Chemopreventive Activities of Sulforaphane and Its Metabolites in Human Hepatoma HepG2 Cells


 ,
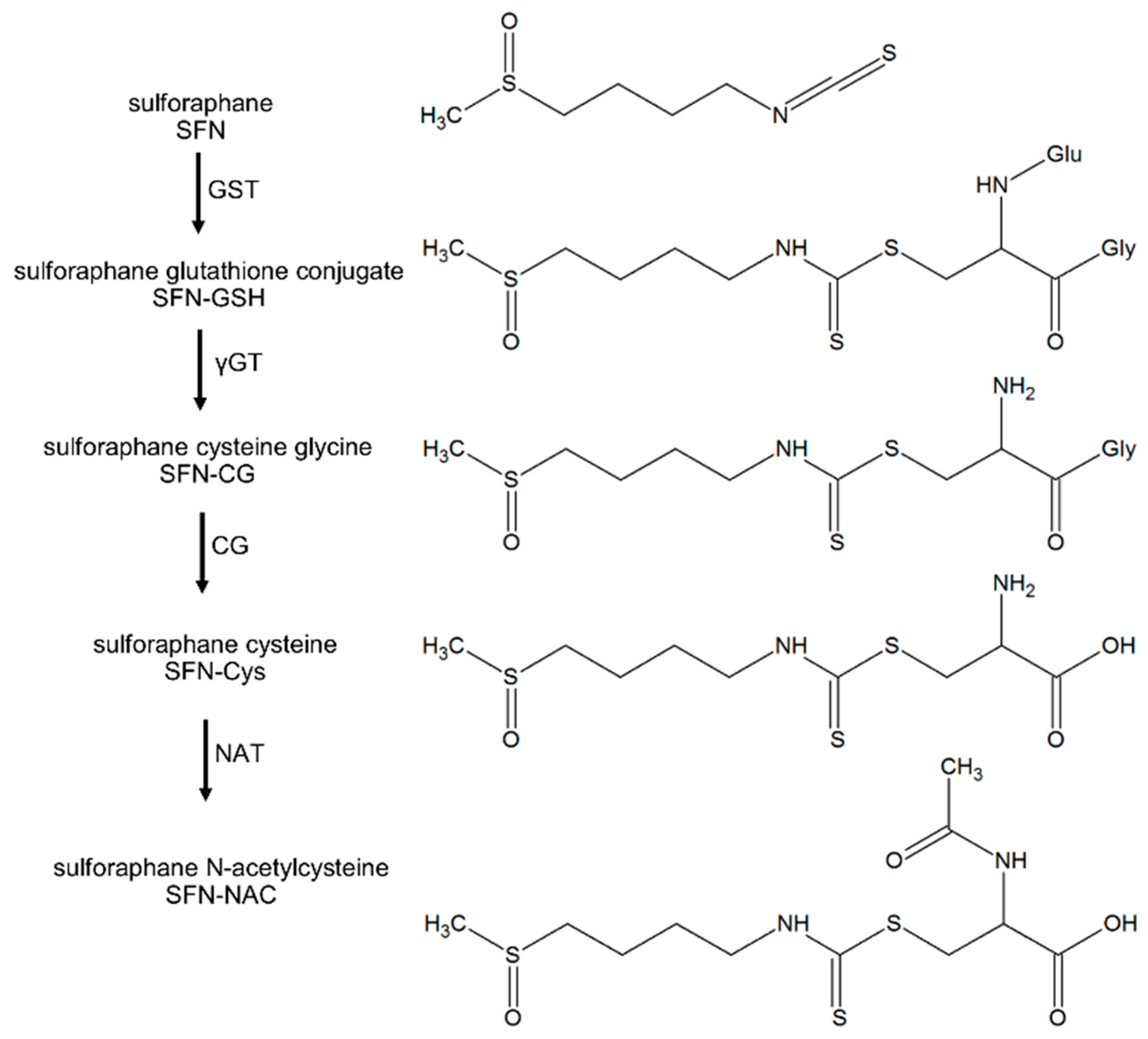
