Vitamin A in Reproduction and Development

Abstract
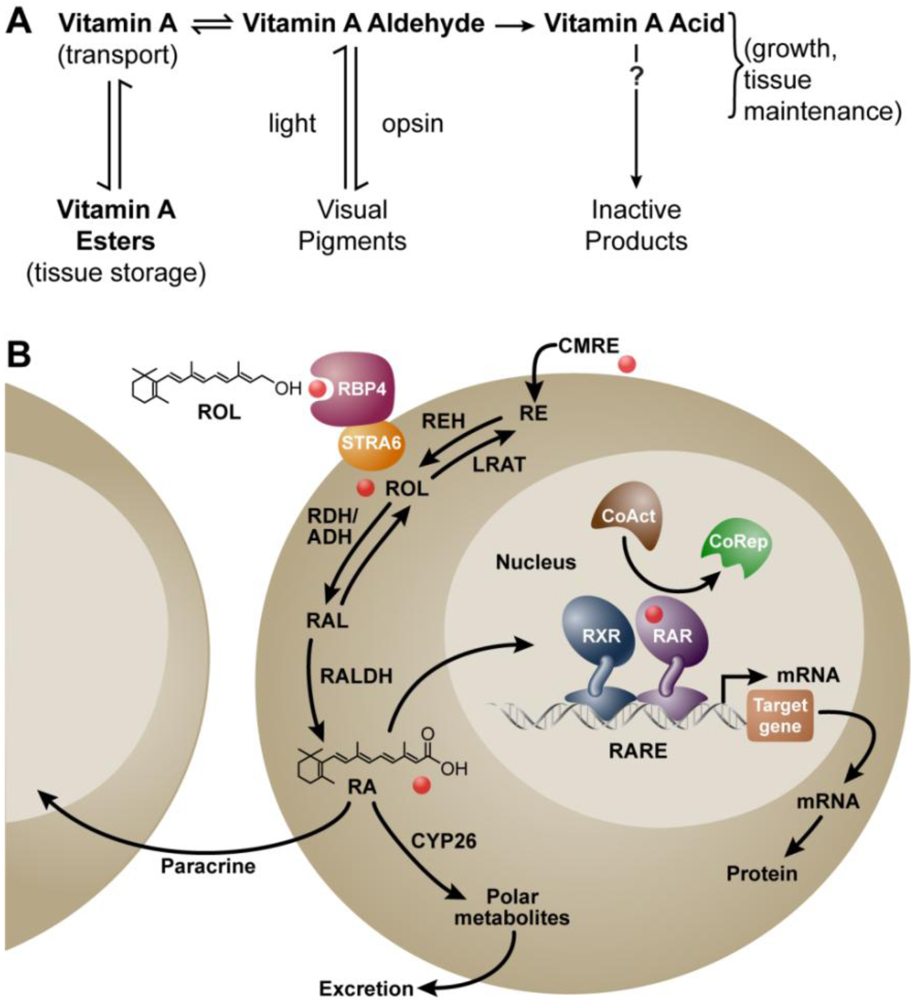
:1. Background
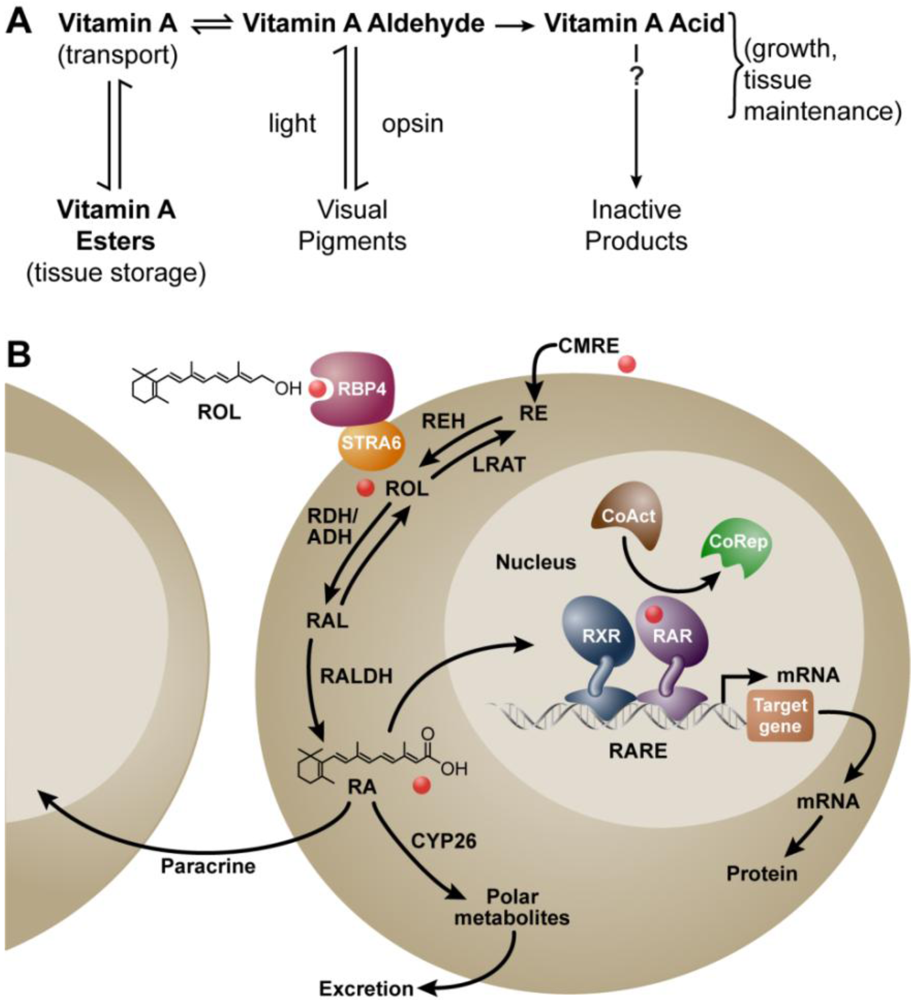
2. Vitamin A and Reproduction
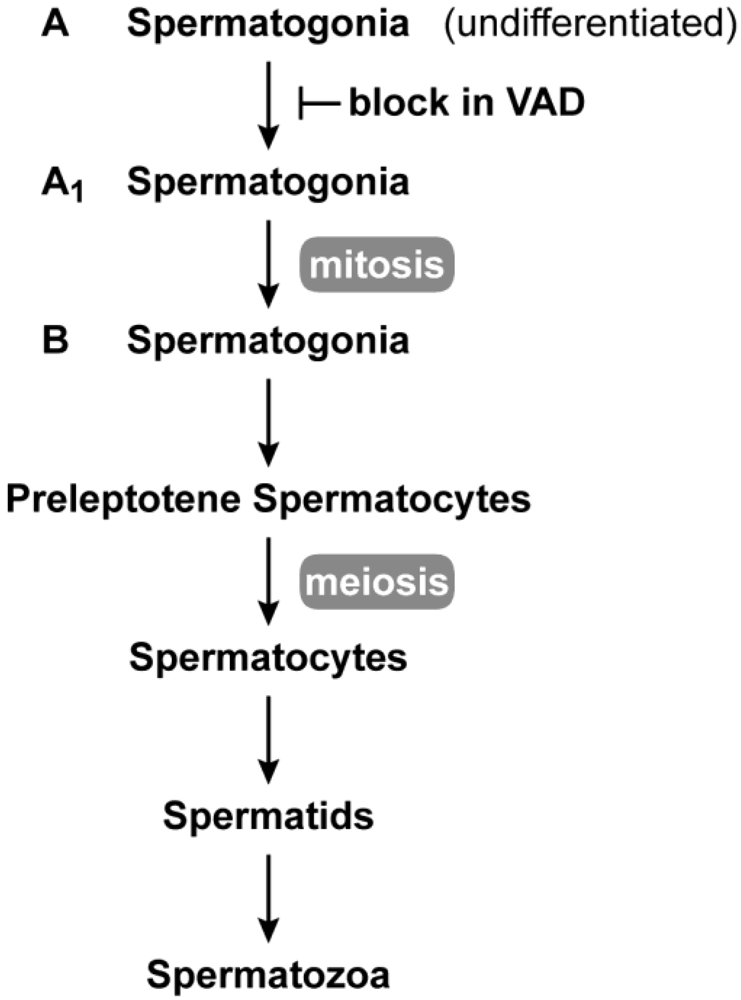
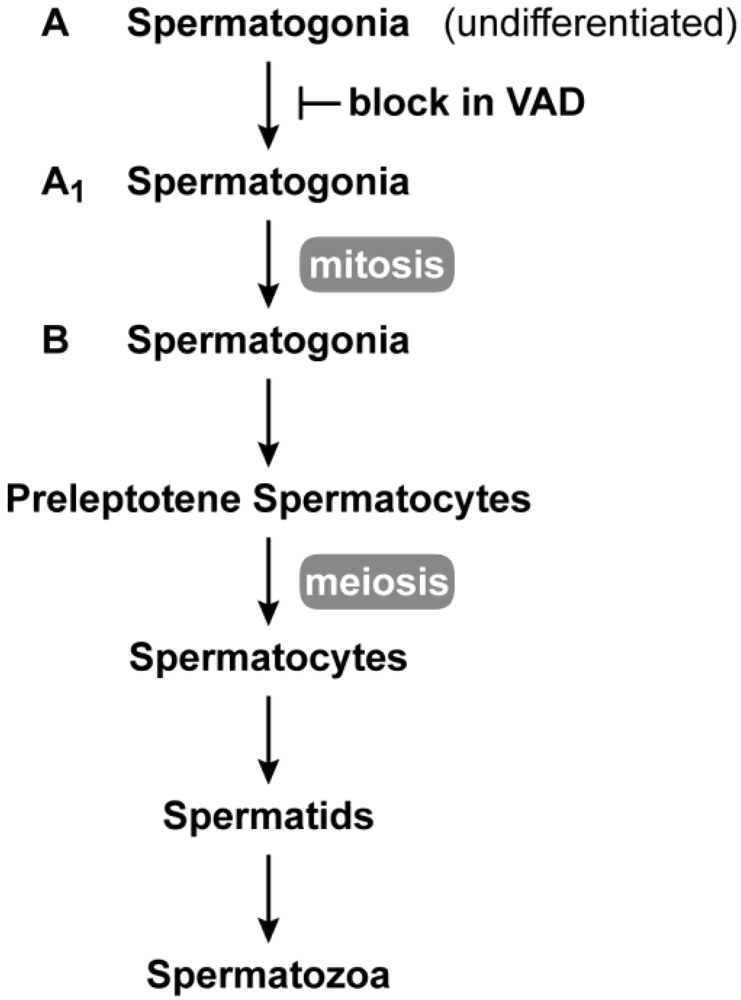
2.1. Male Reproduction
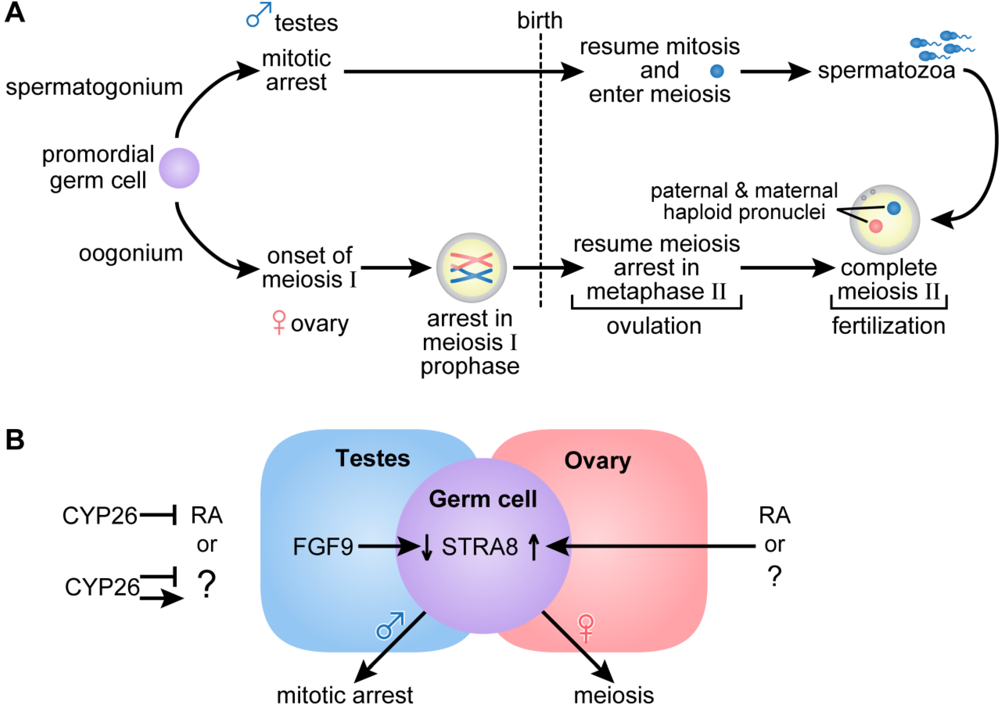
2.2. Female Reproduction
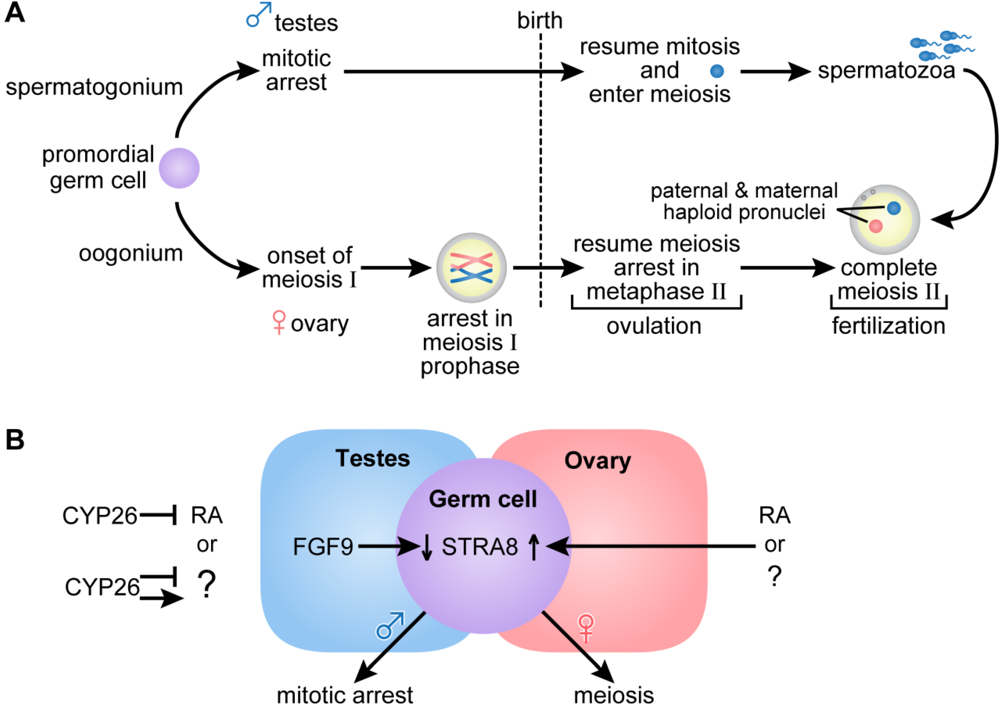
2.3. Germ Cell Development
3. Vitamin A and Embryonic Development
3.1. Embryonic Vitamin A Deficiency Studies

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Late VAD | VAD syndrome | Observed in RAR null mutants | |
|---|---|---|---|
| Ocular | 100% | 49% | Yes |
| Types | |||
| Eversion of retina | 100% | 27% | Yes |
| Fibrous retrolenticular membrane | 100% | 49% | Yes |
| Coloboma | 100% | 18% | Yes |
| Heart-interventricular septal defect | 17% | 4% | Yes |
| Lung-hypoplasia | 100% | 4% | Yes |
| Diaphragmatic hernia | 100% | 31% | Yes |
| Intestinal villi hypoplastic | 83% | Not reported | Not reported |
| Kidney | 100% | 38% | Yes |
| Types | |||
| Kidneys too close or fused | 100% | 20% | Not reported |
| Ectopia | 100% | 4% | Yes |
| Ureter-ectopic termination | 100% | 36% | Yes |
| Undescended testes | 100% | 54% | Not reported |
| Skeletal defects | 100% | Not reported | Yes |
| Glandular defects | 100% | Not reported | Yes |
| Nasal region less developed | 100% | Not reported | Yes |
3.2. Role of the Retinoic Acid Receptors
3.3. Transport of Retinoid from Maternal to Fetal Compartment
3.4. Embryonic RA Synthesis and Catabolism
3.5. RA in the Early Embryo
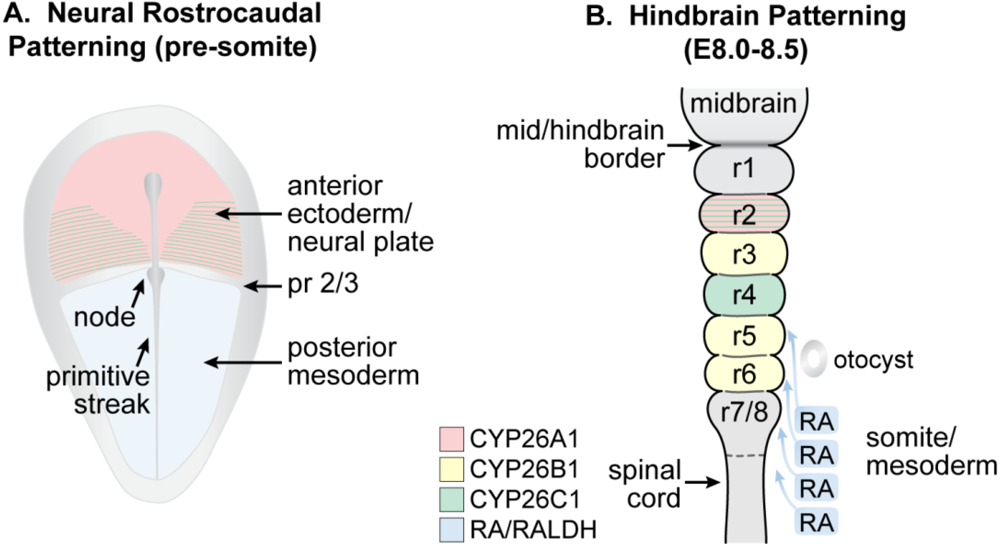
3.6. Early Nervous System Development
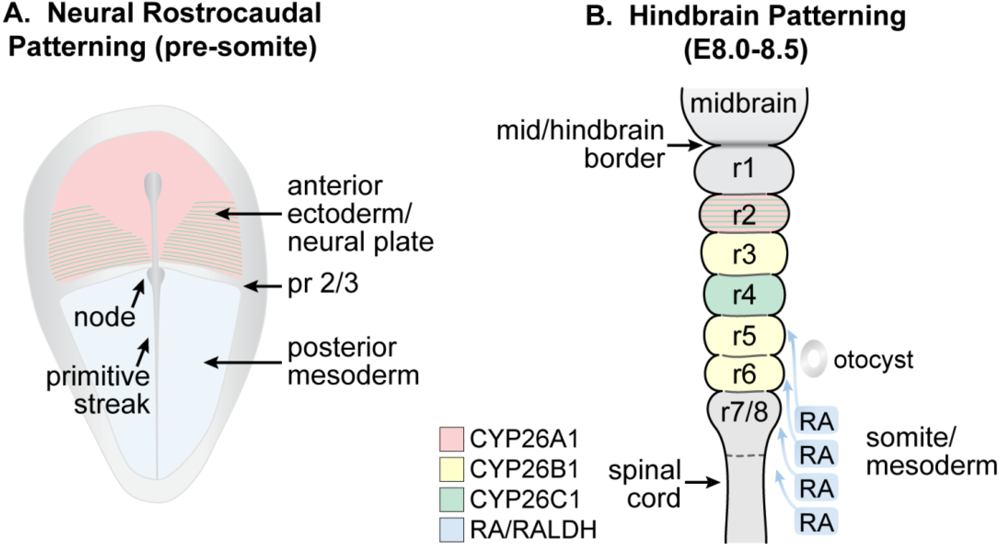
3.7. Spinal Cord and Other Neuronal Development
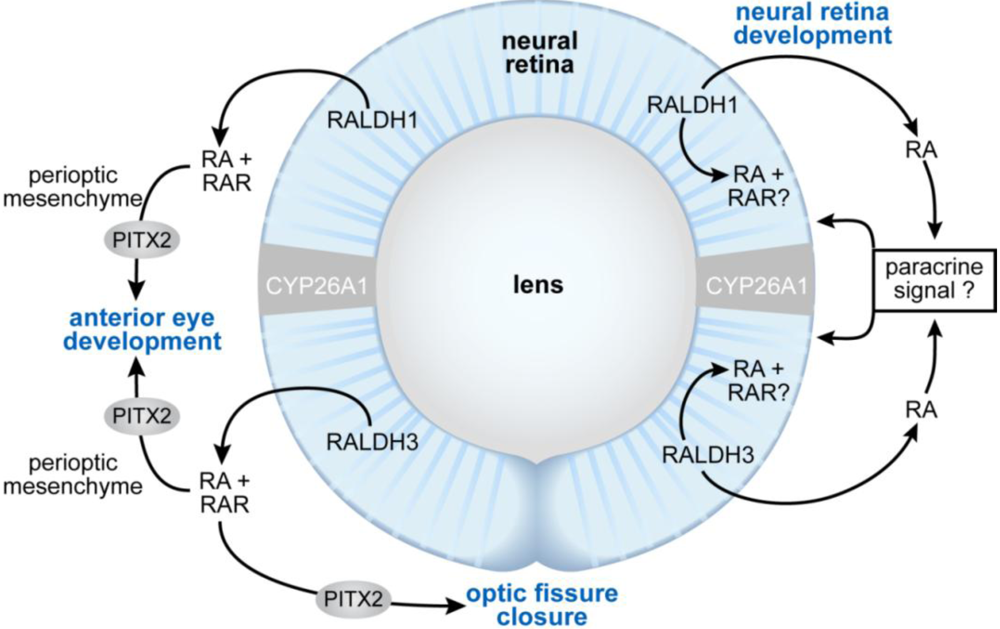
3.8. Eye Development
3.9. Somites and Skeleton
3.10. Heart Development
3.11. Kidney and Urinary Tract Development
3.12. Diaphragm
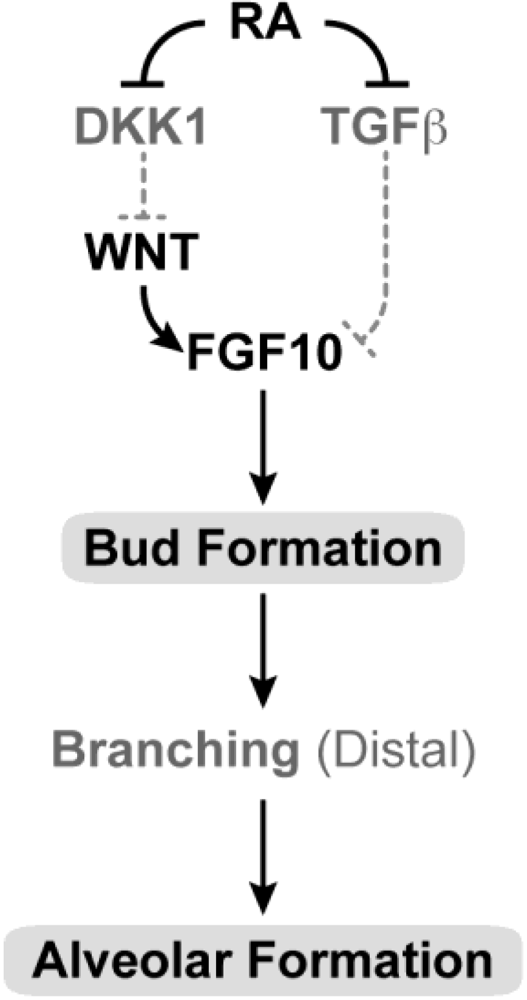
3.13. Lung and Upper Respiratory Tract and Airways

3.14. Pancreas
3.15. Limb Development and Interdigital Cell Death
4. Conclusions
Acknowledgements
References
- McCollum, E.V.; Davis, M. The necessity of certain lipins in the diet during growth. J. Biol. Chem. 1913, 15, 167–175. [Google Scholar]
- Wolbach, S.B.; Howe, P.R. Tissue changes following deprivation of fat-soluble A vitamin. J. Exp. Med. 1925, 42, 753–777. [Google Scholar]
- Evans, H.M. The effects of inadequate vitamin A on the sexual physiology of the female. J. Biol. Chem. 1928, 77, 651–654. [Google Scholar]
- Carpenter, K.J.; Harper, A.E.; Olson, R.E. Experiments That Changed Nutritional Thinking. J. Nutr. 1997, 127, 1017S–1053S. [Google Scholar]
- Moore, T. Vitamin A and carotene: The absence of the liver oil vitamin A from carotene. VI. The conversion of carotene to vitamin A in vivo. Biochem. J. 1930, 24, 692–702. [Google Scholar] [PubMed]
- Karrer, P.; Morf, R.; Schoepp, K. Zur Kenntnis des Vitamins A in Gischtranen. Helv. Chim. Acta 1931, 14, 1431–1436. [Google Scholar]
- Karrer, P. Carotenoids, flavins and vitamin A and B2: Nobel lecture, December 11, 1937. In Nobel Lectures, Chemistry (1922-1941); Elsevier: Amsterdam, The Netherlands, 1966; pp. 433–448. [Google Scholar]
- Arens, J.F.; van Dorp, D.A. Synthesis of some compounds possessing vitamin A activity. Nature 1946, 157, 190–191. [Google Scholar]
- Arens, J.F.; van Dorp, D.A. Activity of vitamin A-acid in the rat. Nature 1946, 158, 622. [Google Scholar]
- van Dorp, D.A.; Arens, J.F. Biological activity of vitamin A acid. Nature 1946, 158, 60. [Google Scholar]
- Dowling, J.E.; Wald, G. The biological function of vitamin A acid. Proc. Natl. Acad. Sci. USA 1960, 46, 587–608. [Google Scholar]
- Moise, A.R.; Noy, N.; Palczewski, K.; Blaner, W.S. Delivery of retinoid-based therapies to target tissues. Biochemistry 2007, 46, 4449–4458. [Google Scholar]
- Pares, X.; Farres, J.; Kedishvili, N.; Duester, G. Medium- and short-chain dehydrogenase/reductase gene and protein families: Medium-chain and short-chain dehydrogenases/reductases in retinoid metabolism. Cell. Mol. Life Sci. 2008, 65, 3936–3949. [Google Scholar]
- Duester, G. Families of retinoid dehydrogenases regulating vitamin A function: Production of visual pigment and retinoic acid. Eur. J. Biochem. 2000, 267, 4315–4324. [Google Scholar]
- Sima, A.; Parisotto, M.; Mader, S.; Bhat, P.V. Kinetic characterization of recombinant mouse retinal dehydrogenase types 3 and 4 for retinal substrates. Biochim. Biophys. Acta 2009, 1790, 1660–1664. [Google Scholar]
- Chambers, D.; Wilson, L.; Maden, M.; Lumsden, A. RALDH-independent generation of retinoic acid during vertebrate embryogenesis by CYP1B1. Development 2007, 134, 1369–1383. [Google Scholar]
- White, J.A.; Beckett-Jones, B.; Guo, Y.D.; Dilworth, F.J.; Bonasoro, J.; Jones, G.; Petkovich, M. cDNA cloning of human retinoic acid-metabolizing enzyme (hP450RAI) identifies a novel family of cytochromes P450. J. Biol. Chem. 1997, 272, 18538–18541. [Google Scholar]
- Fujii, H.; Sato, T.; Kaneko, S.; Gotoh, O.; Fujii-Kuriyama, Y.; Osawa, K.; Kato, S.; Hamada, H. Metabolic inactivation of retinoic acid by a novel P450 differentially expressed in developing mouse embryos. EMBO J. 1997, 16, 4163–4173. [Google Scholar]
- Chithalen, J.V.; Luu, L.; Petkovich, M.; Jones, G. HPLC-MS/MS analysis of the products generated from all-trans-retinoic acid using recombinant human CYP26A. J. Lipid Res. 2002, 43, 1133–1142. [Google Scholar]
- White, J.A.; Ramshaw, H.; Taimi, M.; Stangle, W.; Zhang, A.; Everingham, S.; Creighton, S.; Tam, S.P.; Jones, G.; Petkovich, M. Identification of the human cytochrome P450, P450RAI-2, which is predominantly expressed in the adult cerebellum and is responsible for all-trans-retinoic acid metabolis. Proc. Natl. Acad. Sci. USA 2000, 97, 6403–6408. [Google Scholar]
- Tahayato, A.; Dolle, P.; Petkovich, M. Cyp26C1 encodes a novel retinoic acid-metabolizing enzyme expressed in the hindbrain, inner ear, first branchial arch and tooth buds during murine developmen. Gene Expr. Patterns 2003, 3, 449–454. [Google Scholar]
- Taimi, M.; Helvig, C.; Wisniewski, J.; Ramshaw, H.; White, J.; Amad, M.; Korczak, B.; Petkovich, M. A novel human cytochrome P450, CYP26C1, involved in metabolism of 9-cis and all-trans isomers of retinoic aci. J. Biol. Chem. 2004, 279, 77–85. [Google Scholar]
- White, R.J.; Schilling, T.F. How degrading: Cyp26s in hindbrain development. Dev. Dyn. 2008, 237, 2775–2790. [Google Scholar]
- D’Ambrosio, D.N.; Clugston, R.D.; Blaner, W.S. Vitamin A Metabolism: An Update. Nutrients 2011, 3, 63–103. [Google Scholar]
- Soprano, D.R.; Blaner, W.S. Plasma Retinol-Binding Protein. In The Retinoids: Biology, Chemistry, and Medicine, 2nd; Sporn, M.B., Roberts, A.B., Goodman, D.S., Eds.; Raven Press: New York, NY, USA, 1994; pp. 257–282. [Google Scholar]
- Kawaguchi, R.; Yu, J.; Honda, J.; Hu, J.; Whitelegge, J.; Ping, P.; Wiita, P.; Bok, D.; Sun, H. A membrane receptor for retinol binding protein mediates cellular uptake of vitamin A. Science 2007, 315, 820–825. [Google Scholar]
- Xueping, E.; Zhang, L.; Lu, J.; Tso, P.; Blaner, W.S.; Levin, M.S.; Li, E. Increased neonatal mortality in mice lacking cellular retinol-binding protein II. J. Biol. Chem. 2002, 277, 36617–36623. [Google Scholar]
- Theodosiou, M.; Laudet, V.; Schubert, M. From carrot to clinic: an overview of the retinoic acid signaling pathway. Cell. Mol. Life Sci. 2010, 67, 1423–1445. [Google Scholar]
- Chambon, P. A decade of molecular biology of retinoic acid receptors. FASEB J. 1996, 10, 940–954. [Google Scholar]
- Rochette-Egly, C.; Germain, P. Dynamic and combinatorial control of gene expression by nuclear retinoic acid receptors (RARs). Nucl. Recept. Signal. 2009, 7, e005. [Google Scholar]
- Repa, J.J.; Hanson, K.K.; Clagett-Dame, M. All-trans-retinol is a ligand for the retinoic acid receptors. Proc. Natl. Acad. Sci. USA 1993, 90, 7293–7297. [Google Scholar]
- Mangelsdorf, D.; Umesono, K.; Evans, R.M. The Retinoid Receptors. In The Retinoids: Biology, Chemistry and Medicine, 2nd; Sporn, M.B., Roberts, A.B., Goodman, D.S., Eds.; Raven Press: New York, NY, USA, 1994; pp. 319–350. [Google Scholar]
- Knutson, D.C.; Clagett-Dame, M. atRA Regulation of NEDD9, a gene involved in neurite outgrowth and cell adhesion. Arch. Biochem. Biophys. 2008, 477, 163–174. [Google Scholar]
