Glutathione and Nitric Oxide: Key Team Players in Use and Disuse of Skeletal Muscle

 , , ,
, , , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
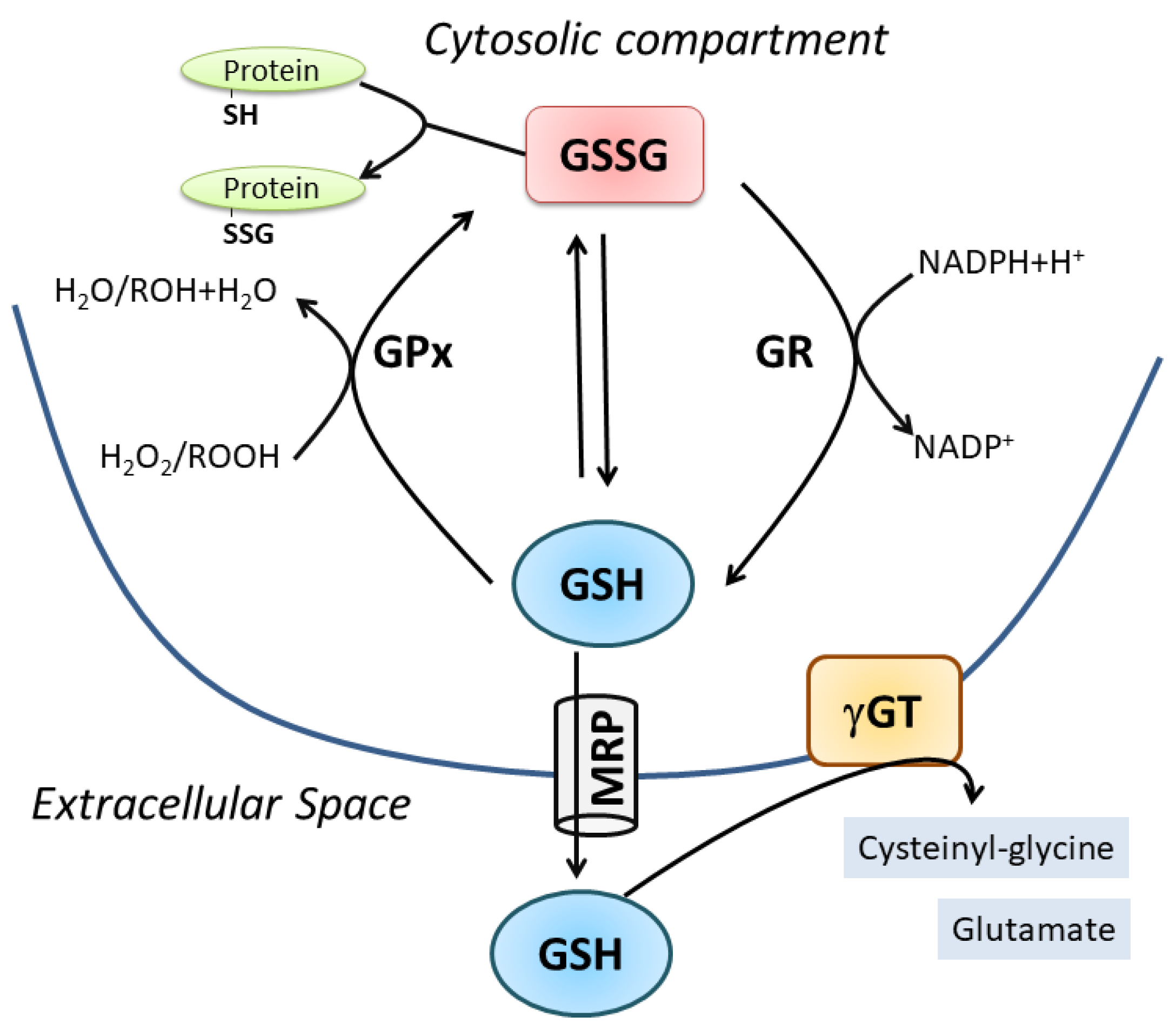
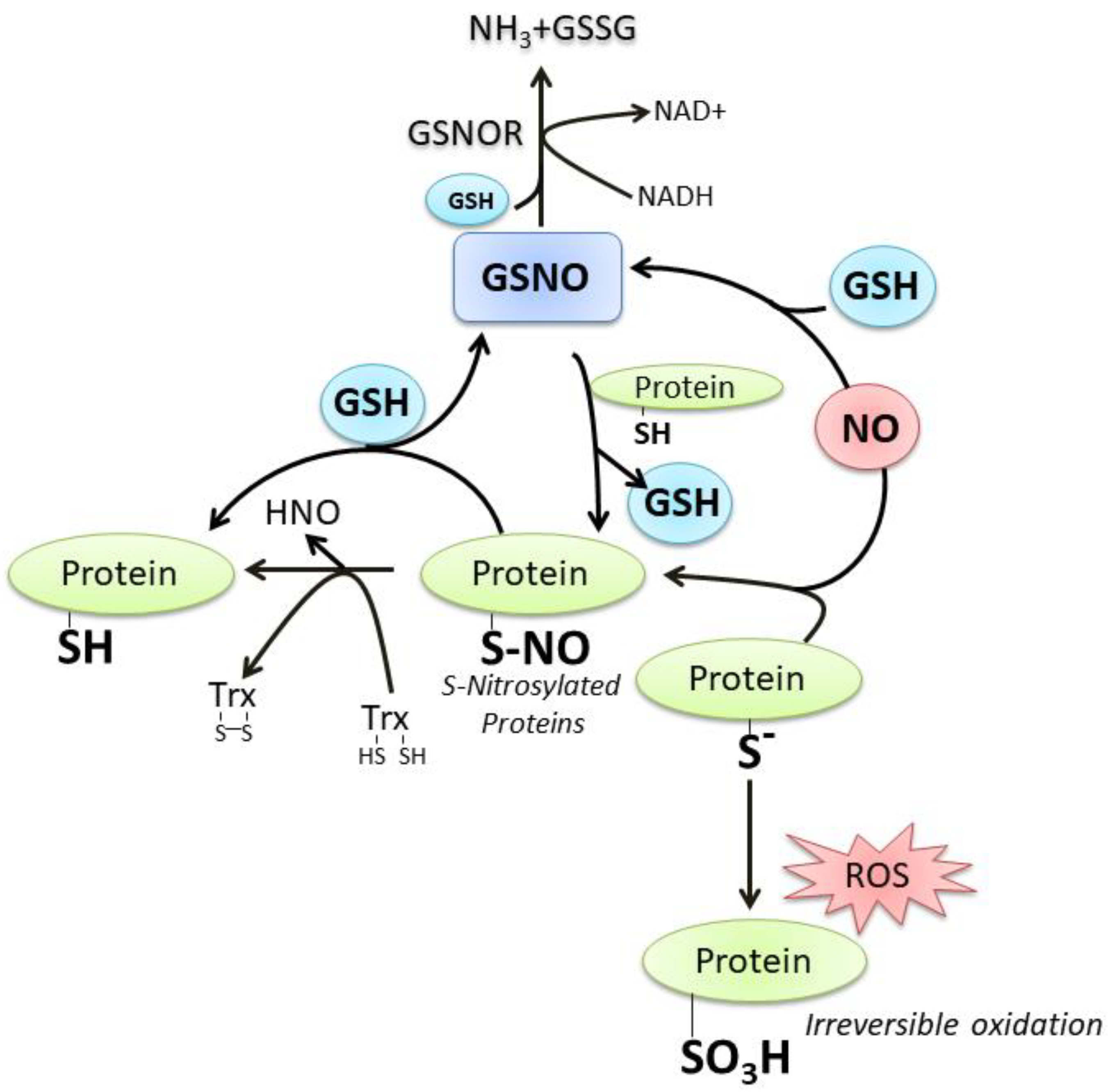
2. Oxidative/Nitrosative-Mediated Signaling and Stress as the Conditions Where NO Mainly Intersects GSH
3. GSH/NO Crosstalk in Skeletal Muscle Physiology
3.1. GSH and NO in Skeletal Muscle Inflammation During Aging
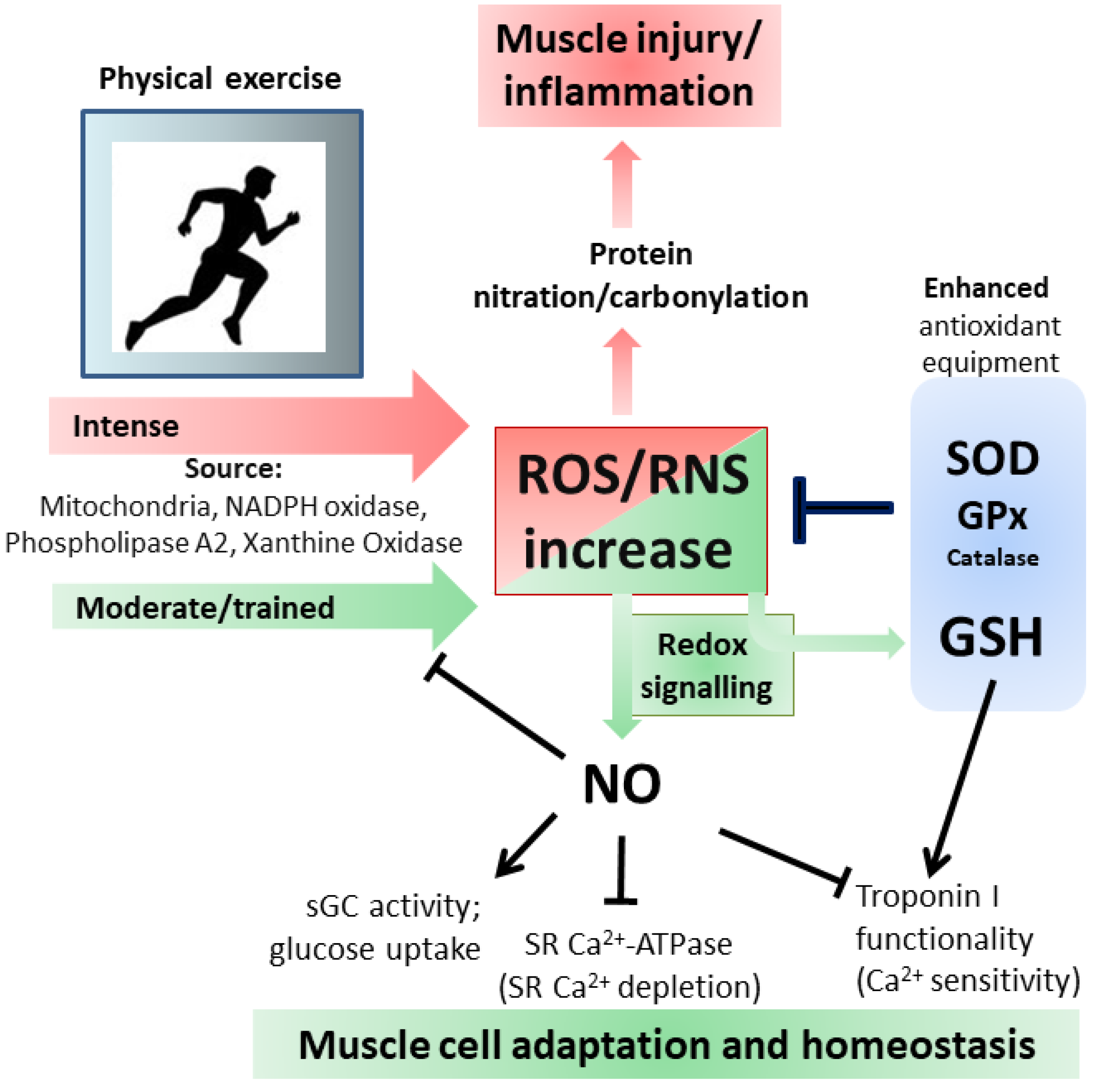
3.2. Inflammation and Exercise
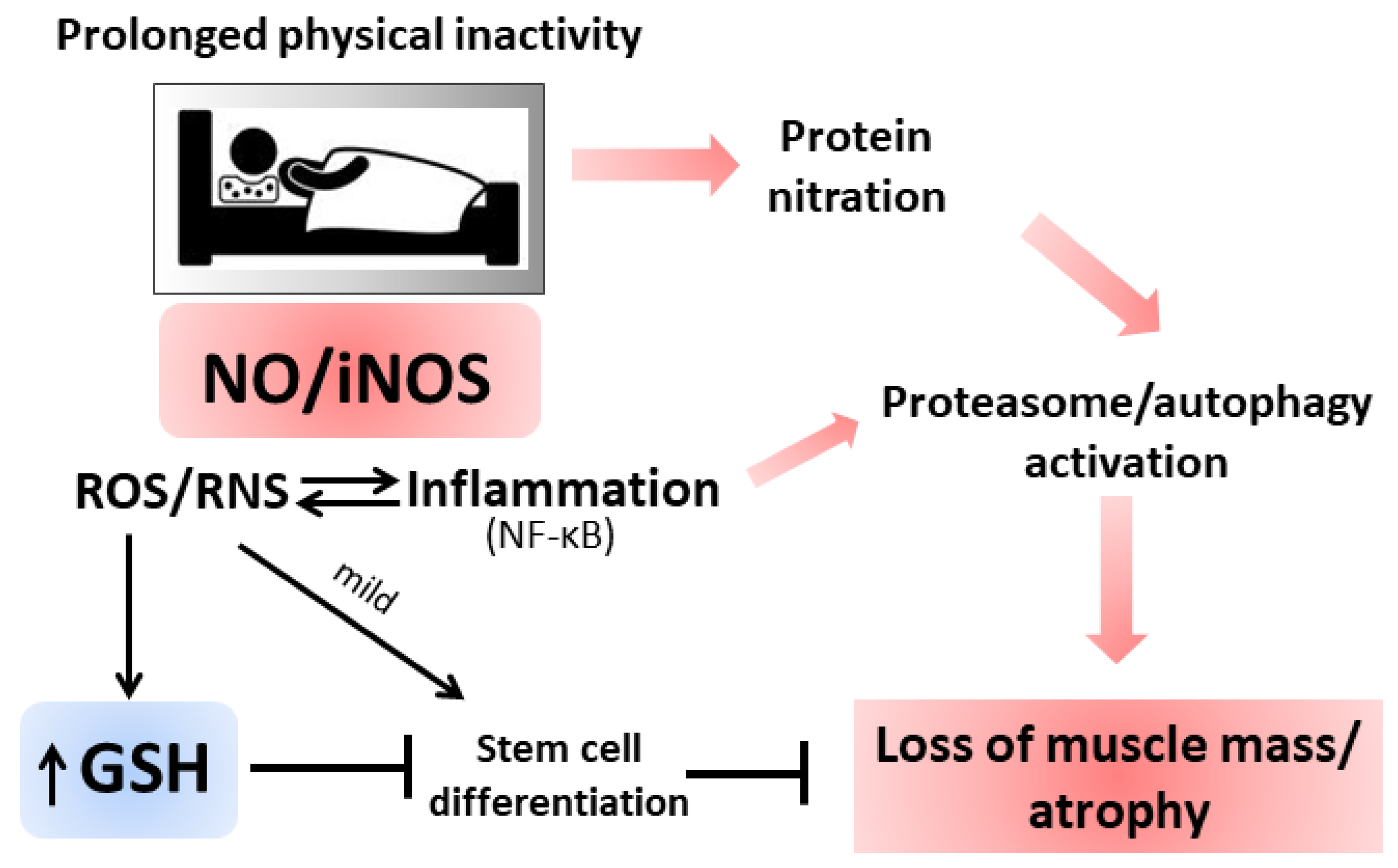
3.3. Inflammation and Atrophy
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Lu, S.C. Regulation of Glutathione Synthesis. Mol. Asp. Med. 2009, 30, 42–59. [Google Scholar] [CrossRef] [PubMed]
- Baldelli, S.; Aquilano, K.; Ciriolo, M.R. Punctum on two different transcription factors regulated by PGC-1α: Nuclear factor erythroid-derived 2-like 2 and nuclear respiratory factor 2. Biochim. Biophys. Acta 2013, 1830, 4137–4146. [Google Scholar] [CrossRef] [PubMed]
- Lu, S.C. Regulation of hepatic glutathione synthesis: Current concepts and controversies. FASEB J. 1999, 13, 1169–1183. [Google Scholar] [CrossRef] [PubMed]
- Xiong, Y.; Uys, J.D.; Tew, K.D.; Townsend, D.M. S-glutathionylation: From molecular mechanisms to health outcomes. Antioxid. Redox Signal. 2011, 15, 233–270. [Google Scholar] [CrossRef] [PubMed]
- Aoyama, K.; Nakaki, T. Impaired glutathione synthesis in neurodegeneration. Int. J. Mol. Sci. 2013, 14, 21021–21044. [Google Scholar] [CrossRef]
- Traverso, N.; Ricciarelli, R.; Nitti, M.; Marengo, B.; Furfaro, A.L.; Pronzato, M.A.; Marinari, U.M.; Domenicotti, C. Role of glutathione in cancer progression and chemoresistance. Oxid. Med. Cell. Longev. 2013, 2013, 972913. [Google Scholar] [CrossRef] [PubMed]
- Ballatori, N.; Krance, S.M.; Notenboom, S.; Shi, S.; Tieu, K.; Hammond, C.L. Glutathione dysregulation and the etiology and progression of human diseases. Biol. Chem. 2009, 390, 191–214. [Google Scholar] [CrossRef]
- Jackson, M.J.; McArdle, A. Age-related changes in skeletal muscle reactive oxygen species generation and adaptive responses to reactive oxygen species. J. Physiol. 2011, 589, 2139–2145. [Google Scholar] [CrossRef]
- Chen, G.; Wang, S.H.; Warner, T.D. Regulation of iNOS mRNA levels in endothelial cells by glutathione, a double-edged sword. Free Radic. Res. 2000, 32, 223–234. [Google Scholar] [CrossRef]
- Zhou, X.J.; Vaziri, N.D.; Wang, X.Q.; Silva, F.G.; Laszik, Z. Nitric oxide synthase expression in hypertension induced by inhibition of glutathione synthase. J. Pharmacol. Exp. Ther. 2002, 300, 762–767. [Google Scholar] [CrossRef]
- Aquilano, K.; Baldelli, S.; Cardaci, S.; Rotilio, G.; Ciriolo, M.R. Nitric oxide is the primary mediator of cytotoxicity induced by GSH depletion in neuronal cells. J. Cell. Sci. 2011, 124, 1043–1054. [Google Scholar] [CrossRef] [PubMed]
- Lundberg, J.O.; Weitzberg, E.; Gladwin, M.T. The nitrate-nitrite-nitric oxide pathway in physiology and therapeutics. Nat. Rev. Drug Discov. 2008, 7, 156–167. [Google Scholar] [CrossRef] [PubMed]