, 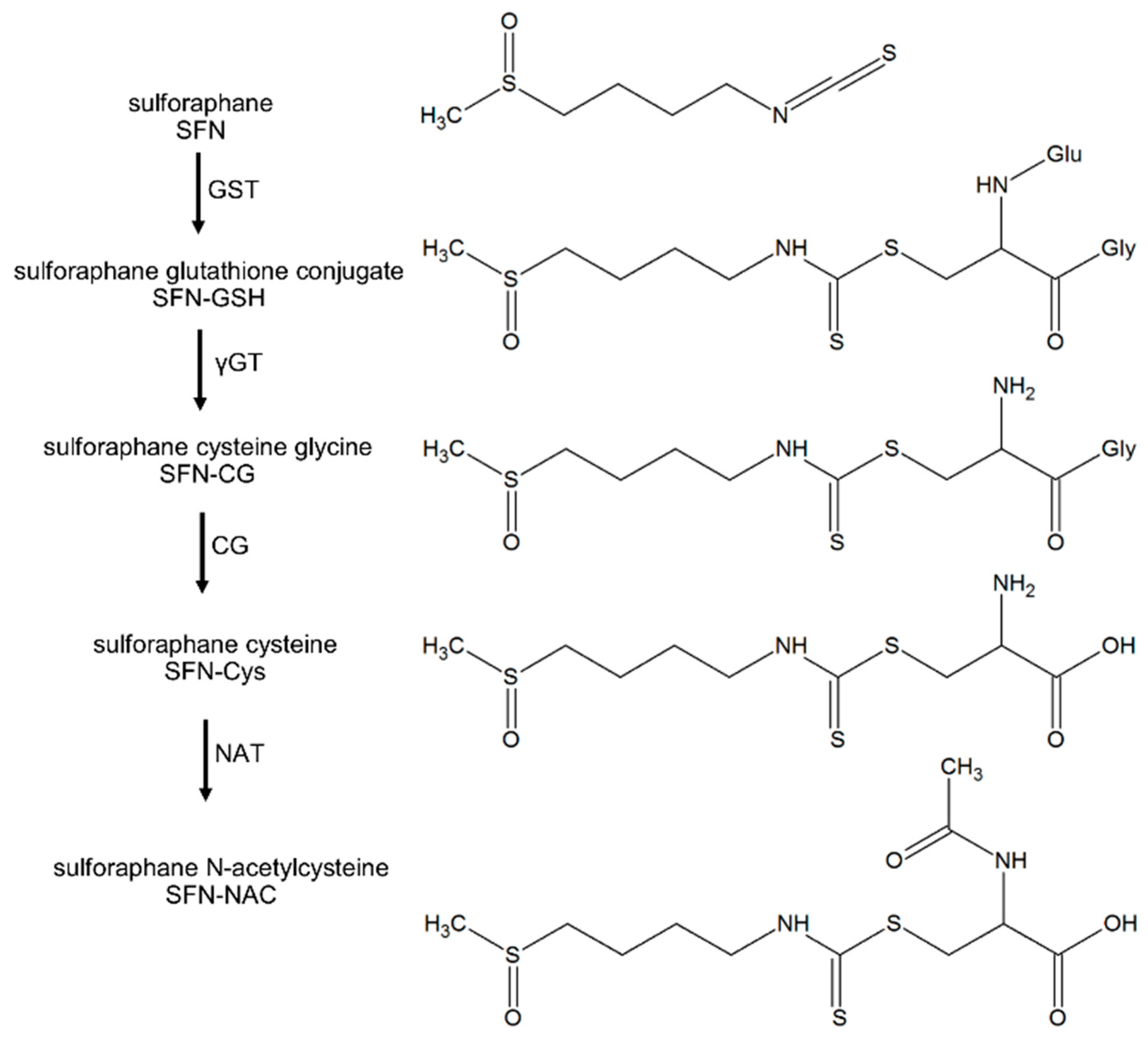{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Experimental Methods
