U.; Galy, B.; Camaschella, C. Two to tango: Regulation of mammalian iron metabolism. Cell 2010, 142, 24–38. [Google Scholar] [CrossRef]
- Peslova, G.; Petrak, J.; Kuzelova, K.; Hrdy, I.; Halada, P.; Kuchel, P.W.; Soe-Lin, S.; Ponka, P.; Sutak, R.; Becker, E.; et al. Hepcidin, the hormone of iron metabolism, is bound specifically to alpha-2-macroglobulin in blood. Blood 2009, 113, 6225–6236. [Google Scholar] [CrossRef]
- Theurl, I.; Theurl, M.; Seifert, M.; Mair, S.; Nairz, M.; Rumpold, H.; Zoller, H.; Bellmann-Weiler, R.; Niederegger, H.; Talasz, H.; et al. Autocrine formation of hepcidin induces iron retention in human monocytes. Blood 2008, 111, 2392–2399. [Google Scholar] [CrossRef]
- Nguyen, N.B.; Callaghan, K.D.; Ghio, A.J.; Haile, D.J.; Yang, F. Hepcidin expression and iron transport in alveolar macrophages. Am. J. Physiol. 2006, 291, L417–L425. [Google Scholar] [CrossRef]
- Merle, U.; Fein, E.; Gehrke, S.G.; Stremmel, W.; Kulaksiz, H. The iron regulatory peptide hepcidin is expressed in the heart and regulated by hypoxia and inflammation. Endocrinology 2007, 148, 2663–2668. [Google Scholar] [CrossRef]
- Kulaksiz, H.; Theilig, F.; Bachmann, S.; Gehrke, S.G.; Rost, D.; Janetzko, A.; Cetin, Y.; Stremmel, W. The iron-regulatory peptide hormone hepcidin: Expression and cellular localization in the mammalian kidney. J. Endocrinol. 2005, 184, 361–370. [Google Scholar] [CrossRef]
- Wang, Q.; Du, F.; Qian, Z.M.; Ge, X.H.; Zhu, L.; Yung, W.H.; Yang, L.; Ke, Y. Lipopolysaccharide induces a significant increase in expression of iron regulatory hormone hepcidin in the cortex and substantia nigra in rat brain. Endocrinology 2008, 149, 3920–3925. [Google Scholar] [CrossRef]
- Bekri, S.; Gual, P.; Anty, R.; Luciani, N.; Dahman, M.; Ramesh, B.; Iannelli, A.; Staccini-Myx, A.; Casanova, D.; Ben Amor, I.; et al. Increased adipose tissue expression of hepcidin in severe obesity is independent from diabetes and nash. Gastroenterology 2006, 131, 788–796. [Google Scholar] [CrossRef]
- Nemeth, E.; Tuttle, M.S.; Powelson, J.; Vaughn, M.B.; Donovan, A.; Ward, D.M.; Ganz, T.; Kaplan, J. Hepcidin regulates cellular iron efflux by binding to ferroportin and inducing its internalization. Science 2004, 306, 2090–2093. [Google Scholar] [CrossRef]
- Qiao, B.; Sugianto, P.; Fung, E.; Del-Castillo-Rueda, A.; Moran-Jimenez, M.J.; Ganz, T.; Nemeth, E. Hepcidin-induced endocytosis of ferroportin is dependent on ferroportin ubiquitination. Cell Metab. 2012, 15, 918–924. [Google Scholar] [CrossRef]
- Ross, S.L.; Tran, L.; Winters, A.; Lee, K.J.; Plewa, C.; Foltz, I.; King, C.; Miranda, L.P.; Allen, J.; Beckman, H.; et al. Molecular mechanism of hepcidin-mediated ferroportin internalization requires ferroportin lysines, not tyrosines or JAK-STAT. Cell Metab. 2012, 15, 905–917. [Google Scholar] [CrossRef]
- Ganz, T.; Nemeth, E. Hepcidin and iron homeostasis. Biochim. Biophys. Acta 2012, 1823, 1434–1443. [Google Scholar] [CrossRef]
- Wolff, F.; Deleers, M.; Melot, C.; Gulbis, B.; Cotton, F. Hepcidin-25: Measurement by LC-MS/MS in serum and urine, reference ranges and urinary fractional excretion. Clin. Chim. Acta 2013, 423, 99–104. [Google Scholar] [CrossRef]
- Steinbicker, A.U.; Bartnikas, T.B.; Lohmeyer, L.K.; Leyton, P.; Mayeur, C.; Kao, S.M.; Pappas, A.E.; Peterson, R.T.; Bloch, D.B.; Yu, P.B.; et al. Perturbation of hepcidin expression by bmp type I receptor deletion induces iron overload in mice. Blood 2011, 118, 4224–4230. [Google Scholar] [CrossRef]
- Steinbicker, A.U.; Sachidanandan, C.; Vonner, A.J.; Yusuf, R.Z.; Deng, D.Y.; Lai, C.S.; Rauwerdink, K.M.; Winn, J.C.; Saez, B.; Cook, C.M.; et al. Inhibition of bone morphogenetic protein signaling attenuates anemia associated with inflammation. Blood 2011, 117, 4915–4923. [Google Scholar] [CrossRef]
- Theurl, I.; Schroll, A.; Nairz, M.; Seifert, M.; Theurl, M.; Sonnweber, T.; Kulaksiz, H.; Weiss, G. Pathways for the regulation of hepcidin expression in anemia of chronic disease and iron deficiency anemia in vivo. Haematologica 2011, 96, 1761–1769. [Google Scholar] [CrossRef]
- Bottcher, Y.; Unbehauen, H.; Kloting, N.; Ruschke, K.; Korner, A.; Schleinitz, D.; Tonjes, A.; Enigk, B.; Wolf, S.; Dietrich, K.; et al. Adipose tissue expression and genetic variants of the bone morphogenetic protein receptor 1A gene (BMPR1A) are associated with human obesity. Diabetes 2009, 58, 2119–2128. [Google Scholar] [CrossRef]
- Theurl, I.; Schroll, A.; Sonnweber, T.; Nairz, M.; Theurl, M.; Willenbacher, W.; Eller, K.; Wolf, D.; Seifert, M.; Sun, C.C.; et al. Pharmacologic inhibition of hepcidin expression reverses anemia of chronic inflammation in rats. Blood 2011, 118, 4977–4984. [Google Scholar] [CrossRef]
