Fructose Rich Diet-Induced High Plasminogen Activator Inhibitor-1 (PAI-1) Production in the Adult Female Rat: Protective Effect of Progesterone

Abstract
:
1. Introduction
2. Experimental Section
2.1. Animals and Experimental Design

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Treatment (i.m. on age 60 days) | Diet (100–120 days of age) | Group (abbreviation) |
|---|---|---|
| Vehicle | Normal | CT-ND |
| P4 | Normal | P4-ND |
| Vehicle | Fructose Rich | CT-FRD |
| P4 | Fructose Rich | P4-FRD |
| Experimentation (on day 120 of age) |
2.2. Peripheral Metabolite Measurements
2.3. Intravenous Glucose Tolerance Test (i.v.-GTT)
2.4. Parametrial Adipose Tissue RNA Isolation and Real-Time Quantitative PCR
2.5. Statistical Analysis
3. Results
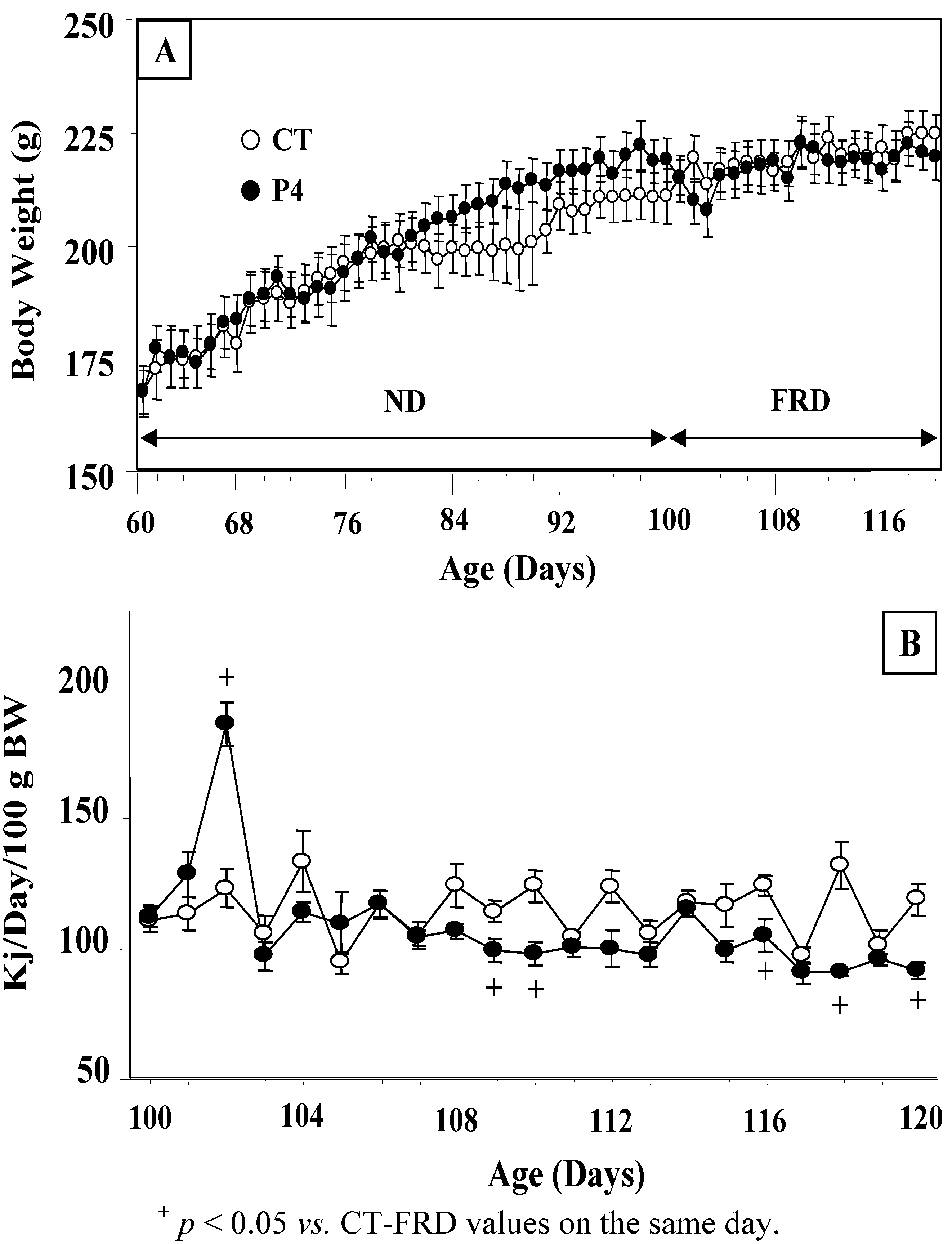
3.1. Effect of Diet on Body Weight, Parametrial Adipose Tissue Mass and Energy Intake in CT and P4-Primed Rats
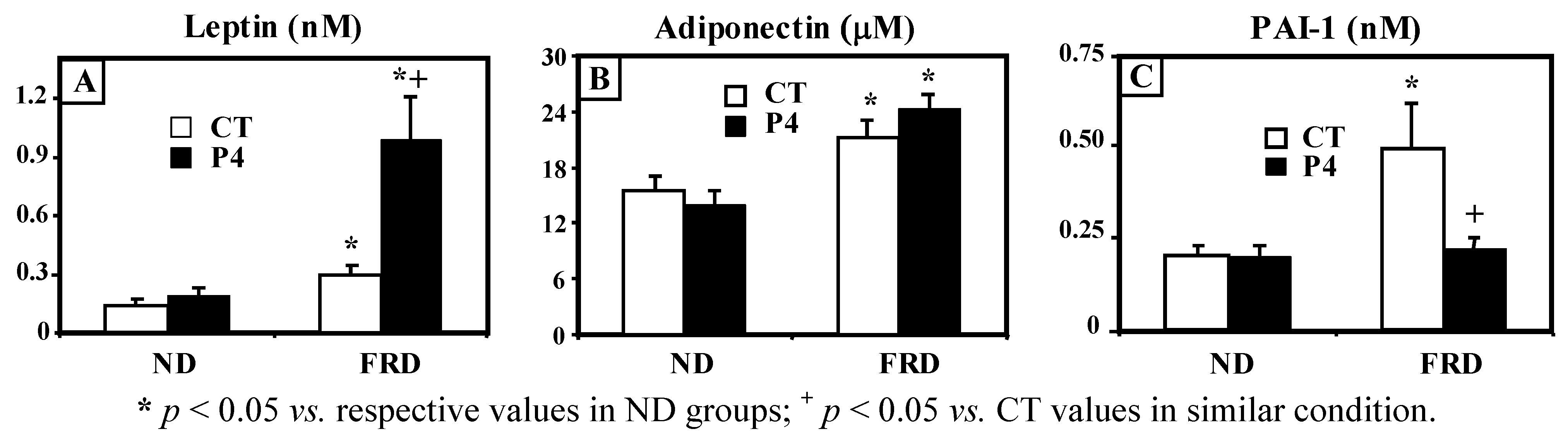
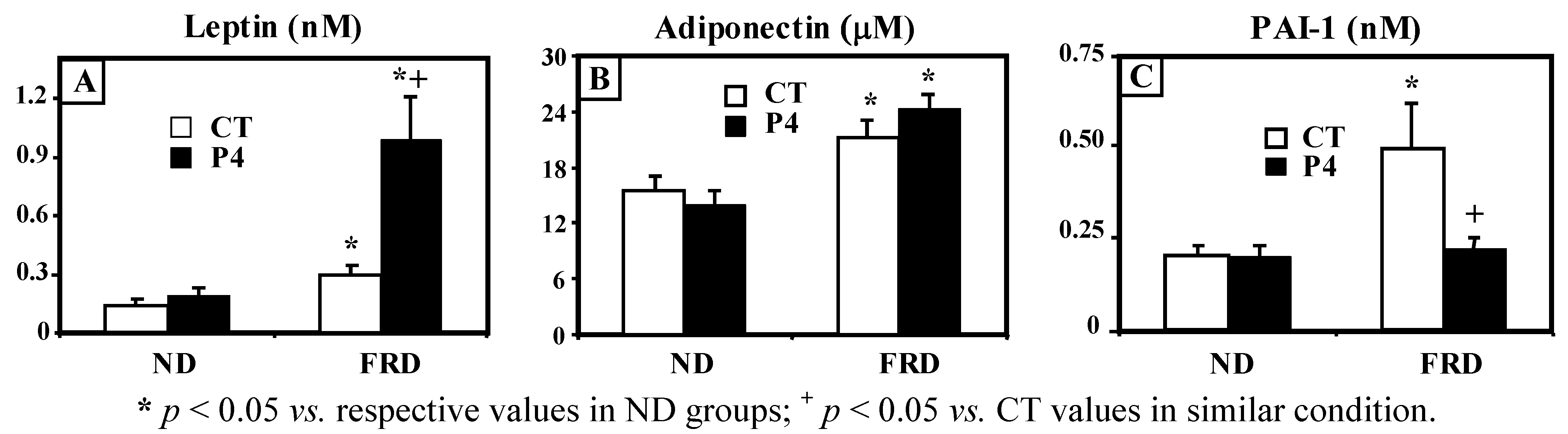
3.2. Effect of FRD Intake on Peripheral Biomarkers in Normal and P4-Primed Rats
| CT | P4 | |
|---|---|---|
| Glucose (mM) | ||
| ND | 6.33 ± 0.28 | 6.01 ± 0.33 |
| FRD | 6.72 ± 0.31 | 7.38 ± 0.36 |
| Insulin (nM) | ||
| ND | 6.17 ± 1.53 | 6.94 ± 0.68 |
| FRD | 5.51 ± 0.61 | 5.39 ± 0.72 |
| Total Cholesterol (mM) | ||
| ND | 1.64 ± 0.13 | 1.58 ± 0.08 |
| FRD | 1.34 ± 0.15 | 1.91 ± 0.12 b,c |
| Triacylglycerol (mM) | ||
| ND | 0.99 ± 0.08 | 1.41 ± 0.09 a |
| FRD | 1.44 ± 0.22 a | 1.97 ± 0.18 c |
| NEFA (mM) | ||
| ND | 0.35 ± 0.04 | 0.53 ± 0.06 a |
| FRD | 0.51 ± 0.05 a | 0.52 ± 0.07 |
| Corticosterone (μM) | ||
| ND | 0.35 ± 0.06 | 0.45 ± 0.08 |
