Augmentation of Glucotoxicity, Oxidative Stress, Apoptosis and Mitochondrial Dysfunction in HepG2 Cells by Palmitic Acid

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Cell Culture, Treatment and Fractionation
2.3. Measurement of Cell Survival
2.4. Measurement of ROS, LPO, Protein Peroxidation, DNA Damage, and Apoptosis
2.5. Measurement of GSH–Redox Metabolism
2.6. Measurement of Catalase and SOD
2.7. Measurement of Cytochrome P450 Activities
2.8. Measurement of Mitochondrial Functions
2.9. Measurement of Mitochondrial Bioenergetics
2.10. Measurement of Hexokinase and Glucose-6-Phosphate Dehydrogenase (G6PDH) Activities
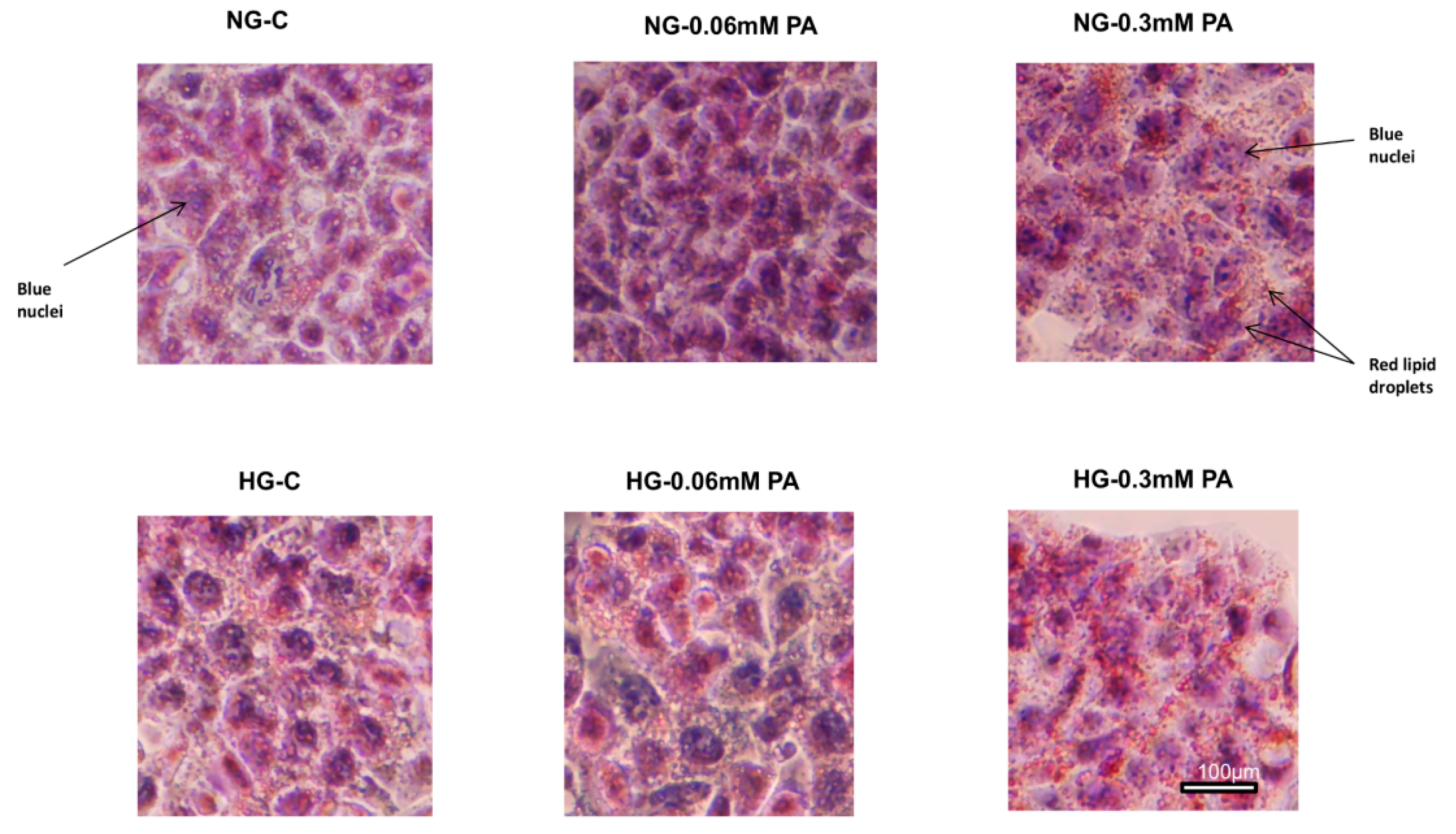
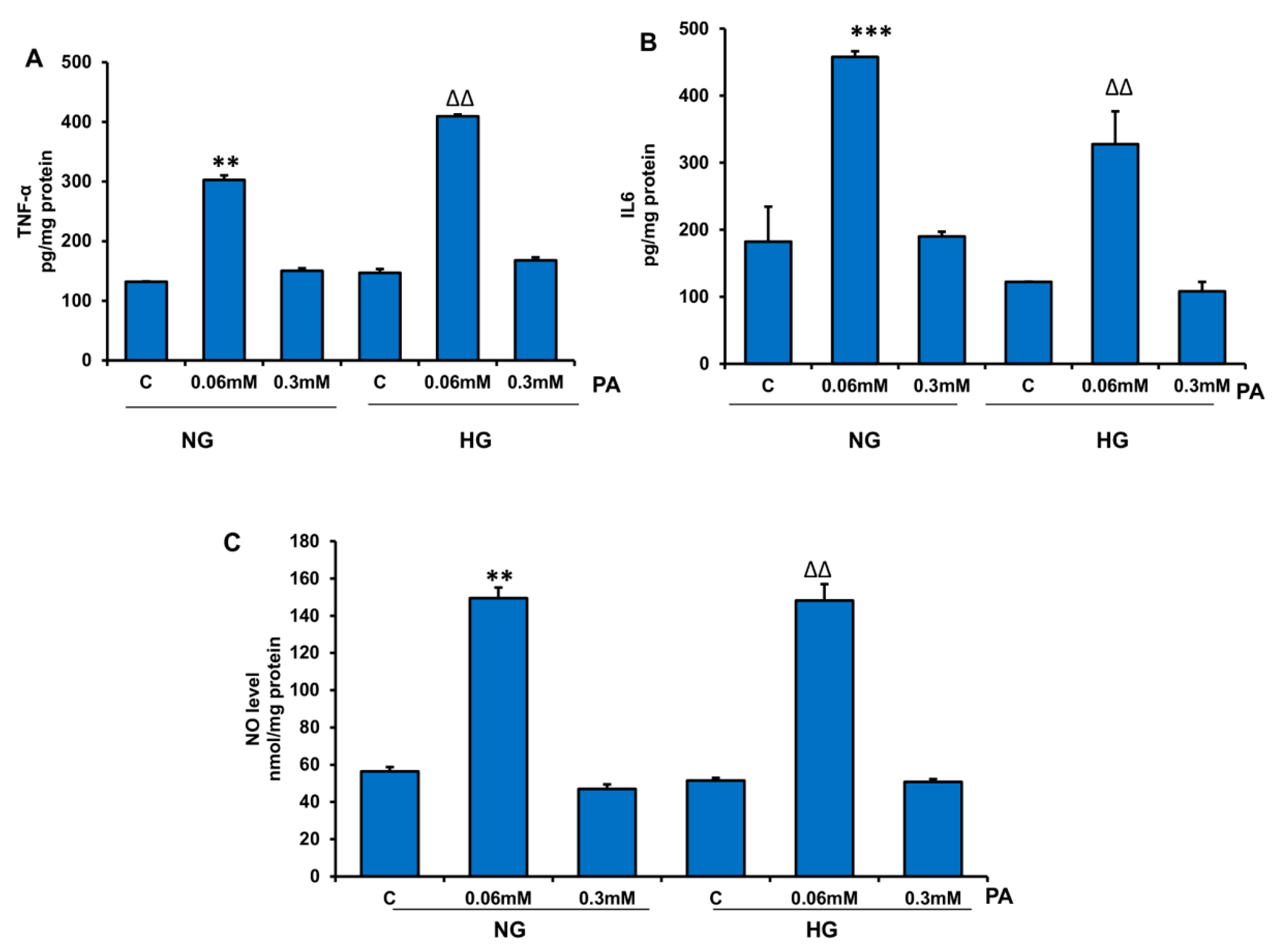
2.11. Measurement of Inflammatory Markers, NO, and Lipid Accumulation
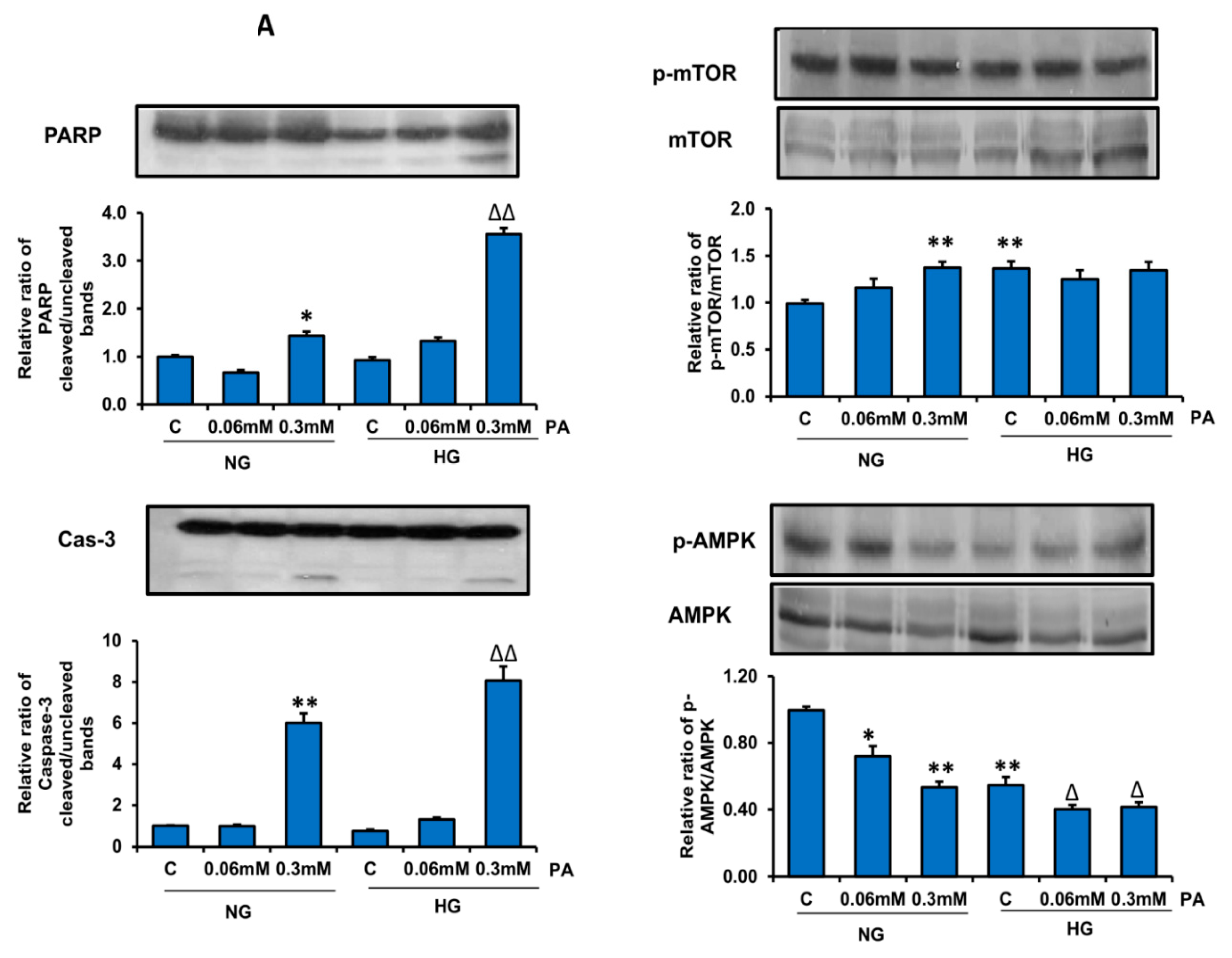
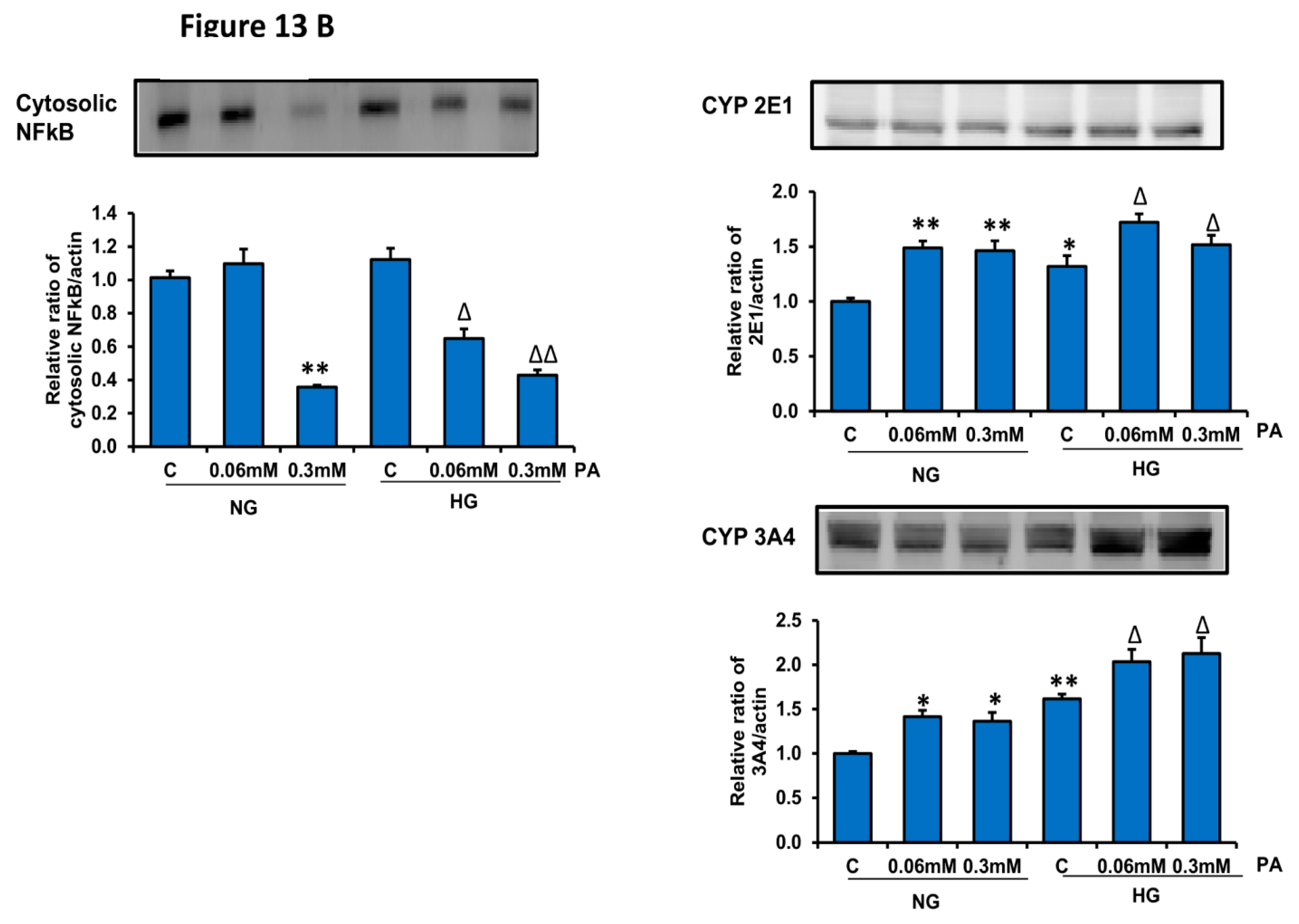
2.12. SDS-PAGE and Western Blot Analysis
2.13. Statistical Analysis
3. Results
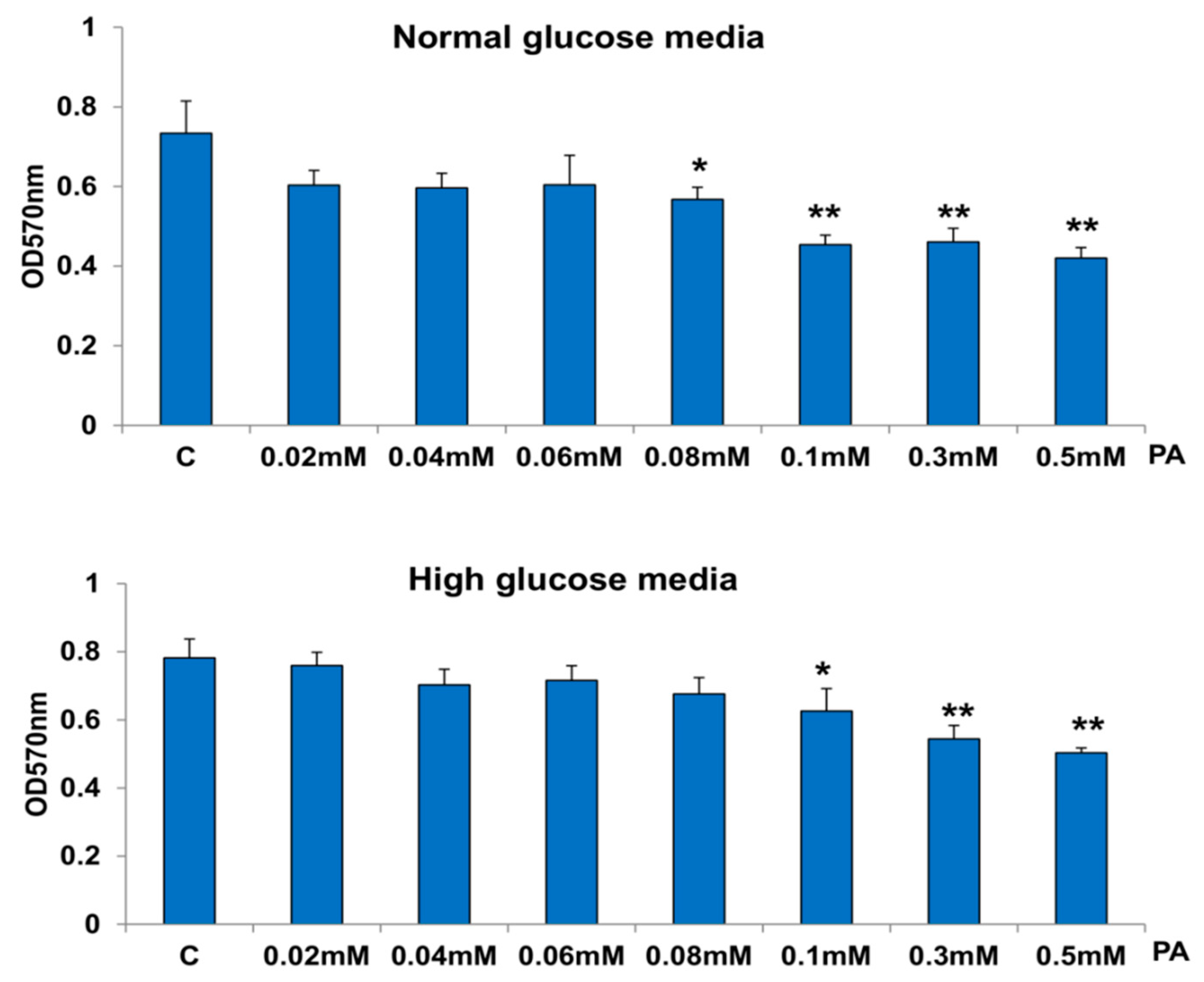
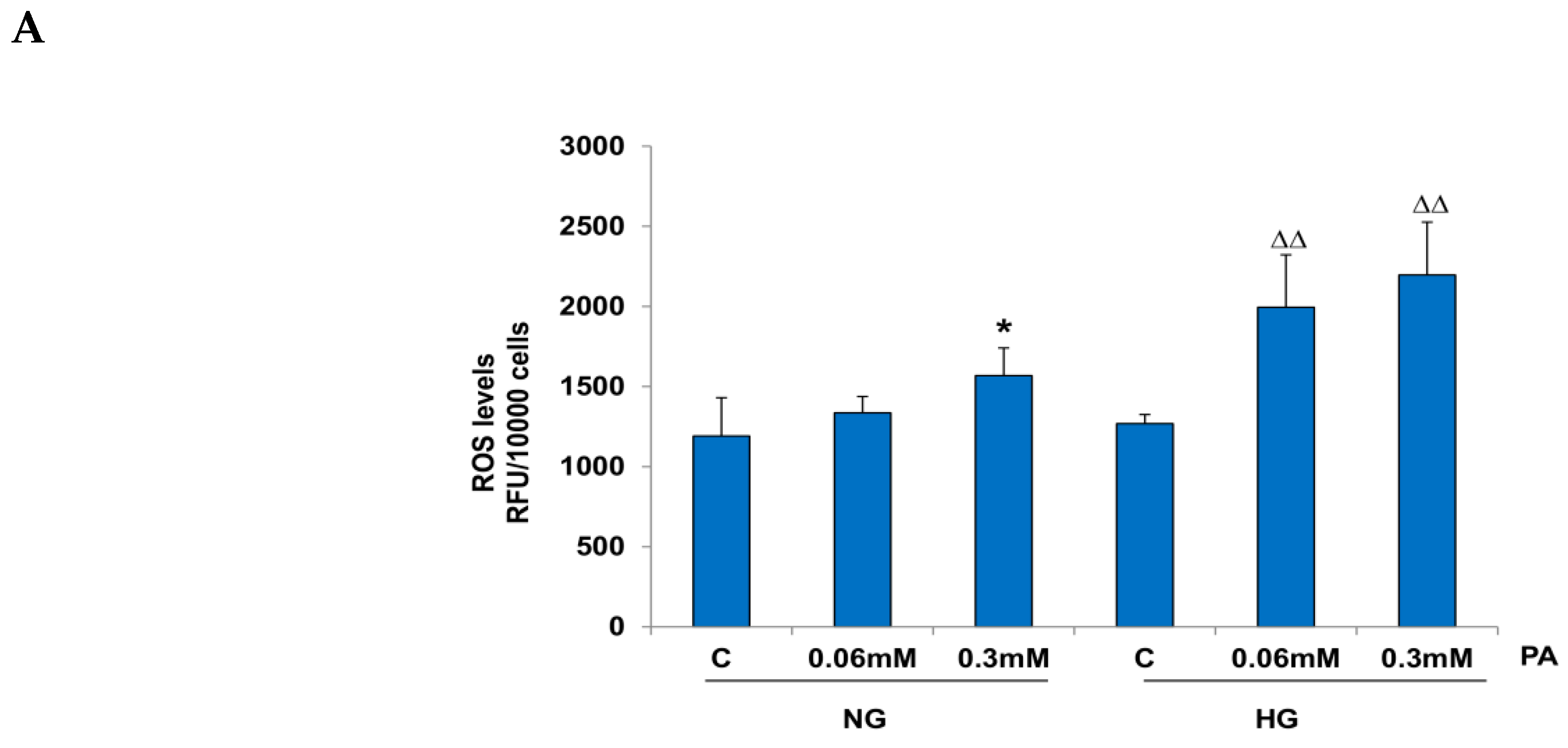
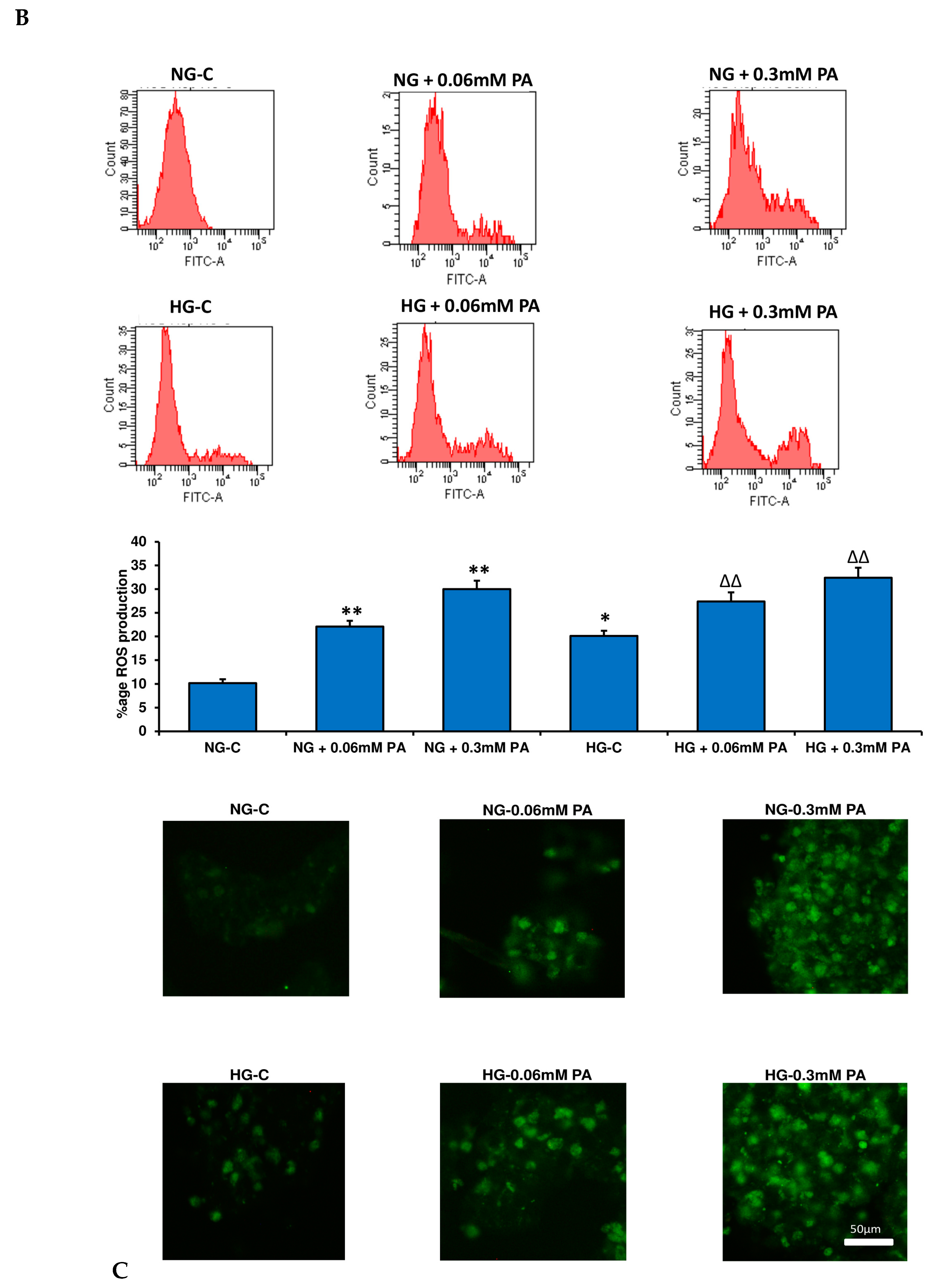
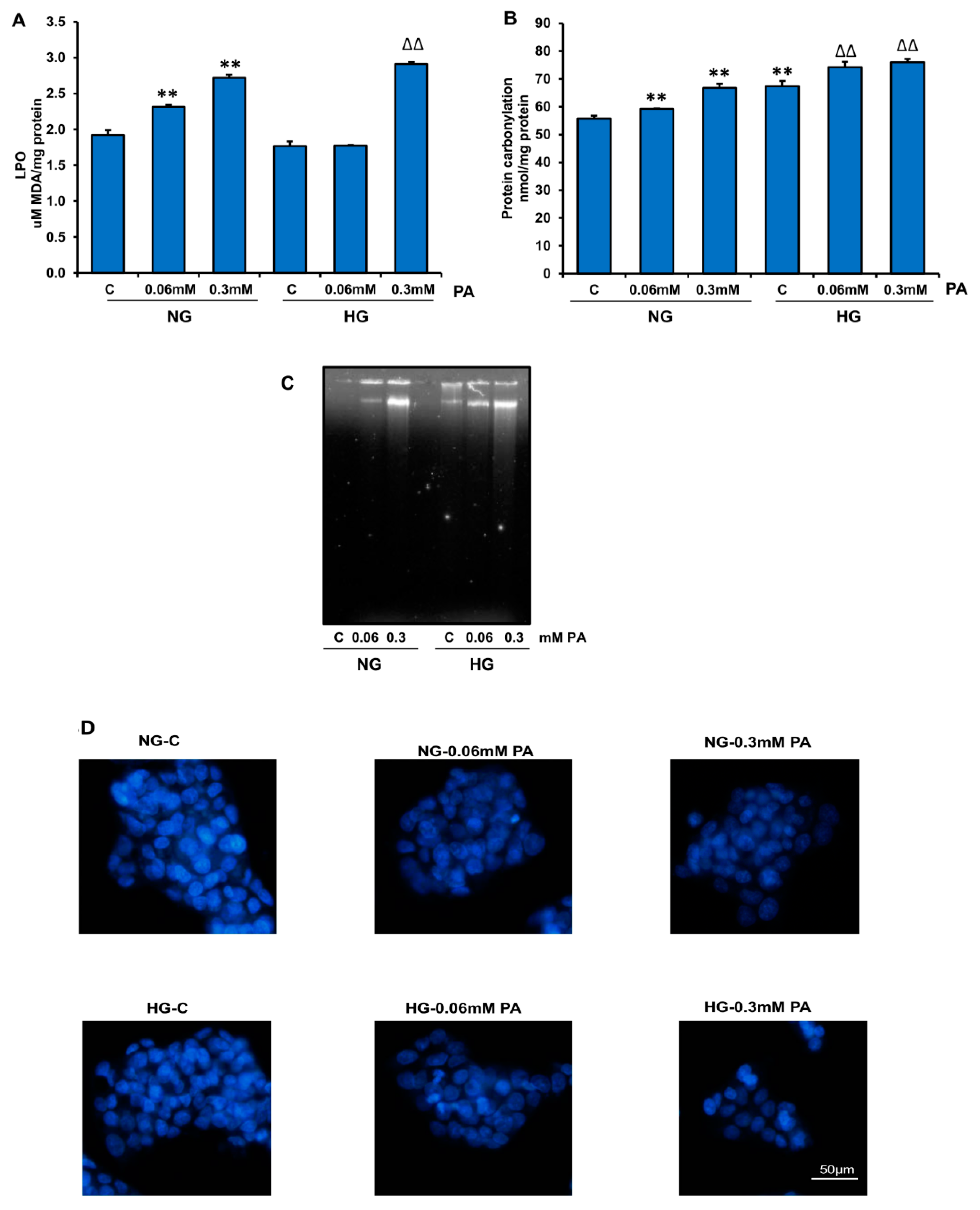
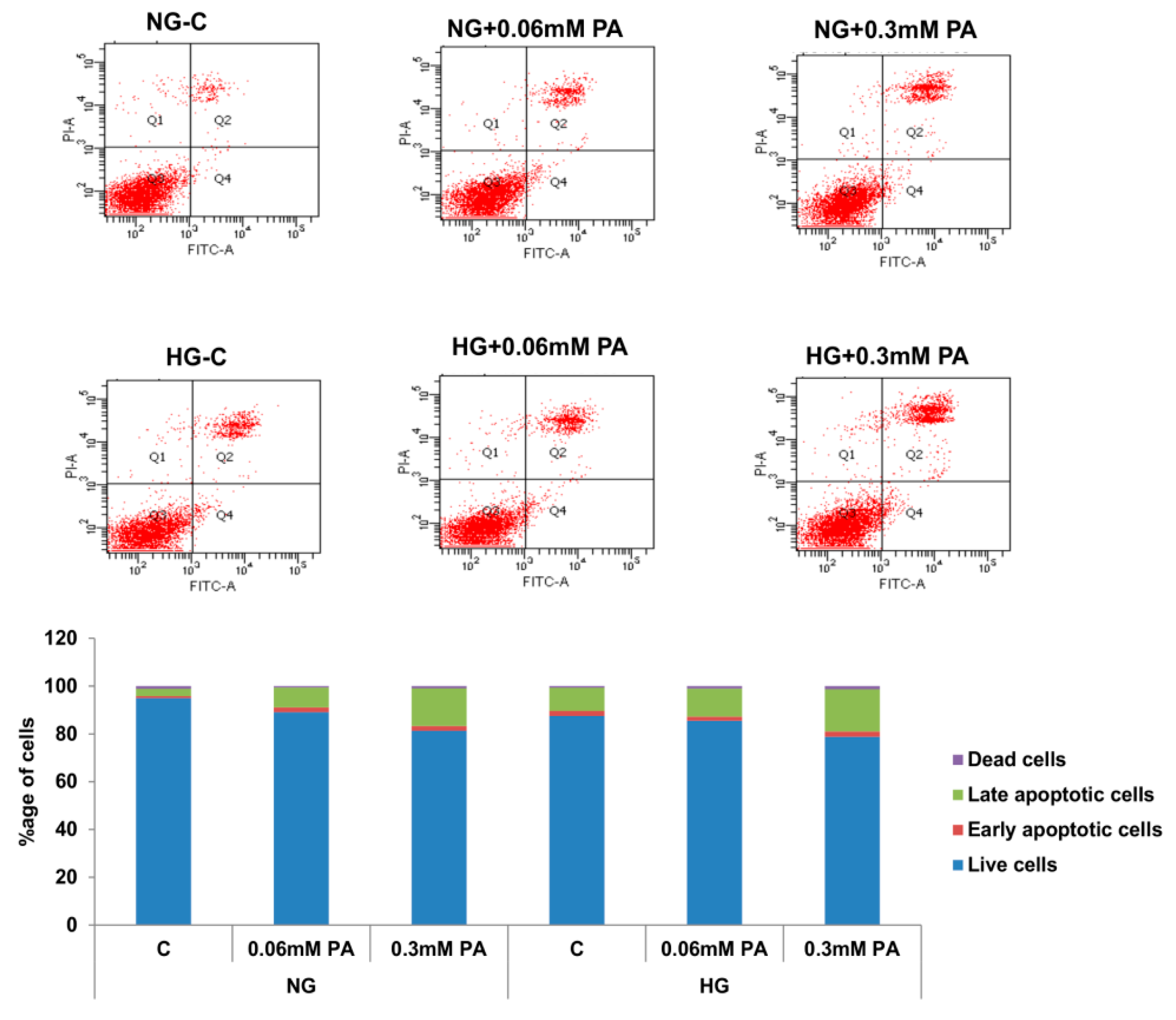
3.1. Effects of HG/HFA on Cell Survival, Oxidative Stress, and Apoptosis
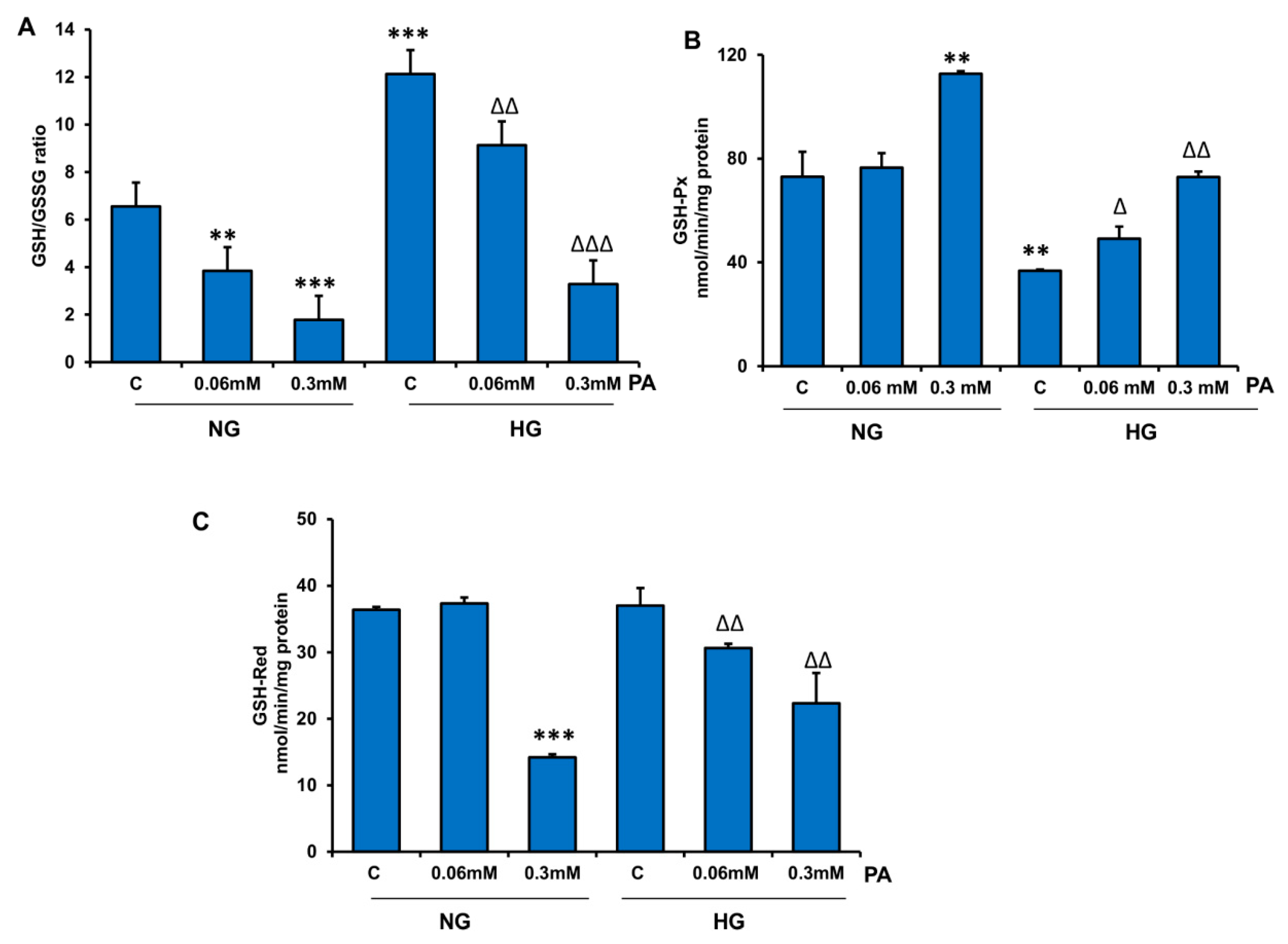
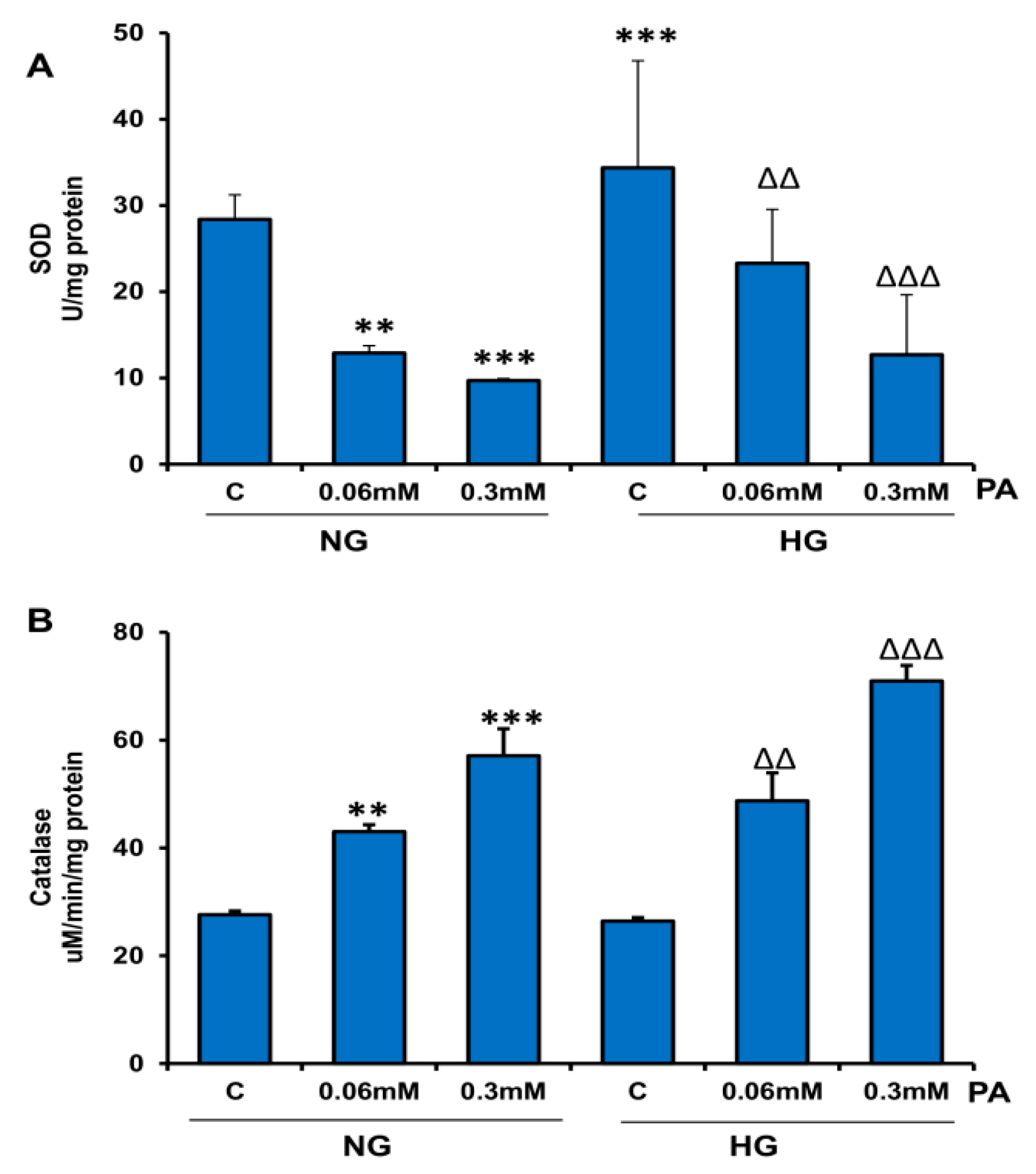
3.2. Effects of HG/HFA on Antioxidant and Redox Metabolism
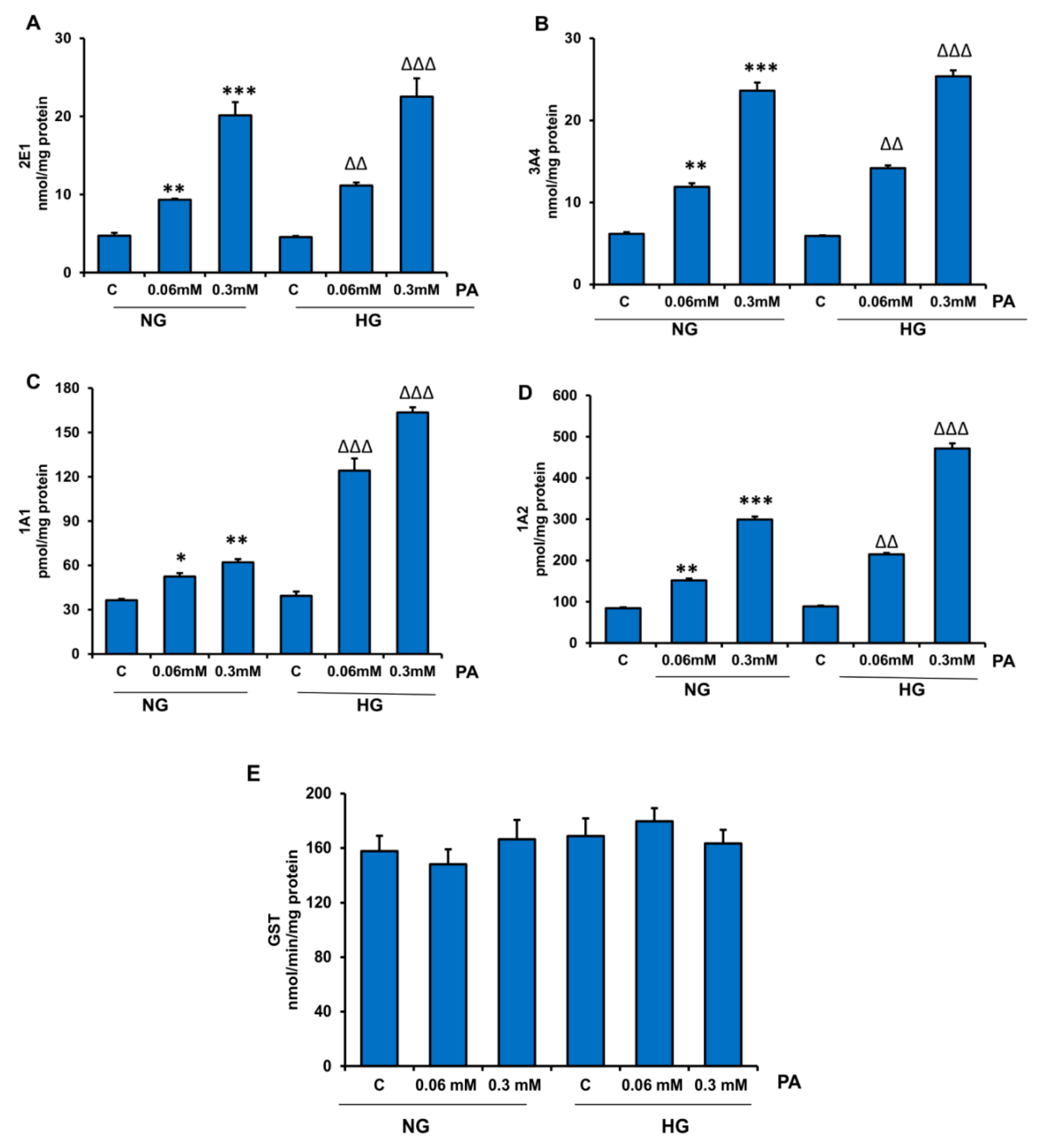
3.3. Effects of HG/HFA on CYP450s and Glutathione S-Transferase (GST) Enzymes
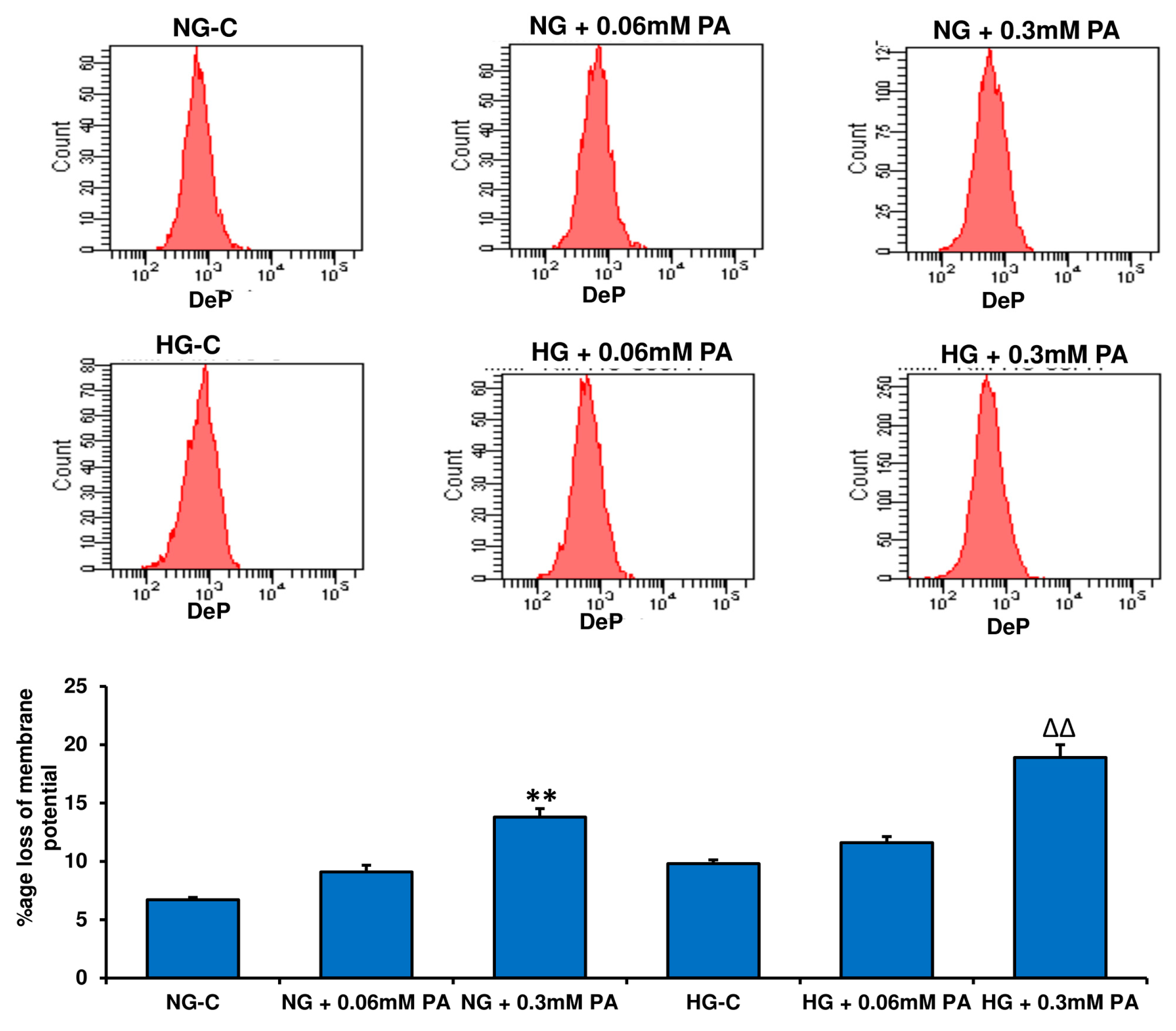
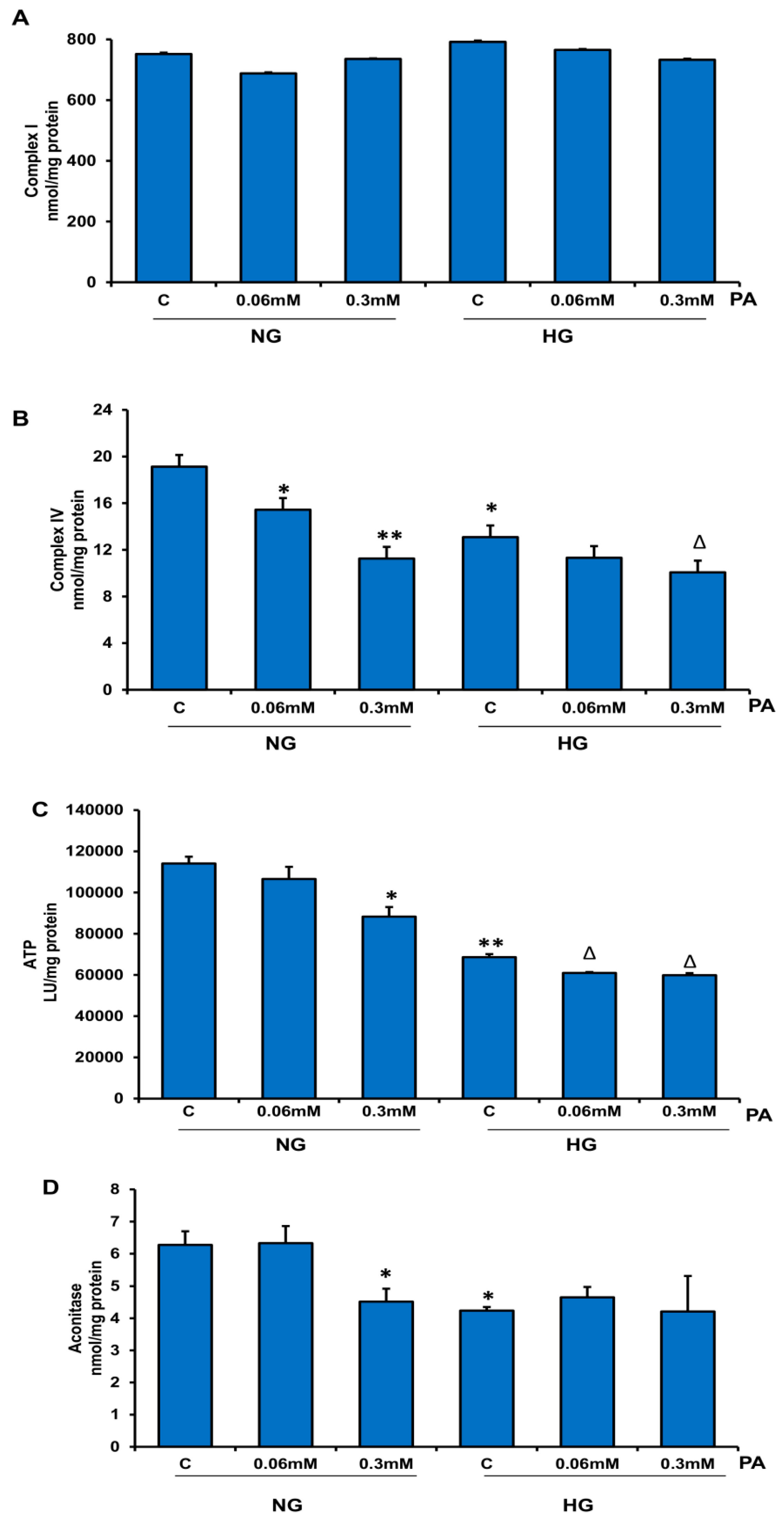
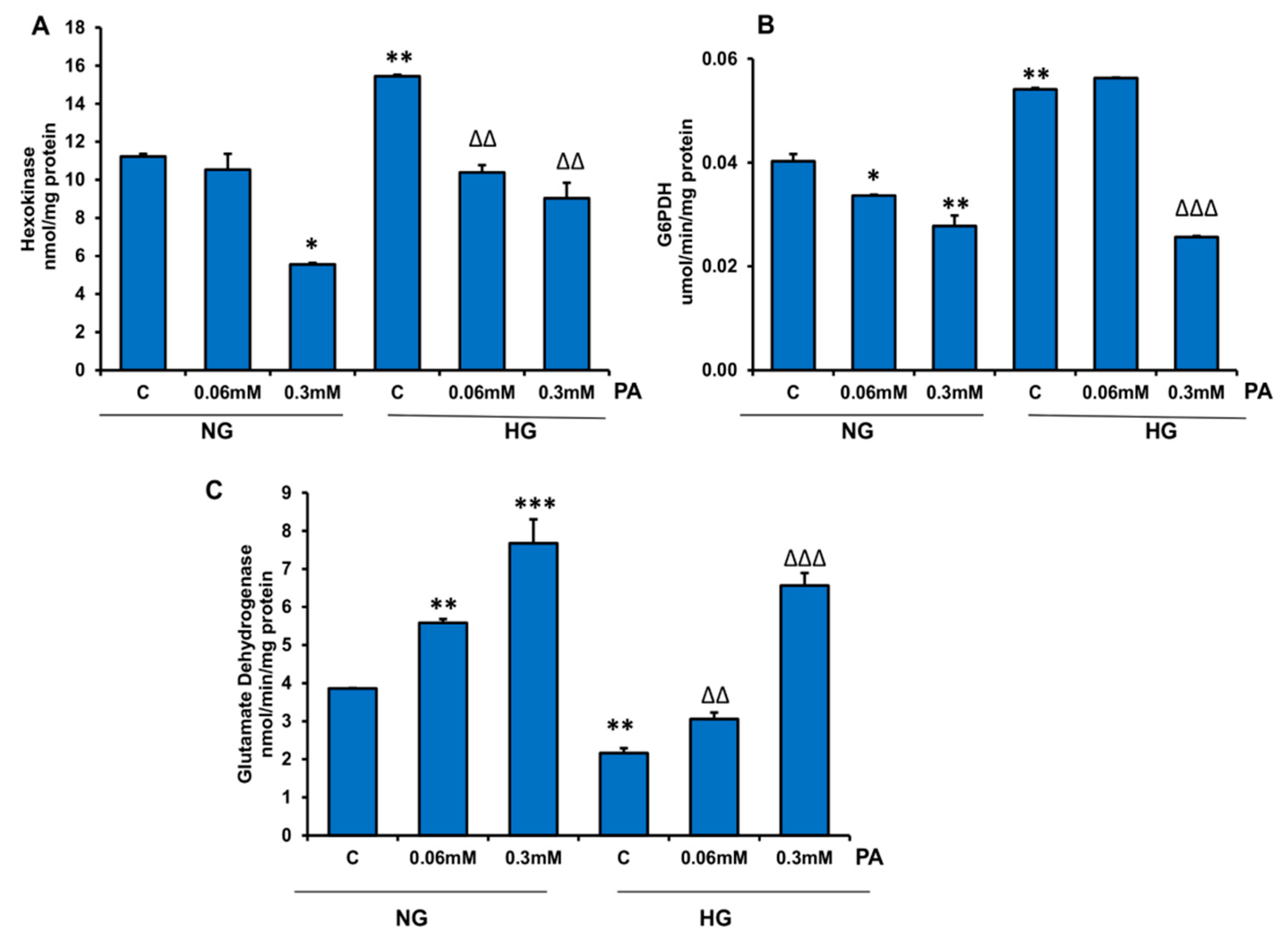
3.4. Effects of HG/HFA on Mitochondrial Functions
4. Discussion
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| PA | palmitic acid |
| NG | normal glucose |
| HG | high glucose |
| HFA | high fatty acid |
| HPA | high palmitic acid |
| MTT | 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide |
| ROS | reactive oxygen species |
| CDNB | 1-chloro 2,4-dinitrobenzene |
| DCFDA | 2′,7′-dichlorofluorescein diacetate |
| LPO | lipid peroxidation |
| NO | nitric oxide |
| GSH | reduced glutathione |
| GSSG | oxidized glutathione |
| GST | glutathione S-transferase |
| GSH-Px | glutathione peroxidase |