- Glass, C.K.; Rosenfeld, M.G. The coregulator exchange in transcriptional functions of nuclear receptors. Genes Dev. 2000, 14, 121–141. [Google Scholar]
- Bastien, J.; Rochette-Egly, C. Nuclear retinoid receptors and the transcription of retinoid-target genes. Gene 2004, 328, 1–16. [Google Scholar]
- Balmer, J.E.; Blomhoff, R. Gene expression regulation by retinoic acid. J. Lipid Res. 2002, 43, 1773–1808. [Google Scholar]
- Blomhoff, R.; Blomhoff, H.K. Overview of retinoid metabolism and function. J. Neurobiol. 2006, 66, 606–630. [Google Scholar]
- Mason, K.E. Differences in testis injury and repair after vitamin A-deficiency, vitamin E-deficiency, and inanitio. Am. J. Anat. 1933, 52, 153–239. [Google Scholar]
- Mitranond, V.; Sobhon, P.; Tosukhowong, P.; Chindaduangrat, W. Cytological changes in the testes of vitamin-A-deficient rats. I. Quantitation of germinal cells in the seminiferous tubules. Acta Anat. (Basel) 1979, 103, 159–168. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.F.; Hembree, W.C. Spermatogenic response to vitamin A in vitamin A deficient rats. Biol. Reprod. 1979, 21, 891–904. [Google Scholar]
- Unni, E.; Rao, M.R.; Ganguly, J. Histological & ultrastructural studies on the effect of vitamin A depletion & subsequent repletion with vitamin A on germ cells & Sertoli cells in rat testis. Indian J. Exp. Biol. 1983, 21, 180–192. [Google Scholar]
- van Pelt, A.M.; de Rooij, D.G. Synchronization of the seminiferous epithelium after vitamin A replacement in vitamin A-deficient mice. Biol. Reprod. 1990, 43, 363–367. [Google Scholar]
- Morales, C.; Griswold, M.D. Retinol-induced stage synchronization in seminiferous tubules of the rat. Endocrinology 1987, 121, 432–434. [Google Scholar]
- van Pelt, A.M.; de Rooij, D.G. The origin of the synchronization of the seminiferous epithelium in vitamin A-deficient rats after vitamin A replacement. Biol. Reprod. 1990, 42, 677–682. [Google Scholar]
- Hogarth, C.A.; Griswold, M.D. The key role of vitamin A in spermatogenesis. J. Clin. Invest. 2010, 120, 956–962. [Google Scholar]
- Matson, C.K.; Murphy, M.W.; Griswold, M.D.; Yoshida, S.; Bardwell, V.J.; Zarkower, D. The mammalian doublesex homolog DMRT1 is a transcriptional gatekeeper that controls the mitosisversus meiosis decision in male germ cells. Dev. Cell 2010, 19, 612–624. [Google Scholar]
- Snyder, E.M.; Small, C.; Griswold, M.D. Retinoic acid availability drives the asynchronous initiation of spermatogonial differentiation in the mouse. Biol. Reprod. 2010, 83, 783–790. [Google Scholar]
- van Pelt, A.M.; de Rooij, D.G. Retinoic acid is able to reinitiate spermatogenesis in vitamin A-deficient rats and high replicate doses support the full development of spermatogenic cells. Endocrinology 1991, 128, 697–704. [Google Scholar]
- Vernet, N.; Dennefeld, C.; Rochette-Egly, C.; Oulad-Abdelghani, M.; Chambon, P.; Ghyselinck, N.B.; Mark, M. Retinoic acid metabolism and signaling pathways in the adult and developing mouse testis. Endocrinology 2006, 147, 96–110. [Google Scholar]
- Ghyselinck, N.B.; Vernet, N.; Dennefeld, C.; Giese, N.; Nau, H.; Chambon, P.; Viville, S.; Mark, M. Retinoids and spermatogenesis: lessons from mutant mice lacking the plasma retinol binding protein. Dev. Dyn. 2006, 235, 1608–1622. [Google Scholar]
- Zhai, Y.; Sperkova, Z.; Napoli, J.L. Cellular expression of retinal dehydrogenase types 1 and 2: effects of vitamin A status on testis mRNA. J. Cell. Physiol. 2001, 186, 220–232. [Google Scholar]
- Livera, G.; Rouiller-Fabre, V.; Pairault, C.; Levacher, C.; Habert, R. Regulation and perturbation of testicular functions by vitamin A. Reproduction 2002, 124, 173–180. [Google Scholar]
- Lohnes, D.; Kastner, P.; Dierich, A.; Mark, M.; LeMeur, M.; Chambon, P. Function of retinoic acid receptor gamma in the mouse. Cell 1993, 73, 643–658. [Google Scholar]
- Lufkin, T.; Lohnes, D.; Mark, M.; Dierich, A.; Gorry, P.; Gaub, M.P.; LeMeur, M.; Chambon, P. High postnatal lethality and testis degeneration in retinoic acid receptor alpha mutant mice. Proc. Natl. Acad. Sci. USA 1993, 90, 7225–7229. [Google Scholar]
- Vernet, N.; Dennefeld, C.; Guillou, F.; Chambon, P.; Ghyselinck, N.B.; Mark, M. Prepubertal testis development relies on retinoic acid but not rexinoid receptors in Sertoli cells. EMBO J. 2006, 25, 5816–5825. [Google Scholar]
- Doyle, T.J.; Braun, K.W.; McLean, D.J.; Wright, R.W.; Griswold, M.D.; Kim, K.H. Potential functions of retinoic acid receptor A in Sertoli cells and germ cells during spermatogenesis. Ann. N. Y. Acad. Sci. 2007, 1120, 114–130. [Google Scholar]
- Chung, S.S.; Choi, C.; Wang, X.; Hallock, L.; Wolgemuth, D.J. Aberrant distribution of junctional complex components in retinoic acid receptor alpha-deficient mice. Microsc. Res. Tech. 2010, 73, 583–596. [Google Scholar]
- Clagett-Dame, M.; DeLuca, H.F. The role of vitamin A in mammalian reproduction and embryonic development. Annu. Rev. Nutr. 2002, 22, 347–381. [Google Scholar]
- Evans, H.M.; Bishop, K.S. On an invariable and characteristic disturbance of reproductive function in animals reared on a diet poor in fat soluble vitamine A. Anat. Rec. 1922, 23, 17–18. [Google Scholar]
- Mason, K.E.; Ellison, E.T. Changes in the vaginal epithelium of the rat after vitamin A-deficiency. J. Nutr. 1935, 9, 735–755. [Google Scholar]
- Warkany, J.; Schraffenberger, E. Congenital malformations induced in rats by maternal vitamin A deficiency. I. Defects of the eye. Arch. Ophthalmol. 1946, 35, 150–169. [Google Scholar]
- Thompson, J.N.; Howell, J.M.; Pitt, G.A. Vitamin a and Reproduction in Rats. Proc. R. Soc. Lond. B Biol. Sci. 1964, 159, 510–535. [Google Scholar]
- White, J.C.; Shankar, V.N.; Highland, M.; Epstein, M.L.; DeLuca, H.F.; Clagett-Dame, M. Defects in embryonic hindbrain development and fetal resorption resulting from vitamin A deficiency in the rat are prevented by feeding pharmacological levels of all-trans-retinoic acid. Proc. Natl. Acad. Sci. USA 1998, 95, 13459–13464. [Google Scholar]
- White, J.C.; Highland, M.; Clagett-Dame, M. Abnormal development of the sinuatrial venous valve and posterior hindbrain may contribute to late fetal resorption of vitamin A-deficient rat embryos. Teratology 2000, 62, 374–384. [Google Scholar]
- White, J.C.; Highland, M.; Kaiser, M.; Clagett-Dame, M. Vitamin A deficiency results in the dose-dependent acquisition of anterior character and shortening of the caudal hindbrain of the rat embryo. Dev. Biol. 2000, 220, 263–284. [Google Scholar]
- Kaiser, M.E.; Merrill, R.A.; Stein, A.C.; Breburda, E.; Clagett-Dame, M. Vitamin A deficiency in the late gastrula stage rat embryo results in a one to two vertebral anteriorization that extends throughout the axial skeleton. Dev. Biol. 2003, 257, 14–29. [Google Scholar]
- See, A.W.; Kaiser, M.E.; White, J.C.; Clagett-Dame, M. A nutritional model of late embryonic vitamin A deficiency produces defects in organogenesis at a high penetrance and reveals new roles for the vitamin in skeletal development. Dev. Biol. 2008, 316, 171–190. [Google Scholar]
- Howell, J.M.; Thompson, J.N.; Pitt, G.A. Histology of the Lesions Produced in the Reproductive Tract of Animals Fed a Diet Deficient in Vitamin A Alcohol but Containing Vitamin A Acid. II. The Female Rat. J. Reprod. Fertil. 1964, 7, 251–258. [Google Scholar] [CrossRef] [PubMed]
- Noback, C.R.; Takahashi, Y.I. Micromorphology of the placenta of rats reared on marginal vitamin-A-deficient diet. Acta Anat. (Basel) 1978, 102, 195–202. [Google Scholar] [CrossRef] [PubMed]
- Koubova, J.; Menke, D.B.; Zhou, Q.; Capel, B.; Griswold, M.D.; Page, D.C. Retinoic acid regulates sex-specific timing of meiotic initiation in mice. Proc. Natl. Acad. Sci. USA 2006, 103, 2474–2479. [Google Scholar]
- Bowles, J.; Knight, D.; Smith, C.; Wilhelm, D.; Richman, J.; Mamiya, S.; Yashiro, K.; Chawengsaksophak, K.; Wilson, M.J.; Rossant, J.; Hamada, H.; Koopman, P. Retinoid signaling determines germ cell fate in mice. Science 2006, 312, 596–600. [Google Scholar]
- Bowles, J.; Koopman, P. Retinoic acid, meiosis and germ cell fate in mammals. Development 2007, 134, 3401–3411. [Google Scholar]
- MacLean, G.; Li, H.; Metzger, D.; Chambon, P.; Petkovich, M. Apoptotic extinction of germ cells in testes of Cyp26b1 knockout mice. Endocrinology 2007, 148, 4560–4567. [Google Scholar]