2.1. Reagents
2.2. Cell Culture
2.3. Cell Viability Assay
2.4. Alkaline Comet Assay
2.5. Colony Formation Assay
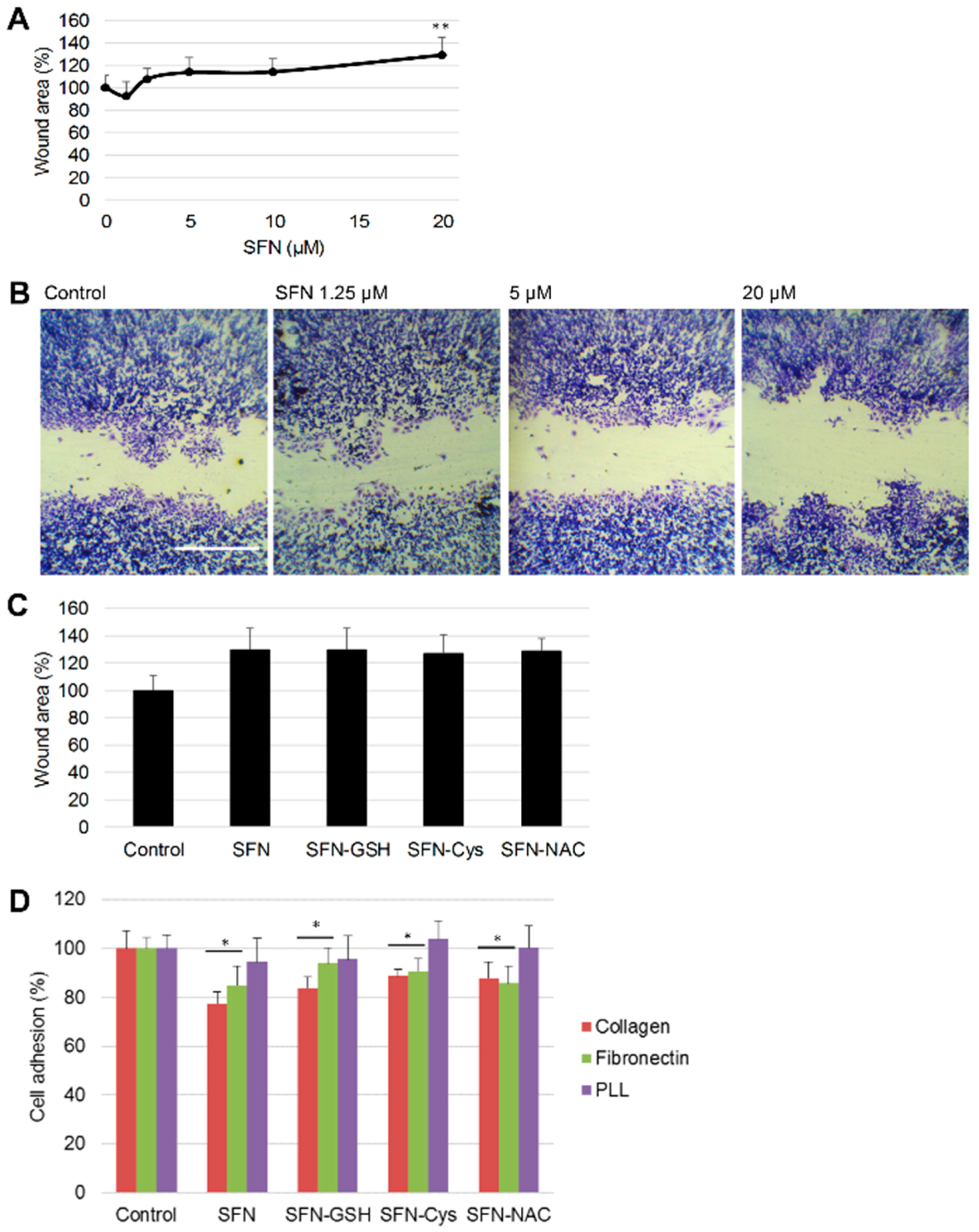
2.6. Wound Assay
2.7. Cell Adhesion Assay
2.8. Tube Formation in a 3D Model
2.9. Western Blotting