| GSH-Red | glutathione reductase |
| SOD | superoxide dismutase |
| CYP | cytochrome P450 |
| MMP | mitochondrial membrane potential |
| GDH | glutamate dehydrogenase |
| HK | hexokinase |
| G6PDH | glucose 6-phosphate dehydrogenase |
| TG | triglyceride |
| NF-kB | nuclear factor kappa-light-chain-enhancer of activated B cells |
| AMPK | AMP-activated protein kinase |
| mTOR | mammalian target of rapamycin |
| TNF-α | tumor necrosis factor alpha |
| IL6 | interleukin 6 |
References
- Robertson, R.P.; Harmon, J.; Tran, P.O.T.; Poitout, V. Beta-cell glucose toxicity, lipotoxicity, and chronic oxidative stress in type 2 diabetes. Diabetes 2004, 53, S119–S124. [Google Scholar] [CrossRef]
- Brownlee, M. Biochemistry and molecular cell biology of diabetic complications. Nature 2001, 414, 813–820. [Google Scholar] [CrossRef] [PubMed]
- Groop, L.C.; Bonadonna, R.C.; Del, P.S.; Ratheiser, K.; Zyck, K.; Ferrannini, E.; DeFronzo, R.A. Glucose and free fatty acid metabolism in non-insulin dependent diabetes mellitus. Evidence for multiple sites of insulin resistance. J. Clin. Investig. 1989, 84, 205–213. [Google Scholar] [CrossRef]
- Groop, L.; Saloranta, C.; Shank, M.; Bonadonna, R.C.; Ferrannini, E.; DeFronzo, R.A. The role of fatty acid metabolism in the pathogenesis of insulin resistance in obesity and non-insulin dependent diabetes mellitus. J. Clin. Endocrinol. Metab. 1991, 72, 96–107. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Barnes, G.T.; Yang, Q.; Tan, G.; Yang, D.; Chou, C.J.; Sole, J.; Nichols, A.; Ross, J.S.; Tartaglia, L.A.; et al. Chronic inflammation in fat plays a crucial role in the development of obesity-related insulin resistance. J. Clin. Investig. 2003, 112, 1821–1830. [Google Scholar] [CrossRef] [PubMed]
- Furukawa, S.; Fujita, T.; Shimabukuro, M.; Iwaki, M.; Yamada, Y.; Nakajima, Y.; Nakayama, O.; Makishima, M.; Matsuda, M.; Shimomura, I. Increased oxidative stress in obesity and its impact on metabolic syndrome. J. Clin. Investig. 2004, 114, 1752–1761. [Google Scholar] [CrossRef] [PubMed]
- Cnop, M. Fatty acids and glucolipotoxicity in the pathogenesis of Type 2 diabetes. Biochem. Soc. Trans. 2008, 36, 348–352. [Google Scholar] [CrossRef] [Green Version]