- Moncada, S.; Higgs, E.A. Molecular mechanisms and therapeutic strategies related to nitric oxide. FASEB J. 1995, 9, 1319–1330. [Google Scholar] [CrossRef] [PubMed]
- Grozdanovic, Z. NO message from muscle. Microsc. Res. Tech. 2001, 55, 148–153. [Google Scholar] [CrossRef] [PubMed]
- Sharma, J.N.; Al-Omran, A.; Parvathy, S.S. Role of nitric oxide in inflammatory diseases. Inflammopharmacology 2007, 15, 252–259. [Google Scholar] [CrossRef]
- Hussain, S.P.; He, P.; Subleski, J.; Hofseth, L.J.; Trivers, G.E.; Mechanic, L.; Hofseth, A.B.; Bernard, M.; Schwank, J.; Nguyen, G.; et al. Nitric Oxide Is a Key Component in Inflammation-Accelerated Tumorigenesis. Cancer Res. 2008, 68, 7130–7136. [Google Scholar] [CrossRef]
- Hughes, M.N. Chemistry of nitric oxide and related species. Meth. Enzymol. 2008, 436, 3–19. [Google Scholar] [PubMed]
- Foster, M.W.; Hess, D.T.; Stamler, J.S. Protein S-nitrosylation in health and disease: A current perspective. Trends Mol. Med. 2009, 15, 391–404. [Google Scholar] [CrossRef]
- Baldelli, S.; Aquilano, K.; Rotilio, G.; Ciriolo, M.R. Glutathione and copper, zinc superoxide dismutase are modulated by overexpression of neuronal nitric oxide synthase. Int. J. Biochem. Cell. Biol. 2008, 40, 2660–2670. [Google Scholar] [CrossRef] [PubMed]
- Moellering, D.; Mc Andrew, J.; Patel, R.P.; Forman, H.J.; Mulcahy, R.T.; Jo, H.; Darley-Usmar, V.M. The induction of GSH synthesis by nanomolar concentrations of NO in endothelial cells: A role for gamma-glutamylcysteine synthetase and gamma-glutamyl transpeptidase. FEBS Lett. 1999, 448, 292–296. [Google Scholar] [CrossRef]
- Filomeni, G.; Rotilio, G.; Ciriolo, M.R. Disulfide relays and phosphorylative cascades: Partners in redox-mediated signaling pathways. Cell. Death Differ. 2005, 12, 1555–1563. [Google Scholar] [CrossRef] [PubMed]
- Vasilaki, A.; Mansouri, A.; Remmen, H.V.; Meulen, J.H.V.D.; Larkin, L.; Richardson, A.G.; McArdle, A.; Faulkner, J.A.; Jackson, M.J. Free radical generation by skeletal muscle of adult and old mice: Effect of contractile activity. Aging Cell. 2006, 5, 109–117. [Google Scholar] [CrossRef] [PubMed]
- Stamler, J.S.; Meissner, G. Physiology of Nitric Oxide in Skeletal Muscle. Physiol. Rev. 2001, 81, 209–237. [Google Scholar] [CrossRef] [PubMed]
- Ji, L.L.; Fu, R.; Mitchell, E.W. Glutathione and antioxidant enzymes in skeletal muscle: Effects of fiber type and exercise intensity. J. Appl. Physiol. 1992, 73, 1854–1859. [Google Scholar] [CrossRef] [PubMed]
- Lew, H.; Pyke, S.; Quintanilha, A. Changes in the glutathione status of plasma, liver and muscle following exhaustive exercise in rats. FEBS Lett. 1985, 185, 262–266. [Google Scholar] [CrossRef]
- Gutteridge, J.M.C.; Halliwell, B. Mini-Review: Oxidative stress, redox stress or redox success? Biochem. Biophys. Res. Commun. 2018, 502, 183–186. [Google Scholar] [CrossRef] [PubMed]