2.10. Knockdown Nrf2 by siRNA
2.11. HPLC Analysis of Intracellular GSH
2.12. Statistics
3. Results
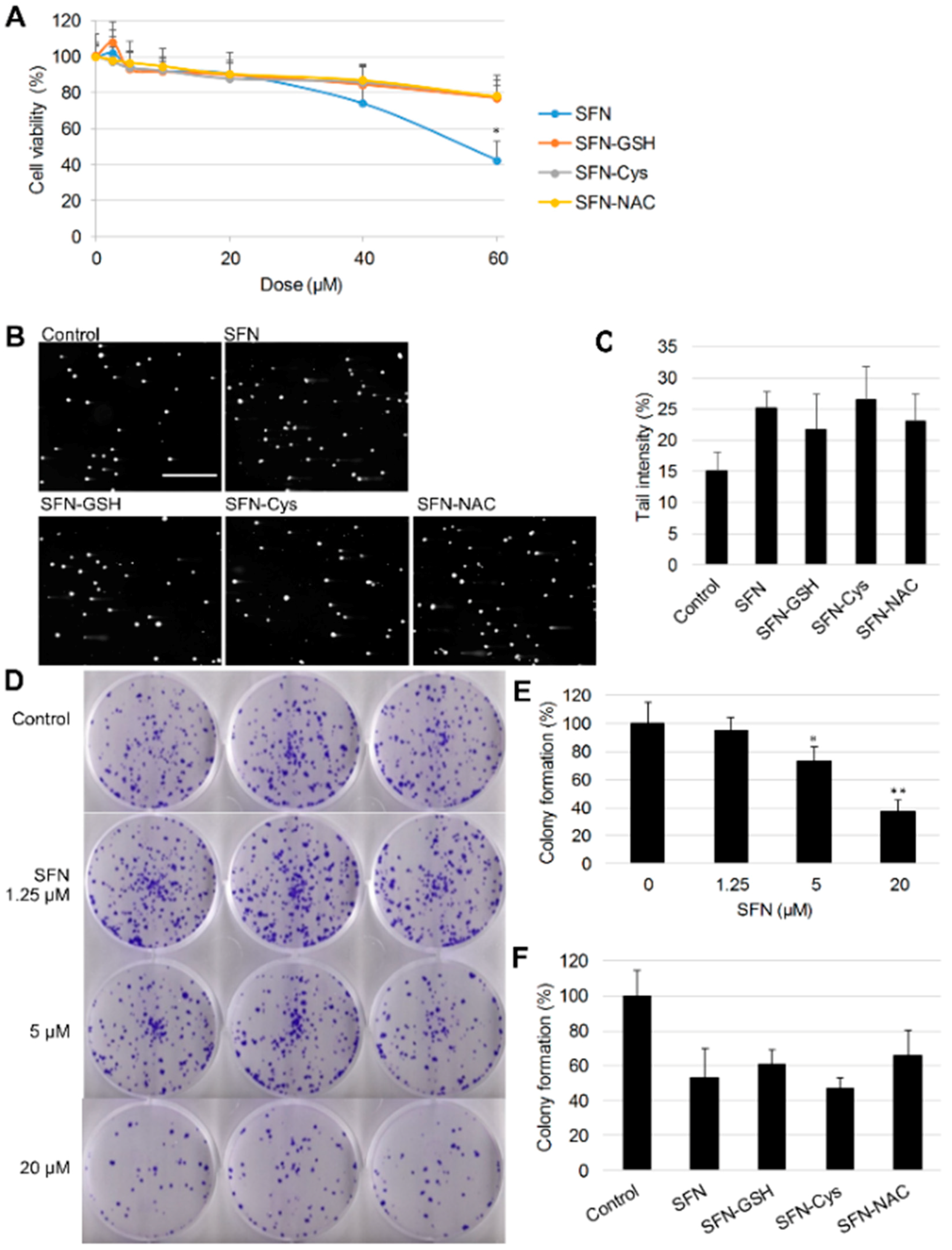