- Kumar, S.; Chatzi, C.; Brade, T.; Cunningham, T.J.; Zhao, X.; Duester, G. Sex-specific timing of meiotic initiation is regulated by Cyp26b1 independent of retinoic acid signalling. Nat. Commun. 2011, 2, 151. [Google Scholar]
- Livera, G.; Rouiller-Fabre, V.; Valla, J.; Habert, R. Effects of retinoids on the meiosis in the fetal rat ovary in culture. Mol. Cell. Endocrinol. 2000, 165, 225–231. [Google Scholar]
- Baltus, A.E.; Menke, D.B.; Hu, Y.C.; Goodheart, M.L.; Carpenter, A.E.; de Rooij, D.G.; Page, D.C. In germ cells of mouse embryonic ovaries, the decision to enter meiosis precedes premeiotic DNA replication. Nat. Genet. 2006, 38, 1430–1434. [Google Scholar]
- Barrios, F.; Filipponi, D.; Pellegrini, M.; Paronetto, M.P.; Di Siena, S.; Geremia, R.; Rossi, P.; De Felici, M.; Jannini, E.A.; Dolci, S. Opposing effects of retinoic acid and FGF9 on Nanos2 expression and meiotic entry of mouse germ cells. J. Cell Sci. 2010, 123, 871–880. [Google Scholar]
- Bowles, J.; Feng, C.W.; Spiller, C.; Davidson, T.L.; Jackson, A.; Koopman, P. FGF9 suppresses meiosis and promotes male germ cell fate in mice. Dev. Cell 2010, 19, 440–449. [Google Scholar]
- Li, H.; Clagett-Dame, M. Vitamin A deficiency blocks the initiation of meiosis of germ cells in the developing rat ovary in vivo. Biol. Reprod. 2009, 81, 996–1001. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Sirbu, I.O.; Mic, F.A.; Molotkova, N.; Molotkov, A.; Kumar, S.; Duester, G. Retinoic acid promotes limb induction through effects on body axis extension but is unnecessary for limb patterning. Curr. Biol. 2009, 19, 1050–1057. [Google Scholar] [PubMed]
- Bowles, J.; Feng, C.W.; Knight, D.; Smith, C.A.; Roeszler, K.N.; Bagheri-Fam, S.; Harley, V.R.; Sinclair, A.H.; Koopman, P. Male-specific expression of Aldh1a1 in mouse and chicken fetal testes: implications for retinoid balance in gonad development. Dev. Dyn. 2009, 238, 2073–2080. [Google Scholar]
- Li, H.; MacLean, G.; Cameron, D.; Clagett-Dame, M.; Petkovich, M. Cyp26b1 expression in murine Sertoli cells is required to maintain male germ cells in an undifferentiated state during embryogenesis. PLoS ONE 2009, 4, e7501. [Google Scholar]
- Li, H.; Palczewski, K.; Baehr, W.; Clagett-Dame, M. Vitamin A deficiency results in meiotic failure and accumulation of undifferentiated spermatogonia in prepubertal mouse testis. Biol. Reprod. 2011, 84, 336–341. [Google Scholar]
- Zhou, Q.; Nie, R.; Li, Y.; Friel, P.; Mitchell, D.; Hess, R.A.; Small, C.; Griswold, M.D. Expression of stimulated by retinoic acid gene 8 (Stra8) in spermatogenic cells induced by retinoic acid: an in vivo study in vitamin A-sufficient postnatal murine testes. Biol. Reprod. 2008, 79, 35–42. [Google Scholar]
- Bellve, A.R.; Millette, C.F.; Bhatnagar, Y.M.; O’Brien, D.A. Dissociation of the mouse testis and characterization of isolated spermatogenic cells. J. Histochem. Cytochem. 1977, 25, 480–494. [Google Scholar]
- Hale, F. Pigs born without eye balls. J. Hered. 1933, 24, 105–106. [Google Scholar]
- Hale, F. The relation of vitamin A to anophthalmos in pigs. Am. J. Ophthalmol. 1935, 18, 1087–1093. [Google Scholar]
- Warkany, J.; Schraffenberger, E. Congenital malformations of the eyes induced in rats by maternal vitamin A deficiency. Proc. Soc. Exp. Biol. Med. 1944, 57, 49–52. [Google Scholar]
- Warkany, J.; Roth, C.B. Congenital malformations induced in rats by maternal vitamin A deficiency. II. Effect of varying the preparatory diet upon the yield of abnormal young. J. Nutr. 1948, 35, 1–11. [Google Scholar]
- Warkany, J.; Roth, C.B.; Wilson, J.G. Multiple congenital malformations: a consideration of etiologic factors. Pediatrics 1948, 1, 462–471. [Google Scholar]
- Wilson, J.G.; Warkany, J. Malformations in the genito-urinary tract induced by maternal vitamin A deficiency in the rat. Am. J. Anat. 1948, 83, 357–407. [Google Scholar]
- Wilson, J.G.; Barch, S. Fetal death and maldevelopment resulting from maternal vitamin A deficiency in the rat. Proc. Soc. Exp. Biol. Med. 1949, 72, 687–693, illust. [Google Scholar] [PubMed]
- Wilson, J.G.; Warkany, J. Aortic-arch and cardiac anomalies in the offspring of vitamin A deficient rats. Am. J. Anat. 1949, 85, 113–155. [Google Scholar]
- Wilson, J.G.; Warkany, J. Cardiac and aortic arch anomalies in the offspring of vitamin A deficient rats correlated with similar human anomalies. Pediatrics 1950, 5, 708–725. [Google Scholar]
- Wilson, J.G.; Roth, C.B.; Warkany, J. An analysis of the syndrome of malformations induced by maternal vitamin A deficiency. Effects of restoration of vitamin A at various times during gestation. Am. J. Anat. 1953, 92, 189–217. [Google Scholar] [CrossRef] [PubMed]
- Dickman, E.D.; Thaller, C.; Smith, S.M. Temporally-regulated retinoic acid depletion produces specific neural crest, ocular and nervous system defects. Development 1997, 124, 3111–3121. [Google Scholar]
- Lohnes, D.; Mark, M.; Mendelsohn, C.; Dolle, P.; Dierich, A.; Gorry, P.; Gansmuller, A.; Chambon, P. Function of the retinoic acid receptors (RARs) during development (I). Craniofacial and skeletal abnormalities in RAR double mutants. Development 1994, 120, 2723–2748. [Google Scholar] [PubMed]
- Mendelsohn, C.; Lohnes, D.; Decimo, D.; Lufkin, T.; LeMeur, M.; Chambon, P.; Mark, M. Function of the retinoic acid receptors (RARs) during development (II). Multiple abnormalities at various stages of organogenesis in RAR double mutants. Development 1994, 120, 2749–2771. [Google Scholar] [PubMed]
- Mark, M.; Ghyselinck, N.B.; Chambon, P. Function of retinoic acid receptors during embryonic development. Nucl. Recept. Signal. 2009, 7, e002. [Google Scholar]
- Dolle, P. Developmental expression of retinoic acid receptors (RARs). Nucl. Recept. Signal. 2009, 7, e006. [Google Scholar]
- Lohnes, D.; Mark, M.; Mendelsohn, C.; Dolle, P.; Decimo, D.; LeMeur, M.; Dierich, A.; Gorry, P.; Chambon, P. Developmental roles of the retinoic acid receptors. J. Steroid Biochem. Mol. Biol. 1995, 53, 475–486. [Google Scholar]
- Mark, M.; Ghyselinck, N.B.; Chambon, P. Function of retinoid nuclear receptors: lessons from genetic and pharmacological dissections of the retinoic acid signaling pathway during mouse embryogenesis. Annu. Rev. Pharmacol. Toxicol. 2006, 46, 451–480. [Google Scholar]
- Warkany, J. Disturbance of embryonic development by maternal vitamin deficiencies. J. Cell. Physiol. 1954, 43, 207–236. [Google Scholar]
- See, A.W.; Clagett-Dame, M. The temporal requirement for vitamin A in the developing eye: mechanism of action in optic fissure closure and new roles for the vitamin in regulating cell proliferation and adhesion in the embryonic retina. Dev. Biol. 2009, 325, 94–105. [Google Scholar]
- Krezel, W.; Dupe, V.; Mark, M.; Dierich, A.; Kastner, P.; Chambon, P. RXR gamma null mice are apparently normal and compound RXR alpha +/−/RXR beta −/−/RXR gamma −/− mutant mice are viable. Proc. Natl. Acad. Sci. USA 1996, 93, 9010–9014. [Google Scholar]
- Mascrez, B.; Ghyselinck, N.B.; Chambon, P.; Mark, M. A transcriptionally silent RXRalpha supports early embryonic morphogenesis and heart development. Proc. Natl. Acad. Sci. USA 2009, 106, 4272–4277. [Google Scholar]
- Mascrez, B.; Mark, M.; Dierich, A.; Ghyselinck, N.B.; Kastner, P.; Chambon, P. The RXRalpha ligand-dependent activation function 2 (AF-2) is important for mouse development. Development 1998, 125, 4691–4707. [Google Scholar]
- Mic, F.A.; Molotkov, A.; Benbrook, D.M.; Duester, G. Retinoid activation of retinoic acid receptor but not retinoid X receptor is sufficient to rescue lethal defect in retinoic acid synthesis. Proc. Natl. Acad. Sci. USA 2003, 100, 7135–7140. [Google Scholar]
- Morriss-Kay, G.M.; Ward, S.J. Retinoids and mammalian development. Int. Rev. Cytol. 1999, 188, 73–131. [Google Scholar]
- Quadro, L.; Hamberger, L.; Gottesman, M.E.; Wang, F.; Colantuoni, V.; Blaner, W.S.; Mendelsohn, C.L. Pathways of vitamin A delivery to the embryo: insights from a new tunable model of embryonic vitamin A deficiency. Endocrinology 2005, 146, 4479–4490. [Google Scholar]
- Soprano, D.R.; Soprano, K.J.; Goodman, D.S. Retinol-binding protein and transthyretin mRNA levels in visceral yolk sac and liver during fetal development in the rat. Proc. Natl. Acad. Sci. USA 1986, 83, 7330–7334. [Google Scholar]
- Sapin, V.; Ward, S.J.; Bronner, S.; Chambon, P.; Dolle, P. Differential expression of transcripts encoding retinoid binding proteins and retinoic acid receptors during placentation of the mouse. Dev. Dyn. 1997, 208, 199–210. [Google Scholar]