- Poitout, V.; Robertson, R.P. Glucolipotoxicity: Fuel excess and beta-cell dysfunction. Endocr. Rev. 2008, 29, 351–366. [Google Scholar] [CrossRef] [PubMed]
- Warensjö, E.; Risérus, U.; Vessby, B. Fatty acid composition of serum lipids predicts the development of the metabolic syndrome in men. Diabetologia 2005, 48, 1999–2005. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, L.; Guo, X.; Wang, O.; Zhang, H.; Zhou, F.; Liu, J.; Ji, B. Fructose and glucose combined with free fatty acids induce metabolic disorders in HepG2 cell: A new model to study the impacts of high-fructose/sucrose and high-fat diets in vitro. Mol. Nutr. Food Res. 2016, 60, 909–921. [Google Scholar] [CrossRef]
- Arner, P. Insulin resistance in type 2 diabetes: Role of fatty acids. Diabetes/Metab. Res. Rev. 2002, 18, S5–S9. [Google Scholar] [CrossRef]
- Hirabara, S.M.; Curi, R.; Maechler, P. Saturated fatty acid-induced insulin resistance is associated with mitochondrial dysfunction in skeletal muscle cells. J. Cell. Physiol. 2010, 222, 187–194. [Google Scholar] [CrossRef]
- Yao, H.-R.; Liu, J.; Plumeri, D.; Cao, Y.-B.; He, T.; Lin, L.; Li, Y.; Jiang, Y.-Y.; Li, J.; Shang, J. Lipotoxicity in HepG2 cells triggered by free fatty acids. Am. J. Transl. Res. 2011, 3, 284–291. [Google Scholar] [PubMed]
- Ricchi, M.; Odoardi, M.R.; Carulli, L.; Anzivino, C.; Ballestri, S.; Pinetti, A.; Fantoni, L.I.; Marra, F.; Bertolotti, M.; Banni, S.; et al. Differential effect of oleic and palmitic acid on lipid accumulation and apoptosis in cultured hepatocytes. J. Gastroenterol. Hepatol. 2009, 24, 830–840. [Google Scholar] [CrossRef]
- Mei, S.; Ni, H.-M.; Manley, S.; Bockus, A.; Kassel, K.M.; Luyendyk, J.P.; Copple, B.L.; Ding, W.-X. Differential Roles of Unsaturated and Saturated Fatty Acids on Autophagy and Apoptosis in Hepatocytes. J. Pharmacol. Exp. Ther. 2011, 339, 487–498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maedler, K.; Oberholzer, J.; Bucher, P.; Spinas, G.A.; Donath, M.Y. Monounsaturated fatty acids prevent the deleterious effects of palmitate and high glucose on human pancreatic beta-cell turnover and function. Diabetes 2003, 52, 726–733. [Google Scholar] [CrossRef] [PubMed]