| FRD | 0.33 ± 0.09 | 0.43 ± 0.09 |
| Progesterone (nM) | ||
| ND | 37.34 ± 8.14 | 48.31 ± 14.22 |
| FRD | 47.16 ± 9.51 | 31.36 ± 7.89 |
| Estradiol (nM) | ||
| ND | 0.19 ± 0.03 | 0.20 ± 0.04 |
| FRD | 0.22 ± 0.02 | 0.26 ± 0.03 |
| Testosterone (nM) | ||
| ND | 0.86 ± 0.06 | 0.98 ± 0.05 |
| FRD | 0.95 ± 0.08 | 0.91 ± 0.17 |
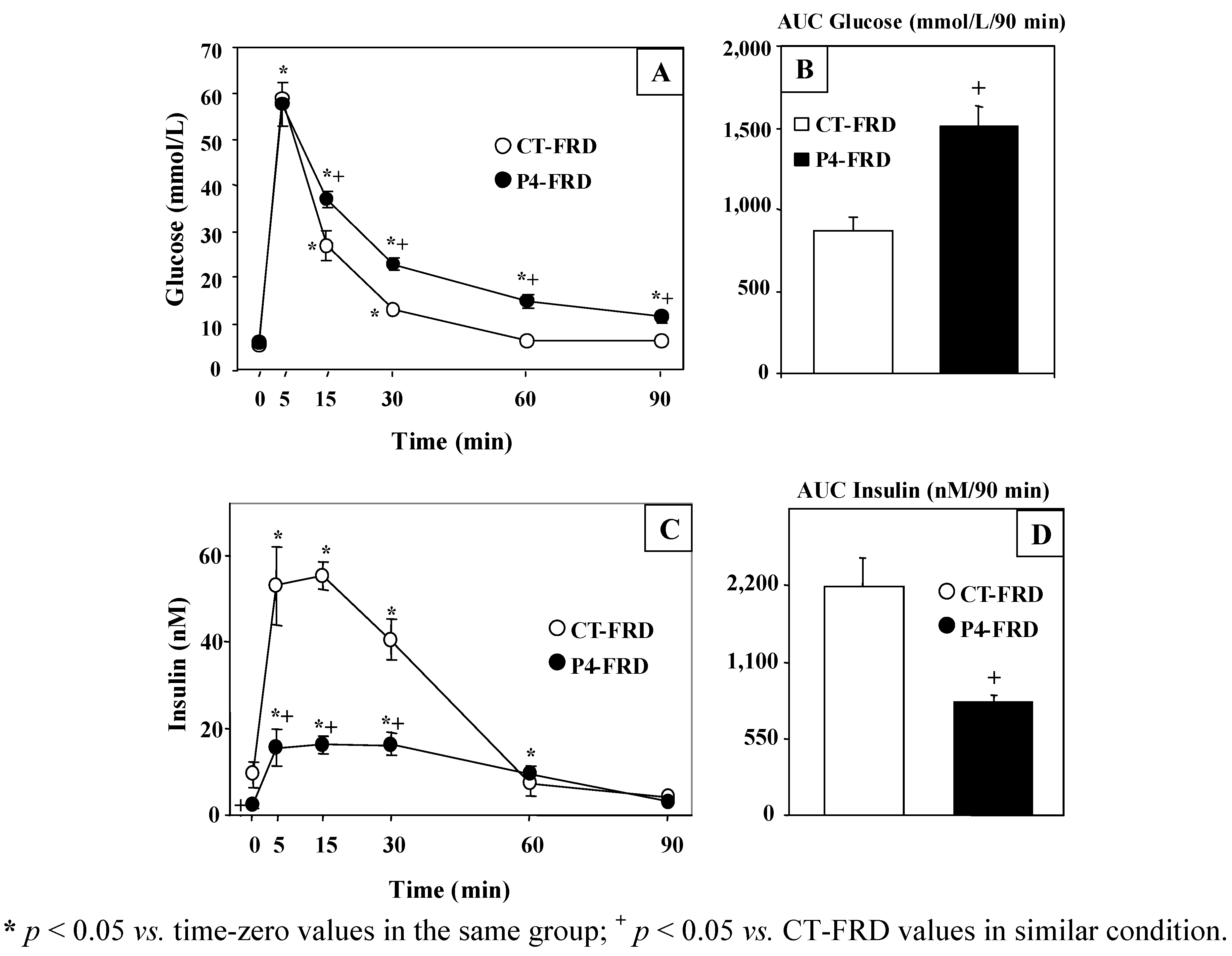
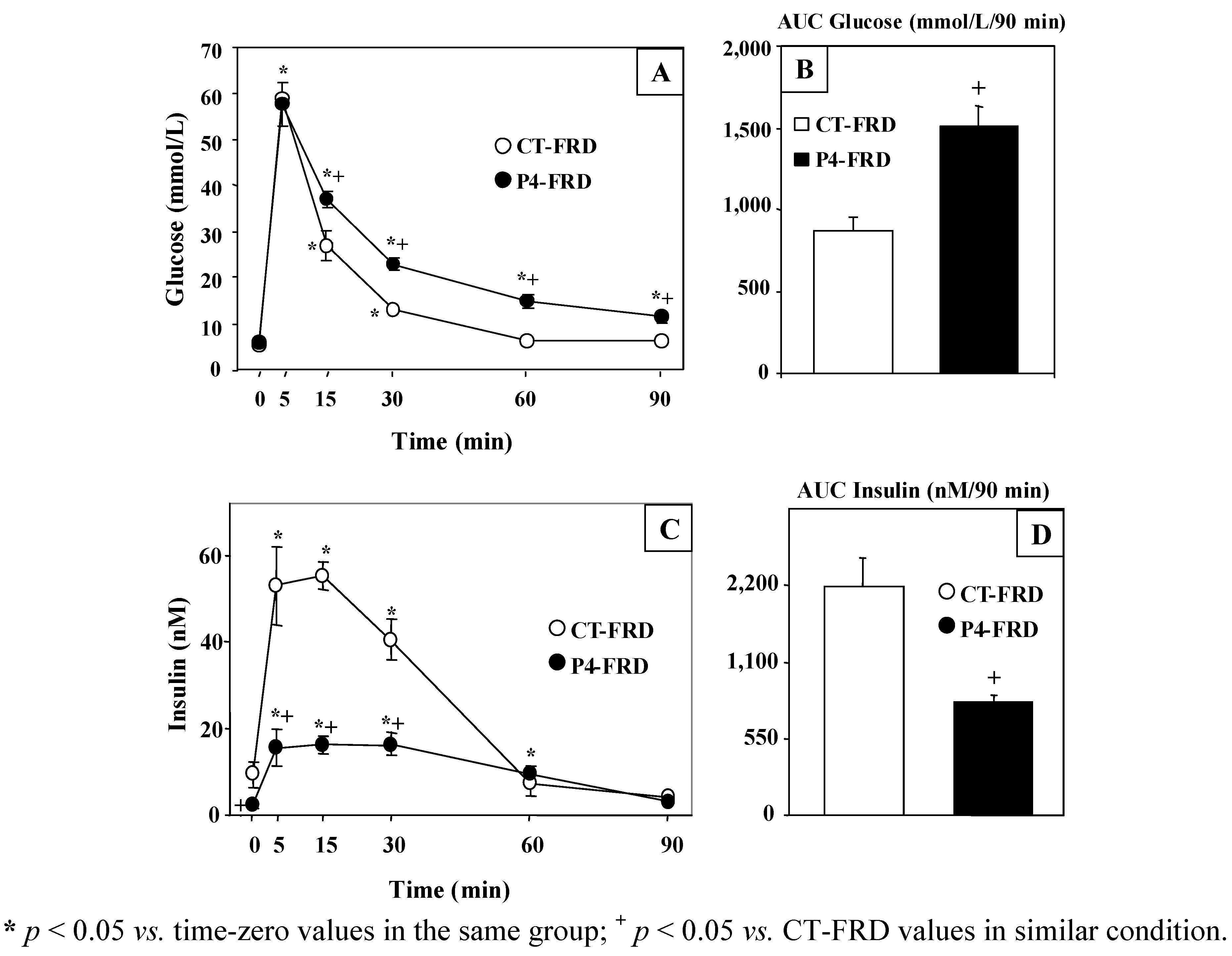
3.3. Changes in Plasma Glucose and Insulin Concentrations throughout the i.v.-GTT
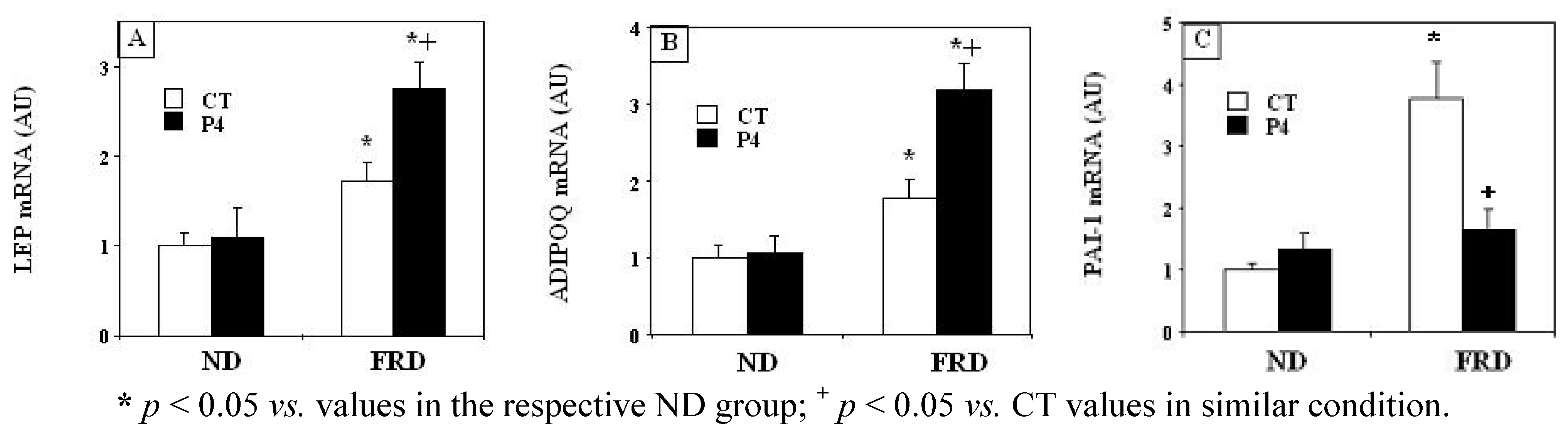
3.4. Effects of P4 Treatment and FRD on PMAT Genes
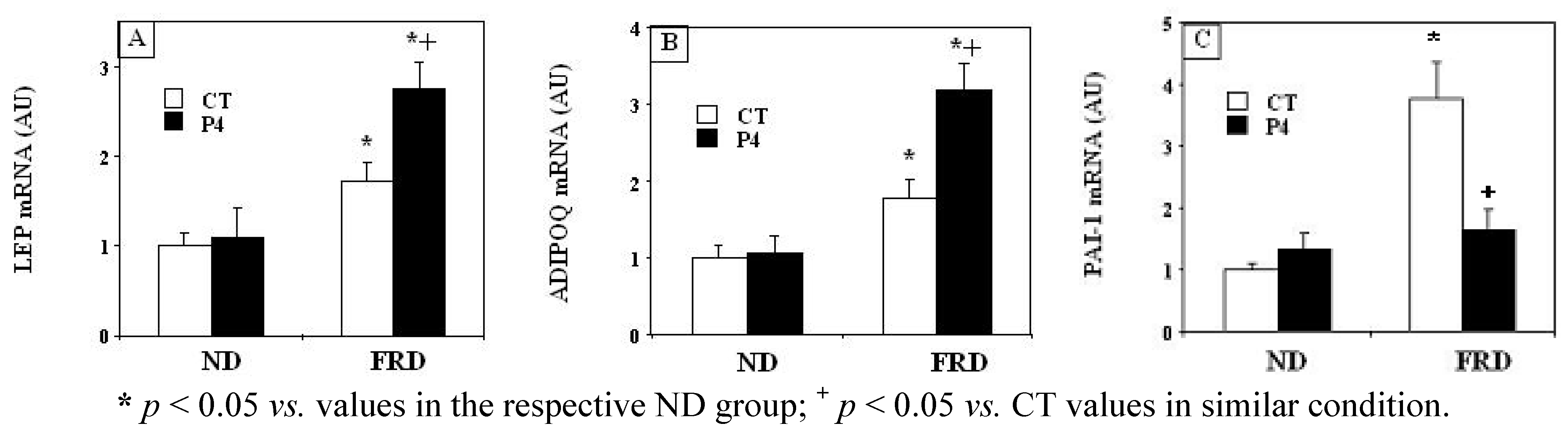
4. Discussion
5. Conclusion
Acknowledgments
Conflict of Interest
References
- Elliott, S.S.; Keim, N.L.; Stern, J.S.; Teff, K.; Havel, P.J. Fructose, weight gain, and the insulin resistance syndrome. Am. J. Clin. Nutr. 2002, 76, 911–922. [Google Scholar]
- Bray, G.A.; Nielsen, S.J.; Popkin, B.M. Consumption of high-fructose corn syrup in beverages may play a role in the epidemic of obesity. Am. J. Clin. Nutr. 2004, 79, 537–543. [Google Scholar]
- Kohen-Avramoglu, R.; Theriault, A.; Adeli, K. Emergence of the metabolic syndrome in childhood: An epidemiological overview and mechanistic link to dyslipidemia. Clin. Biochem. 2003, 36, 413–420. [Google Scholar] [CrossRef]
- Verma, S.; Bhanot, S.; Yao, L.; McNeill, J.H. Vascular insulin resistance in fructose-hypertensive rats. Eur. J. Pharmacol. 1997, 322, R1–R2. [Google Scholar] [CrossRef]
- Alzamendi, A.; Castrogiovanni, D.; Ortega, H.H.; Gaillard, R.C.; Giovambattista, A.; Spinedi, E. Parametrial adipose tissue and metabolic dysfunctions induced by fructose-rich diet in normal and neonatal-androgenized adult female rats. Obesity 2010, 18, 441–448. [Google Scholar] [CrossRef]
