Dietary Influence on Body Fluid Acid-Base and Volume Balance: The Deleterious “Norm” Furthers and Cloaks Subclinical Pathophysiology

{kind=link}
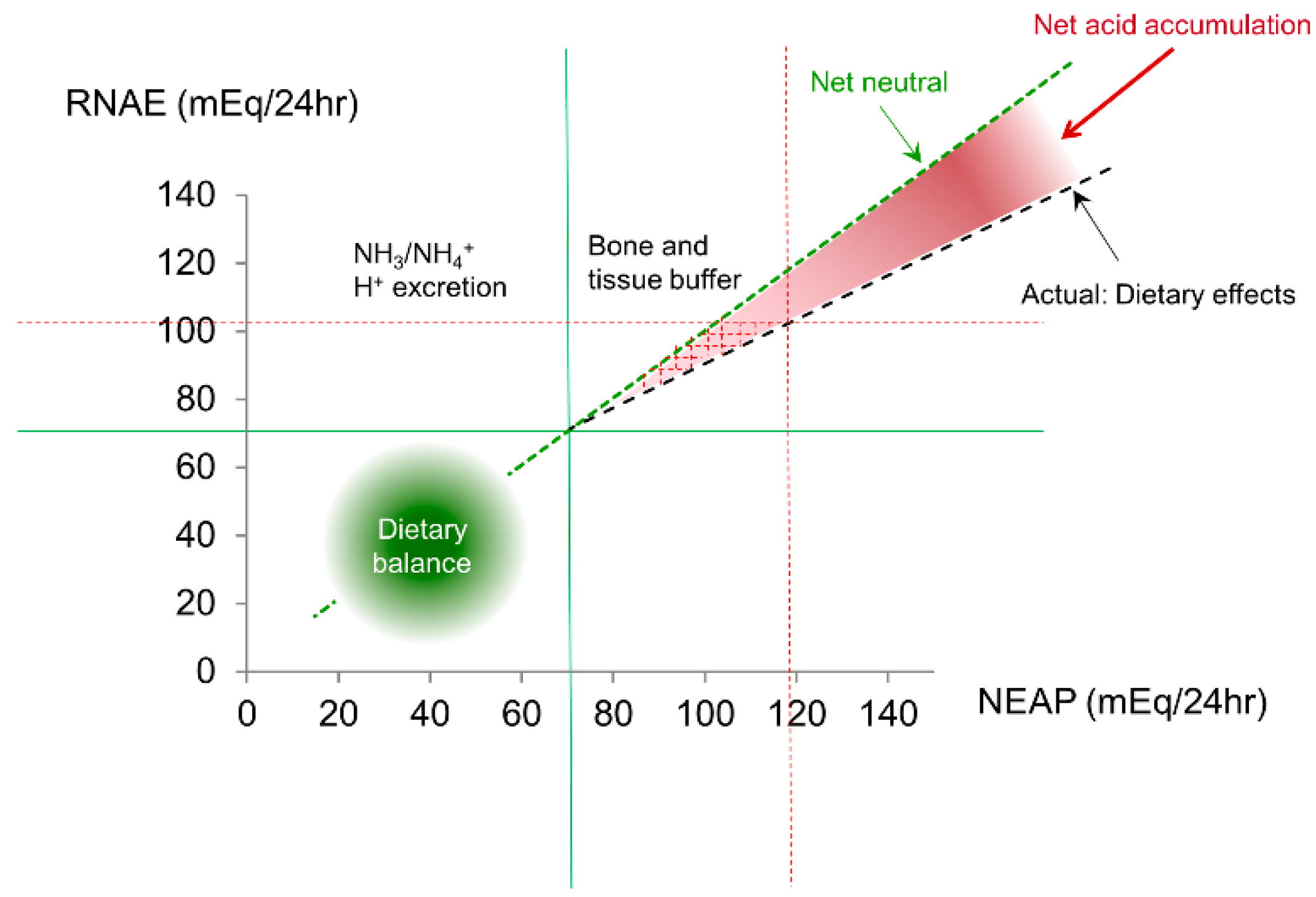{kind=link}
Abstract
:1. Introduction
2. Changes in Key Dietary Nutrient Compositions Since the Ancestral and Premodern Diet
3. Role of the Kidneys in the Active Maintenance of Body-Fluid Balance
3.1. Acid-Base and Potassium Balance
3.2. Salt and Water Balance
4. Maladaptations to the Modern Diet and Health Implications
4.1. Net Acid Retention
4.2. Inadequate Dietary Potassium (K+)
4.3. Salt Overconsumption and Insufficient Hydration
4.4. Excess Urea Production and Metabolism
5. Prevention of Acid-Base and Electrolyte Maladaptive Responses, the Power of Dietary Modification
6. Summary
Conflicts of Interest
References
- Scientific report of the 2015 Dietary Guideline Advisory Committee part A. Available online: https://health.gov/dietaryguidelines/2015-scientific-report/ (accessed on 30 May 2018).