- Chavez, J.A.; Summers, S.A. Lipid oversupply, selective insulin resistance, and lipotoxicity: Molecular mechanisms. Biochim. Biophys. Acta 2010, 1801, 252–265. [Google Scholar] [CrossRef] [Green Version]
- Hotamisligil, G.S. Endoplasmic Reticulum Stress and the Inflammatory Basis of Metabolic Disease. Cell 2010, 140, 900–917. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grishko, V.; Rachek, L.; Musiyenko, S.; LeDoux, S.P.; Wilson, G.L. Involvement of mtDNA damage in free fatty acid-induced apoptosis. Free Radic. Biol. Med. 2005, 38, 755–762. [Google Scholar] [CrossRef] [PubMed]
- Brookheart, R.T.; Michel, C.I.; Scaffer, J.E. As a matter of fat. Cell Metab. 2009, 10, 9–12. [Google Scholar] [CrossRef]
- Mir, S.U.R.; George, N.M.; Zahoor, L.; Harms, R.; Guinn, Z.; Sarvetnick, N.E. Inhibition of autophagic turnover in β- cells by fatty acids and glucose leads to apoptotic cell death. J. Biol. Chem. 2015, 290, 6071–6085. [Google Scholar] [CrossRef] [PubMed]
- Weikel, K.A.; Cacicedo, J.M.; Ruderman, N.B.; Ido, Y. Glucose and palmitate uncouple AMPK from autophagy in human aortic endothelial cells. Am. J. Physiol. Cell Physiol. 2014, 308, C249–C263. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Min, Q.; Ouyang, C.; Lee, J.; He, C.; Zou, M.-H.; Xie, Z. AMPK activation prevents excess nutrient-induced hepatic lipid accumulation by inhibiting mTORC1 signaling and endoplasmic reticulum stress response. Biochim. Biophys. Acta 2014, 1842, 1844–1854. [Google Scholar] [CrossRef] [Green Version]
- Cacicedo, J.M.; Benjachareowong, S.; Chou, E.; Ruderman, N.B.; Ido, Y. Palmitate-induced apoptosis in cultured bovine retinal periccytes: Roles of NAD (P)H oxidase, oxidative stress, and ceramide. Diabetes 2005, 54, 1838–1845. [Google Scholar] [CrossRef] [PubMed]
- Raza, H.; John, A. Streptozotocin-Induced Cytotoxicity, Oxidative Stress and Mitochondrial Dysfunction in Human Hepatoma HepG2 Cells. Int. J. Mol. Sci. 2012, 13, 5751–5767. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raza, H.; John, A.; Shafarin, J. Potentiation of LPS-Induced Apoptotic Cell Death in Human Hepatoma HepG2 Cells by Aspirin via ROS and Mitochondrial Dysfunction: Protection by N-Acetyl Cysteine. PLoS ONE 2016, 11, e0159750. [Google Scholar] [CrossRef]