- Rebolledo, O.R.; Marra, C.A.; Raschia, A.; Rodríguez, S.; Gagliardino, J.J. Abdominal adipose tissue: Early metabolic dysfunction associated to insulin resistance and oxidative stress induced by an unbalanced diet. Horm. Metab. Res. 2008, 40, 794–800. [Google Scholar] [CrossRef]
- Alzamendi, A.; Giovambattista, A.; Raschia, A.; Madrid, V.; Gaillard, R.C.; Rebolledo, O.; Gagliardino, J.J.; Spinedi, E. Fructose-rich diet-induced abdominal adipose tissue endocrine dysfunction in normal male rats. Endocrine 2009, 35, 227–232. [Google Scholar] [CrossRef]
- Delbosc, S.; Paizanis, E.; Magous, R.; Araiz, C.; Dimo, T.; Cristol, J.P.; Cros, G.; Azay, J. Involvement of oxidative stress and NADPH oxidase activation in the development of cardiovascular complications in a model of insulin resistance, the fructose-fed rat. Atherosclerosis 2005, 179, 43–49. [Google Scholar] [CrossRef]
- Kumtepe, Y.; Borekci, B.; Karaca, M.; Salman, S.; Alp, H.H.; Suleyman, H. Effect of acute and chronic administration of progesterone, estrogen, FSH and LH on oxidant and antioxidant parameters in rat gastric tissue. Chem. Biol. Interact. 2009, 182, 1–6. [Google Scholar]
- Behl, C.; Trapp, T.; Skutella, T.; Holsboer, F. Protection against oxidative stress-induced neuronal cell death—A novel role for RU486. Eur. J. Neurosci. 1997, 9, 912–920. [Google Scholar] [CrossRef]
- Perelló, M.; Castrogiovanni, D.; Giovambattista, A.; Gaillard, R.C.; Spinedi, E. Impairment in insulin sensitivity after early androgenization in the post-pubertal female rat. Life Sci. 2007, 80, 1792–1798. [Google Scholar] [CrossRef]