- Quadro, L.; Hamberger, L.; Gottesman, M.E.; Colantuoni, V.; Ramakrishnan, R.; Blaner, W.S. Transplacental delivery of retinoid: the role of retinol-binding protein and lipoprotein retinyl ester. Am. J. Physiol. Endocrinol. Metab 2004, 286, E844–E851. [Google Scholar]
- Bavik, C.; Ward, S.J.; Chambon, P. Developmental abnormalities in cultured mouse embryos deprived of retinoic by inhibition of yolk-sac retinol binding protein synthesis. Proc. Natl. Acad. Sci. USA 1996, 93, 3110–3114. [Google Scholar]
- Farese, R.V., Jr.; Cases, S.; Ruland, S.L.; Kayden, H.J.; Wong, J.S.; Young, S.G.; Hamilton, R.L. A novel function for apolipoprotein B: lipoprotein synthesis in the yolk sac is critical for maternal-fetal lipid transport in mice. J. Lipid Res. 1996, 37, 347–360. [Google Scholar]
- Sapin, V.; Chaib, S.; Blanchon, L.; Alexandre-Gouabau, M.C.; Lemery, D.; Charbonne, F.; Gallot, D.; Jacquetin, B.; Dastugue, B.; Azais-Braesco, V. Esterification of vitamin A by the human placenta involves villous mesenchymal fibroblasts. Pediatr. Res. 2000, 48, 565–572. [Google Scholar]
- Kochhar, D.M. Teratogenic activity of retinoic acid. Acta Pathol. Microbiol. Scand. 1967, 70, 398–404. [Google Scholar]
- Conlon, R.A. Retinoic acid and pattern formation in vertebrates. Trends Genet. 1995, 11, 314–319. [Google Scholar]
- Shenefelt, R.E. Morphogenesis of malformations in hamsters caused by retinoic acid: relation to dose and stage at treatment. Teratology 1972, 5, 103–118. [Google Scholar]
- Collins, M.D.; Mao, G.E. Teratology of retinoids. Annu. Rev. Pharmacol. Toxicol. 1999, 39, 399–430. [Google Scholar]
- Reijntjes, S.; Blentic, A.; Gale, E.; Maden, M. The control of morphogen signalling: regulation of the synthesis and catabolism of retinoic acid in the developing embryo. Dev. Biol. 2005, 285, 224–237. [Google Scholar]
- Sandell, L.L.; Sanderson, B.W.; Moiseyev, G.; Johnson, T.; Mushegian, A.; Young, K.; Rey, J.P.; Ma, J.X.; Staehling-Hampton, K.; Trainor, P.A. RDH10 is essential for synthesis of embryonic retinoic acid and is required for limb, craniofacial, and organ developmen. Genes Dev. 2007, 21, 1113–1124. [Google Scholar]
- Niederreither, K.; Subbarayan, V.; Dolle, P.; Chambon, P. Embryonic retinoic acid synthesis is essential for early mouse post-implantation development. Nat. Genet. 1999, 21, 444–448. [Google Scholar]
- Reijntjes, S.; Zile, M.H.; Maden, M. The expression of Stra6 and Rdh10 in the avian embryo and their contribution to the generation of retinoid signatures. Int. J. Dev. Biol. 2010, 54, 1267–1275. [Google Scholar]
- Strate, I.; Min, T.H.; Iliev, D.; Pera, E.M. Retinol dehydrogenase 10 is a feedback regulator of retinoic acid signalling during axis formation and patterning of the central nervous system. Development 2009, 136, 461–472. [Google Scholar]
- Ang, H.L.; Deltour, L.; Hayamizu, T.F.; Zgombic-Knight, M.; Duester, G. Retinoic acid synthesis in mouse embryos during gastrulation and craniofacial development linked to class IV alcohol dehydrogenase gene expression. J. Biol. Chem. 1996, 271, 9526–9534. [Google Scholar]
- Ang, H.L.; Deltour, L.; Zgombic-Knight, M.; Wagner, M.A.; Duester, G. Expression patterns of class I and class IV alcohol dehydrogenase genes in developing epithelia suggest a role for alcohol dehydrogenase in local retinoic acid synthesis. Alcohol. Clin. Exp. Res. 1996, 20, 1050–1064. [Google Scholar]
- Molotkov, A.; Deltour, L.; Foglio, M.H.; Cuenca, A.E.; Duester, G. Distinct retinoid metabolic functions for alcohol dehydrogenase genes Adh1 and Adh4 in protection against vitamin A toxicity or deficiency revealed in double null mutant mice. J. Biol. Chem. 2002, 277, 13804–13811. [Google Scholar]
- Molotkov, A.; Fan, X.; Deltour, L.; Foglio, M.H.; Martras, S.; Farres, J.; Pares, X.; Duester, G. Stimulation of retinoic acid production and growth by ubiquitously expressed alcohol dehydrogenase Adh3. Proc. Natl. Acad. Sci. USA 2002, 99, 5337–5342. [Google Scholar]
- Mic, F.A.; Haselbeck, R.J.; Cuenca, A.E.; Duester, G. Novel retinoic acid generating activities in the neural tube and heart identified by conditional rescue of Raldh2 null mutant mice. Development 2002, 129, 2271–2282. [Google Scholar]
- Dupe, V.; Matt, N.; Garnier, J.M.; Chambon, P.; Mark, M.; Ghyselinck, N.B. A newborn lethal defect due to inactivation of retinaldehyde dehydrogenase type 3 is prevented by maternal retinoic acid treatment. Proc. Natl. Acad. Sci. USA 2003, 100, 14036–14041. [Google Scholar]
- Mic, F.A.; Molotkov, A.; Molotkova, N.; Duester, G. Raldh2 expression in optic vesicle generates a retinoic acid signal needed for invagination of retina during optic cup formation. Dev. Dyn. 2004, 231, 270–277. [Google Scholar]
- Fan, X.; Molotkov, A.; Manabe, S.; Donmoyer, C.M.; Deltour, L.; Foglio, M.H.; Cuenca, A.E.; Blaner, W.S.; Lipton, S.A.; Duester, G. Targeted disruption of Aldh1a1 (Raldh1) provides evidence for a complex mechanism of retinoic acid synthesis in the developing retina. Mol. Cell. Biochem. 2003, 23, 4637–4648. [Google Scholar]
- Matt, N.; Dupe, V.; Garnier, J.M.; Dennefeld, C.; Chambon, P.; Mark, M.; Ghyselinck, N.B. Retinoic acid-dependent eye morphogenesis is orchestrated by neural crest cells. Development 2005, 132, 4789–4800. [Google Scholar]
- Molotkov, A.; Molotkova, N.; Duester, G. Retinoic acid guides eye morphogenetic movements via paracrine signaling but is unnecessary for retinal dorsoventral patterning. Development 2006, 133, 1901–1910. [Google Scholar]
- Duester, G. Retinoic acid synthesis and signaling during early organogenesis. Cell 2008, 134, 921–931. [Google Scholar]
- Buters, J.T.; Sakai, S.; Richter, T.; Pineau, T.; Alexander, D.L.; Savas, U.; Doehmer, J.; Ward, J.M.; Jefcoate, C.R.; Gonzalez, F.J. Cytochrome P450 CYP1B1 determines susceptibility to 7,12-dimethylbenz[a]anthracene-induced lymphomas. Proc. Natl. Acad. Sci. USA 1999, 96, 1977–1982. [Google Scholar]
- Feng, L.; Hernandez, R.E.; Waxman, J.S.; Yelon, D.; Moens, C.B. Dhrs3a regulates retinoic acid biosynthesis through a feedback inhibition mechanism. Dev. Biol. 2010, 338, 1–14. [Google Scholar]
- Abu-Abed, S.; Dolle, P.; Metzger, D.; Beckett, B.; Chambon, P.; Petkovich, M. The retinoic acid-metabolizing enzyme, CYP26A1, is essential for normal hindbrain patterning, vertebral identity, and development of posterior structu. Genes Dev. 2001, 15, 226–240. [Google Scholar]
- Sakai, Y.; Meno, C.; Fujii, H.; Nishino, J.; Shiratori, H.; Saijoh, Y.; Rossant, J.; Hamada, H. The retinoic acid-inactivating enzyme CYP26 is essential for establishing an uneven distribution of retinoic acid along the anterio-posterior axis within the mouse embryo. Genes Dev. 2001, 15, 213–225. [Google Scholar]
- Pennimpede, T.; Cameron, D.A.; Maclean, G.A.; Li, H.; Abu-Abed, S.; Petkovich, M. The role of CYP26 enzymes in defining appropriate retinoic acid exposure during embryogenesis. Birth Defects Res. A Clin. Mol. Teratol. 2010, 88, 883–894. [Google Scholar] [CrossRef] [PubMed]
- Yashiro, K.; Zhao, X.; Uehara, M.; Yamashita, K.; Nishijima, M.; Nishino, J.; Saijoh, Y.; Sakai, Y.; Hamada, H. Regulation of retinoic acid distribution is required for proximodistal patterning and outgrowth of the developing mouse limb. Dev. Cell 2004, 6, 411–422. [Google Scholar]
- MacLean, G.; Dolle, P.; Petkovich, M. Genetic disruption of CYP26B1 severely affects development of neural crest derived head structures, but does not compromise hindbrain patterning. Dev. Dyn. 2009, 238, 732–745. [Google Scholar]
- Uehara, M.; Yashiro, K.; Mamiya, S.; Nishino, J.; Chambon, P.; Dolle, P.; Sakai, Y. CYP26A1 and CYP26C1 cooperatively regulate anterior-posterior patterning of the developing brain and the production of migratory cranial neural crest cells in the mouse. Dev. Biol. 2007, 302, 399–411. [Google Scholar]
- Uehara, M.; Yashiro, K.; Takaoka, K.; Yamamoto, M.; Hamada, H. Removal of maternal retinoic acid by embryonic CYP26 is required for correct Nodal expression during early embryonic patterning. Genes Dev. 2009, 23, 1689–1698. [Google Scholar]