- Ridnour, L.A.; Thomas, D.D.; Mancardi, D.; Espey, M.G.; Miranda, K.M.; Paolocci, N.; Feelisch, M.; Fukuto, J.; Wink, D.A. The chemistry of nitrosative stress induced by nitric oxide and reactive nitrogen oxide species. Putting perspective on stressful biological situations. Biol. Chem. 2005, 385, 1–10. [Google Scholar] [CrossRef]
- Stuehr, D.J.; Haque, M.M. Nitric oxide synthase enzymology in the 20 years after the Nobel Prize. Br. J. Pharmac. 2019, 176, 177–188. [Google Scholar] [CrossRef] [PubMed]
- Förstermann, U.; Closs, E.I.; Pollock, J.S.; Nakane, M.; Schwarz, P.; Gath, I.; Kleinert, H. Nitric oxide synthase isozymes. Characterization, purification, molecular cloning, and functions. Hypertension 1994, 23, 1121–1131. [Google Scholar] [CrossRef]
- Larsson, B.; Phillips, S.C. Isolation and Characterization of a Novel, Human Neuronal Nitric Oxide Synthase cDNA. Biochem. Biophys. Res. Commun. 1998, 251, 898–902. [Google Scholar] [CrossRef]
- Silvagno, F.; Xia, H.; Bredt, D.S. Neuronal Nitric-oxide Synthase-, an Alternatively Spliced Isoform Expressed in Differentiated Skeletal Muscle. J. Biol. Chem. 1996, 271, 11204–11208. [Google Scholar] [CrossRef] [PubMed]
- McNeill, E.; Crabtree, M.J.; Sahgal, N.; Patel, J.; Chuaiphichai, S.; Iqbal, A.J.; Hale, A.B.; Greaves, D.R.; Channon, K.M. Regulation of iNOS function and cellular redox state by macrophage Gch1 reveals specific requirements for tetrahydrobiopterin in NRF2 activation. Free Rad. Biol. Med. 2015, 79, 206–216. [Google Scholar] [CrossRef] [PubMed]
- Elfering, S.L.; Sarkela, T.M.; Giulivi, C. Biochemistry of Mitochondrial Nitric-oxide Synthase. J. Biol. Chem. 2002, 277, 38079–38086. [Google Scholar] [CrossRef]
- Aguirre, E.; López-Bernardo, E.; Cadenas, S. Functional evidence for nitric oxide production by skeletal-muscle mitochondria from lipopolysaccharide-treated mice. Mitochondrion 2012, 12, 126–131. [Google Scholar] [CrossRef] [PubMed]
- Villanueva, C.; Giulivi, C. Subcellular and cellular locations of nitric-oxide synthase isoforms as determinants of health and disease. Free Rad. Biol. Med. 2010, 49, 307. [Google Scholar] [CrossRef]
- Bartesaghi, S.; Radi, R. Fundamentals on the biochemistry of peroxynitrite and protein tyrosine nitration. Redox Biol. 2018, 14, 618–625. [Google Scholar] [CrossRef]
- Tejero, J.; Hunt, A.P.; Santolini, J.; Lehnert, N.; Stuehr, D.J. Mechanism and regulation of ferrous heme-nitric oxide (NO) oxidation in NO synthases. J. Biol. Chem. 2019, 294, 7904–7916. [Google Scholar] [CrossRef] [PubMed]
- Gusarov, I.; Nudler, E. Protein S-Nitrosylation: Enzymatically Controlled, but Intrinsically Unstable, Post-translational Modification. Mol. Cell. 2018, 69, 351–353. [Google Scholar] [CrossRef]
- Martínez-Ruiz, A.; Lamas, S. S-nitrosylation: A potential new paradigm in signal transduction. Cardiovasc. Res. 2004, 62, 43–52. [Google Scholar] [CrossRef]
- Hess, D.T.; Matsumoto, A.; Kim, S.-O.; Marshall, H.E.; Stamler, J.S. Protein S nitrosylation: Purview and parameters. Nat. Rev. Mol. Cell. Biol. 2005, 6, 150–166. [Google Scholar] [CrossRef]
- Shinyashiki, M.; Pan, C.-J.G.; Lopez, B.E.; Fukuto, J.M. Inhibition of the yeast metal reductase heme protein fre1 by nitric oxide (NO): A model for inhibition of NADPH oxidase by NO. Free Radic. Biol. Med. 2004, 37, 713–723. [Google Scholar] [CrossRef] [PubMed]