- Millen, B.E.; Abrams, S.; Adams-Campbell, L.; Anderson, C.A.; Brenna, J.T.; Campbell, W.W.; Clinton, S.; Hu, F.; Nelson, M.; Neuhouser, M.L.; et al. The 2015 Dietary Guidelines Advisory Committee Scientific Report: Development and Major Conclusions. Adv. Nutr. 2016, 7, 438–444. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferreira-Pego, C.; Guelinckx, I.; Moreno, L.A.; Kavouras, S.A.; Gandy, J.; Martinez, H.; Bardosono, S.; Abdollahi, M.; Nasseri, E.; Jarosz, A.; et al. Total fluid intake and its determinants: Cross-sectional surveys among adults in 13 countries worldwide. Eur. J. Nutr. 2015, 54 (Suppl. 2), 35–43. [Google Scholar] [CrossRef] [PubMed]
- Iglesia, I.; Guelinckx, I.; De Miguel-Etayo, P.M.; Gonzalez-Gil, E.M.; Salas-Salvado, J.; Kavouras, S.A.; Gandy, J.; Martinez, H.; Bardosono, S.; Abdollahi, M.; et al. Total fluid intake of children and adolescents: Cross-sectional surveys in 13 countries worldwide. Eur. J. Nutr. 2015, 54 (Suppl. 2), 57–67. [Google Scholar] [CrossRef] [PubMed]
- Kurtz, I.; Maher, T.; Hulter, H.N.; Schambelan, M.; Sebastian, A. Effect of diet on plasma acid-base composition in normal humans. Kidney Int. 1983, 24, 670–680. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. Guideline: Potassium Intake for Adults and Children; WHO: Geneva, Switzerland, 2012. [Google Scholar]
- David, L.A.; Maurice, C.F.; Carmody, R.N.; Gootenberg, D.B.; Button, J.E.; Wolfe, B.E.; Ling, A.V.; Devlin, A.S.; Varma, Y.; Fischbach, M.A.; et al. Diet rapidly and reproducibly alters the human gut microbiome. Nature 2014, 505, 559–563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hietavala, E.M.; Ihalainen, J.K.; Frassetto, L.A.; Schumann, M.; Eklund, D.; Pitkanen, H.; Hakkinen, K.; Mero, A.A. Effects of 12-Week Low or Moderate Dietary Acid Intake on Acid-Base Status and Kidney Function at Rest and during Submaximal Cycling. Nutrients 2018, 10, 323. [Google Scholar] [CrossRef] [PubMed]
- Remer, T.; Dimitriou, T.; Manz, F. Dietary potential renal acid load and renal net acid excretion in healthy, free-living children and adolescents. Am. J. Clin. Nutr. 2003, 77, 1255–1260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frassetto, L.A.; Todd, K.M.; Morris, R.C., Jr.; Sebastian, A. Estimation of net endogenous noncarbonic acid production in humans from diet potassium and protein contents. Am. J. Clin. Nutr. 1998, 68, 576–583. [Google Scholar] [CrossRef] [PubMed]
- Sebastian, A.; Frassetto, L.A.; Sellmeyer, D.E.; Merriam, R.L.; Morris, R.C., Jr. Estimation of the net acid load of the diet of ancestral preagricultural Homo sapiens and their hominid ancestors. Am. J. Clin. Nutr. 2002, 76, 1308–1316. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Cann, M.J.; Litvin, T.N.; Iourgenko, V.; Sinclair, M.L.; Levin, L.R.; Buck, J. Soluble adenylyl cyclase as an evolutionarily conserved bicarbonate sensor. Science 2000, 289, 625–628. [Google Scholar] [CrossRef] [PubMed]
- Dhondup, T.; Qian, Q. Acid-Base and Electrolyte Disorders in Patients with and without Chronic Kidney Disease: An Update. Kidney Dis. 2017, 3, 136–148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schulze Blasum, B.; Schroter, R.; Neugebauer, U.; Hofschroer, V.; Pavenstadt, H.; Ciarimboli, G.; Schlatter, E.; Edemir, B. The kidney-specific expression of genes can be modulated by the extracellular osmolality. FASEB J. 2016, 30, 3588–3597. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rakova, N.; Kitada, K.; Lerchl, K.; Dahlmann, A.; Birukov, A.; Daub, S.; Kopp, C.; Pedchenko, T.; Zhang, Y.; Beck, L.; et al. Increased salt consumption induces body water conservation and decreases fluid intake. J. Clin. Investig. 2017, 127, 1932–1943. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kitada, K.; Daub, S.; Zhang, Y.; Klein, J.D.; Nakano, D.; Pedchenko, T.; Lantier, L.; LaRocque, L.M.; Marton, A.; Neubert, P.; et al. High salt intake reprioritizes osmolyte and energy metabolism for body fluid conservation. J. Clin. Investig. 2017, 127, 1944–1959. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qian, Q. Salt, water and nephron: Mechanisms of action and link to hypertension and chronic kidney disease. Nephron 2018, in press. [Google Scholar]