- Ulven, S.M.; Gundersen, T.E.; Weedon, M.S.; Landaas, V.O.; Sakhi, A.K.; Fromm, S.H.; Geronimo, B.A.; Moskaug, J.O.; Blomhoff, R. Identification of endogenous retinoids, enzymes, binding proteins, and receptors during early postimplantation development in mouse: important role of retinal dehydrogenase type 2 in synthesis of all-trans-retinoic acid. Dev. Biol. 2000, 220, 379–391. [Google Scholar] [CrossRef] [PubMed]
- Rossant, J.; Zirngibl, R.; Cado, D.; Shago, M.; Giguere, V. Expression of a retinoic acid response element-hsplacZ transgene defines specific domains of transcriptional activity during mouse embryogenesis. Genes Dev. 1991, 5, 1333–1344. [Google Scholar]
- Niederreither, K.; McCaffery, P.; Drager, U.C.; Chambon, P.; Dolle, P. Restricted expression and retinoic acid-induced downregulation of the retinaldehyde dehydrogenase type 2 (RALDH-2) gene during mouse development. Mech. Dev. 1997, 62, 67–78. [Google Scholar]
- Ribes, V.; Le Roux, I.; Rhinn, M.; Schuhbaur, B.; Dolle, P. Early mouse caudal development relies on crosstalk between retinoic acid, Shh and Fgf signalling pathways. Development 2009, 136, 665–676. [Google Scholar]
- Lai, L.; Bohnsack, B.L.; Niederreither, K.; Hirschi, K.K. Retinoic acid regulates endothelial cell proliferation during vasculogenesis. Development 2003, 130, 6465–6474. [Google Scholar]
- Bohnsack, B.L.; Hirschi, K.K. Red light, green light: signals that control endothelial cell proliferation during embryonic vascular development. Cell Cycle 2004, 3, 1506–1511. [Google Scholar]
- Bohnsack, B.L.; Lai, L.; Dolle, P.; Hirschi, K.K. Signaling hierarchy downstream of retinoic acid that independently regulates vascular remodeling and endothelial cell proliferation. Genes Dev. 2004, 18, 1345–1358. [Google Scholar]
- Satre, M.A.; Kochhar, D.M. Elevations in the endogenous levels of the putative morphogen retinoic acid in embryonic mouse limb-buds associated with limb dysmorphogenesis. Dev. Biol. 1989, 133, 529–536. [Google Scholar]
- Horton, C.; Maden, M. Endogenous distribution of retinoids during normal development and teratogenesis in the mouse embryo. Dev. Dyn. 1995, 202, 312–323. [Google Scholar]
- Scott, W.J., Jr.; Walter, R.; Tzimas, G.; Sass, J.O.; Nau, H.; Collins, M.D. Endogenous status of retinoids and their cytosolic binding proteins in limb buds of chick vs. mouse embryos. Dev. Biol. 1994, 165, 397–409. [Google Scholar] [CrossRef] [PubMed]
- Vermot, J.; Fraulob, V.; Dolle, P.; Niederreither, K. Expression of enzymes synthesizing (aldehyde dehydrogenase 1 and reinaldehyde dehydrogenase 2) and metabolizaing (Cyp26) retinoic acid in the mouse female reproductive system. Endocrinology 2000, 141, 3638–3645. [Google Scholar]
- Clagett-Dame, M.; McNeill, E.M.; Muley, P.D. Role of all-trans retinoic acid in neurite outgrowth and axonal elongation. J. Neurobiol. 2006, 66, 739–756. [Google Scholar]
- Maden, M. Retinoic acid in the development, regeneration and maintenance of the nervous system. Nat. Rev. Neurosci. 2007, 8, 755–765. [Google Scholar]
- Glover, J.C.; Renaud, J.S.; Rijli, F.M. Retinoic acid and hindbrain patterning. J. Neurobiol. 2006, 66, 705–725. [Google Scholar]
- Maden, M. Retinoids and spinal cord development. J. Neurobiol. 2006, 66, 726–738. [Google Scholar]
- Gavalas, A.; Krumlauf, R. Retinoid signalling and hindbrain patterning. Curr. Opin. Genet. Dev. 2000, 10, 380–386. [Google Scholar]
- Maden, M.; Gale, E.; Kostetskii, I.; Zile, M. Vitamin A-deficient quail embryos have half a hindbrain and other neural defects. Curr. Biol. 1996, 6, 417–426. [Google Scholar]
- Niederreither, K.; Vermot, J.; Schuhbaur, B.; Chambon, P.; Dolle, P. Retinoic acid synthesis and hindbrain patterning in the mouse embryo. Development 2000, 127, 75–85. [Google Scholar]
- Begemann, G.; Schilling, T.F.; Rauch, G.J.; Geisler, R.; Ingham, P.W. The zebrafish neckless mutation reveals a requirement for raldh2 in mesodermal signals that pattern the hindbrain. Development 2001, 128, 3081–3094. [Google Scholar]
- Grandel, H.; Lun, K.; Rauch, G.J.; Rhinn, M.; Piotrowski, T.; Houart, C.; Sordino, P.; Kuchler, A.M.; Schulte-Merker, S.; Geisler, R.; Holder, N.; Wilson, S.W.; Brand, M. Retinoic acid signalling in the zebrafish embryo is necessary during pre-segmentation stages to pattern the anterior-posterior axis of the CNS and to induce a pectoral fin bud. Development 2002, 129, 2851–2865. [Google Scholar]
- Wendling, O.; Ghyselinck, N.B.; Chambon, P.; Mark, M. Roles of retinoic acid receptors in early embryonic morphogenesis and hindbrain patterning. Development 2001, 128, 2031–2038. [Google Scholar]
- Dupe, V.; Ghyselinck, N.B.; Wendling, O.; Chambon, P.; Mark, M. Key roles of retinoic acid receptors alpha and beta in the patterning of the caudal hindbrain, pharyngeal arches and otocyst in the mouse. Development 1999, 126, 5051–5059. [Google Scholar]
- Linville, A.; Radtke, K.; Waxman, J.S.; Yelon, D.; Schilling, T.F. Combinatorial roles for zebrafish retinoic acid receptors in the hindbrain, limbs and pharyngeal arches. Dev. Biol. 2009, 325, 60–70. [Google Scholar]
- Dupe, V.; Lumsden, A. Hindbrain patterning involves graded responses to retinoic acid signalling. Development 2001, 128, 2199–2208. [Google Scholar]
- Sirbu, I.O.; Duester, G. Retinoic-acid signalling in node ectoderm and posterior neural plate directs left-right patterning of somitic mesoderm. Nat. Cell Biol. 2006, 8, 271–277. [Google Scholar]
- MacLean, G.; Abu-Abed, S.; Dolle, P.; Tahayato, A.; Chambon, P.; Petkovich, M. Cloning of a novel retinoic-acid metabolizing cytochrome P450, Cyp26B1, and comparative expression analysis with Cyp26A1 during early murine developmen. Mech. Dev. 2001, 107, 195–201. [Google Scholar]
- Sirbu, I.O.; Gresh, L.; Barra, J.; Duester, G. Shifting boundaries of retinoic acid activity control hindbrain segmental gene expression. Development 2005, 132, 2611–2622. [Google Scholar]
- Hernandez, R.E.; Putzke, A.P.; Myers, J.P.; Margaretha, L.; Moens, C.B. Cyp26 enzymes generate the retinoic acid response pattern necessary for hindbrain development. Development 2007, 134, 177–187. [Google Scholar]
- Ribes, V.; Fraulob, V.; Petkovich, M.; Dolle, P. The oxidizing enzyme CYP26a1 tightly regulates the availability of retinoic acid in the gastrulating mouse embryo to ensure proper head development and vasculogenesis. Dev. Dyn. 2007, 236, 644–653. [Google Scholar]
- Koide, T.; Downes, M.; Chandraratna, R.A.; Blumberg, B.; Umesono, K. Active repression of RAR signaling is required for head formation. Genes Dev. 2001, 15, 2111–2121. [Google Scholar]
- Schneider, R.A.; Hu, D.; Rubenstein, J.L.; Maden, M.; Helms, J.A. Local retinoid signaling coordinates forebrain and facial morphogenesis by maintaining FGF8 and SHH. Development 2001, 128, 2755–2767. [Google Scholar]
- Halilagic, A.; Zile, M.H.; Studer, M. A novel role for retinoids in patterning the avian forebrain during presomite stages. Development 2003, 130, 2039–2050. [Google Scholar]
- Halilagic, A.; Ribes, V.; Ghyselinck, N.B.; Zile, M.H.; Dolle, P.; Studer, M. Retinoids control anterior and dorsal properties in the developing forebrain. Dev. Biol. 2007, 303, 362–375. [Google Scholar]
- Marklund, M.; Sjodal, M.; Beehler, B.C.; Jessell, T.M.; Edlund, T.; Gunhaga, L. Retinoic acid signalling specifies intermediate character in the developing telencephalon. Development 2004, 131, 4323–4332. [Google Scholar]
- Molotkova, N.; Molotkov, A.; Duester, G. Role of retinoic acid during forebrain development begins late when Raldh3 generates retinoic acid in the ventral subventricular zone. Dev. Biol. 2007, 303, 601–610. [Google Scholar]
- Luo, T.; Wagner, E.; Drager, U.C. Integrating retinoic acid signaling with brain function. Dev. Psychol. 2009, 45, 139–150. [Google Scholar]
- Zhang, J.; Smith, D.; Yamamoto, M.; Ma, L.; McCaffery, P. The meninges is a source of retinoic acid for the late-developing hindbrain. J. Neurosci. 2003, 23, 7610–7620. [Google Scholar]
- Yamamoto, M.; Fujinuma, M.; Hirano, S.; Hayakawa, Y.; Clagett-Dame, M.; Zhang, J.; McCaffery, P. Retinoic acid influences the development of the inferior olivary nucleus in the rodent. Dev. Biol. 2005, 280, 421–433. [Google Scholar]
- Smith, D.; Wagner, E.; Koul, O.; McCaffery, P.; Drager, U.C. Retinoic acid synthesis for the developing telencephalon. Cereb. Cortex 2001, 11, 894–905. [Google Scholar]
- Siegenthaler, J.A.; Ashique, A.M.; Zarbalis, K.; Patterson, K.P.; Hecht, J.H.; Kane, M.A.; Folias, A.E.; Choe, Y.; May, S.R.; Kume, T.; Napoli, J.L.; Peterson, A.S.; Pleasure, S.J. Retinoic acid from the meninges regulates cortical neuron generation. Cell 2009, 139, 597–609. [Google Scholar]