- Ródenas, J.; Mitjavila, M.T.; Carbonell, T. Nitric oxide inhibits superoxide production by inflammatory polymorphonuclear leukocytes. Am. J. Physiol. Cell. Phys. 1998, 274, 827–830. [Google Scholar] [CrossRef] [PubMed]
- Henderson, S.A.; Lee, P.H.; Aeberhard, E.E.; Adams, J.W.; Ignarro, L.J.; Murphy, W.J.; Sherman, M.P. Nitric oxide reduces early growth response-1 gene expression in rat lung macrophages treated with interferon-gamma and lipopolysaccharide. J. Biol. Chem. 1994, 269, 25239–25242. [Google Scholar] [PubMed]
- Aquilano, K.; Baldelli, S.; Pagliei, B.; Cannata, S.M.; Rotilio, G.; Ciriolo, M.R. p53 Orchestrates the PGC-1α-Mediated Antioxidant Response Upon Mild Redox and Metabolic Imbalance. Antioxid. Redox Signal. 2013, 18, 386. [Google Scholar] [CrossRef] [PubMed]
- Baldelli, S.; Ciriolo, M.R. Altered S-nitrosylation of p53 is responsible for impaired antioxidant response in skeletal muscle during aging. Aging 2016, 8, 3450. [Google Scholar] [CrossRef] [PubMed]
- Tan, B.L.; Norhaizan, M.E.; Liew, W.-P.-P.; Sulaiman Rahman, H. Antioxidant and Oxidative Stress: A Mutual Interplay in Age-Related Diseases. Front. Pharmacol. 2018, 9. [Google Scholar] [CrossRef] [PubMed]
- Lück, G.; Hoch, W.; Hopf, C.; Blottner, D. Nitric Oxide Synthase (NOS-1) Coclustered With Agrin-Induced AChR-Specializations on Cultured Skeletal Myotubes. Mol. Cell. Neurosci. 2000, 16, 269–281. [Google Scholar] [CrossRef]
- Loureiro, A.C.C.; do Rêgo-Monteiro, I.C.; Louzada, R.A.; Ortenzi, V.H.; de Aguiar, A.P.; de Abreu, E.S.; Cavalcanti-de-Albuquerque, J.P.A.; Hecht, F.; de Oliveira, A.C.; Ceccatto, V.M.; et al. Differential Expression of NADPH Oxidases Depends on Skeletal Muscle Fiber Type in Rats. Oxid. Med. Cell. Longev. 2016, 2016. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Shin, H.S.; Shireman, P.K.; Vasilaki, A.; Van Remmen, H.; Csete, M.E. Glutathione-peroxidase-1 null muscle progenitor cells are globally defective. Free Radic. Biol. Med. 2006, 41, 1174–1184. [Google Scholar] [CrossRef]
- Powers, S.K.; Jackson, M.J. Exercise-Induced Oxidative Stress: Cellular Mechanisms and Impact on Muscle Force Production. Physiol. Rev. 2008, 88, 1243–1276. [Google Scholar] [CrossRef] [PubMed]
- Hellsten, Y.; Apple, F.S.; Sjödin, B. Effect of sprint cycle training on activities of antioxidant enzymes in human skeletal muscle. J. Appl. Physiol. 1996, 81, 1484–1487. [Google Scholar] [CrossRef] [PubMed]
- Gnaiger, E. Capacity of oxidative phosphorylation in human skeletal muscle: New perspectives of mitochondrial physiology. Int. J. Biochem. Cell. Biol. 2009, 41, 1837–1845. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Li, C.G.; Qi, Z.; Cui, D.; Ding, S. Acute Exercise Induced Mitochondrial H2O2 Production in Mouse Skeletal Muscle: Association with p66Shc and FOXO3a Signaling and Antioxidant Enzymes. Available online: https://www.hindawi.com/journals/omcl/2015/536456/ (accessed on 6 September 2019).