- Scott, G.J.; Ray, M.K.; Ward, T.; McCann, K.; Peddada, S.; Jiang, F.X.; Mishina, Y. Abnormal glucose metabolism in heterozygous mutant mice for a type I receptor required for BMP signaling. Genesis 2009, 47, 385–391. [Google Scholar] [CrossRef]
- Xia, Y.; Babitt, J.L.; Sidis, Y.; Chung, R.T.; Lin, H.Y. Hemojuvelin regulates hepcidin expression via a selective subset of BMP ligands and receptors independently of neogenin. Blood 2008, 111, 5195–5204. [Google Scholar] [CrossRef]
- Yu, P.B.; Beppu, H.; Kawai, N.; Li, E.; Bloch, K.D. Bone morphogenetic protein (BMP) type II receptor deletion reveals BMP ligand-specific gain of signaling in pulmonary artery smooth muscle cells. J. Biol. Chem. 2005, 280, 24443–24450. [Google Scholar] [CrossRef]
- Yu, P.B.; Deng, D.Y.; Beppu, H.; Hong, C.C.; Lai, C.; Hoyng, S.A.; Kawai, N.; Bloch, K.D. Bone morphogenetic protein (BMP) type II receptor is required for BMP-mediated growth arrest and differentiation in pulmonary artery smooth muscle cells. J. Biol. Chem. 2008, 283, 3877–3888. [Google Scholar]
- Ikeda, Y.; Tajima, S.; Izawa-Ishizawa, Y.; Kihira, Y.; Ishizawa, K.; Tomita, S.; Tsuchiya, K.; Tamaki, T. Estrogen regulates hepcidin expression via GPR30-BMP6-dependent signaling in hepatocytes. PLoS One 2012, 7, e40465. [Google Scholar] [CrossRef]
- Meynard, D.; Kautz, L.; Darnaud, V.; Canonne-Hergaux, F.; Coppin, H.; Roth, M.P. Lack of the bone morphogenetic protein BMP6 induces massive iron overload. Nat. Genet. 2009, 41, 478–481. [Google Scholar] [CrossRef]
- Meynard, D.; Vaja, V.; Sun, C.C.; Corradini, E.; Chen, S.; Lopez-Otin, C.; Grgurevic, L.; Hong, C.C.; Stirnberg, M.; Gutschow, M.; et al. Regulation of TMPRSS6 by BMP6 and iron in human cells and mice. Blood 2011, 118, 747–756. [Google Scholar] [CrossRef] [Green Version]
- Patel, N.; Masaratana, P.; Diaz-Castro, J.; Latunde-Dada, G.O.; Qureshi, A.; Lockyer, P.; Jacob, M.; Arno, M.; Matak, P.; Mitry, R.R.; et al. Bmper protein is a negative regulator of hepcidin and is up-regulated in hypotransferrinemic mice. J. Biol. Chem. 2012, 287, 4099–4106. [Google Scholar] [CrossRef]
- Santos, P.C.; Krieger, J.E.; Pereira, A.C. Molecular diagnostic and pathogenesis of hereditary hemochromatosis. Int. J. Mol. Sci. 2012, 13, 1497–1511. [Google Scholar] [CrossRef]
- Andrews, N.C. The iron transporter DMT1. Int. J. Biochem. Cell Biol. 1999, 31, 991–994. [Google Scholar] [CrossRef]
- McKie, A.T.; Barrow, D.; Latunde-Dada, G.O.; Rolfs, A.; Sager, G.; Mudaly, E.; Mudaly, M.; Richardson, C.; Barlow, D.; Bomford, A.; et al. An iron-regulated ferric reductase associated with the absorption of dietary iron. Science 2001, 291, 1755–1759. [Google Scholar] [CrossRef]
- Donovan, A.; Lima, C.A.; Pinkus, J.L.; Pinkus, G.S.; Zon, L.I.; Robine, S.; Andrews, N.C. The iron exporter ferroportin/Slc40a1 is essential for iron homeostasis. Cell Metab. 2005, 1, 191–200. [Google Scholar] [CrossRef]
- Lymboussaki, A.; Pignatti, E.; Montosi, G.; Garuti, C.; Haile, D.J.; Pietrangelo, A. The role of the iron responsive element in the control of ferroportin1/IREG1/MTP1 gene expression. J. Hepatol. 2003, 39, 710–715. [Google Scholar] [CrossRef]
- McGregor, J.A.; Shayeghi, M.; Vulpe, C.D.; Anderson, G.J.; Pietrangelo, A.; Simpson, R.J.; McKie, A.T. Impaired iron transport activity of ferroportin 1 in hereditary iron overload. J. Membr. Biol. 2005, 206, 3–7. [Google Scholar] [CrossRef]
- Casanovas, G.; Spasic, M.V.; Casu, C.; Rivella, S.; Strelau, J.; Unsicker, K.; Muckenthaler, M.U. The murine growth differentiation factor 15 is not essential for systemic iron homeostasis in phlebotomized mice. Haematologica 2012, 98, 444–447. [Google Scholar]
- Goodnough, J.B.; Ramos, E.; Nemeth, E.; Ganz, T. Inhibition of hepcidin transcription by growth factors. Hepatology 2012, 56, 291–299. [Google Scholar] [CrossRef]
- Tanno, T.; Bhanu, N.V.; Oneal, P.A.; Goh, S.H.; Staker, P.; Lee, Y.T.; Moroney, J.W.; Reed, C.H.; Luban, N.L.; Wang, R.H.; et al. High levels of GDF15 in thalassemia suppress expression of the iron regulatory protein hepcidin. Nat. Med. 2007, 13, 1096–1101. [Google Scholar] [CrossRef]
- Nicolas, G.; Bennoun, M.; Devaux, I.; Beaumont, C.; Grandchamp, B.; Kahn, A.; Vaulont, S. Lack of hepcidin gene expression and severe tissue iron overload in upstream stimulatory factor 2 (USF2) knockout mice. Proc. Natl. Acad. Sci. USA 2001, 98, 8780–8785. [Google Scholar] [CrossRef]
- Viatte, L.; Lesbordes-Brion, J.C.; Lou, D.Q.; Bennoun, M.; Nicolas, G.; Kahn, A.; Canonne-Hergaux, F.; Vaulont, S. Deregulation of proteins involved in iron metabolism in hepcidin-deficient mice. Blood 2005, 105, 4861–4864. [Google Scholar] [CrossRef]
- Pigeon, C.; Ilyin, G.; Courselaud, B.; Leroyer, P.; Turlin, B.; Brissot, P.; Loreal, O. A new mouse liver-specific gene, encoding a protein homologous to human antimicrobial peptide hepcidin, is overexpressed during iron overload. J. Biol. Chem. 2001, 276, 7811–7819. [Google Scholar]