- Lorenzo, M.; Roncero, C.; Fabregat, I.; Benito, M. Hormonal regulation of rat foetal lipogenesis in brown-adipocyte primary cultures. Biochem. J. 1988, 251, 617–620. [Google Scholar]
- Roncero, C.; Lorenzo, M.; Benito, M. Regulation of rat foetal lipogenesis in brown adipose tissue in vivo and in isolated brown adipocytes during the last day of, and after prolonged, gestation. Biochem. J. 1987, 243, 617–620. [Google Scholar]
- Shirling, D.; Ashby, J.P.; Baird, J.D. Effect of progesterone on lipid metabolism in the intact rat. J. Endocrinol. 1981, 90, 285–294. [Google Scholar] [CrossRef]
- Sandberg, M.; Borg, L.A. Steroid effects on intracellular degradation of insulin and crinophagy in isolated pancreatic islets. Mol. Cell. Endocrinol. 2007, 277, 35–41. [Google Scholar] [CrossRef]
- Palomar-Morales, M.; Morimoto, S.; Mendoza-Rodríguez, C.A.; Cerbón, M.A. The protective effect of testosterone on streptozotocin-induced apoptosis in beta cells is sex specific. Pancreas 2010, 39, 193–200. [Google Scholar] [CrossRef]
- Straub, S.G.; Sharp, G.W.; Meglasson, M.D.; de Souza, C.-J. Progesterone inhibits insulin secretion by a membrane delimited, non-genomic action. Biosci. Rep. 2001, 21, 653–666. [Google Scholar] [CrossRef]
- Ordóñez, P.; Moreno, M.; Alonso, A.; Fernández, R.; Díaz, F.; González, C. Insulin sensitivity in streptozotocin-induced diabetic rats treated with different doses of 17beta-oestradiol or progesterone. Exp. Physiol. 2007, 92, 241–249. [Google Scholar]