- Wilson, V.; Olivera-Martinez, I.; Storey, K.G. Stem cells, signals and vertebrate body axis extension. Development 2009, 136, 1591–1604. [Google Scholar]
- Molotkova, N.; Molotkov, A.; Sirbu, I.O.; Duester, G. Requirement of mesodermal retinoic acid generated by Raldh2 for posterior neural transformation. Mech. Dev. 2005, 122, 145–155. [Google Scholar]
- Diez del Corral, R.; Olivera-Martinez, I.; Goriely, A.; Gale, E.; Maden, M.; Storey, K. Opposing FGF and retinoid pathways control ventral neural pattern, neuronal differentiation, and segmentation during body axis extensio. Neuron 2003, 40, 65–79. [Google Scholar]
- Diez del Corral, R.; Storey, K.G. Opposing FGF and retinoid pathways: a signalling switch that controls differentiation and patterning onset in the extending vertebrate body axis. Bioessays 2004, 26, 857–869. [Google Scholar]
- Pierani, A.; Brenner-Morton, S.; Chiang, C.; Jessell, T.M. A sonic hedgehog-independent, retinoid-activated pathway of neurogenesis in the ventral spinal cord. Cell 1999, 97, 903–915. [Google Scholar]
- Novitch, B.G.; Wichterle, H.; Jessell, T.M.; Sockanathan, S. A requirement for retinoic acid-mediated transcriptional activation in ventral neural patterning and motor neuron specification. Neuron 2003, 40, 81–95. [Google Scholar]
- Sockanathan, S.; Perlmann, T.; Jessell, T.M. Retinoid receptor signaling in postmitotic motor neurons regulates rostrocaudal positional identity and axonal projection pattern. Neuron 2003, 40, 97–111. [Google Scholar]
- Vermot, J.; Schuhbaur, B.; Le Mouellic, H.; McCaffery, P.; Garnier, J.M.; Hentsch, D.; Brulet, P.; Niederreither, K.; Chambon, P.; Dolle, P.; Le Roux, I. Retinaldehyde dehydrogenase 2 and Hoxc8 are required in the murine brachial spinal cord for the specification of Lim1+ motoneurons and the correct distribution of Islet1+ motoneurons. Development 2005, 132, 1611–1621. [Google Scholar]
- Misra, M.; Shah, V.; Carpenter, E.; McCaffery, P.; Lance-Jones, C. Restricted patterns of Hoxd10 and Hoxd11 set segmental differences in motoneuron subtype complement in the lumbosacral spinal cord. Dev. Biol. 2009, 330, 54–72. [Google Scholar]
- Rodriguez-Tebar, A.; Rohrer, H. Retinoic acid induces NGF-dependent survival response and high-affinity NGF receptors in immature chick sympathetic neurons. Development 1991, 112, 813–820. [Google Scholar]
- Plum, L.A.; Clagett-Dame, M. All-trans retinoic acid stimulates and maintains neurite outgrowth in nerve growth factor-supported developing chick embryonic sympathetic neurons. Dev. Biol. 1996, 205, 52–63. [Google Scholar]
- Plum, L.A.; Parada, L.F.; Tsoulfas, P.; Clagett-Dame, M. Retinoic acid combined with neurotrophin-3 enhances the survival and neurite outgrowth of embryonic sympathetic neurons. Exp. Biol. Med. 2001, 226, 766–775. [Google Scholar]
- Merrill, R.A.; Plum, L.A.; Kaiser, M.E.; Clagett-Dame, M. A mammalian homolog of unc-53 is regulated by all-trans retinoic acid in neuroblastoma cells and embryos. Proc. Natl. Acad. Sci. USA 2002, 99, 3422–3427. [Google Scholar]
- Merrill, R.A.; Ahrens, J.M.; Kaiser, M.E.; Federhart, K.S.; Poon, V.Y.; Clagett-Dame, M. All-trans retinoic acid-responsive genes identified in the human SH-SY5Y neuroblastoma cell line and their regulated expression in the nervous system of early embryos. Biol. Chem. 2004, 385, 605–614. [Google Scholar]
- Merrill, R.A.; See, A.W.; Wertheim, M.L.; Clagett-Dame, M. Crk-associated substrate (Cas) family member, NEDD9, is regulated in human neuroblastoma cells and in the embryonic hindbrain by all-trans retinoic aci. Dev. Dyn. 2004, 231, 564–575. [Google Scholar]
- Muley, P.D.; McNeill, E.M.; Marzinke, M.A.; Knobel, K.M.; Barr, M.M.; Clagett-Dame, M. The atRA-responsive gene neuron navigator 2 functions in neurite outgrowth and axonal elongation. Dev. Neurobiol. 2008, 68, 1441–1453. [Google Scholar]
- McNeill, E.M.; Roos, K.P.; Moechars, D.; Clagett-Dame, M. Nav2 is necessary for cranial nerve development and blood pressure regulation. Neural Dev. 2010, 5, 6. [Google Scholar]
- Wagner, E.; McCaffery, P.; Drager, U.C. Retinoic acid in the formation of the dorsoventral retina and its central projections. Dev. Biol. 2000, 222, 460–470. [Google Scholar]
- Drager, U.C.; Li, H.; Wagner, E.; McCaffery, P. Retinoic acid synthesis and breakdown in the developing mouse retina. Prog. Brain Res. 2001, 131, 579–587. [Google Scholar]
- Mic, F.A.; Molotkov, A.; Fan, X.; Cuenca, A.E.; Duester, G. RALDH3, a retinaldehyde dehydrogenase that generates retinoic acid, is expressed in the ventral retina, otic vesicle and olfactory pit during mouse development. Mech. Dev. 2000, 97, 227–230. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Wagner, E.; McCaffery, P.; Smith, D.; Andreadis, A.; Drager, U.C. A retinoic acid synthesizing enzyme in ventral retina and telencephalon of the embryonic mouse. Mech. Dev. 2000, 95, 283–289. [Google Scholar]
- McCaffery, P.; Tempst, P.; Lara, G.; Drager, U.C. Aldehyde dehydrogenase is a positional marker in the retina. Development 1991, 112, 693–702. [Google Scholar]
- Mori, M.; Ghyselinck, N.B.; Chambon, P.; Mark, M. Systematic immunolocalization of retinoid receptors in developing and adult mouse eyes. Invest. Ophthalmol. Vis. Sci. 2001, 42, 1312–1318. [Google Scholar]
- Ghyselinck, N.B.; Dupe, V.; Dierich, A.; Messaddeq, N.; Garnier, J.M.; Rochette-Egly, C.; Chambon, P.; Mark, M. Role of the retinoic acid receptor beta (RARbeta) during mouse development. Int. J. Dev. Biol. 1997, 41, 425–447. [Google Scholar]
- Kastner, P.; Grondona, J.M.; Mark, M.; Gansmuller, A.; LeMeur, M.; Decimo, D.; Vonesch, J.L.; Dolle, P.; Chambon, P. Genetic analysis of RXR alpha developmental function: convergence of RXR and RAR signaling pathways in heart and eye morphogenesis. Cell 1994, 78, 987–1003. [Google Scholar]
- Kastner, P.; Mark, M.; Ghyselinck, N.; Krezel, W.; Dupe, V.; Grondona, J.M.; Chambon, P. Genetic evidence that the retinoid signal is transduced by heterodimeric RXR/RAR functional units during mouse development. Development 1997, 124, 313–326. [Google Scholar]
- Matt, N.; Ghyselinck, N.B.; Pellerin, I.; Dupe, V. Impairing retinoic acid signalling in the neural crest cells is sufficient to alter entire eye morphogenesis. Dev. Biol. 2008, 320, 140–148. [Google Scholar]
- Cammas, L.; Trensz, F.; Jellali, A.; Ghyselinck, N.B.; Roux, M.J.; Dolle, P. Retinoic acid receptor (RAR)-alpha is not critically required for mediating retinoic acid effects in the developing mouse retina. Invest. Ophthalmol. Vis. Sci. 2010, 51, 3281–3290. [Google Scholar]
- Kumar, S.; Duester, G. Retinoic acid signaling in perioptic mesenchyme represses Wnt signaling via induction of Pitx2 and Dkk2. Dev. Biol. 2010, 340, 67–74. [Google Scholar]
- Gage, P.J.; Suh, H.; Camper, S.A. Dosage requirement of Pitx2 for development of multiple organs. Development 1999, 126, 4643–4651. [Google Scholar]
- Zacharias, A.L.; Gage, P.J. Canonical Wnt/beta-catenin signaling is required for maintenance but not activation of Pitx2 expression in neural crest during eye development. Dev. Dyn. 2010, 239, 3215–3225. [Google Scholar]
- Aulehla, A.; Pourquie, O. Signaling gradients during paraxial mesoderm development. Cold Spring Harb. Perspect. Biol. 2010, 2, a000869. [Google Scholar]
- Dubrulle, J.; McGrew, M.J.; Pourquie, O. FGF signaling controls somite boundary position and regulates segmentation clock control of spatiotemporal Hox gene activation. Cell 2001, 106, 219–232. [Google Scholar]
- Sawada, A.; Shinya, M.; Jiang, Y.J.; Kawakami, A.; Kuroiwa, A.; Takeda, H. Fgf/MAPK signalling is a crucial positional cue in somite boundary formation. Development 2001, 128, 4873–4880. [Google Scholar]
- Tenin, G.; Wright, D.; Ferjentsik, Z.; Bone, R.; McGrew, M.J.; Maroto, M. The chick somitogenesis oscillator is arrested before all paraxial mesoderm is segmented into somites. BMC Dev. Biol. 2010, 10, 24. [Google Scholar]
- Kieny, M.; Mauger, A.; Sengel, P. Early regionalization of somitic mesoderm as studied by the development of axial skeleton of the chick embryo. Dev. Biol. 1972, 28, 142–161. [Google Scholar]
- Gruss, P.; Kessel, M. Axial specification in higher vertebrates. Curr. Opin. Genet. Dev. 1991, 1, 204–210. [Google Scholar]