3.1. Cytotoxicity, Genotoxicity and Tumorigenicity of SFN vs. Its Metabolites
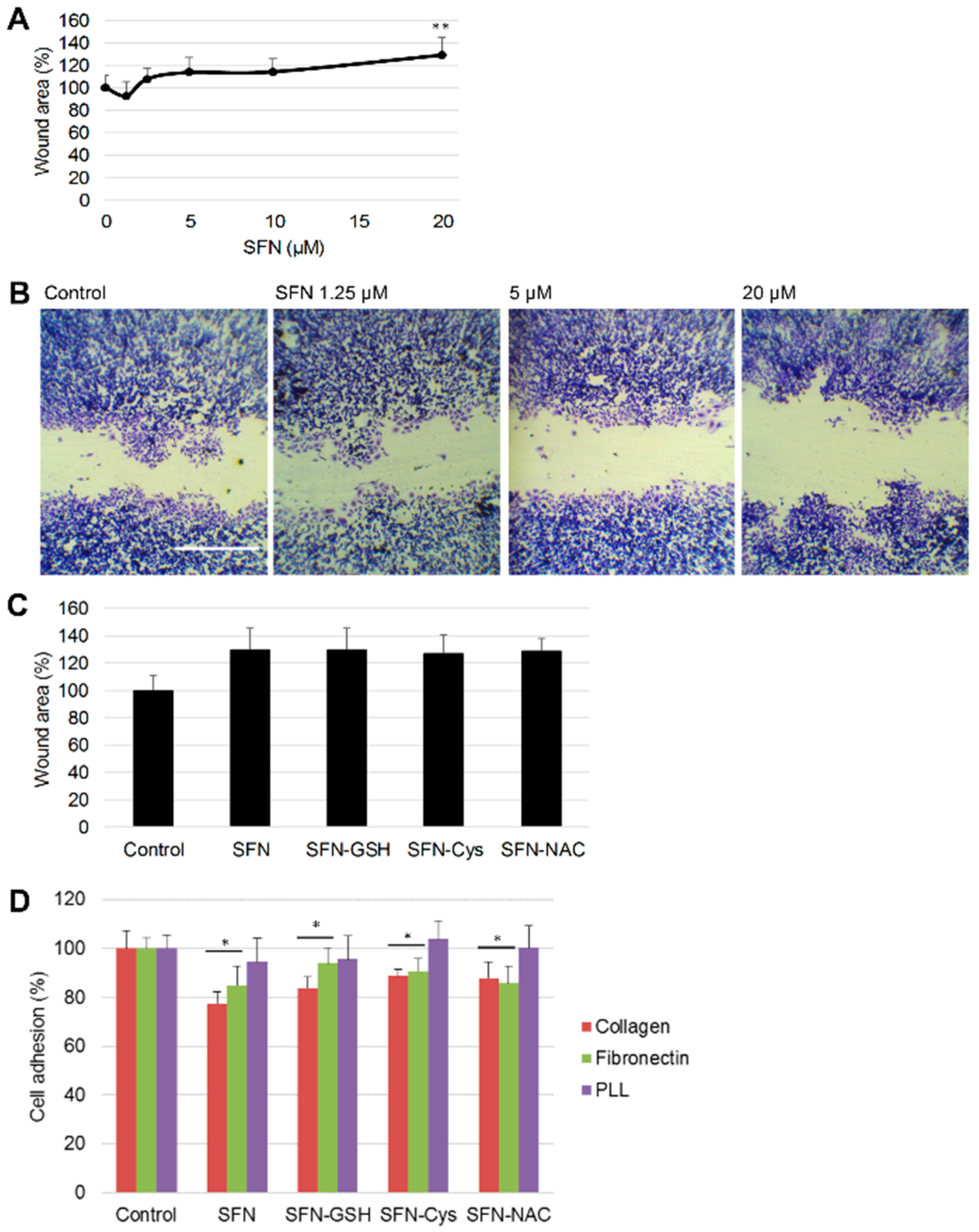