- Nicolas, G.; Viatte, L.; Bennoun, M.; Beaumont, C.; Kahn, A.; Vaulont, S. Hepcidin, a new iron regulatory peptide. Blood Cells Mol. Dis. 2002, 29, 327–335. [Google Scholar] [CrossRef]
- Nemeth, E.; Valore, E.V.; Territo, M.; Schiller, G.; Lichtenstein, A.; Ganz, T. Hepcidin, a putative mediator of anemia of inflammation, is a type II acute-phase protein. Blood 2003, 101, 2461–2463. [Google Scholar] [CrossRef]
- Lee, P.; Peng, H.; Gelbart, T.; Beutler, E. The IL-6- and lipopolysaccharide-induced transcription of hepcidin in HFE-, transferrin receptor 2-, and beta 2-microglobulin-deficient hepatocytes. Proc. Natl. Acad. Sci. USA 2004, 101, 9263–9265. [Google Scholar] [CrossRef]
- Corradini, E.; Garuti, C.; Montosi, G.; Ventura, P.; Andriopoulos, B., Jr.; Lin, H.Y.; Pietrangelo, A.; Babitt, J.L. Bone morphogenetic protein signaling is impaired in an hfe knockout mouse model of hemochromatosis. Gastroenterology 2009, 137, 1489–1497. [Google Scholar] [CrossRef]
- Ryan, J.D.; Ryan, E.; Fabre, A.; Lawless, M.W.; Crowe, J. Defective bone morphogenic protein signaling underlies hepcidin deficiency in HFE hereditary hemochromatosis. Hepatology 2010, 52, 1266–1273. [Google Scholar] [CrossRef]
- Bolondi, G.; Garuti, C.; Corradini, E.; Zoller, H.; Vogel, W.; Finkenstedt, A.; Babitt, J.L.; Lin, H.Y.; Pietrangelo, A. Altered hepatic BMP signaling pathway in human hfe hemochromatosis. Blood Cells Mol. Dis. 2010, 45, 308–312. [Google Scholar] [CrossRef]
- Feder, J.N.; Gnirke, A.; Thomas, W.; Tsuchihashi, Z.; Ruddy, D.A.; Basava, A.; Dormishian, F.; Domingo, R., Jr.; Ellis, M.C.; Fullan, A.; et al. A novel MHC class I-like gene is mutated in patients with hereditary haemochromatosis. Nat. Genet. 1996, 13, 399–408. [Google Scholar] [CrossRef]
- D’Alessio, F.; Hentze, M.W.; Muckenthaler, M.U. The hemochromatosis proteins HFE, TFR2, and HJV form a membrane-associated protein complex for hepcidin regulation. J. Hepatol. 2012, 57, 1052–1060. [Google Scholar] [CrossRef]
- Verga Falzacappa, M.V.; Casanovas, G.; Hentze, M.W.; Muckenthaler, M.U. A bone morphogenetic protein (BMP)-responsive element in the hepcidin promoter controls HFE2-mediated hepatic hepcidin expression and its response to IL-6 in cultured cells. J. Mol. Med. (Berl.) 2008, 86, 531–540. [Google Scholar] [CrossRef]
- Coppin, H.; Darnaud, V.; Kautz, L.; Meynard, D.; Aubry, M.; Mosser, J.; Martinez, M.; Roth, M.P. Gene expression profiling of Hfe−/− liver and duodenum in mouse strains with differing susceptibilities to iron loading: Identification of transcriptional regulatory targets of hfe and potential hemochromatosis modifiers. Genome Biol. 2007, 8, R221. [Google Scholar] [CrossRef]
- Hentze, M.W.; Muckenthaler, M.U.; Andrews, N.C. Balancing acts: Molecular control of mammalian iron metabolism. Cell 2004, 117, 285–297. [Google Scholar] [CrossRef]
- Babitt, J.L.; Huang, F.W.; Wrighting, D.M.; Xia, Y.; Sidis, Y.; Samad, T.A.; Campagna, J.A.; Chung, R.T.; Schneyer, A.L.; Woolf, C.J.; et al. Bone morphogenetic protein signaling by hemojuvelin regulates hepcidin expression. Nat. Genet. 2006, 38, 531–539. [Google Scholar] [CrossRef]
- Pietrangelo, A.; Dierssen, U.; Valli, L.; Garuti, C.; Rump, A.; Corradini, E.; Ernst, M.; Klein, C.; Trautwein, C. Stat3 is required for IL-6-GP130-dependent activation of hepcidin in vivo. Gastroenterology 2007, 132, 294–300. [Google Scholar] [CrossRef]
- Verga Falzacappa, M.V.; Vujic Spasic, M.; Kessler, R.; Stolte, J.; Hentze, M.W.; Muckenthaler, M.U. STAT3 mediates hepatic hepcidin expression and its inflammatory stimulation. Blood 2007, 109, 353–358. [Google Scholar] [CrossRef]
- Weiss, G.; Goodnough, L.T. Anemia of chronic disease. N. Engl. J. Med. 2005, 352, 1011–1023. [Google Scholar] [CrossRef]
- Wrighting, D.M.; Andrews, N.C. Interleukin-6 induces hepcidin expression through STAT3. Blood 2006, 108, 3204–3209. [Google Scholar] [CrossRef]
- Enns, C.A.; Ahmed, R.; Zhang, A.S. Neogenin interacts with matriptase-2 to facilitate hemojuvelin cleavage. J. Biol. Chem. 2012, 287, 35104–35117. [Google Scholar] [CrossRef]
- Lee, D.H.; Zhou, L.J.; Zhou, Z.; Xie, J.X.; Jung, J.U.; Liu, Y.; Xi, C.X.; Mei, L.; Xiong, W.C. Neogenin inhibits HJV secretion and regulates BMP-induced hepcidin expression and iron homeostasis. Blood 2010, 115, 3136–3145. [Google Scholar] [CrossRef]
- Muckenthaler, M.; Roy, C.N.; Custodio, A.O.; Minana, B.; deGraaf, J.; Montross, L.K.; Andrews, N.C.; Hentze, M.W. Regulatory defects in liver and intestine implicate abnormal hepcidin and Cybrd1 expression in mouse hemochromatosis. Nat. Genet. 2003, 34, 102–107. [Google Scholar] [CrossRef]
- Knutson, M.; Wessling-Resnick, M. Iron metabolism in the reticuloendothelial system. Crit. Rev. Biochem. Mol. Biol. 2003, 38, 61–88. [Google Scholar] [CrossRef]