- Kessel, M. Respecification of vertebral identities by retinoic acid. Development 1992, 115, 487–501. [Google Scholar]
- Kessel, M.; Gruss, P. Homeotic transformations of murine vertebrae and concomitant alteration of Hox codes induced by retinoic acid. Cell 1991, 67, 89–104. [Google Scholar]
- Marshall, H.; Nonchev, S.; Sham, M.H.; Muchamore, I.; Lumsden, A.; Krumlauf, R. Retinoic acid alters hindbrain Hox code and induces transformation of rhombomeres 2/3 into a 4/5 identity. Nature 1992, 360, 737–741. [Google Scholar]
- Hall, B.K.; Horstadius, S. The Neural Crest; Oxford University Press: New York, NY, USA, 1988; pp. 1–303. [Google Scholar]
- Dupe, V.; Pellerin, I. Retinoic acid receptors exhibit cell-autonomous functions in cranial neural crest cells. Dev. Dyn. 2009, 238, 2701–2711. [Google Scholar]
- Dersch, H.; Zile, M.H. Induction of normal cardiovascular development in the vitamin A-deprived quail embryo by natural retinoids. Dev. Biol. 1993, 160, 424–433. [Google Scholar]
- Zile, M.H. Vitamin A-Not for Your Eyes Only: Requirement for Heart Formation Begins Early in Embryogenesis. Nutrients 2010, 2, 532–550. [Google Scholar]
- Niederreither, K.; Vermot, J.; Messaddeq, N.; Schuhbaur, B.; Chambon, P.; Dolle, P. Embryonic retinoic acid synthesis is essential for heart morphogenesis in the mouse. Development 2001, 128, 1019–1031. [Google Scholar]
- Wagner, M.; Siddiqui, M.A. Signal transduction in early heart development (II): ventricular chamber specification, trabeculation, and heart valve formation. Exp. Biol. Med. (Maywood) 2007, 232, 866–880. [Google Scholar] [PubMed]
- Hoover, L.L.; Burton, E.G.; Brooks, B.A.; Kubalak, S.W. The expanding role for retinoid signaling in heart development. Sci. World J. 2008, 8, 194–211. [Google Scholar]
- Lin, S.C.; Dolle, P.; Ryckebusch, L.; Noseda, M.; Zaffran, S.; Schneider, M.D.; Niederreither, K. Endogenous retinoic acid regulates cardiac progenitor differentiation. Proc. Natl. Acad. Sci. USA 2010, 107, 9234–9239. [Google Scholar]
- Rochais, F.; Mesbah, K.; Kelly, R.G. Signaling pathways controlling second heart field development. Circ. Res. 2009, 104, 933–942. [Google Scholar]
- Lelievre-Pegorier, M.; Vilar, J.; Ferrier, M.L.; Moreau, E.; Freund, N.; Gilbert, T.; Merlet-Benichou, C. Mild vitamin A deficiency leads to inborn nephron deficit in the rat. Kidney Int. 1998, 54, 1455–1462. [Google Scholar]
- Mendelsohn, C.; Batourina, E.; Fung, S.; Gilbert, T.; Dodd, J. Stromal cells mediate retinoid-dependent functions essential for renal development. Development 1999, 126, 1139–1148. [Google Scholar]
- Batourina, E.; Gim, S.; Bello, N.; Shy, M.; Clagett-Dame, M.; Srinivas, S.; Costantini, F.; Mendelsohn, C. Vitamin A controls epithelial/mesenchymal interactions through Ret expression. Nat. Genet. 2001, 27, 74–78. [Google Scholar]
- Niederreither, K.; Fraulob, V.; Garnier, J.M.; Chambon, P.; Dolle, P. Differential expression of retinoic acid-synthesizing (RALDH) enzymes during fetal development and organ differentiation in the mouse. Mech. Dev. 2002, 110, 165–171. [Google Scholar]
- Schuchardt, A.; D’Agati, V.; Larsson-Blomberg, L.; Costantini, F.; Pachnis, V. Defects in the kidney and enteric nervous system of mice lacking the tyrosine kinase receptor Ret. Nature 1994, 367, 380–383. [Google Scholar]
- Schuchardt, A.; D’Agati, V.; Pachnis, V.; Costantini, F. Renal agenesis and hypodysplasia in ret-k- mutant mice result from defects in ureteric bud development. Development 1996, 122, 1919–1929. [Google Scholar]
- Batourina, E.; Choi, C.; Paragas, N.; Bello, N.; Hensle, T.; Costantini, F.D.; Schuchardt, A.; Bacallao, R.L.; Mendelsohn, C.L. Distal ureter morphogenesis depends on epithelial cell remodeling mediated by vitamin A and Ret. Nat. Genet. 2002, 32, 109–115. [Google Scholar]
- Rosselot, C.; Spraggon, L.; Chia, I.; Batourina, E.; Riccio, P.; Lu, B.; Niederreither, K.; Dolle, P.; Duester, G.; Chambon, P.; Costantini, F.; Gilbert, T.; Molotkov, A.; Mendelsohn, C. Non-cell-autonomous retinoid signaling is crucial for renal development. Development 2010, 137, 283–292. [Google Scholar]
- Hoy, W.E.; Hughson, M.D.; Bertram, J.F.; Douglas-Denton, R.; Amann, K. Nephron number, hypertension, renal disease, and renal failu. J. Am. Soc. Nephrol. 2005, 16, 2557–2564. [Google Scholar]
- Brenner, B.M.; Mackenzie, H.S. Nephron mass as a risk factor for progression of renal disease. Kidney Int. Suppl. 1997, 63, S124–S127. [Google Scholar]
- Poladia, D.P.; Kish, K.; Kutay, B.; Bauer, J.; Baum, M.; Bates, C.M. Link between reduced nephron number and hypertension: studies in a mutant mouse model. Pediatr. Res. 2006, 59, 489–493. [Google Scholar]
- Keller, G.; Zimmer, G.; Mall, G.; Ritz, E.; Amann, K. Nephron number in patients with primary hypertension. New Engl. J. Med. 2003, 348, 101–108. [Google Scholar]
- Makrakis, J.; Zimanyi, M.A.; Black, M.J. Retinoic acid enhances nephron endowment in rats exposed to maternal protein restriction. Pediatr. Nephrol. 2007, 22, 1861–1867. [Google Scholar]
- Torfs, C.P.; Curry, C.J.; Bateson, T.F.; Honore, L.H. A population-based study of congenital diaphragmatic hernia. Teratology 1992, 46, 555–565. [Google Scholar]
- Beurskens, L.W.; Tibboel, D.; Lindemans, J.; Duvekot, J.J.; Cohen-Overbeek, T.E.; Veenma, D.C.; de Klein, A.; Greer, J.J.; Steegers-Theunissen, R.P. Retinol status of newborn infants is associated with congenital diaphragmatic hernia. Pediatrics 2010, 126, 712–720. [Google Scholar]
- Pasutto, F.; Sticht, H.; Hammersen, G.; Gillessen-Kaesbach, G.; Fitzpatrick, D.R.; Nurnberg, G.; Brasch, F.; Schirmer-Zimmermann, H.; Tolmie, J.L.; Chitayat, D.; et al. Mutations in STRA6 cause a broad spectrum of malformations including anophthalmia, congenital heart defects, diaphragmatic hernia, alveolar capillary dysplasia, lung hypoplasia, and mental retarda. Am. J. Hum. Genet. 2007, 80, 550–560. [Google Scholar]
- Goumy, C.; Gouas, L.; Marceau, G.; Coste, K.; Veronese, L.; Gallot, D.; Sapin, V.; Vago, P.; Tchirkov, A. Retinoid pathway and congenital diaphragmatic hernia: hypothesis from the analysis of chromosomal abnormalities. Fetal Diagn. Ther. 2010, 28, 129–139. [Google Scholar]
- Clugston, R.D.; Klattig, J.; Englert, C.; Clagett-Dame, M.; Martinovic, J.; Benachi, A.; Greer, J.J. Teratogen-induced, dietary and genetic models of congenital diaphragmatic hernia share a common mechanism of pathogenesis. Am. J. Pathol. 2006, 169, 1541–1549. [Google Scholar]
- Mey, J.; Babiuk, R.P.; Clugston, R.; Zhang, W.; Greer, J.J. Retinal dehydrogenase-2 is inhibited by compounds that induce congenital diaphragmatic hernias in rodents. Am. J. Pathol. 2003, 162, 673–679. [Google Scholar]
- Clugston, R.D.; Zhang, W.; Alvarez, S.; de Lera, A.R.; Greer, J.J. Understanding abnormal retinoid signaling as a causative mechanism in congenital diaphragmatic hernia. Am. J. Respir. Cell Mol. Biol. 2010, 42, 276–285. [Google Scholar]
- Hind, M.; Corcoran, J.; Maden, M. Temporal/spatial expression of retinoid binding proteins and RAR isoforms in the postnatal lung. Am. J. Physiol. Lung Cell. Mol. Physiol. 2002, 282, L468–L476. [Google Scholar]
- Malpel, S.; Mendelsohn, C.; Cardoso, W.V. Regulation of retinoic acid signaling during lung morphogenesis. Development 2000, 127, 3057–3067. [Google Scholar]
- Mollard, R.; Viville, S.; Ward, S.J.; Decimo, D.; Chambon, P.; Dolle, P. Tissue-specific expression of retinoic acid receptor isoform transcripts in the mouse embryo. Mech. Dev. 2000, 94, 223–232. [Google Scholar]
- Wang, Z.; Dolle, P.; Cardoso, W.V.; Niederreither, K. Retinoic acid regulates morphogenesis and patterning of posterior foregut derivatives. Dev. Biol. 2006, 297, 433–445. [Google Scholar]
- Desai, T.J.; Chen, F.; Lu, J.; Qian, J.; Niederreither, K.; Dolle, P.; Chambon, P.; Cardoso, W.V. Distinct roles for retinoic acid receptors alpha and beta in early lung morphogenesis. Dev. Biol. 2006, 291, 12–24. [Google Scholar]
- Hind, M.; Gilthorpe, A.; Stinchcombe, S.; Maden, M. Retinoid induction of alveolar regeneration: from mice to man? Thorax 2009, 64, 451–457. [Google Scholar] [CrossRef] [PubMed]
- Mollard, R.; Ghyselinck, N.B.; Wendling, O.; Chambon, P.; Mark, M. Stage-dependent responses of the developing lung to retinoic acid signaling. Int. J. Dev. Biol. 2000, 44, 457–462. [Google Scholar]