- Al-Nahdi, A.M.; John, A.; Raza, H. Cytoprotective Effects of N-Acetylcysteine on Streptozotocin- Induced Oxidative Stress and Apoptosis in RIN-5F Pancreatic β-Cells. Cell. Physiol. Biochem. 2018, 51, 201–216. [Google Scholar] [CrossRef]
- Al Nahdi, A.M.T.; John, A.; Raza, H. Elucidation of Molecular Mechanisms of Streptozotocin-Induced Oxidative Stress, Apoptosis, and Mitochondrial Dysfunction in Rin-5F Pancreatic β-Cells. Oxidative Med. Cell. Longev. 2017, 2017, 7054272. [Google Scholar] [CrossRef]
- Al-Nahdi, A.M.T.; John, A.; Raza, H. Streptozotocin-induced molecular and metabolic targets in pancreatic beta-cell toxicity. Hamdan Med. J. 2019, 12, 65–71. [Google Scholar]
- Paglia, D.E.; Valentine, W.N. Studies on the quantitative and qualitative characterization of erythrocyte glutathione peroxidase. J. Lab. Clin. Med. 1967, 70, 158–169. [Google Scholar]
- Smith, I.K.; Vierheller, T.L.; Thorne, C.A. Assay of glutathione reductase in crude tissue homogenates using 5,5′-dithiobis(2-nitrobenzoic acid). Anal. Biochem. 1988, 175, 408–413. [Google Scholar] [CrossRef]
- Habig, W.H.; Pabst, M.J.; Jakoby, W.B. Glutathione S-transferases. The first enzymatic step in mercapturic acid formation. J. Biol. Chem. 1974, 249, 7130–7139. [Google Scholar] [PubMed]
- Czygan, P.; Greim, H.; Garro, A.J.; Hutterer, F.; Schaffner, F.; Popper, H.; Rosenthal, O.; Cooper, D.Y. Microsomal metabolism of dimethylnitrosamine and the cytochrome P-450 dependency of its activation to a mutagen. Cancer Res. 1973, 33, 2983–2986. [Google Scholar]
- Wang, R.W.; Newton, D.J.; Scheri, T.D.; Lu, A.Y. Human cytochrome P450 3A4-Catalyzed testosterone 6b-hydroxylation and erythromycin N- demethylation: Competition during catalysis. Drug Metab. Dispos. 1997, 25, 502–507. [Google Scholar] [PubMed]
- Pohl, R.J.; Fouts, J.R. A rapid method for assaying the metabolism of 7-ethoxyresorufin by microsomal subcellular fractions. Anal. Biochem. 1980, 107, 150–155. [Google Scholar] [CrossRef]
- Nerurkar, P.V.; Park, S.S.; Thomas, P.E.; Nims, R.W.; Lubet, R.A. Methoxyresorufin and benzyloxyresorufin: Substrates preferentially metabolized by cytochromes P4501A2 AND 2B, respectively, in the rat and mouse. Biochem. Pharmacol. 1993, 46, 933–943. [Google Scholar] [CrossRef]