- Enhorning, S.; Christensson, A.; Melander, O. Plasma copeptin as a predictor of kidney disease. Nephrol. Dial. Transplant. 2018. [Google Scholar] [CrossRef] [PubMed]
- Hill, N.R.; Fatoba, S.T.; Oke, J.L.; Hirst, J.A.; O’Callaghan, C.A.; Lasserson, D.S.; Hobbs, F.D. Global Prevalence of Chronic Kidney Disease—A Systematic Review and Meta-Analysis. PLoS ONE 2016, 11, e0158765. [Google Scholar] [CrossRef] [PubMed]
- Kopp, C.; Linz, P.; Dahlmann, A.; Hammon, M.; Jantsch, J.; Muller, D.N.; Schmieder, R.E.; Cavallaro, A.; Eckardt, K.U.; Uder, M.; et al. 23Na magnetic resonance imaging-determined tissue sodium in healthy subjects and hypertensive patients. Hypertension 2013, 61, 635–640. [Google Scholar] [CrossRef] [PubMed]
- Nikpey, E.; Karlsen, T.V.; Rakova, N.; Titze, J.M.; Tenstad, O.; Wiig, H. High-Salt Diet Causes Osmotic Gradients and Hyperosmolality in Skin Without Affecting Interstitial Fluid and Lymph. Hypertension 2017, 69, 660–668. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schneider, M.P.; Raff, U.; Kopp, C.; Scheppach, J.B.; Toncar, S.; Wanner, C.; Schlieper, G.; Saritas, T.; Floege, J.; Schmid, M.; et al. Skin Sodium Concentration Correlates with Left Ventricular Hypertrophy in CKD. J. Am. Soc. Nephrol. 2017, 28, 1867–1876. [Google Scholar] [CrossRef] [PubMed]
- Smit, E.; Nieto, F.J.; Crespo, C.J.; Mitchell, P. Estimates of animal and plant protein intake in US adults: Results from the Third National Health and Nutrition Examination Survey, 1988–1991. J. Am. Diet. Assoc. 1999, 99, 813–820. [Google Scholar] [CrossRef]
- Frassetto, L.A.; Morris, R.C., Jr.; Sebastian, A. Effect of age on blood acid-base composition in adult humans: Role of age-related renal functional decline. Am. J. Physiol. 1996, 271, F1114–F1122. [Google Scholar] [CrossRef] [PubMed]
- Wesson, D.E.; Pruszynski, J.; Cai, W.; Simoni, J. Acid retention with reduced glomerular filtration rate increases urine biomarkers of kidney and bone injury. Kidney Int. 2017, 91, 914–927. [Google Scholar] [CrossRef] [PubMed]
- Zha, Y.; Qian, Q. Protein Nutrition and Malnutrition in CKD and ESRD. Nutrients 2017, 9, 208. [Google Scholar] [CrossRef] [PubMed]
- Goraya, N.; Simoni, J.; Sager, L.N.; Pruszynski, J.; Wesson, D.E. Acid retention in chronic kidney disease is inversely related to GFR. Am. J. Physiol. Ren. Physiol. 2018, 314, F985–F991. [Google Scholar] [CrossRef] [PubMed]
- Wesson, D.E.; Simoni, J.; Broglio, K.; Sheather, S. Acid retention accompanies reduced GFR in humans and increases plasma levels of endothelin and aldosterone. Am. J. Physiol. Ren. Physiol. 2011, 300, F830–F837. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- So, R.; Song, S.; Lee, J.E.; Yoon, H.J. The Association between Renal Hyperfiltration and the Sources of Habitual Protein Intake and Dietary Acid Load in a General Population with Preserved Renal Function: The KoGES Study. PLoS ONE 2016, 11, e0166495. [Google Scholar] [CrossRef] [PubMed]
- Ko, B.J.; Chang, Y.; Ryu, S.; Kim, E.M.; Lee, M.Y.; Hyun, Y.Y.; Lee, K.B. Dietary acid load and chronic kidney disease in elderly adults: Protein and potassium intake. PLoS ONE 2017, 12, e0185069. [Google Scholar] [CrossRef] [PubMed]
- Haring, B.; Selvin, E.; Liang, M.; Coresh, J.; Grams, M.E.; Petruski-Ivleva, N.; Steffen, L.M.; Rebholz, C.M. Dietary Protein Sources and Risk for Incident Chronic Kidney Disease: Results From the Atherosclerosis Risk in Communities (ARIC) Study. J. Ren. Nutr. 2017, 27, 233–242. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, T.; Crews, D.C.; Wesson, D.E.; Tilea, A.; Saran, R.; Burrows, N.R.; Williams, D.E.; Powe, N.R. Dietary acid load and chronic kidney disease among adults in the United States. BMC Nephrol. 2014, 15, 137. [Google Scholar] [CrossRef] [PubMed]
- Mirmiran, P.; Yuzbashian, E.; Bahadoran, Z.; Asghari, G.; Azizi, F. Dietary Acid-Base Load and Risk of Chronic Kidney Disease in Adults: Tehran Lipid and Glucose Study. Iran. J. Kidney Dis. 2016, 10, 119–125. [Google Scholar] [PubMed]