- Leeuwenburgh, C.; Ji, L.L. Alteration of Glutathione and Antioxidant Status with Exercise in Unfed and Refed Rats. J. Nutr. 1996, 126, 1833–1843. [Google Scholar] [PubMed]
- Saoi, M.; Percival, M.; Nemr, C.; Li, A.; Gibala, M.; Britz-McKibbin, P. Characterization of the Human Skeletal Muscle Metabolome for Elucidating the Mechanisms of Bicarbonate Ingestion on Strenuous Interval Exercise. Anal. Chem. 2019, 91, 4709–4718. [Google Scholar] [CrossRef]
- Steinbacher, P.; Eckl, P. Impact of Oxidative Stress on Exercising Skeletal Muscle. Biomolecules 2015, 5, 356–377. [Google Scholar] [CrossRef] [PubMed]
- Pastore, A.; Piemonte, F. Protein Glutathionylation in Cardiovascular Diseases. Int. J. Mol. Sci. 2013, 14, 20845–20876. [Google Scholar] [CrossRef]
- Alegre-Cebollada, J.; Kosuri, P.; Giganti, D.; Eckels, E.; Rivas-Pardo, J.A.; Hamdani, N.; Warren, C.M.; Solaro, R.J.; Linke, W.A.; Fernández, J.M. S-Glutathionylation of Cryptic Cysteines Enhances Titin Elasticity by Blocking Protein Folding. Cell 2014, 156, 1235–1246. [Google Scholar] [CrossRef]
- Mollica, J.P.; Dutka, T.L.; Merry, T.L.; Lamboley, C.R.; McConell, G.K.; McKenna, M.J.; Murphy, R.M.; Lamb, G.D. S-Glutathionylation of troponin I (fast) increases contractile apparatus Ca2+ sensitivity in fast-twitch muscle fibres of rats and humans. J. Physiol. 2012, 590, 1443–1463. [Google Scholar] [CrossRef]
- Dutka, T.L.; Mollica, J.P.; Lamboley, C.R.; Weerakkody, V.C.; Greening, D.W.; Posterino, G.S.; Murphy, R.M.; Lamb, G.D. S-nitrosylation and S-glutathionylation of Cys134 on troponin I have opposing competitive actions on Ca2+ sensitivity in rat fast-twitch muscle fibers. Am. J. Physiol. Cell. Physiol. 2016, 312, 316–327. [Google Scholar] [CrossRef]
- Gill, R.M.; O’Brien, M.; Young, A.; Gardiner, D.; Mailloux, R.J. Protein S-glutathionylation lowers superoxide/hydrogen peroxide release from skeletal muscle mitochondria through modification of complex I and inhibition of pyruvate uptake. PLoS ONE 2018, 13, e0192801. [Google Scholar] [CrossRef]
- De Palma, C.; Morisi, F.; Pambianco, S.; Assi, E.; Touvier, T.; Russo, S.; Perrotta, C.; Romanello, V.; Carnio, S.; Cappello, V.; et al. Deficient nitric oxide signalling impairs skeletal muscle growth and performance: Involvement of mitochondrial dysregulation. Skelet. Muscle 2014, 4, 22. [Google Scholar] [CrossRef] [PubMed]
- Mészáros, L.G.; Minarovic, I.; Zahradnikova, A. Inhibition of the skeletal muscle ryanodine receptor calcium release channel by nitric oxide. FEBS Lett. 1996, 380, 49–52. [Google Scholar] [CrossRef]
- Maréchal, G.; Gailly, P. Effects of nitric oxide on the contraction of skeletal muscle. Cell. Mol. Life Sci. 1999, 55, 1088–1102. [Google Scholar] [CrossRef] [PubMed]
- Xiyuan, Z.; Fink, R.H.A.; Mosqueira, M. NO-sGC Pathway Modulates Ca2+ Release and Muscle Contraction in Zebrafish Skeletal Muscle. Front Physiol. 2017, 8, 607. [Google Scholar] [CrossRef] [PubMed]
- Ishii, T.; Sunami, O.; Saitoh, N.; Nishio, H.; Takeuchi, T.; Hata, F. Inhibition of skeletal muscle sarcoplasmic reticulum Ca2+-ATPase by nitric oxide. FEBS Lett. 1998, 440, 218–222. [Google Scholar] [CrossRef]
- Poderoso, J.J.; Carreras, M.C.; Lisdero, C.; Riobó, N.; Schöpfer, F.; Boveris, A. Nitric oxide inhibits electron transfer and increases superoxide radical production in rat heart mitochondria and submitochondrial particles. Arch. Biochem. Biophys. 1996, 328, 85–92. [Google Scholar] [CrossRef]
- Iglesias, D.E.; Bombicino, S.S.; Valdez, L.B.; Boveris, A. Nitric oxide interacts with mitochondrial complex III producing antimycin-like effects. Free Radic. Biol. Med. 2015, 89, 602–613. [Google Scholar] [CrossRef] [PubMed]
- Cooper, C.E.; Brown, G.C. The inhibition of mitochondrial cytochrome oxidase by the gases carbon monoxide, nitric oxide, hydrogen cyanide and hydrogen sulfide: Chemical mechanism and physiological significance. J. Bioenerg. Biomembr. 2008, 40, 533–539. [Google Scholar] [CrossRef]
- Lira, V.A.; Soltow, Q.A.; Long, J.H.D.; Betters, J.L.; Sellman, J.E.; Criswell, D.S. Nitric oxide increases GLUT4 expression and regulates AMPK signaling in skeletal muscle. Am. J. Physiol. Endocrinol. Metab. 2007, 293, 1062–1068. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Hiam, D.; Hong, Y.-H.; Zulli, A.; Hayes, A.; Rattigan, S.; McConell, G.K. Nitric oxide is required for the insulin sensitizing effects of contraction in mouse skeletal muscle. J. Physiol. 2017, 595, 7427–7439. [Google Scholar] [CrossRef]
- Lira, V.A.; Brown, D.L.; Lira, A.K.; Kavazis, A.N.; Soltow, Q.A.; Zeanah, E.H.; Criswell, D.S. Nitric oxide and AMPK cooperatively regulate PGC-1 in skeletal muscle cells. J. Physiol. 2010, 588, 3551–3566. [Google Scholar] [CrossRef] [PubMed]
- Lira, V.A.; Benton, C.R.; Yan, Z.; Bonen, A. PGC-1alpha regulation by exercise training and its influences on muscle function and insulin sensitivity. Am. J. Physiol. Endocrinol. Metab. 2010, 299, 145–161. [Google Scholar] [CrossRef] [PubMed]
- Jäger, S.; Handschin, C.; St-Pierre, J.; Spiegelman, B.M. AMP-activated protein kinase (AMPK) action in skeletal muscle via direct phosphorylation of PGC-1alpha. Proc. Natl. Acad. Sci. USA 2007, 104, 12017–12022. [Google Scholar] [CrossRef] [PubMed]