- Birch-Machin, M.A.; Turnbull, D.M. Chapter 5 Assaying mitochondrial respiratory complex activity in mitochondria isolated from human cells and tissues. Methods Cell Biol. 2001, 65, 97–117. [Google Scholar]
- Raza, H.; John, A.; Shafarin, J. NAC Attenuates LPS-Induced Toxicity in Aspirin-Sensitized Mouse Macrophages via Suppression of Oxidative Stress and Mitochondrial Dysfunction. PLoS ONE 2014, 9, e103379. [Google Scholar] [CrossRef]
- Pittenger, M.F. Multilineage Potential of Adult Human Mesenchymal Stem Cells. Science 1999, 284, 143–147. [Google Scholar] [CrossRef] [Green Version]
- Iori, E.; Vinvi, B.; Murphy, E.; Marescotti, M.C.; Avogara, A.; Ahluwalia, A. Glucose and fatty acid metabolism in a 3 tissue in-vitro model challenged with normo- and hyperglycemia. PLoS ONE 2012, 7, e34704. [Google Scholar] [CrossRef]
- Barlow, J.; Jensen, V.H.; Jastroch, M.; Affourtit, C. Palmitae-induced impairment of glucose-stimulated insulin secretion preceds mitochondrial dysfunction in mouse pancreatic islets. Biochem. J. 2016, 473, 487–496. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, S.; Takamura, T.; Matsuzawa-Nagata, N.; Takayama, H.; Misu, H.; Noda, H.; Nabemoto, S.; Kurita, S.; Ota, T.; Ando, H.; et al. Palmitate Induces Insulin Resistance in H4IIEC3 Hepatocytes through Reactive Oxygen Species Produced by Mitochondria. J. Biol. Chem. 2009, 284, 14809–14818. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inoguchi, T.; Li, P.; Umeda, F.; Yu, H.Y.; Kakimoto, M.; Imamura, M.; Aoki, T.; Etoh, T.; Hashimoto, T.; Naruse, M.; et al. High glucose level and fress fatty acid stimulate reactive oxygen species production through protein kinase C-dependent activation of NAD9P)H oxidase in cultured vascular cells. Diabetes 2000, 49, 1939–1945. [Google Scholar] [CrossRef] [PubMed]
- Garcia, R.I.; Solis-Munoz, P.; Fernandez-Moreira, D.; Munoz-Yague, T.; Solis-Herruzo, J.A. In vitro treatment of HepG2 cells with saturated fatty acids reproduces mitochondrial dysfunction found in metabolic steatohepatitis. Dis. Models Mech. 2015, 8, 183–191. [Google Scholar] [CrossRef]
- Raza, H.; Prabu, S.K.; Robin, M.-A.; Avadhani, N.G. Elevated mitochondrial cytochrome P450 2E1 and glutathione S-transferase A4-4 in streptozotocin-induced diabetic rats: Tissue-specific variations and roles in oxidative stress. Diabetes 2004, 53, 185–194. [Google Scholar] [CrossRef]