- Rebholz, C.M.; Coresh, J.; Grams, M.E.; Steffen, L.M.; Anderson, C.A.; Appel, L.J.; Crews, D.C. Dietary Acid Load and Incident Chronic Kidney Disease: Results from the ARIC Study. Am. J. Nephrol. 2015, 42, 427–435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, M.; Jung, S.J.; Yoon, S.; Yun, J.M.; Yoon, H.J. Association between the markers of metabolic acid load and higher all-cause and cardiovascular mortality in a general population with preserved renal function. Hypertens. Res. 2015, 38, 433–438. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Akesson, A.; Orsini, N.; Hakansson, N.; Wolk, A.; Carrero, J.J. Modest U-Shaped Association between Dietary Acid Load and Risk of All-Cause and Cardiovascular Mortality in Adults. J. Nutr. 2016, 146, 1580–1585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Sun, S.R.; Yap, J.Q.; Chen, J.H.; Qian, Q. 0.9% saline is neither normal nor physiological. J. Zhejiang Univ. Sci. B 2016, 17, 181–187. [Google Scholar] [CrossRef] [PubMed]
- Kinoshita, H.; Fujimoto, S.; Nakazato, M.; Yokota, N.; Date, Y.; Yamaguchi, H.; Hisanaga, S.; Eto, T. Urine and plasma levels of uroguanylin and its molecular forms in renal diseases. Kidney Int. 1997, 52, 1028–1034. [Google Scholar] [CrossRef] [PubMed]
- Wall, S.M.; Lazo-Fernandez, Y. The role of pendrin in renal physiology. Annu. Rev. Physiol. 2015, 77, 363–378. [Google Scholar] [CrossRef] [PubMed]
- Sindic, A.; Schlatter, E. Cellular effects of guanylin and uroguanylin. J. Am. Soc. Nephrol. 2006, 17, 607–616. [Google Scholar] [CrossRef] [PubMed]
- Rozenfeld, J.; Tal, O.; Kladnitsky, O.; Adler, L.; Efrati, E.; Carrithers, S.L.; Alper, S.L.; Zelikovic, I. The pendrin anion exchanger gene is transcriptionally regulated by uroguanylin: A novel enterorenal link. Am. J. Physiol. Ren. Physiol. 2012, 302, F614–F624. [Google Scholar] [CrossRef] [PubMed]
- Frassetto, L.A.; Morris, R.C., Jr.; Sebastian, A. Dietary sodium chloride intake independently predicts the degree of hyperchlore mic metabolic acidosis in healthy humans consuming a net acid-producing diet. Am. J. Physiol. Ren. Physiol. 2007, 293, F521–F525. [Google Scholar] [CrossRef] [PubMed]
- Esche, J.; Shi, L.; Sanchez-Guijo, A.; Hartmann, M.F.; Wudy, S.A.; Remer, T. Higher diet-dependent renal acid load associates with higher glucocorticoid secretion and potentially bioactive free glucocorticoids in healthy children. Kidney Int. 2016, 90, 325–333. [Google Scholar] [CrossRef] [PubMed]
- Shi, L.; Sanchez-Guijo, A.; Hartmann, M.F.; Schonau, E.; Esche, J.; Wudy, S.A.; Remer, T. Higher glucocorticoid secretion in the physiological range is associated with lower bone strength at the proximal radius in healthy children: Importance of protein intake adjustment. J. Bone Miner. Res. 2015, 30, 240–248. [Google Scholar] [CrossRef] [PubMed]
- Brungger, M.; Hulter, H.N.; Krapf, R. Effect of chronic metabolic acidosis on the growth hormone/IGF-1 endocrine axis: New cause of growth hormone insensitivity in humans. Kidney Int. 1997, 51, 216–221. [Google Scholar] [CrossRef] [PubMed]
- Jia, T.; Gama Axelsson, T.; Heimburger, O.; Barany, P.; Lindholm, B.; Stenvinkel, P.; Qureshi, A.R. IGF-1 and survival in ESRD. Clin. J. Am. Soc. Nephrol. 2014, 9, 120–127. [Google Scholar] [CrossRef] [PubMed]
- Williams, R.S.; Kozan, P.; Samocha-Bonet, D. The role of dietary acid load and mild metabolic acidosis in insulin resistance in humans. Biochimie 2016, 124, 171–177. [Google Scholar] [CrossRef] [PubMed]
- Dobre, M.; Gaussoin, S.A.; Bates, J.T.; Chonchol, M.B.; Cohen, D.L.; Hostetter, T.H.; Raphael, K.L.; Taylor, A.A.; Lerner, A.J.; Wright, J.T., Jr.; et al. Serum Bicarbonate Concentration and Cognitive Function in Hypertensive Adults. Clin. J. Am. Soc. Nephrol. 2018, 13, 596–603. [Google Scholar] [CrossRef] [PubMed]
- Qian, Q. Inflammation: A Key Contributor to the Genesis and Progression of Chronic Kidney Disease. Contrib. Nephrol. 2017, 191, 72–83. [Google Scholar] [PubMed]