- Merry, T.L.; Ristow, M. Nuclear factor erythroid-derived 2-like 2 (NFE2L2, Nrf2) mediates exercise-induced mitochondrial biogenesis and the anti-oxidant response in mice. J. Physiol. 2016, 594, 5195–5207. [Google Scholar] [CrossRef] [PubMed]
- Aquilano, K.; Baldelli, S.; Ciriolo, M.R. Nuclear recruitment of neuronal nitric-oxide synthase by α-syntrophin is crucial for the induction of mitochondrial biogenesis. J. Biol. Chem. 2014, 289, 365–378. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Vanhoutte, P.M.; Leung, S.W.S. Vascular nitric oxide: Beyond eNOS. J. Pharmacol. Sci. 2015, 129, 83–94. [Google Scholar] [CrossRef] [PubMed]
- Lee-Young, R.S.; Ayala, J.E.; Hunley, C.F.; James, F.D.; Bracy, D.P.; Kang, L.; Wasserman, D.H. Endothelial nitric oxide synthase is central to skeletal muscle metabolic regulation and enzymatic signaling during exercise in vivo. Am. J. Physiol. Regul. Integr. Comparat. Physiol. 2010, 298, 1399–1408. [Google Scholar] [CrossRef]
- Batthyány, C.; Bartesaghi, S.; Mastrogiovanni, M.; Lima, A.; Demicheli, V.; Radi, R. Tyrosine-Nitrated Proteins: Proteomic and Bioanalytical Aspects. Antioxid. Redox Signal. 2017, 26, 313–328. [Google Scholar] [CrossRef] [PubMed]
- Feng, J.; Navratil, M.; Thompson, L.V.; Arriaga, E.A. Principal component analysis reveals age-related and muscle-type-related differences in protein carbonyl profiles of muscle mitochondria. J. Gerontol. A Biol. Sci. Med. Sci. 2008, 63, 1277–1288. [Google Scholar] [CrossRef]
- Murakami, H.; Guillet, C.; Tardif, N.; Salles, J.; Migné, C.; Boirie, Y.; Walrand, S. Cumulative 3-nitrotyrosine in specific muscle proteins is associated with muscle loss during aging. Exp. Gerontol. 2012, 47, 129–135. [Google Scholar] [CrossRef]
- Cakatay, U.; Telci, A.; Kayali, R.; Tekeli, F.; Akçay, T.; Sivas, A. Relation of aging with oxidative protein damage parameters in the rat skeletal muscle. Clin. Biochem. 2003, 36, 51–55. [Google Scholar] [CrossRef]
- Sinha-Hikim, I.; Sinha-Hikim, A.P.; Parveen, M.; Shen, R.; Goswami, R.; Tran, P.; Crum, A.; Norris, K.C. Long-term supplementation with a cystine-based antioxidant delays loss of muscle mass in aging. J. Gerontol. A Biol. Sci. Med. Sci. 2013, 68, 749–759. [Google Scholar] [CrossRef] [PubMed]
- Petry, E.R.; Cruzat, V.F.; Heck, T.G.; Leite, J.S.M.; Homem de Bittencourt, P.I.; Tirapegui, J. Alanyl-glutamine and glutamine plus alanine supplements improve skeletal redox status in trained rats: Involvement of heat shock protein pathways. Life Sci. 2014, 94, 130–136. [Google Scholar] [CrossRef] [PubMed]
- Leite, J.S.M.; Raizel, R.; Hypólito, T.M.; Rosa, T.D.S.; Cruzat, V.F.; Tirapegui, J. l-glutamine and l-alanine supplementation increase glutamine-glutathione axis and muscle HSP-27 in rats trained using a progressive high-intensity resistance exercise. Appl. Physiol. Nutr. Metab. 2016, 41, 842–849. [Google Scholar] [CrossRef] [PubMed]
- Michailidis, Y.; Karagounis, L.G.; Terzis, G.; Jamurtas, A.Z.; Spengos, K.; Tsoukas, D.; Chatzinikolaou, A.; Mandalidis, D.; Stefanetti, R.J.; Papassotiriou, I.; et al. Thiol-based antioxidant supplementation alters human skeletal muscle signaling and attenuates its inflammatory response and recovery after intense eccentric exercise. Am. J. Clin. Nutr. 2013, 98, 233–245. [Google Scholar] [CrossRef] [PubMed]
- Venditti, P.; Napolitano, G.; Barone, D.; Di Meo, S. Vitamin E supplementation modifies adaptive responses to training in rat skeletal muscle. Free Radic. Res. 2014, 48, 1179–1189. [Google Scholar] [CrossRef] [PubMed]
- Morrison, D.; Hughes, J.; Della Gatta, P.A.; Mason, S.; Lamon, S.; Russell, A.P.; Wadley, G.D. Vitamin C and E supplementation prevents some of the cellular adaptations to endurance-training in humans. Free Radic. Biol. Med. 2015, 89, 852–862. [Google Scholar] [CrossRef]
- Gomez-Cabrera, M.C.; Salvador-Pascual, A.; Cabo, H.; Ferrando, B.; Viña, J. Redox modulation of mitochondriogenesis in exercise. Does antioxidant supplementation blunt the benefits of exercise training? Free Radic. Biol. Med. 2015, 86, 37–46. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Jiao, H.; Zhao, J.; Wang, X.; Lin, H. L-Arginine Enhances Protein Synthesis by Phosphorylating mTOR (Thr 2446) in a Nitric Oxide-Dependent Manner in C2C12 Cells. Oxid. Med. Cell. Longev. 2018, 2018, 7569127. [Google Scholar] [CrossRef]
- Ham, D.J.; Gleeson, B.G.; Chee, A.; Baum, D.M.; Caldow, M.K.; Lynch, G.S.; Koopman, R. L-Citrulline Protects Skeletal Muscle Cells from Cachectic Stimuli through an iNOS-Dependent Mechanism. PLoS ONE 2015, 10, e0141572. [Google Scholar] [CrossRef]
- Franceschi, C.; Campisi, J. Chronic inflammation (inflammaging) and its potential contribution to age-associated diseases. J. Gerontol. A Biol. Sci. Med. Sci. 2014, 69, 4–9. [Google Scholar] [CrossRef] [PubMed]
- Draganidis, D.; Karagounis, L.G.; Athanailidis, I.; Chatzinikolaou, A.; Jamurtas, A.Z.; Fatouros, I.G. Inflammaging and Skeletal Muscle: Can Protein Intake Make a Difference? J. Nutr. 2016, 146, 1940–1952. [Google Scholar] [CrossRef] [PubMed]
- Szentesi, P.; Csernoch, L.; Dux, L.; Keller-Pintér, A. Changes in Redox Signaling in the Skeletal Muscle with Aging. Available online: https://www.hindawi.com/journals/omcl/2019/4617801/ (accessed on 9 September 2019).