3.2. Effect of SFN vs. Its Metabolites on Cancer Cell Migration
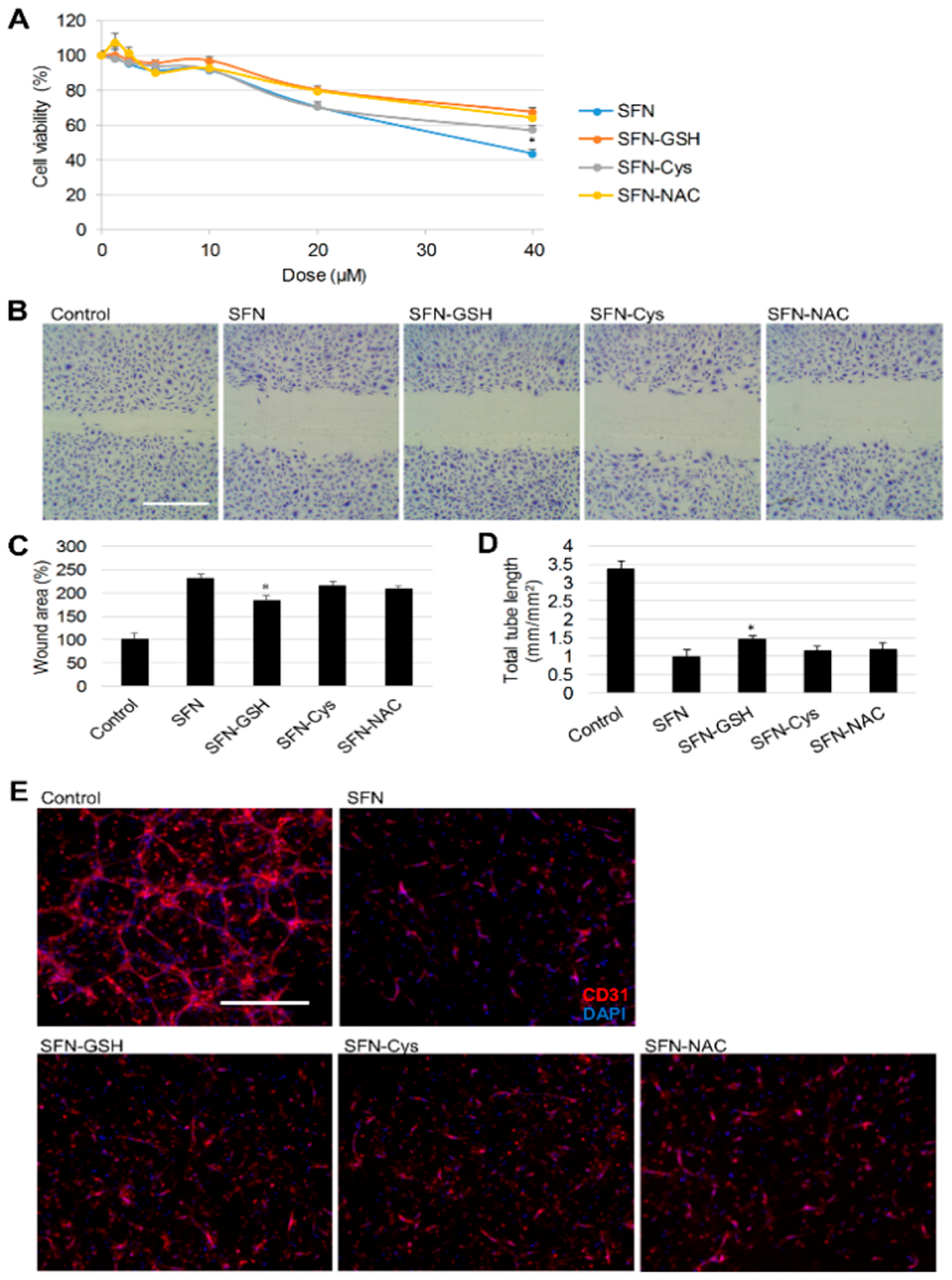
3.3. Effect of SFN vs. Its