- Kieneker, L.M.; Eisenga, M.F.; Joosten, M.M.; de Boer, R.A.; Gansevoort, R.T.; Kootstra-Ros, J.E.; Navis, G.; Bakker, S.J. Plasma potassium, diuretic use and risk of developing chronic kidney disease in a predominantly White population. PLoS ONE 2017, 12, e0174686. [Google Scholar] [CrossRef] [PubMed]
- Blumenthal, J.A.; Babyak, M.A.; Hinderliter, A.; Watkins, L.L.; Craighead, L.; Lin, P.H.; Caccia, C.; Johnson, J.; Waugh, R.; Sherwood, A. Effects of the DASH diet alone and in combination with exercise and weight loss on blood pressure and cardiovascular biomarkers in men and women with high blood pressure: The ENCORE study. Arch. Intern. Med. 2010, 170, 126–135. [Google Scholar] [CrossRef] [PubMed]
- Young, D.B.; Lin, H.; McCabe, R.D. Potassium’s cardiovascular protective mechanisms. Am. J. Physiol. 1995, 268, R825–R837. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Byon, C.H.; Yang, Y.; Bradley, W.E.; Dell’Italia, L.J.; Sanders, P.W.; Agarwal, A.; Wu, H.; Chen, Y. Dietary potassium regulates vascular calcification and arterial stiffness. JCI Insight 2017, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jackson, S.L.; Cogswell, M.E.; Zhao, L.; Terry, A.L.; Wang, C.Y.; Wright, J.; Coleman King, S.M.; Bowman, B.; Chen, T.C.; Merritt, R.; et al. Association between Urinary Sodium and Potassium Excretion and Blood Pressure among Adults in the United States: National Health and Nutrition Examination Survey, 2014. Circulation 2018, 137, 237–246. [Google Scholar] [CrossRef] [PubMed]
- Kieneker, L.M.; Bakker, S.J.; de Boer, R.A.; Navis, G.J.; Gansevoort, R.T.; Joosten, M.M. Low potassium excretion but not high sodium excretion is associated with increased risk of developing chronic kidney disease. Kidney Int. 2016, 90, 888–896. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.; McFann, K.; Chonchol, M.; de Boer, I.H.; Kendrick, J. Association between dietary sodium and potassium intake with chronic kidney disease in US adults: A cross-sectional study. Am. J. Nephrol. 2013, 37, 526–533. [Google Scholar] [CrossRef] [PubMed]
- Leonberg-Yoo, A.K.; Tighiouart, H.; Levey, A.S.; Beck, G.J.; Sarnak, M.J. Urine Potassium Excretion, Kidney Failure, and Mortality in CKD. Am. J. Kidney Dis. 2017, 69, 341–349. [Google Scholar] [CrossRef] [PubMed]
- Smyth, A.; Dunkler, D.; Gao, P.; Teo, K.K.; Yusuf, S.; O’donnell, M.J.; Mann, J.F.; Clase, C.M. The relationship between estimated sodium and potassium excretion and subsequent renal outcomes. Kidney Int. 2014, 86, 1205–1212. [Google Scholar] [CrossRef] [PubMed]
- Ribeiro, S.C.; Figueiredo, A.E.; Barretti, P.; Pecoits-Filho, R.; de Moraes, T.P. Low Serum Potassium Levels Increase the Infectious-Caused Mortality in Peritoneal Dialysis Patients: A Propensity-Matched Score Study. PLoS ONE 2015, 10, e0127453. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Kang, E.; Yoo, K.D.; Choi, Y.; Kim, D.K.; Joo, K.W.; Yang, S.H.; Kim, Y.L.; Kang, S.W.; Yang, C.W.; et al. Lower serum potassium associated with increased mortality in dialysis patients: A nationwide prospective observational cohort study in Korea. PLoS ONE 2017, 12, e0171842. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. Guideline: Sodium Intake for Adults and Children; WHO: Geneva, Switzerland, 2012. [Google Scholar]
- Sumida, K.; Molnar, M.Z.; Potukuchi, P.K.; George, K.; Thomas, F.; Lu, J.L.; Yamagata, K.; Kalantar-Zadeh, K.; Kovesdy, C.P. Changes in Albuminuria and Subsequent Risk of Incident Kidney Disease. Clin. J. Am. Soc. Nephrol. 2017, 12, 1941–1949. [Google Scholar] [CrossRef] [PubMed]
- Kannel, W.B.; Stampfer, M.J.; Castelli, W.P.; Verter, J. The prognostic significance of proteinuria: The Framingham study. Am. Heart J. 1984, 108, 1347–1352. [Google Scholar] [CrossRef]