- González, C.; Alonso, A.; Alvarez, N.; Díaz, F.; Martínez, M.; Fernández, S.; Patterson, A.M. Role of 17beta-estradiol and/or progesterone on insulin sensitivity in the rat: Implications during pregnancy. J. Endocrinol. 2000, 166, 283–291. [Google Scholar] [CrossRef]
- Sutter-Dub, M.T.; Dazey, B.; Hamdan, E.; Vergnaud, M.T. Progesterone and insulin-resistance: Studies of progesterone action on glucose transport, lipogenesis and lipolysis in isolated fat cells of the female rat. J. Endocrinol. 1981, 88, 455–462. [Google Scholar] [CrossRef]
- Ashby, J.P.; Shirling, D.; Baird, J.D. Effect of progesterone on the secretion and peripheral action of insulin and glucagon in the intact rat. J. Endocrinol. 1981, 88, 49–55. [Google Scholar] [CrossRef]
- Castrogiovanni, D.; Perelló, M.; Gaillard, R.C.; Spinedi, E. Modulatory role of testosterone in plasma leptin turnover in rats. Endocrine 2003, 22, 203–210. [Google Scholar] [CrossRef]
- Perelló, M.; Castrogiovanni, D.; Moreno, G.; Gaillard, R.C.; Spinedi, E. Neonatal hypothalamic androgenization in the female rat induces changes in peripheral insulin sensitivity and adiposity function at adulthood. Neur. Endocrinol. Lett. 2003, 24, 241–248. [Google Scholar]
- Spinedi, E.; Suescun, M.O.; Hadid, R.; Daneva, T.; Gaillard, R.C. Effects of gonadectomy and sex hormone therapy on the endotoxin-stimulated hypothalamo-pituitary-adrenal axis: Evidence for a neuroendocrine-immunological sexual dimorphism. Endocrinology 1992, 131, 2430–2436. [Google Scholar] [CrossRef]