- Chen, F.; Cao, Y.; Qian, J.; Shao, F.; Niederreither, K.; Cardoso, W.V. A retinoic acid-dependent network in the foregut controls formation of the mouse lung primordium. J. Clin. Invest. 2010, 120, 2040–2048. [Google Scholar]
- Wongtrakool, C.; Malpel, S.; Gorenstein, J.; Sedita, J.; Ramirez, M.I.; Underhill, T.M.; Cardoso, W.V. Down-regulation of retinoic acid receptor alpha signaling is required for sacculation and type I cell formation in the developing lung. J. Biol. Chem. 2003, 278, 46911–46918. [Google Scholar]
- Massaro, D.; Massaro, G.D. Retinoids, alveolus formation, and alveolar deficiency: clinical implication. Am. J. Respir. Cell Mol. Biol. 2003, 28, 271–274. [Google Scholar]
- Massaro, G.D.; Massaro, D. Retinoic acid treatment partially rescues failed septation in rats and in mice. Am. J. Physiol. Lung Cell. Mol. Physiol. 2000, 278, L955–L960. [Google Scholar]
- Massaro, G.D.; Massaro, D. Postnatal treatment with retinoic acid increases the number of pulmonary alveoli in rats. Am. J. Physiol. 1996, 270, L305–L310. [Google Scholar]
- Massaro, G.D.; Massaro, D.; Chambon, P. Retinoic acid receptor-alpha regulates pulmonary alveolus formation in mice after, but not during, perinatal perio. Am. J. Physiol. Lung Cell. Mol. Physiol. 2003, 284, L431–L433. [Google Scholar]
- Massaro, G.D.; Massaro, D.; Chan, W.Y.; Clerch, L.B.; Ghyselinck, N.; Chambon, P.; Chandraratna, R.A. Retinoic acid receptor-beta: an endogenous inhibitor of the perinatal formation of pulmonary alveoli. Physiol. Genomics 2000, 4, 51–57. [Google Scholar]
- McGowan, S.; Jackson, S.K.; Jenkins-Moore, M.; Dai, H.H.; Chambon, P.; Snyder, J.M. Mice bearing deletions of retinoic acid receptors demonstrate reduced lung elastin and alveolar numbers. Am. J. Respir. Cell Mol. Biol. 2000, 23, 162–167. [Google Scholar]
- Checkley, W.; West, K.P., Jr.; Wise, R.A.; Baldwin, M.R.; Wu, L.; LeClerq, S.C.; Christian, P.; Katz, J.; Tielsch, J.M.; Khatry, S.; Sommer, A. Maternal vitamin A supplementation and lung function in offspring. New Engl. J. Med. 2010, 362, 1784–1794. [Google Scholar]
- Martin, M.; Gallego-Llamas, J.; Ribes, V.; Kedinger, M.; Niederreither, K.; Chambon, P.; Dolle, P.; Gradwohl, G. Dorsal pancreas agenesis in retinoic acid-deficient Raldh2 mutant mice. Dev. Biol. 2005, 284, 399–411. [Google Scholar]
- Molotkov, A.; Molotkova, N.; Duester, G. Retinoic acid generated by Raldh2 in mesoderm is required for mouse dorsal endodermal pancreas development. Dev. Dyn. 2005, 232, 950–957. [Google Scholar]
- Stafford, D.; Hornbruch, A.; Mueller, P.R.; Prince, V.E. A conserved role for retinoid signaling in vertebrate pancreas development. Dev. Genes Evol. 2004, 214, 432–441. [Google Scholar]
- Stafford, D.; Prince, V.E. Retinoic acid signaling is required for a critical early step in zebrafish pancreatic development. Curr. Biol. 2002, 12, 1215–1220. [Google Scholar]
- Chen, Y.; Pan, F.C.; Brandes, N.; Afelik, S.; Solter, M.; Pieler, T. Retinoic acid signaling is essential for pancreas development and promotes endocrine at the expense of exocrine cell differentiation in Xenopus. Dev. Biol. 2004, 271, 144–160. [Google Scholar]
- Alexa, K.; Choe, S.K.; Hirsch, N.; Etheridge, L.; Laver, E.; Sagerstrom, C.G. Maternal and zygotic aldh1a2 activity is required for pancreas development in zebrafish. PLoS ONE 2009, 4, e8261. [Google Scholar]
- Pan, F.C.; Chen, Y.; Bayha, E.; Pieler, T. Retinoic acid-mediated patterning of the pre-pancreatic endoderm in Xenopus operates via direct and indirect mechanisms. Mech. Dev. 2007, 124, 518–531. [Google Scholar]
- Tulachan, S.S.; Doi, R.; Kawaguchi, Y.; Tsuji, S.; Nakajima, S.; Masui, T.; Koizumi, M.; Toyoda, E.; Mori, T.; Ito, D.; Kami, K.; Fujimoto, K.; Imamura, M. All-trans retinoic acid induces differentiation of ducts and endocrine cells by mesenchymal/epithelial interactions in embryonic pancreas. Diabetes 2003, 52, 76–84. [Google Scholar]
- Ahlgren, U.; Jonsson, J.; Edlund, H. The morphogenesis of the pancreatic mesenchyme is uncoupled from that of the pancreatic epithelium in IPF1/PDX1-deficient mice. Development 1996, 122, 1409–1416. [Google Scholar]
- Jonsson, J.; Carlsson, L.; Edlund, T.; Edlund, H. Insulin-promoter-factor 1 is required for pancreas development in mice. Nature 1994, 371, 606–609. [Google Scholar]
- Offield, M.F.; Jetton, T.L.; Labosky, P.A.; Ray, M.; Stein, R.W.; Magnuson, M.A.; Hogan, B.L.; Wright, C.V. PDX-1 is required for pancreatic outgrowth and differentiation of the rostral duodenum. Development 1996, 122, 983–995. [Google Scholar]
- Kinkel, M.D.; Sefton, E.M.; Kikuchi, Y.; Mizoguchi, T.; Ward, A.B.; Prince, V.E. Cyp26 enzymes function in endoderm to regulate pancreatic field size. Proc. Natl. Acad. Sci. USA 2009, 106, 7864–7869. [Google Scholar]
- Gittes, G.K. Developmental biology of the pancreas: a comprehensive review. Dev. Biol. 2009, 326, 4–35. [Google Scholar]
- Pearl, E.J.; Bilogan, C.K.; Mukhi, S.; Brown, D.D.; Horb, M.E. Xenopus pancreas development. Dev. Dyn. 2009, 238, 1271–1286. [Google Scholar]
- Stratford, T.; Horton, C.; Maden, M. Retinoic acid is required for the initiation of outgrowth in the chick limb bud. Curr. Biol. 1996, 6, 1124–1133. [Google Scholar]
- Stratford, T.; Logan, C.; Zile, M.; Maden, M. Abnormal anteroposterior and dorsoventral patterning of the limb bud in the absence of retinoids. Mech. Dev. 1999, 81, 115–125. [Google Scholar]
- Niederreither, K.; Vermot, J.; Schuhbaur, B.; Chambon, P.; Dolle, P. Embryonic retinoic acid synthesis is required for forelimb growth and anteroposterior patterning in the mouse. Development 2002, 129, 3563–3574. [Google Scholar]
- Mic, F.A.; Sirbu, I.O.; Duester, G. Retinoic acid synthesis controlled by Raldh2 is required early for limb bud initiation and then later as a proximodistal signal during apical ectodermal ridge formation. J. Biol. Chem. 2004, 279, 26698–26706. [Google Scholar]
- Gibert, Y.; Gajewski, A.; Meyer, A.; Begemann, G. Induction and prepatterning of the zebrafish pectoral fin bud requires axial retinoic acid signaling. Development 2006, 133, 2649–2659. [Google Scholar]
- Mercader, N.; Leonardo, E.; Piedra, M.E.; Martinez, A.C.; Ros, M.A.; Torres, M. Opposing RA and FGF signals control proximodistal vertebrate limb development through regulation of Meis genes. Development 2000, 127, 3961–3970. [Google Scholar]
- Benazet, J.D.; Zeller, R. Vertebrate limb development: moving from classical morphogen gradients to an integrated 4-dimensional patterning system. Cold Spring Harb. Perspect. Biol. 2009, 1, a001339. [Google Scholar]
- Zeller, R.; Lopez-Rios, J.; Zuniga, A. Vertebrate limb bud development: moving towards integrative analysis of organogenesis. Nat. Rev. Genet. 2009, 10, 845–858. [Google Scholar]
- Lewandoski, M.; Mackem, S. Limb development: the rise and fall of retinoic acid. Curr. Biol. 2009, 19, R558–R561. [Google Scholar]
- Dupe, V.; Ghyselinck, N.B.; Thomazy, V.; Nagy, L.; Davies, P.J.; Chambon, P.; Mark, M. Essential roles of retinoic acid signaling in interdigital apoptosis and control of BMP-7 expression in mouse autopods. Dev. Biol. 1999, 208, 30–43. [Google Scholar]
- Zhao, X.; Brade, T.; Cunningham, T.J.; Duester, G. Retinoic acid controls expression of tissue remodeling genes Hmgn1 and Fgf18 at the digit-interdigit junction. Dev. Dyn. 2010, 239, 665–671. [Google Scholar]
- Rodriguez-Leon, J.; Merino, R.; Macias, D.; Ganan, Y.; Santesteban, E.; Hurle, J.M. Retinoic acid regulates programmed cell death through BMP signalling. Nat. Cell Biol. 1999, 1, 125–126. [Google Scholar]
- Hernandez-Martinez, R.; Castro-Obregon, S.; Covarrubias, L. Progressive interdigital cell death: regulation by the antagonistic interaction between fibroblast growth factor 8 and retinoic acid. Development 2009, 136, 3669–3678. [Google Scholar]
© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Clagett-Dame, M.; Knutson, D. Vitamin A in Reproduction and Development. Nutrients 2011, 3, 385-428. https://doi.org/10.3390/nu3040385
Clagett-Dame M, Knutson D. Vitamin A in Reproduction and Development. Nutrients. 2011; 3(4):385-428. https://doi.org/10.3390/nu3040385
Chicago/Turabian StyleClagett-Dame, Margaret, and Danielle Knutson. 2011. "Vitamin A in Reproduction and Development" Nutrients 3, no. 4: 385-428. https://doi.org/10.3390/nu3040385
APA StyleClagett-Dame, M., & Knutson, D. (2011). Vitamin A in Reproduction and Development. Nutrients, 3(4), 385-428. https://doi.org/10.3390/nu3040385