- Montalbetti, N.; Simonin, A.; Kovacs, G.; Hediger, M.A. Mammalian iron transporters: Families SLC11 and SLC40. Mol. Aspects Med. 2013, 34, 270–287. [Google Scholar] [CrossRef]
- Wharton, K.A.; Serpe, M. Fine-tuned shuttles for bone morphogenetic proteins. Curr. Opin. Genet. Dev. 2013, in press. [Google Scholar]
- Vujic Spasic, M.; Sparla, R.; Mleczko-Sanecka, K.; Migas, M.C.; Breitkopf-Heinlein, K.; Dooley, S.; Vaulont, S.; Fleming, R.E.; Muckenthaler, M.U. SMAD6 and SMAD7 are co-regulated with hepcidin in mouse models of iron overload. Biochim. Biophys. Acta 2012, 1832, 76–84. [Google Scholar]
- Sakamori, R.; Takehara, T.; Tatsumi, T.; Shigekawa, M.; Hikita, H.; Hiramatsu, N.; Kanto, T.; Hayashi, N. STAT3 signaling within hepatocytes is required for anemia of inflammation in vivo. J. Gastroenterol. 2010, 45, 244–248. [Google Scholar] [CrossRef]
- Chen, J.; Enns, C.A. Hereditary hemochromatosis and transferrin receptor 2. Biochim. Biophys. Acta 2012, 1820, 256–263. [Google Scholar] [CrossRef]
- Corradini, E.; Rozier, M.; Meynard, D.; Odhiambo, A.; Lin, H.Y.; Feng, Q.; Migas, M.C.; Britton, R.S.; Babitt, J.L.; Fleming, R.E. Iron regulation of hepcidin despite attenuated smad1,5,8 signaling in mice without transferrin receptor 2 or HFE. Gastroenterology 2011, 141, 1907–1914. [Google Scholar] [CrossRef] [Green Version]
- Kawabata, H.; Yang, R.; Hirama, T.; Vuong, P.T.; Kawano, S.; Gombart, A.F.; Koeffler, H.P. Molecular cloning of transferrin receptor 2. A new member of the transferrin receptor-like family. J. Biol. Chem. 1999, 274, 20826–20832. [Google Scholar]
- Goswami, T.; Andrews, N.C. Hereditary hemochromatosis protein, HFE, interaction with transferrin receptor 2 suggests a molecular mechanism for mammalian iron sensing. J. Biol. Chem. 2006, 281, 28494–28498. [Google Scholar] [CrossRef]
- Finberg, K.E.; Heeney, M.M.; Campagna, D.R.; Aydinok, Y.; Pearson, H.A.; Hartman, K.R.; Mayo, M.M.; Samuel, S.M.; Strouse, J.J.; Markianos, K.; et al. Mutations in TMPRSS6 cause iron-refractory iron deficiency anemia (IRIDA). Nat. Genet. 2008, 40, 569–571. [Google Scholar] [CrossRef]
- Beutler, E.; van Geet, C.; te Loo, D.M.; Gelbart, T.; Crain, K.; Truksa, J.; Lee, P.L. Polymorphisms and mutations of human TMPRSS6 in iron deficiency anemia. Blood Cells Mol. Dis. 2010, 44, 16–21. [Google Scholar] [CrossRef]
- Silvestri, L.; Pagani, A.; Nai, A.; de Domenico, I.; Kaplan, J.; Camaschella, C. The serine protease matriptase-2 (TMPRSS6) inhibits hepcidin activation by cleaving membrane hemojuvelin. Cell Metab. 2008, 8, 502–511. [Google Scholar] [CrossRef]
- Tanno, T.; Porayette, P.; Sripichai, O.; Noh, S.J.; Byrnes, C.; Bhupatiraju, A.; Lee, Y.T.; Goodnough, J.B.; Harandi, O.; Ganz, T.; et al. Identification of TWSG1 as a second novel erythroid regulator of hepcidin expression in murine and human cells. Blood 2009, 114, 181–186. [Google Scholar] [CrossRef]
- Andriopoulos, B., Jr.; Corradini, E.; Xia, Y.; Faasse, S.A.; Chen, S.; Grgurevic, L.; Knutson, M.D.; Pietrangelo, A.; Vukicevic, S.; Lin, H.Y.; et al. BMP6 is a key endogenous regulator of hepcidin expression and iron metabolism. Nat. Genet. 2009, 41, 482–487. [Google Scholar] [CrossRef]
- Otu, H.H.; Naxerova, K.; Ho, K.; Can, H.; Nesbitt, N.; Libermann, T.A.; Karp, S.J. Restoration of liver mass after injury requires proliferative and not embryonic transcriptional patterns. J. Biol. Chem. 2007, 282, 11197–11204. [Google Scholar] [CrossRef]
- Yu, P.B.; Hong, C.C.; Sachidanandan, C.; Babitt, J.L.; Deng, D.Y.; Hoyng, S.A.; Lin, H.Y.; Bloch, K.D.; Peterson, R.T. Dorsomorphin inhibits bmp signals required for embryogenesis and iron metabolism. Nat. Chem. Biol. 2008, 4, 33–41. [Google Scholar] [CrossRef]
- Mleczko-Sanecka, K.; Casanovas, G.; Ragab, A.; Breitkopf, K.; Muller, A.; Boutros, M.; Dooley, S.; Hentze, M.W.; Muckenthaler, M.U. SMAD7 controls iron metabolism as a potent inhibitor of hepcidin expression. Blood 2010, 115, 2657–2665. [Google Scholar] [CrossRef]
- Castoldi, M.; Muckenthaler, M.U. Regulation of iron homeostasis by microRNAs. Cell. Mol. Life Sci. 2012, 69, 3945–3952. [Google Scholar] [CrossRef]
- Castoldi, M.; Vujic Spasic, M.; Altamura, S.; Elmen, J.; Lindow, M.; Kiss, J.; Stolte, J.; Sparla, R.; D’Alessandro, L.A.; Klingmuller, U.; et al. The liver-specific microRNA miR-122 controls systemic iron homeostasis in mice. J. Clin. Investig. 2011, 121, 1386–1396. [Google Scholar] [CrossRef]
- Zumbrennen-Bullough, K.; Wu, Q.; Chen, W.; Babitt, J. MicroRNA-130a Downregulates Hepcidin Expression during Iron Deficiency by Targeting ALK2. In Proceedings of Fifth Congress of the International BioIron Society (IBIS), Biennial World Meeting (BioIron 2013), London, UK, 14–18 April 2013.