- Carrero, J.J.; Grams, M.E.; Sang, Y.; Arnlov, J.; Gasparini, A.; Matsushita, K.; Qureshi, A.R.; Evans, M.; Barany, P.; Lindholm, B.; et al. Albuminuria changes are associated with subsequent risk of end-stage renal disease and mortality. Kidney Int. 2017, 91, 244–251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meijer, E.; Bakker, S.J.; Halbesma, N.; de Jong, P.E.; Struck, J.; Gansevoort, R.T. Copeptin, a surrogate marker of vasopressin, is associated with microalbuminuria in a large population cohort. Kidney Int. 2010, 77, 29–36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ponte, B.; Pruijm, M.; Ackermann, D.; Vuistiner, P.; Guessous, I.; Ehret, G.; Alwan, H.; Youhanna, S.; Paccaud, F.; Mohaupt, M.; et al. Copeptin is associated with kidney length, renal function, and prevalence of simple cysts in a population-based study. J. Am. Soc. Nephrol. 2015, 26, 1415–1425. [Google Scholar] [CrossRef] [PubMed]
- Alvarez-Aguilar, C.; Arroyo-Ramirez, E.; Gomez-Garcia, A.; Alvarez-Paredes, A.R.; Rodriguez-Orozco, A.R.; Flores-Guajardo, G.; Rangel-Lopez, A. BNP predicts mortality of cardiovascular disease in patients with end-stage renal disease treated. Rev. Med. Inst. Mex. Seguro Soc. 2018, 55 (Suppl. 2), S158–S166. [Google Scholar]
- York, M.K.; Gupta, D.K.; Reynolds, C.F.; Farber-Eger, E.; Wells, Q.S.; Bachmann, K.N.; Xu, M.; Harrell, F.E., Jr.; Wang, T.J. B-Type Natriuretic Peptide Levels and Mortality in Patients with and without Heart Failure. J. Am. Coll. Cardiol. 2018, 71, 2079–2088. [Google Scholar] [CrossRef] [PubMed]
- Christ-Crain, M.; Fenske, W. Copeptin in the diagnosis of vasopressin-dependent disorders of fluid homeostasis. Nat. Rev. Endocrinol. 2016, 12, 168–176. [Google Scholar] [CrossRef] [PubMed]
- Sabat, R.; Ouyang, W.; Wolk, K. Therapeutic opportunities of the IL-22-IL-22R1 system. Nat. Rev. Drug Discov. 2014, 13, 21–38. [Google Scholar] [CrossRef] [PubMed]
- Wilck, N.; Matus, M.G.; Kearney, S.M.; Olesen, S.W.; Forslund, K.; Bartolomaeus, H.; Haase, S.; Mahler, A.; Balogh, A.; Marko, L.; et al. Salt-responsive gut commensal modulates TH17 axis and disease. Nature 2017, 551, 585–589. [Google Scholar] [CrossRef] [PubMed]
- Faraco, G.; Brea, D.; Garcia-Bonilla, L.; Wang, G.; Racchumi, G.; Chang, H.; Buendia, I.; Santisteban, M.M.; Segarra, S.G.; Koizumi, K.; et al. Dietary salt promotes neurovascular and cognitive dysfunction through a gut-initiated TH17 response. Nat. Neurosci. 2018, 21, 240–249. [Google Scholar] [CrossRef] [PubMed]
- Maughan, R.J. Impact of mild dehydration on wellness and on exercise performance. Eur. J. Clin. Nutr. 2003, 57 (Suppl. 2), S19–S23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siener, R.; Hesse, A. Fluid intake and epidemiology of urolithiasis. Eur. J. Clin. Nutr. 2003, 57 (Suppl. 2), S47–S51. [Google Scholar] [CrossRef] [PubMed]
- Thornton, S.N. Increased Hydration Can Be Associated with Weight Loss. Front. Nutr. 2016, 3, 18. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.J.; Grantham, J.J.; Wetmore, J.B. The medicinal use of water in renal disease. Kidney Int. 2013, 84, 45–53. [Google Scholar] [CrossRef] [PubMed]
- Vaziri, N.D.; Zhao, Y.Y.; Pahl, M.V. Altered intestinal microbial flora and impaired epithelial barrier structure and function in CKD: The nature, mechanisms, consequences and potential treatment. Nephrol. Dial. Transplant. 2016, 31, 737–746. [Google Scholar] [CrossRef] [PubMed]
- Ramezani, A.; Massy, Z.A.; Meijers, B.; Evenepoel, P.; Vanholder, R.; Raj, D.S. Role of the Gut Microbiome in Uremia: A Potential Therapeutic Target. Am. J. Kidney Dis. 2016, 67, 483–498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Velasquez, M.T.; Ramezani, A.; Manal, A.; Raj, D.S. Trimethylamine N-Oxide: The Good, the Bad and the Unknown. Toxins 2016, 8, 326. [Google Scholar] [CrossRef] [PubMed]
- Vanholder, R.; Gryp, T.; Glorieux, G. Urea and chronic kidney disease: The comeback of the century? (in uraemia research). Nephrol. Dial. Transplant. 2018, 33, 4–12. [Google Scholar] [CrossRef] [PubMed]