- Rigamonti, E.; Touvier, T.; Clementi, E.; Manfredi, A.A.; Brunelli, S.; Rovere-Querini, P. Requirement of inducible nitric oxide synthase for skeletal muscle regeneration after acute damage. J. Immunol. 2013, 190, 1767–1777. [Google Scholar] [CrossRef] [PubMed]
- Buck, M.; Chojkier, M. Muscle wasting and dedifferentiation induced by oxidative stress in a murine model of cachexia is prevented by inhibitors of nitric oxide synthesis and antioxidants. EMBO J. 1996, 15, 1753–1765. [Google Scholar] [CrossRef] [PubMed]
- De Palma, C.; Clementi, E. Nitric oxide in myogenesis and therapeutic muscle repair. Mol. Neurobiol. 2012, 46, 682–692. [Google Scholar] [CrossRef] [PubMed]
- Pizza, F.X.; Hernandez, I.J.; Tidball, J.G. Nitric oxide synthase inhibition reduces muscle inflammation and necrosis in modified muscle use. J. Leukoc. Biol. 1998, 64, 427–433. [Google Scholar] [CrossRef] [PubMed]
- Shinozaki, S.; Chang, K.; Sakai, M.; Shimizu, N.; Yamada, M.; Tanaka, T.; Nakazawa, H.; Ichinose, F.; Yamada, Y.; Ishigami, A.; et al. Inflammatory stimuli induce inhibitory S-nitrosylation of the deacetylase SIRT1 to increase acetylation and activation of p53 and p65. Sci. Signal. 2014, 7, 106. [Google Scholar] [CrossRef] [PubMed]
- Limongi, D.; Baldelli, S.; Checconi, P.; Marcocci, M.; De Chiara, G.; Fraternale, A.; Magnani, M.; Ciriolo, M.R.; Palamara, A.T. GSH-C4 Acts as Anti-inflammatory Drug in Different Models of Canonical and Cell Autonomous Inflammation Through NFκB Inhibition. Front. Immunol. 2019, 10, 155. [Google Scholar] [CrossRef]
- Aquilano, K.; Baldelli, S.; La Barbera, L.; Lettieri Barbato, D.; Tatulli, G.; Ciriolo, M.R. Adipose triglyceride lipase decrement affects skeletal muscle homeostasis during aging through FAs-PPARα-PGC-1α antioxidant response. Oncotarget 2016, 7, 23019–23032. [Google Scholar] [CrossRef]
- Jocken, J.W.E.; Smit, E.; Goossens, G.H.; Essers, Y.P.G.; van Baak, M.A.; Mensink, M.; Saris, W.H.M.; Blaak, E.E. Adipose triglyceride lipase (ATGL) expression in human skeletal muscle is type I (oxidative) fiber specific. Histochem. Cell. Biol. 2008, 129, 535–538. [Google Scholar] [CrossRef]
- Vegliante, R.; Di Leo, L.; Ciccarone, F.; Ciriolo, M.R. Hints on ATGL implications in cancer: Beyond bioenergetic clues. Cell. Death Dis. 2018, 9, 316. [Google Scholar] [CrossRef] [PubMed]
- Delerive, P.; De Bosscher, K.; Besnard, S.; Vanden Berghe, W.; Peters, J.M.; Gonzalez, F.J.; Fruchart, J.C.; Tedgui, A.; Haegeman, G.; Staels, B. Peroxisome proliferator-activated receptor alpha negatively regulates the vascular inflammatory gene response by negative cross-talk with transcription factors NF-kappaB and AP-1. J. Biol. Chem. 1999, 274, 32048–32054. [Google Scholar] [CrossRef] [PubMed]
- Shelar, S.B.; Narasimhan, M.; Shanmugam, G.; Litovsky, S.H.; Gounder, S.S.; Karan, G.; Arulvasu, C.; Kensler, T.W.; Hoidal, J.R.; Darley-Usmar, V.M.; et al. Disruption of nuclear factor (erythroid-derived-2)-like 2 antioxidant signaling: A mechanism for impaired activation of stem cells and delayed regeneration of skeletal muscle. FASEB J. 2016, 30, 1865–1879. [Google Scholar] [CrossRef] [PubMed]
- Kitaoka, Y.; Tamura, Y.; Takahashi, K.; Takeda, K.; Takemasa, T.; Hatta, H. Effects of Nrf2 deficiency on mitochondrial oxidative stress in aged skeletal muscle. Physiol. Rep. 2019, 7, e13998. [Google Scholar] [CrossRef] [PubMed]
- Piccirillo, R. Exercise-Induced Myokines With Therapeutic Potential for Muscle Wasting. Front. Physiol. 2019, 10, 287. [Google Scholar] [CrossRef] [PubMed]
- Giudice, J.; Taylor, J.M. Muscle as a paracrine and endocrine organ. Curr. Opin. Pharmacol. 2017, 34, 49–55. [Google Scholar] [CrossRef] [PubMed]
- Andrich, D.E.; Melbouci, L.; Ou, Y.; Auclair, N.; Mercier, J.; Grenier, J.-C.; Lira, F.S.; Barreiro, L.B.; Danialou, G.; Comtois, A.-S.; et al. A Short-Term High-Fat Diet Alters Glutathione Levels and IL-6 Gene Expression in Oxidative Skeletal Muscles of Young Rats. Front Physiol. 2019, 10, 372. [Google Scholar] [CrossRef]
- Vassilakopoulos, T.; Karatza, M.-H.; Katsaounou, P.; Kollintza, A.; Zakynthinos, S.; Roussos, C. Antioxidants attenuate the plasma cytokine response to exercise in humans. J. Appl. Physiol. 2003, 94, 1025–1032. [Google Scholar] [CrossRef]
- Steensberg, A.; Keller, C.; Hillig, T.; Frøsig, C.; Wojtaszewski, J.F.P.; Pedersen, B.K.; Pilegaard, H.; Sander, M. Nitric oxide production is a proximal signaling event controlling exercise-induced mRNA expression in human skeletal muscle. FASEB J. 2007, 21, 2683–2694. [Google Scholar] [CrossRef]
- Fehrenbach, E.; Schneider, M.E. Trauma-Induced Systemic Inflammatory Response versus Exercise-Induced Immunomodulatory Effects. Sports Med. 2006, 36, 373–384. [Google Scholar] [CrossRef]
- Mofarrahi, M.; Brandes, R.P.; Gorlach, A.; Hanze, J.; Terada, L.S.; Quinn, M.T.; Mayaki, D.; Petrof, B.; Hussain, S.N.A. Regulation of proliferation of skeletal muscle precursor cells by NADPH oxidase. Antioxid. Redox Signal. 2008, 10, 559–574. [Google Scholar] [CrossRef] [PubMed]
- Richard-Bulteau, H.; Serrurier, B.; Crassous, B.; Banzet, S.; Peinnequin, A.; Bigard, X.; Koulmann, N. Recovery of skeletal muscle mass after extensive injury: Positive effects of increased contractile activity. Am. J. Physiol. Cell. Physiol. 2008, 294, 467–476. [Google Scholar] [CrossRef] [PubMed]
- Neufer, P.D.; Bamman, M.M.; Muoio, D.M.; Bouchard, C.; Cooper, D.M.; Goodpaster, B.H.; Booth, F.W.; Kohrt, W.M.; Gerszten, R.E.; Mattson, M.P.; et al. Understanding the Cellular and Molecular Mechanisms of Physical Activity-Induced Health Benefits. Cell. Metab. 2015, 22, 4–11. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, S.; Khazaei, M.; Moien-Afshari, F.; Ang, L.S.; Granville, D.J.; Verchere, C.B.; Dunn, S.R.; McCue, P.; Mizisin, A.; Sharma, K.; et al. Moderate exercise attenuates caspase-3 activity, oxidative stress, and inhibits progression of diabetic renal disease in db/db mice. Am. J. Physiol. Renal Physiol. 2009, 296, F700–F708. [Google Scholar] [CrossRef] [PubMed]