- Raza, H.; John, A.; Howarth, F.C. Increased Metabolic Stress in Zucker Diabetic Fatty Rat Kidney and Pancreas. Cell. Physiol. Biochem. 2013, 32, 1610–1620. [Google Scholar] [CrossRef]
- Raza, H.; John, A.; Howarth, F.C. Alterations in Glutathione Redox Metabolism, Oxidative Stress, and Mitochondrial Function in the Left Ventricle of Elderly Zucker Diabetic Fatty Rat Heart. Int. J. Mol. Sci. 2012, 13, 16241–16254. [Google Scholar] [CrossRef] [Green Version]
- Raza, H.; John, A.; Shafarin, J.; Howarth, F.C. Exercise-induced alterations in pancreatic oxidative stress and mitochondrial function in type 2 diabetic Goto-Kakizaki rats. Physiol. Rep. 2016, 4, e12751. [Google Scholar] [CrossRef] [Green Version]
- Fu, J.; Qui, Q.; Yang, B.; Hou, Y.; Wang, H.; Xu, Y.; Wang, D.; Zhang, Q.; Pi, J. The impairment of glucose-stimulated insulin secretion in pancreatic B-cells caused by prolonged glucotoxicity and lipotoxicity is associated with elevated adaptive antioxidant response. Food Chem. Toxicol. 2017, 100, 161–167. [Google Scholar] [CrossRef] [PubMed]
- Kumar, B.; Kowluru, A.; Kowluru, R.A. Lipotoxicity Augments Glucotoxicity-Induced Mitochondrial Damage in the Development of Diabetic Retinopathy. Investig. Opthalmology Vis. Sci. 2015, 56, 2985–2992. [Google Scholar] [CrossRef] [Green Version]
- Tu, Q.-Q.; Zheng, R.-Y.; Li, J.; Hu, L.; Chang, Y.-X.; Li, L.; Li, M.-H.; Wang, R.-Y.; Huang, D.-D.; Wu, M.-C.; et al. Palmitic acid induces autophagy in hepatocytes via JNK2 activation. Acta Pharmacol. Sin. 2014, 35, 504–512. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Z.; Liew, C.W.; Handy, D.E.; Zhang, Y.; Leopold, J.A.; Hu, J.; Guo, L.; Kulkarni, R.N.; Loscalzo, J.; Stanton, R.C. High glucose inhibits glucose-6-phosphate dehydrogenase, leading to increased oxidative stress and β-cell apoptosis. FASEB J. 2010, 24, 1497–1505. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.-P.; Shin, J.-H.; Seo, S.-H.; Kim, S.-G.; Lee, S.H.; Shin, E.-H. Effects of Antioxidants in Reducing Accumulation of Fat in Hepatocyte. Int. J. Mol. Sci. 2018, 19, 2563. [Google Scholar] [CrossRef] [PubMed]