- Lakhal, S.; Schodel, J.; Townsend, A.R.; Pugh, C.W.; Ratcliffe, P.J.; Mole, D.R. Regulation of type II transmembrane serine proteinase TMPRSS6 by hypoxia-inducible factors: New link between hypoxia signaling and iron homeostasis. J. Biol. Chem. 2011, 286, 4090–4097. [Google Scholar]
- Maurer, E.; Gutschow, M.; Stirnberg, M. Matriptase-2 (TMPRSS6) is directly up-regulated by hypoxia inducible factor-1: Identification of a hypoxia-responsive element in the TMPRSS6 promoter region. Biol. Chem. 2012, 393, 535–540. [Google Scholar]
- Zhang, A.S.; Anderson, S.A.; Wang, J.; Yang, F.; DeMaster, K.; Ahmed, R.; Nizzi, C.P.; Eisenstein, R.S.; Tsukamoto, H.; Enns, C.A. Suppression of hepatic hepcidin expression in response to acute iron deprivation is associated with an increase of matriptase-2 protein. Blood 2011, 117, 1687–1699. [Google Scholar] [CrossRef]
- Chen, W.; Huang, F.W.; de Renshaw, T.B.; Andrews, N.C. Skeletal muscle hemojuvelin is dispensable for systemic iron homeostasis. Blood 2011, 117, 6319–6325. [Google Scholar] [CrossRef]
- Gkouvatsos, K.; Wagner, J.; Papanikolaou, G.; Sebastiani, G.; Pantopoulos, K. Conditional disruption of mouse HFE2 gene: Maintenance of systemic iron homeostasis requires hepatic but not skeletal muscle hemojuvelin. Hepatology 2011, 54, 1800–1807. [Google Scholar] [CrossRef]
- Armitage, A.E.; Eddowes, L.A.; Gileadi, U.; Cole, S.; Spottiswoode, N.; Selvakumar, T.A.; Ho, L.P.; Townsend, A.R.; Drakesmith, H. Hepcidin regulation by innate immune and infectious stimuli. Blood 2011, 118, 4129–4139. [Google Scholar] [CrossRef]
- Finberg, K.E. Regulation of systemic iron homeostasis. Curr. Opin. Hematol. 2013, 20, 208–214. [Google Scholar] [CrossRef]
- Wang, R.H.; Li, C.; Xu, X.; Zheng, Y.; Xiao, C.; Zerfas, P.; Cooperman, S.; Eckhaus, M.; Rouault, T.; Mishra, L.; et al. A role of SMAD4 in iron metabolism through the positive regulation of hepcidin expression. Cell Metab. 2005, 2, 399–409. [Google Scholar] [CrossRef]
- Prince, O.D.; Langdon, J.M.; Layman, A.J.; Prince, I.C.; Sabogal, M.; Mak, H.H.; Berger, A.E.; Cheadle, C.; Chrest, F.J.; Yu, Q.; et al. Late stage erythroid precursor production is impaired in mice with chronic inflammation. Haematologica 2012, 97, 1648–1656. [Google Scholar] [CrossRef]
- Fattori, E.; Cappelletti, M.; Costa, P.; Sellitto, C.; Cantoni, L.; Carelli, M.; Faggioni, R.; Fantuzzi, G.; Ghezzi, P.; Poli, V. Defective inflammatory response in interleukin 6-deficient mice. J. Exp. Med. 1994, 180, 1243–1250. [Google Scholar] [CrossRef]
- Roy, C.N.; Custodio, A.O.; de Graaf, J.; Schneider, S.; Akpan, I.; Montross, L.K.; Sanchez, M.; Gaudino, A.; Hentze, M.W.; Andrews, N.C.; et al. An HFE-dependent pathway mediates hyposideremia in response to lipopolysaccharide-induced inflammation in mice. Nat. Genet. 2004, 36, 481–485. [Google Scholar] [CrossRef]
- Wallace, D.F.; McDonald, C.J.; Ostini, L.; Subramaniam, V.N. Blunted hepcidin response to inflammation in the absence of HFE and transferrin receptor 2. Blood 2011, 117, 2960–2966. [Google Scholar] [CrossRef]
- Besson-Fournier, C.; Latour, C.; Kautz, L.; Bertrand, J.; Ganz, T.; Roth, M.P.; Coppin, H. Induction of activin B by inflammatory stimuli up-regulates expression of the iron-regulatory peptide hepcidin through Smad1/5/8 signaling. Blood 2012, 120, 431–439. [Google Scholar] [CrossRef]
- Sasu, B.J.; Cooke, K.S.; Arvedson, T.L.; Plewa, C.; Ellison, A.R.; Sheng, J.; Winters, A.; Juan, T.; Li, H.; Begley, C.G.; et al. Antihepcidin antibody treatment modulates iron metabolism and is effective in a mouse model of inflammation-induced anemia. Blood 2010, 115, 3616–3624. [Google Scholar] [CrossRef]
- Pak, M.; Lopez, M.A.; Gabayan, V.; Ganz, T.; Rivera, S. Suppression of hepcidin during anemia requires erythropoietic activity. Blood 2006, 108, 3730–3735. [Google Scholar] [CrossRef]
- Piperno, A.; Galimberti, S.; Mariani, R.; Pelucchi, S.; Ravasi, G.; Lombardi, C.; Bilo, G.; Revera, M.; Giuliano, A.; Faini, A.; et al. Modulation of hepcidin production during hypoxia-induced erythropoiesis in humans in vivo: Data from the HIGHCARE project. Blood 2011, 117, 2953–2959. [Google Scholar] [CrossRef]
- Talbot, N.P.; Lakhal, S.; Smith, T.G.; Privat, C.; Nickol, A.H.; Rivera-Ch, M.; Leon-Velarde, F.; Dorrington, K.L.; Mole, D.R.; Robbins, P.A. Regulation of hepcidin expression at high altitude. Blood 2012, 119, 857–860. [Google Scholar] [CrossRef]
- Liu, Q.; Davidoff, O.; Niss, K.; Haase, V.H. Hypoxia-inducible factor regulates hepcidin via erythropoietin-induced erythropoiesis. J. Clin. Investig. 2012, 122, 4635–4644. [Google Scholar] [CrossRef]
- Braliou, G.G.; Verga Falzacappa, M.V.; Chachami, G.; Casanovas, G.; Muckenthaler, M.U.; Simos, G. 2-Oxoglutarate-dependent oxygenases control hepcidin gene expression. J. Hepatol. 2008, 48, 801–810. [Google Scholar] [CrossRef]
- Lee, P.; Hsu, M.H.; Welser-Alves, J.; Peng, H. Severe microcytic anemia but increased erythropoiesis in mice lacking hfe or TFR2 and TMPRSS6. Blood Cells Mol. Dis. 2012, 48, 173–178. [Google Scholar] [CrossRef]