- Alzamendi, A.; Castrogiovanni, D.; Gaillard, R.C.; Spinedi, E.; Giovambattista, A. Increased male offspring’s risk of metabolic-neuroendocrine dysfunction and overweight after fructose-rich diet intake by the lactating mother. Endocrinology 2010, 151, 4214–4223. [Google Scholar] [CrossRef]
- Chomczynski, P.; Sacchi, N. Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal. Biochem. 1987, 162, 156–159. [Google Scholar]
- Zar, J.H. Biostatistical Analysis; Prentice-Hall, Inc.: Englewood Cliffs, NJ, USA, 1974. [Google Scholar]
- Lacasa, D.; Le Liepvre, X.; Ferre, P.; Dugail, I. Progesterone stimulates adipocyte determination and differentiation 1/sterol regulatory element-binding protein 1c gene expression. Potential mechanism for the lipogenic effect of progesterone in adipose tissue. J. Biol. Chem. 2001, 276, 11512–11516. [Google Scholar]
- Furukawa, S.; Fujita, T.; Shimabukuro, M.; Iwaki, M.; Yamada, Y.; Nakajima, Y.; Nakayama, O.; Makishima, M.; Matsuda, M.; Shimomura, I. Increased oxidative stress in obesity and its impact on metabolic syndrome. J. Clin. Invest. 2004, 114, 1752–1761. [Google Scholar]
- Brownlee, M. The pathobiology of diabetic complications: A unifying mechanism. Diabetes 2005, 54, 1615–1625. [Google Scholar] [CrossRef]
- Francini, F.; Castro, M.C.; Schinella, G.; García, M.E.; Maiztegui, B.; Raschia, M.A.; Gagliardino, J.J.; Massa, M.L. Changes induced by a fructose-rich diet on hepatic metabolism and the antioxidant system. Life Sci. 2010, 86, 965–971. [Google Scholar] [CrossRef]
- Walder, K.; Filippis, A.; Clark, S.; Zimmet, P.; Collier, G.R. Leptin inhibits insulin binding in isolated rat adipocytes. J. Endocrinol. 1997, 155, R5–R7. [Google Scholar]