- De Santis, S.; Cavalcanti, E.; Mastronardi, M.; Jirillo, E.; Chieppa, M. Nutritional Keys for Intestinal Barrier Modulation. Front. Immunol. 2015, 6, 612. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, M. Interaction between food substances and the intestinal epithelium. Biosci. Biotechnol. Biochem. 2010, 74, 232–241. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Goel, R.; Kumar, A.; Qi, Y.; Lobaton, G.; Hosaka, K.; Mohammed, M.; Handberg, E.M.; Richards, E.M.; Pepine, C.J.; et al. Imbalance of gut microbiome and intestinal epithelial barrier dysfunction in patients with high blood pressure. Clin. Sci. 2018, 132, 701–718. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santisteban, M.M.; Qi, Y.; Zubcevic, J.; Kim, S.; Yang, T.; Shenoy, V.; Cole-Jeffrey, C.T.; Lobaton, G.O.; Stewart, D.C.; Rubiano, A.; et al. Hypertension-Linked Pathophysiological Alterations in the Gut. Circ. Res. 2017, 120, 312–323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pluznick, J. A novel SCFA receptor, the microbiota, and blood pressure regulation. Gut Microbes 2014, 5, 202–207. [Google Scholar] [CrossRef] [PubMed]
- Stern, J.M.; Moazami, S.; Qiu, Y.; Kurland, I.; Chen, Z.; Agalliu, I.; Burk, R.; Davies, K.P. Evidence for a distinct gut microbiome in kidney stone formers compared to non-stone formers. Urolithiasis 2016, 44, 399–407. [Google Scholar] [CrossRef] [PubMed]
- Vaziri, N.D.; Yuan, J.; Norris, K. Role of urea in intestinal barrier dysfunction and disruption of epithelial tight junction in chronic kidney disease. Am. J. Nephrol. 2013, 37, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Aaron, K.J.; Sanders, P.W. Role of dietary salt and potassium intake in cardiovascular health and disease: A review of the evidence. Mayo Clin. Proc. 2013, 88, 987–995. [Google Scholar] [CrossRef] [PubMed]
- Aburto, N.J.; Hanson, S.; Gutierrez, H.; Hooper, L.; Elliott, P.; Cappuccio, F.P. Effect of increased potassium intake on cardiovascular risk factors and disease: Systematic review and meta-analyses. BMJ 2013, 346, f1378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, B.; Bauman, A.; Gale, J.; Banks, E.; Kritharides, L.; Ding, D. Fruit and vegetable consumption and all-cause mortality: Evidence from a large Australian cohort study. Int. J. Behav. Nutr. Phys. Act. 2016, 13, 9. [Google Scholar] [CrossRef] [PubMed]
- Leenders, M.; Boshuizen, H.C.; Ferrari, P.; Siersema, P.D.; Overvad, K.; Tjonneland, A.; Olsen, A.; Boutron-Ruault, M.C.; Dossus, L.; Dartois, L.; et al. Fruit and vegetable intake and cause-specific mortality in the EPIC study. Eur. J. Epidemiol. 2014, 29, 639–652. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Ouyang, Y.; Liu, J.; Zhu, M.; Zhao, G.; Bao, W.; Hu, F.B. Fruit and vegetable consumption and mortality from all causes, cardiovascular disease, and cancer: Systematic review and dose-response meta-analysis of prospective cohort studies. BMJ 2014, 349, g4490. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwingshackl, L.; Schwedhelm, C.; Hoffmann, G.; Lampousi, A.M.; Knuppel, S.; Iqbal, K.; Bechthold, A.; Schlesinger, S.; Boeing, H. Food groups and risk of all-cause mortality: A systematic review and meta-analysis of prospective studies. Am. J. Clin. Nutr. 2017, 105, 1462–1473. [Google Scholar] [CrossRef] [PubMed]
- Geleijnse, J.M.; Kok, F.J.; Grobbee, D.E. Blood pressure response to changes in sodium and potassium intake: A metaregression analysis of randomised trials. J. Hum. Hypertens. 2003, 17, 471–480. [Google Scholar] [CrossRef] [PubMed]
- Whelton, P.K.; He, J.; Cutler, J.A.; Brancati, F.L.; Appel, L.J.; Follmann, D.; Klag, M.J. Effects of oral potassium on blood pressure. Meta-analysis of randomized controlled clinical trials. JAMA 1997, 277, 1624–1632. [Google Scholar] [CrossRef] [PubMed]
- Ceglia, L.; Harris, S.S.; Abrams, S.A.; Rasmussen, H.M.; Dallal, G.E.; Dawson-Hughes, B. Potassium bicarbonate attenuates the urinary nitrogen excretion that accompanies an increase in dietary protein and may promote calcium absorption. J. Clin. Endocrinol. Metab. 2009, 94, 645–653. [Google Scholar] [CrossRef] [PubMed]
- Frassetto, L.; Morris, R.C., Jr.; Sebastian, A. Potassium bicarbonate reduces urinary nitrogen excretion in postmenopausal women. J. Clin. Endocrinol. Metab. 1997, 82, 254–259. [Google Scholar] [CrossRef] [PubMed]
- Lemann, J., Jr.; Gray, R.W.; Pleuss, J.A. Potassium bicarbonate, but not sodium bicarbonate, reduces urinary calcium excretion and improves calcium balance in healthy men. Kidney Int. 1989, 35, 688–695. [Google Scholar] [CrossRef] [PubMed]