- Ding, W.-X. Uncoupling AMPK from autophagy: A foe that hinders the beneficial effects of metformin treatment on metabolic syndrome-associated atherosclerosis? Focus on “Glucose and palmitate uncouple AMPK from autophagy in human aortic endothelial cells”. Am. J. Physiol. Cell Physiol. 2015, 308, C246–C248. [Google Scholar] [CrossRef] [PubMed]
- Hinchy, E.C.; Gruszczyk, A.V.; Willows, R.; Navaratnam, N.; Hall, A.R.; Bates, G.; Bright, T.P.; Krieg, T.; Carling, D.; Murphy, M.P. Mitochondria-derived ROS activate AMP-activated protein kinase (AMPK) indirectly. J. Biol. Chem. 2018, 293, 17208–17217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lindholm, C.R.; Ertel, R.L.; Bauwens, J.D.; Schmuck, E.G.; Mulligan, J.C.; Saupe, K.W. A high fat diet decreases AMPK activity in multiple tissues in the absence of hyperglycemia or systemic inflammation in rats. J. Physiol. Biochem. 2013, 69, 165–175. [Google Scholar] [CrossRef]
- Hu, N.; Hu, M.; Duan, R.; Liu, C.; Guo, H.; Zhang, M.; Yu, Y.; Wang, X.; Liu, L.; Liu, X. Increased levels of fatty acids contributed to induction of hepatic CYP3A4 activity induced by diabetes—In vitro evidence from HepG2 cell and Fa2N-4 cell lines. J. Pharmacol. Sci. 2014, 124, 433–444. [Google Scholar] [CrossRef]















© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alnahdi, A.; John, A.; Raza, H. Augmentation of Glucotoxicity, Oxidative Stress, Apoptosis and Mitochondrial Dysfunction in HepG2 Cells by Palmitic Acid. Nutrients 2019, 11, 1979. https://doi.org/10.3390/nu11091979
Alnahdi A, John A, Raza H. Augmentation of Glucotoxicity, Oxidative Stress, Apoptosis and Mitochondrial Dysfunction in HepG2 Cells by Palmitic Acid. Nutrients. 2019; 11(9):1979. https://doi.org/10.3390/nu11091979
Chicago/Turabian StyleAlnahdi, Arwa, Annie John, and Haider Raza. 2019. "Augmentation of Glucotoxicity, Oxidative Stress, Apoptosis and Mitochondrial Dysfunction in HepG2 Cells by Palmitic Acid" Nutrients 11, no. 9: 1979. https://doi.org/10.3390/nu11091979
APA StyleAlnahdi, A., John, A., & Raza, H. (2019). Augmentation of Glucotoxicity, Oxidative Stress, Apoptosis and Mitochondrial Dysfunction in HepG2 Cells by Palmitic Acid. Nutrients, 11(9), 1979. https://doi.org/10.3390/nu11091979