- Berg, A.H.; Combs, T.P.; Du, X.; Brownlee, M.; Scherer, P.E. The adipocyte-secreted protein Acrp30 enhances hepatic insulin action. Nat. Med. 2001, 7, 947–953. [Google Scholar] [CrossRef]
- Withers, D.J.; White, M. Perspective: The insulin signaling system—A common link in the pathogenesis of type 2 diabetes. Endocrinology 2000, 141, 1917–1921. [Google Scholar] [CrossRef]
- Krotkiewski, M.; Björntorp, P. The effect of progesterone and of insulin administration on regional adipose tissue cellularity in the rat. Acta Physiol. Scand. 1976, 96, 122–127. [Google Scholar] [CrossRef]
- Mueck, A.O.; Lippert, C.; Seeger, H.; Wallwiener, D. Effects of tibolone on human breast cancer cells and human vascular coronary cells. Arch. Gynecol. Obstet. 2003, 267, 139–144. [Google Scholar] [CrossRef]
- Winkler, U.H.; Altkemper, R.; Kwee, B.; Helmond, F.A.; Coelingh Bennink, H.J. Effects of tibolone and continuous combined hormone replacement therapy on parameters in the clotting cascade: A multicenter, double-blind, randomized study. Fertil. Steril. 2000, 74, 10–19. [Google Scholar] [CrossRef]
- Koh, K.K.; Han, S.H.; Shin, M.S.; Ahn, J.Y.; Lee, Y.; Shin, E.K. Significant differential effects of lower doses of hormone therapy or tibolone on markers of cardiovascular disease in post menopausal women: A randomized, double-blind, crossover study. Eur. Heart J. 2005, 26, 1362–1368. [Google Scholar] [CrossRef]
- Willeit, J.; Kiechl, S. Biology of arterial atheroma. Cerebrovasc. Dis. 2000, 10, 1–8. [Google Scholar] [CrossRef]
- Liu, Y.X. Plasminogen activator/plasminogen activator inhibitors in ovarian physiology. Front. Biosci. 2004, 9, 3356–3373. [Google Scholar] [CrossRef]