- Troutt, J.S.; Rudling, M.; Persson, L.; Stahle, L.; Angelin, B.; Butterfield, A.M.; Schade, A.E.; Cao, G.; Konrad, R.J. Circulating human hepcidin-25 concentrations display a diurnal rhythm, increase with prolonged fasting, and are reduced by growth hormone administration. Clin. Chem. 2012, 58, 1225–1232. [Google Scholar] [CrossRef]
- Hou, Y.; Zhang, S.; Wang, L.; Li, J.; Qu, G.; He, J.; Rong, H.; Ji, H.; Liu, S. Estrogen regulates iron homeostasis through governing hepatic hepcidin expression via an estrogen response element. Gene 2012, 511, 398–403. [Google Scholar] [CrossRef]
- Guo, W.; Bachman, E.; Li, M.; Roy, C.N.; Blusztajn, J.; Wong, S.; Chan, S.Y.; Serra, C.; Jasuja, R.; Travison, T.G.; et al. Testosterone administration inhibits hepcidin transcription and is associated with increased iron incorporation into red blood cells. Aging Cell 2013, 12, 280–291. [Google Scholar] [CrossRef]
- Aigner, E.; Felder, T.K.; Oberkofler, H.; Hahne, P.; Auer, S.; Soyal, S.; Stadlmayr, A.; Schwenoha, K.; Pirich, C.; Hengster, P.; et al. Glucose acts as a regulator of serum iron by increasing serum hepcidin concentrations. J. Nutr. Biochem. 2013, 24, 112–117. [Google Scholar] [CrossRef]
- Hallberg, L. Iron requirements and bioavailability of dietary iron. Experientia 1983, 44, 223–244. [Google Scholar]
- Hallberg, L.; Hulthen, L.; Garby, L. Iron stores in man in relation to diet and iron requirements. Eur. J. Clin. Nutr. 1998, 52, 623–631. [Google Scholar]
- Hunt, J.R. Bioavailability of iron, zinc, and other trace minerals from vegetarian diets. Am. J. Clin. Nutr. 2003, 78, 633S–639S. [Google Scholar]
- Yip, R. Chapter 30. Iron. In Present Knowledge in Nutrition; ILSI Press: Washington, DC, USA, 2001; pp. 311–328. [Google Scholar]
- Lonnerdal, B. Soybean ferritin: Implications for iron status of vegetarians. Am. J. Clin. Nutr. 2009, 89, 1680S–1685S. [Google Scholar] [CrossRef]
- Cook, J.D. Adaptation in iron metabolism. Am. J. Clin. Nutr. 1990, 51, 301–308. [Google Scholar]
- Polin, V.; Coriat, R.; Perkins, G.; Dhooge, M.; Abitbol, V.; Leblanc, S.; Prat, F.; Chaussade, S. Iron deficiency: From diagnosis to treatment. Dig. Liver Dis. 2013. [Google Scholar] [CrossRef]
- Sharma, J.B.; Jain, S.; Mallika, V.; Singh, T.; Kumar, A.; Arora, R.; Murthy, N.S. A prospective, partially randomized study of pregnancy outcomes and hematologic responses to oral and intramuscular iron treatment in moderately anemic pregnant women. Am. J. Clin. Nutr. 2004, 79, 116–122. [Google Scholar]
- Pena-Rosas, J.P.; De-Regil, L.M.; Dowswell, T.; Viteri, F.E. Daily oral iron supplementation during pregnancy. Cochrane Database Syst. Rev. 2012, 12, CD004736. [Google Scholar]
- Heath, C.; Strauss, M.; Castle, W. Quantitative aspects of iron deficiency in hypochromic anemia. (The parenteral administration of iron). J. Clin. Investig. 1932, 11, 1293–1312. [Google Scholar] [CrossRef]
- Righetti, A.A.; Adiossan, L.G.; Ouattara, M.; Glinz, D.; Hurrell, R.F.; N’Goran, E.K.; Wegmuller, R.; Utzinger, J. Dynamics of anemia in relation to parasitic infections, micronutrient status, and increasing age in South-Central Côte d’ivoire. J. Infect. Dis. 2013, 207, 1604–1615. [Google Scholar] [CrossRef]
- Haji, K.A.; Khatib, B.O.; Smith, S.; Ali, A.S.; Devine, G.J.; Coetzee, M.; Majambere, S. Challenges for malaria elimination in Zanzibar: Pyrethroid resistance in malaria vectors and poor performance of long-lasting insecticide nets. Parasit. Vectors 2013, 6, 82. [Google Scholar] [CrossRef] [Green Version]
- Drakesmith, H.; Prentice, A. Viral infection and iron metabolism. Nat. Rev. 2008, 6, 541–552. [Google Scholar] [CrossRef]
- Zhang, X.; Rovin, B.H. Beyond anemia: Hepcidin, monocytes and inflammation. Biol. Chem. 2013, 394, 231–238. [Google Scholar]
- Denic, S.; Agarwal, M.M. Nutritional iron deficiency: An evolutionary perspective. Nutrition 2007, 23, 603–614. [Google Scholar] [CrossRef]
- Brugnara, C. Iron deficiency and erythropoiesis: New diagnostic approaches. Clin. Chem. 2003, 49, 1573–1578. [Google Scholar] [CrossRef]
- Clark, S.F. Iron deficiency anemia. Nutr. Clin. Pract. 2008, 23, 128–141. [Google Scholar] [CrossRef]
- Swanson, C.A. Iron intake and regulation: Implications for iron deficiency and iron overload. Alcohol 2003, 30, 99–102. [Google Scholar] [CrossRef]
- Weinstein, D.A.; Roy, C.N.; Fleming, M.D.; Loda, M.F.; Wolfsdorf, J.I.; Andrews, N.C. Inappropriate expression of hepcidin is associated with iron refractory anemia: Implications for the anemia of chronic disease. Blood 2002, 100, 3776–3781. [Google Scholar] [CrossRef]
- Roy, C.N. Anemia of inflammation. Hematology Am. Soc. Hematol. Educ. Program 2010, 2010, 276–280. [Google Scholar] [CrossRef]
- Weiss, G.; Gordeuk, V.R. Benefits and risks of iron therapy for chronic anaemias. Eur. J. Clin. Investig. 2005, 35, 36–45. [Google Scholar] [CrossRef]
- Hohlbaum, A.; Gille, H.; Christian, J.; Allerdsodrfer, A.; Jaworski, J.; Burrows, J.; Rattenstetter, B.; Kolodziejczyk, M.; Olwill, S.; Audoly, L. Iron Mobilization and Pharmacodynamic Marker Measurements in Non-Human Primates Following Administration of PRS-080, a Novel and Highly Specific Anti-Hepcidin Therapeutic. In Proceedings of Fifth Congress of the International BioIron Society (IBIS), Biennial World Meeting (BioIron 2013), London, UK, 14–18 April 2013.