- Liberman, K.; Forti, L.N.; Beyer, I.; Bautmans, I. The effects of exercise on muscle strength, body composition, physical functioning and the inflammatory profile of older adults: A systematic review. Curr. Opin. Clin. Nutr. Metab. Care 2017, 20, 30–53. [Google Scholar] [CrossRef]
- Clark, B.C. In vivo alterations in skeletal muscle form and function after disuse atrophy. Med. Sci. Sports Exerc. 2009, 41, 1869–1875. [Google Scholar] [CrossRef] [PubMed]
- Londhe, P.; Guttridge, D.C. Inflammation induced loss of skeletal muscle. Bone 2015, 80, 131–142. [Google Scholar] [CrossRef]
- Talbert, E.E.; Smuder, A.J.; Min, K.; Kwon, O.S.; Powers, S.K. Calpain and caspase-3 play required roles in immobilization-induced limb muscle atrophy. J. Appl. Physiol. 2013, 114, 1482–1489. [Google Scholar] [CrossRef]
- Wang, J.; Leung, K.-S.; Chow, S.K.-H.; Cheung, W.-H. Inflammation and age-associated skeletal muscle deterioration (sarcopaenia). J. Orthop. Translat. 2017, 10, 94–101. [Google Scholar] [CrossRef]
- Wåhlin-Larsson, B.; Wilkinson, D.J.; Strandberg, E.; Hosford-Donovan, A.; Atherton, P.J.; Kadi, F. Mechanistic Links Underlying the Impact of C-Reactive Protein on Muscle Mass in Elderly. Cell. Physiol. Biochem. 2017, 44, 267–278. [Google Scholar] [CrossRef]
- Muller, F.L.; Song, W.; Jang, Y.C.; Liu, Y.; Sabia, M.; Richardson, A.; Van Remmen, H. Denervation-induced skeletal muscle atrophy is associated with increased mitochondrial ROS production. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2007, 293, 1159–1168. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.F.; Sanchez, B.J.; Hall, D.T.; Tremblay, A.-M.K.; Di Marco, S.; Gallouzi, I.-E. STAT3 promotes IFNγ/TNFα-induced muscle wasting in an NF-κB-dependent and IL-6-independent manner. EMBO Mol. Med. 2017, 9, 622–637. [Google Scholar] [CrossRef] [PubMed]
- Di Marco, S.; Mazroui, R.; Dallaire, P.; Chittur, S.; Tenenbaum, S.A.; Radzioch, D.; Marette, A.; Gallouzi, I.-E. NF-kappa B-mediated MyoD decay during muscle wasting requires nitric oxide synthase mRNA stabilization, HuR protein, and nitric oxide release. Mol. Cell. Biol. 2005, 25, 6533–6545. [Google Scholar] [CrossRef] [PubMed]
- Bae, S.-K.; Cha, H.-N.; Ju, T.-J.; Kim, Y.-W.; Kim, H.S.; Kim, Y.-D.; Dan, J.-M.; Kim, J.-Y.; Kim, S.-D.; Park, S.-Y. Deficiency of inducible nitric oxide synthase attenuates immobilization-induced skeletal muscle atrophy in mice. J. Appl. Physiol. 2012, 113, 114–123. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Li, H.; Malhotra, S.; Kumar, A. Nuclear Factor-kappa B Signaling in Skeletal Muscle Atrophy. J. Mol. Med. 2008, 86, 1113–1126. [Google Scholar] [CrossRef] [PubMed]
- Agostini, F.; Dalla Libera, L.; Rittweger, J.; Mazzucco, S.; Jurdana, M.; Mekjavic, I.B.; Pisot, R.; Gorza, L.; Narici, M.; Biolo, G. Effects of inactivity on human muscle glutathione synthesis by a double-tracer and single-biopsy approach. J. Physiol. 2010, 588, 5089–5104. [Google Scholar] [CrossRef]
- Derbre, F.; Ferrando, B.; Gomez-Cabrera, M.C.; Sanchis-Gomar, F.; Martinez-Bello, V.E.; Olaso-Gonzalez, G.; Diaz, A.; Gratas-Delamarche, A.; Cerda, M.; Viña, J. Inhibition of Xanthine Oxidase by Allopurinol Prevents Skeletal Muscle Atrophy: Role of p38 MAPKinase and E3 Ubiquitin Ligases. PLoS ONE 2012, 7, e46668. [Google Scholar] [CrossRef]
- Suzuki, N.; Motohashi, N.; Uezumi, A.; Fukada, S.; Yoshimura, T.; Itoyama, Y.; Aoki, M.; Miyagoe-Suzuki, Y.; Takeda, S. NO production results in suspension-induced muscle atrophy through dislocation of neuronal NOS. J. Clin. Investig. 2007, 117, 2468–2476. [Google Scholar] [CrossRef]
- Lomonosova, Y.N.; Kalamkarov, G.R.; Bugrova, A.E.; Shevchenko, T.F.; Kartashkina, N.L.; Lysenko, E.A.; Shvets, V.I.; Nemirovskaya, T.L. Protective effect of L-Arginine administration on proteins of unloaded m. soleus. Biochem. Mosc. 2011, 76, 571–580. [Google Scholar] [CrossRef]
- Tripathi, D.N.; Chowdhury, R.; Trudel, L.J.; Tee, A.R.; Slack, R.S.; Walker, C.L.; Wogan, G.N. Reactive nitrogen species regulate autophagy through ATM-AMPK-TSC2-mediated suppression of mTORC1. Proc. Natl. Acad. Sci. USA 2013, 110, 2950–2957. [Google Scholar] [CrossRef]
- Zhang, J.S.; Kraus, W.E.; Truskey, G.A. Stretch-induced nitric oxide modulates mechanical properties of skeletal muscle cells. Am. J. Physiol. Cell. Physiol. 2004, 287, 292–299. [Google Scholar] [CrossRef] [PubMed]
- Sellman, J.E.; DeRuisseau, K.C.; Betters, J.L.; Lira, V.A.; Soltow, Q.A.; Selsby, J.T.; Criswell, D.S. In vivo inhibition of nitric oxide synthase impairs upregulation of contractile protein mRNA in overloaded plantaris muscle. J. Appl. Physiol. 2006, 100, 258–265. [Google Scholar] [CrossRef] [PubMed]
- Tatsumi, R.; Hattori, A.; Ikeuchi, Y.; Anderson, J.E.; Allen, R.E. Release of Hepatocyte Growth Factor from Mechanically Stretched Skeletal Muscle Satellite Cells and Role of pH and Nitric Oxide. Mol. Biol. Cell. 2002, 13, 2909–2918. [Google Scholar] [CrossRef] [PubMed]
- Tatsumi, R.; Sheehan, S.M.; Iwasaki, H.; Hattori, A.; Allen, R.E. Mechanical stretch induces activation of skeletal muscle satellite cells in vitro. Exp. Cell. Res. 2001, 267, 107–114. [Google Scholar] [CrossRef]




© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Baldelli, S.; Ciccarone, F.; Limongi, D.; Checconi, P.; Palamara, A.T.; Ciriolo, M.R. Glutathione and Nitric Oxide: Key Team Players in Use and Disuse of Skeletal Muscle. Nutrients 2019, 11, 2318. https://doi.org/10.3390/nu11102318
Baldelli S, Ciccarone F, Limongi D, Checconi P, Palamara AT, Ciriolo MR. Glutathione and Nitric Oxide: Key Team Players in Use and Disuse of Skeletal Muscle. Nutrients. 2019; 11(10):2318. https://doi.org/10.3390/nu11102318
Chicago/Turabian StyleBaldelli, Sara, Fabio Ciccarone, Dolores Limongi, Paola Checconi, Anna Teresa Palamara, and Maria Rosa Ciriolo. 2019. "Glutathione and Nitric Oxide: Key Team Players in Use and Disuse of Skeletal Muscle" Nutrients 11, no. 10: 2318. https://doi.org/10.3390/nu11102318
APA StyleBaldelli, S., Ciccarone, F., Limongi, D., Checconi, P., Palamara, A. T., & Ciriolo, M. R. (2019). Glutathione and Nitric Oxide: Key Team Players in Use and Disuse of Skeletal Muscle. Nutrients, 11(10), 2318. https://doi.org/10.3390/nu11102318