- Schneider, J.G.; Tompkins, C.; Blumenthal, R.S.; Mora, S. The metabolic syndrome in women. Cardiol. Rev. 2006, 14, 286–291. [Google Scholar] [CrossRef]
- Go, V.L.; Nguyen, C.T.; Harris, D.M.; Lee, W.N. Nutrient-gene interaction: Metabolic genotype-phenotype relationship. J. Nutr. 2005, 135, 3016S–3020S. [Google Scholar]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Castrogiovanni, D.; Alzamendi, A.; Ongaro, L.; Giovambattista, A.; Gaillard, R.C.; Spinedi, E. Fructose Rich Diet-Induced High Plasminogen Activator Inhibitor-1 (PAI-1) Production in the Adult Female Rat: Protective Effect of Progesterone. Nutrients 2012, 4, 1137-1150. https://doi.org/10.3390/nu4081137
Castrogiovanni D, Alzamendi A, Ongaro L, Giovambattista A, Gaillard RC, Spinedi E. Fructose Rich Diet-Induced High Plasminogen Activator Inhibitor-1 (PAI-1) Production in the Adult Female Rat: Protective Effect of Progesterone. Nutrients. 2012; 4(8):1137-1150. https://doi.org/10.3390/nu4081137
Chicago/Turabian StyleCastrogiovanni, Daniel, Ana Alzamendi, Luisina Ongaro, Andrés Giovambattista, Rolf C. Gaillard, and Eduardo Spinedi. 2012. "Fructose Rich Diet-Induced High Plasminogen Activator Inhibitor-1 (PAI-1) Production in the Adult Female Rat: Protective Effect of Progesterone" Nutrients 4, no. 8: 1137-1150. https://doi.org/10.3390/nu4081137
APA StyleCastrogiovanni, D., Alzamendi, A., Ongaro, L., Giovambattista, A., Gaillard, R. C., & Spinedi, E. (2012). Fructose Rich Diet-Induced High Plasminogen Activator Inhibitor-1 (PAI-1) Production in the Adult Female Rat: Protective Effect of Progesterone. Nutrients, 4(8), 1137-1150. https://doi.org/10.3390/nu4081137