- Park, M.; So, R.; Joo, K.W.; Yoon, H.J. Association between lower serum bicarbonate and renal hyperfiltration in the general population with preserved renal function: A cross-sectional study. BMC Nephrol. 2016, 17, 3. [Google Scholar] [CrossRef] [PubMed]
- Lew, Q.J.; Jafar, T.H.; Koh, H.W.; Jin, A.; Chow, K.Y.; Yuan, J.M.; Koh, W.P. Red Meat Intake and Risk of ESRD. J. Am. Soc. Nephrol. 2017, 28, 304–312. [Google Scholar] [CrossRef] [PubMed]
- Buehlmeier, J.; Remer, T.; Frings-Meuthen, P.; Maser-Gluth, C.; Heer, M. Glucocorticoid activity and metabolism with NaCl-induced low-grade metabolic acidosis and oral alkalization: Results of two randomized controlled trials. Endocrine 2016, 52, 139–147. [Google Scholar] [CrossRef] [PubMed]
- Benton, D.; Burgess, N. The effect of the consumption of water on the memory and attention of children. Appetite 2009, 53, 143–146. [Google Scholar] [CrossRef] [PubMed]
- Edmonds, C.J.; Burford, D. Should children drink more water?: The effects of drinking water on cognition in children. Appetite 2009, 52, 776–779. [Google Scholar] [CrossRef] [PubMed]
- Edmonds, C.J.; Jeffes, B. Does having a drink help you think? 6-7-Year-old children show improvements in cognitive performance from baseline to test after having a drink of water. Appetite 2009, 53, 469–472. [Google Scholar] [CrossRef] [PubMed]
- Kuwabara, M.; Hisatome, I.; Roncal-Jimenez, C.A.; Niwa, K.; Andres-Hernando, A.; Jensen, T.; Bjornstad, P.; Milagres, T.; Cicerchi, C.; Song, Z.; et al. Increased Serum Sodium and Serum Osmolarity Are Independent Risk Factors for Developing Chronic Kidney Disease; 5 Year Cohort Study. PLoS ONE 2017, 12, e0169137. [Google Scholar] [CrossRef] [PubMed]
- Clark, W.F.; Sontrop, J.M.; Huang, S.H.; Gallo, K.; Moist, L.; House, A.A.; Cuerden, M.S.; Weir, M.A.; Bagga, A.; Brimble, S.; et al. Effect of Coaching to Increase Water Intake on Kidney Function Decline in Adults With Chronic Kidney Disease: The CKD WIT Randomized Clinical Trial. JAMA 2018, 319, 1870–1879. [Google Scholar] [CrossRef] [PubMed]
- Jessri, M.; Lou, W.Y.; L’Abbe, M.R. The 2015 Dietary Guidelines for Americans is associated with a more nutrient-dense diet and a lower risk of obesity. Am. J. Clin. Nutr. 2016, 104, 1378–1392. [Google Scholar] [CrossRef] [PubMed]
- Voortman, T.; Kiefte-de Jong, J.C.; Ikram, M.A.; Stricker, B.H.; van Rooij, F.J.A.; Lahousse, L.; Tiemeier, H.; Brusselle, G.G.; Franco, O.H.; Schoufour, J.D. Adherence to the 2015 Dutch dietary guidelines and risk of non-communicable diseases and mortality in the Rotterdam Study. Eur. J. Epidemiol. 2017, 32, 993–1005. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kromhout, D.; Spaaij, C.J.; de Goede, J.; Weggemans, R.M. The 2015 Dutch food-based dietary guidelines. Eur. J. Clin. Nutr. 2016, 70, 869–878. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wakasugi, M.; Kazama, J.; Narita, I.; Iseki, K.; Fujimoto, S.; Moriyama, T.; Yamagata, K.; Konta, T.; Tsuruya, K.; Asahi, K.; et al. Association between Overall Lifestyle Changes and the Incidence of Proteinuria: A Population-based, Cohort Study. Intern. Med. 2017, 56, 1475–1484. [Google Scholar] [CrossRef] [PubMed] [Green Version]

© 2018 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Qian, Q. Dietary Influence on Body Fluid Acid-Base and Volume Balance: The Deleterious “Norm” Furthers and Cloaks Subclinical Pathophysiology. Nutrients 2018, 10, 778. https://doi.org/10.3390/nu10060778
Qian Q. Dietary Influence on Body Fluid Acid-Base and Volume Balance: The Deleterious “Norm” Furthers and Cloaks Subclinical Pathophysiology. Nutrients. 2018; 10(6):778. https://doi.org/10.3390/nu10060778
Chicago/Turabian StyleQian, Qi. 2018. "Dietary Influence on Body Fluid Acid-Base and Volume Balance: The Deleterious “Norm” Furthers and Cloaks Subclinical Pathophysiology" Nutrients 10, no. 6: 778. https://doi.org/10.3390/nu10060778
APA StyleQian, Q. (2018). Dietary Influence on Body Fluid Acid-Base and Volume Balance: The Deleterious “Norm” Furthers and Cloaks Subclinical Pathophysiology. Nutrients, 10(6), 778. https://doi.org/10.3390/nu10060778