- Schwoebel, F.; van Eijk, L.T.; Zboralski, D.; Sell, S.; Buchner, K.; Maasch, C.; Purschke, W.G.; Humphrey, M.; Zollner, S.; Eulberg, D.; et al. The effects of the anti-hepcidin spiegelmer NOX-H94 on inflammation-induced anemia in cynomolgus monkeys. Blood 2013, 121, 2311–2315. [Google Scholar] [CrossRef]
- Garcia-Manero, G. Myelodysplastic syndromes: 2012 Update on diagnosis, risk-stratification, and management. Am. J. Hematol. 2012, 87, 692–701. [Google Scholar] [CrossRef]
- Gulbis, B.; Eleftheriou, A.; Angastiniotis, M.; Ball, S.; Surralles, J.; Castella, M.; Heimpel, H.; Hill, A.; Corrons, J.L. Epidemiology of rare anaemias in europe. Adv. Exp. Med. Biol. 2010, 686, 375–396. [Google Scholar] [CrossRef]
- Iolascon, A.; de Falco, L. Mutations in the gene encoding DMT1: Clinical presentation and treatment. Semin. Hematol. 2009, 46, 358–370. [Google Scholar] [CrossRef]
- Bardou-Jacquet, E.; Island, M.L.; Jouanolle, A.M.; Detivaud, L.; Fatih, N.; Ropert, M.; Brissot, E.; Mosser, A.; Maisonneuve, H.; Brissot, P.; et al. A novel N491s mutation in the human SLC11A2 gene impairs protein trafficking and in association with the G212V mutation leads to microcytic anemia and liver iron overload. Blood Cells Mol. Dis. 2011, 47, 243–248. [Google Scholar] [CrossRef]
- Sendamarai, A.K.; Ohgami, R.S.; Fleming, M.D.; Lawrence, C.M. Structure of the membrane proximal oxidoreductase domain of human Steap3, the dominant ferrireductase of the erythroid transferrin cycle. Proc. Natl. Acad. Sci. USA 2008, 105, 7410–7415. [Google Scholar] [CrossRef]
- Zhang, A.S.; Sheftel, A.D.; Ponka, P. The anemia of “Haemoglobin-deficit” (hbd/hbd) mice is caused by a defect in transferrin cycling. Exp. Hematol. 2006, 34, 593–598. [Google Scholar] [CrossRef]
- Troadec, M.B.; Warner, D.; Wallace, J.; Thomas, K.; Spangrude, G.J.; Phillips, J.; Khalimonchuk, O.; Paw, B.H.; Ward, D.M.; Kaplan, J. Targeted deletion of the mouse mitoferrin1 gene: From anemia to protoporphyria. Blood 2011, 117, 5494–5502. [Google Scholar] [CrossRef]
- Ye, H.; Jeong, S.Y.; Ghosh, M.C.; Kovtunovych, G.; Silvestri, L.; Ortillo, D.; Uchida, N.; Tisdale, J.; Camaschella, C.; Rouault, T.A. Glutaredoxin 5 deficiency causes sideroblastic anemia by specifically impairing heme biosynthesis and depleting cytosolic iron in human erythroblasts. J. Clin. Investig. 2010, 120, 1749–1761. [Google Scholar] [CrossRef]
- Bergmann, A.K.; Campagna, D.R.; McLoughlin, E.M.; Agarwal, S.; Fleming, M.D.; Bottomley, S.S.; Neufeld, E.J. Systematic molecular genetic analysis of congenital sideroblastic anemia: Evidence for genetic heterogeneity and identification of novel mutations. Pediatr. Blood Cancer 2010, 54, 273–278. [Google Scholar]
- Allikmets, R.; Raskind, W.H.; Hutchinson, A.; Schueck, N.D.; Dean, M.; Koeller, D.M. Mutation of a putative mitochondrial iron transporter gene (ABC7) in X-linked sideroblastic anemia and ataxia (XLSA/A). Hum. Mol. Genet. 1999, 8, 743–749. [Google Scholar] [CrossRef]
- Savary, S.; Allikmets, R.; Denizot, F.; Luciani, M.F.; Mattei, M.G.; Dean, M.; Chimini, G. Isolation and chromosomal mapping of a novel ATP-binding cassette transporter conserved in mouse and human. Genomics 1997, 41, 275–278. [Google Scholar] [CrossRef]
- Balwani, M.; Desnick, R.J. The porphyrias: Advances in diagnosis and treatment. Blood 2012, 120, 4496–4504. [Google Scholar] [CrossRef]
- Camaschella, C.; Campanella, A.; de Falco, L.; Boschetto, L.; Merlini, R.; Silvestri, L.; Levi, S.; Iolascon, A. The human counterpart of zebrafish shiraz shows sideroblastic-like microcytic anemia and iron overload. Blood 2007, 110, 1353–1358. [Google Scholar] [CrossRef]
- Camaschella, C.; Poggiali, E. Inherited disorders of iron metabolism. Curr. Opin. Pediatr. 2011, 23, 14–20. [Google Scholar] [CrossRef]
- Lawler, P.R.; Filion, K.B.; Dourian, T.; Atallah, R.; Garfinkle, M.; Eisenberg, M.J. Anemia and mortality in acute coronary syndromes: A systematic review and meta-analysis. Am. Heart J. 2013, 165, 143–153. [Google Scholar] [CrossRef]
- Datz, C.; Felder, T.K.; Niederseer, D.; Aigner, E. Iron homeostasis in the metabolic syndrome. Eur. J. Clin. Investig. 2013, 43, 215–224. [Google Scholar] [CrossRef]
- Pietrangelo, A. Hereditary hemochromatosis: Pathogenesis, diagnosis, and treatment. Gastroenterology 2011, 139, 393–408. [Google Scholar] [CrossRef]
- Babitt, J.L.; Lin, H.Y. The molecular pathogenesis of hereditary hemochromatosis. Semin. Liver Dis. 2011, 31, 280–292. [Google Scholar] [CrossRef]
- Musallam, K.M.; Cappellini, M.D.; Wood, J.C.; Taher, A.T. Iron overload in non-transfusion-dependent thalassemia: A clinical perspective. Blood Rev. 2012, 26, S16–S19. [Google Scholar] [CrossRef]
- Adams, R.L.; Bird, R.J. Safety and efficacy of deferasirox in the management of transfusion-dependent patients with myelodysplastic syndrome and aplastic anaemia: A perspective review. Ther. Adv. Hematol. 2013, 4, 93–102. [Google Scholar] [CrossRef]
- Thuret, I. Post-transfusional iron overload in the haemoglobinopathies. C. B. Biol. 2013, 336, 164–172. [Google Scholar] [CrossRef]
- Whitington, P.F. Gestational alloimmune liver disease and neonatal hemochromatosis. Semin. Liver Dis. 2012, 32, 325–332. [Google Scholar]
- Pietrangelo, A. Iron in NASH, chronic liver diseases and HCC: How much iron is too much? J. Hepatol. 2009, 50, 249–251. [Google Scholar] [CrossRef]
- Nelson, J.E.; Wilson, L.; Brunt, E.M.; Yeh, M.M.; Kleiner, D.E.; Unalp-Arida, A.; Kowdley, K.V. Relationship between the pattern of hepatic iron deposition and histological severity in nonalcoholic fatty liver disease. Hepatology 2011, 53, 448–457. [Google Scholar] [CrossRef]
- Lambrecht, R.W.; Sterling, R.K.; Naishadham, D.; Stoddard, A.M.; Rogers, T.; Morishima, C.; Morgan, T.R.; Bonkovsky, H.L. Iron levels in hepatocytes and portal tract cells predict progression and outcome of patients with advanced chronic hepatitis C. Gastroenterology 2011, 140, 1490–1500. [Google Scholar] [CrossRef]
- Ruivard, M.; Laine, F.; Ganz, T.; Olbina, G.; Westerman, M.; Nemeth, E.; Rambeau, M.; Mazur, A.; Gerbaud, L.; Tournilhac, V.; et al. Iron absorption in dysmetabolic iron overload syndrome is decreased and correlates with increased plasma hepcidin. J. Hepatol. 2009, 50, 1219–1225. [Google Scholar] [CrossRef]
- Martinelli, N.; Traglia, M.; Campostrini, N.; Biino, G.; Corbella, M.; Sala, C.; Busti, F.; Masciullo, C.; Manna, D.; Previtali, S.; et al. Increased serum hepcidin levels in subjects with the metabolic syndrome: A population study. PLoS One 2012, 7, e48250. [Google Scholar] [CrossRef]
- Valenti, L.; Dongiovanni, P.; Motta, B.M.; Swinkels, D.W.; Bonara, P.; Rametta, R.; Burdick, L.; Frugoni, C.; Fracanzani, A.L.; Fargion, S. Serum hepcidin and macrophage iron correlate with MCP-1 release and vascular damage in patients with metabolic syndrome alterations. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 683–690. [Google Scholar] [CrossRef]
- Altamura, S.; Muckenthaler, M.U. Iron toxicity in diseases of aging: Alzheimer’s disease, Parkinson’s disease and atherosclerosis. J. Alzheimers Dis. 2009, 16, 879–895. [Google Scholar]
- Liu, B.; Moloney, A.; Meehan, S.; Morris, K.; Thomas, S.E.; Serpell, L.C.; Hider, R.; Marciniak, S.J.; Lomas, D.A.; Crowther, D.C. Iron promotes the toxicity of amyloid beta peptide by impeding its ordered aggregation. J. Biol. Chem. 2011, 286, 4248–4256. [Google Scholar] [CrossRef]
- Lee, H.P.; Zhu, X.; Liu, G.; Chen, S.G.; Perry, G.; Smith, M.A.; Lee, H.G. Divalent metal transporter, iron, and Parkinson’s disease: A pathological relationship. Cell Res. 2010, 20, 397–399. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Steinbicker, A.U.; Muckenthaler, M.U. Out of Balance—Systemic Iron Homeostasis in Iron-Related Disorders. Nutrients 2013, 5, 3034-3061. https://doi.org/10.3390/nu5083034
Steinbicker AU, Muckenthaler MU. Out of Balance—Systemic Iron Homeostasis in Iron-Related Disorders. Nutrients. 2013; 5(8):3034-3061. https://doi.org/10.3390/nu5083034
Chicago/Turabian StyleSteinbicker, Andrea U., and Martina U. Muckenthaler. 2013. "Out of Balance—Systemic Iron Homeostasis in Iron-Related Disorders" Nutrients 5, no. 8: 3034-3061. https://doi.org/10.3390/nu5083034
APA StyleSteinbicker, A. U., & Muckenthaler, M. U. (2013). Out of Balance—Systemic Iron Homeostasis in Iron-Related Disorders. Nutrients, 5(8), 3034-3061. https://doi.org/10.3390/nu5083034