Metabolites on Angiogenesis
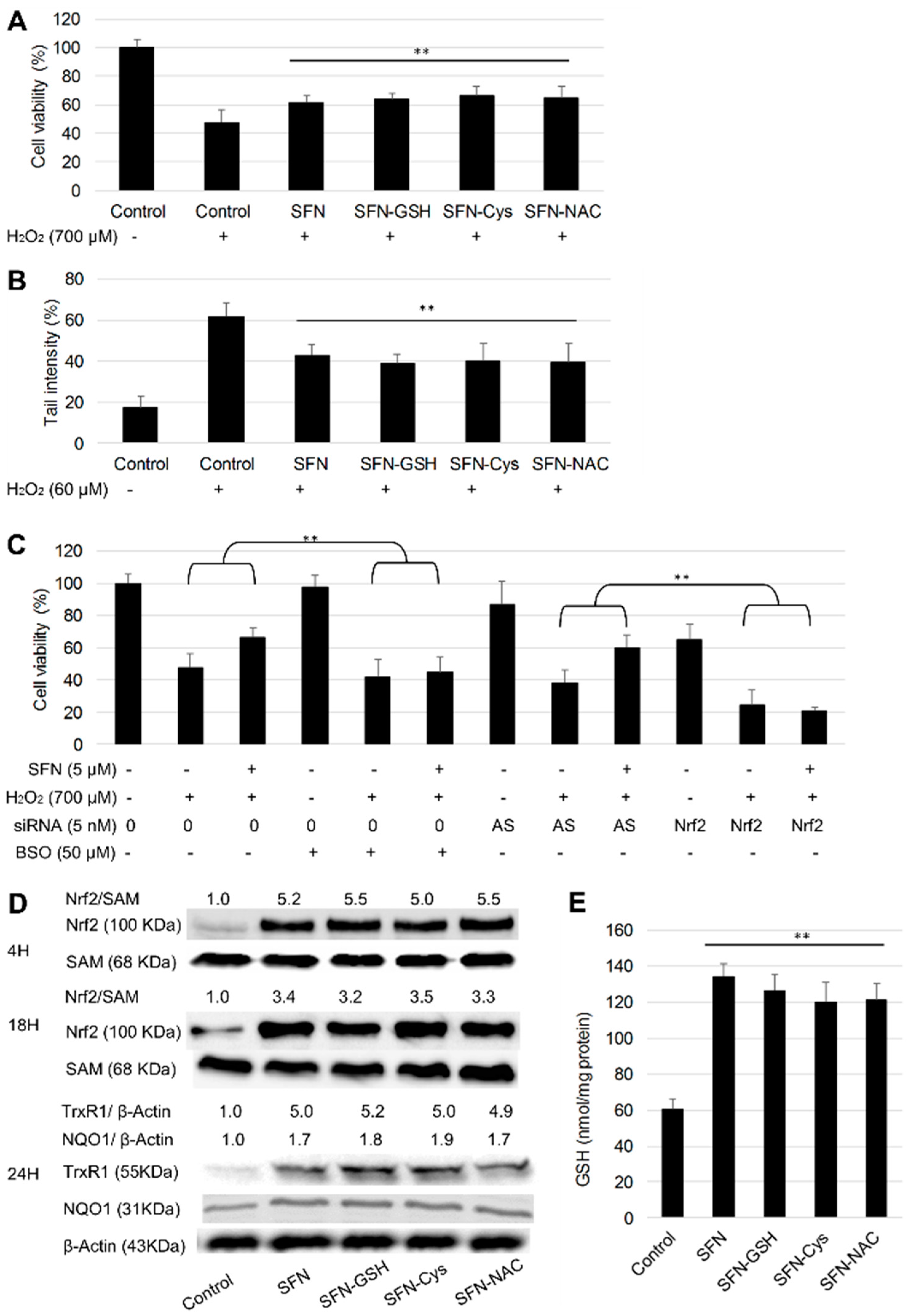
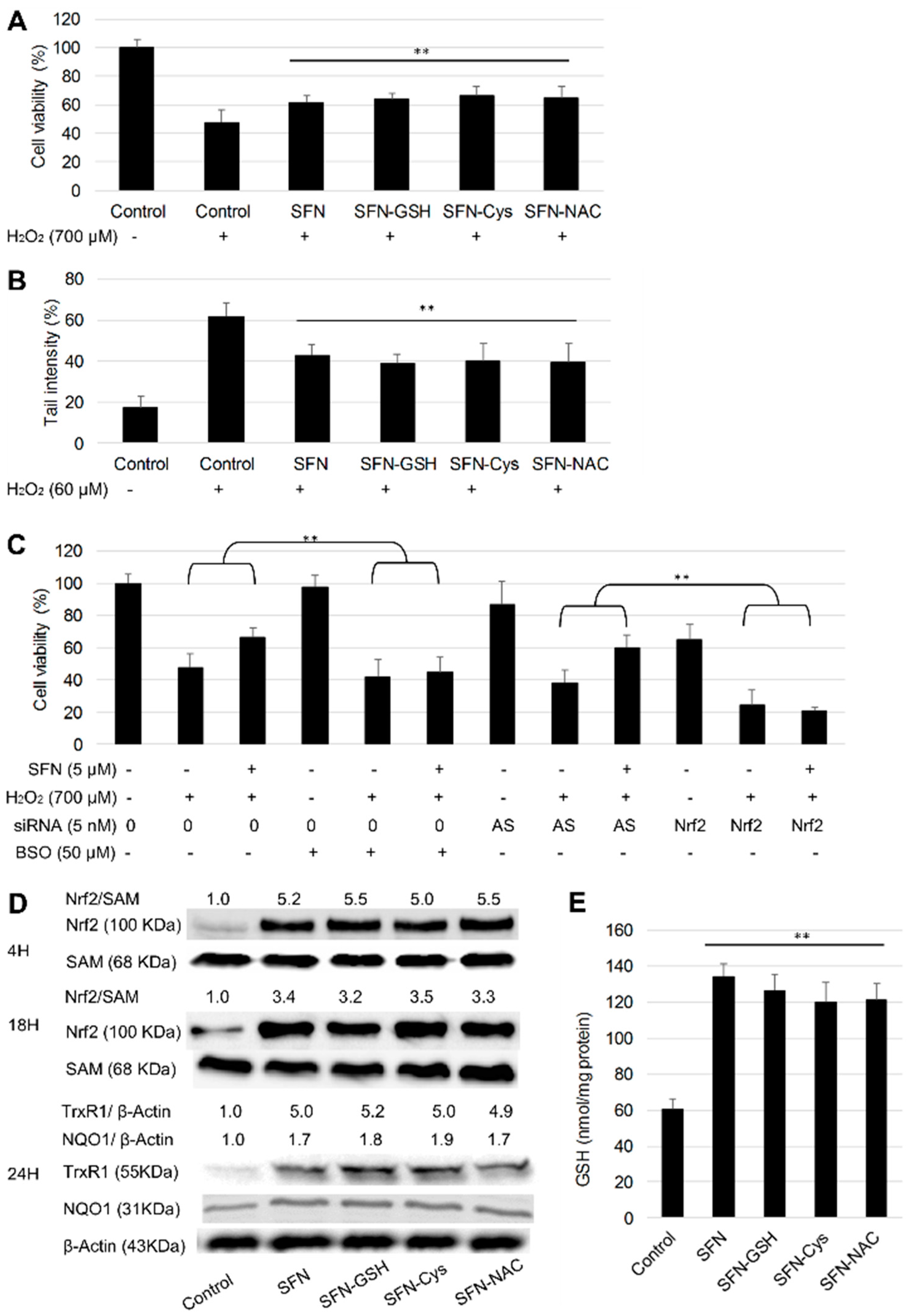
3.4. Protective Effect of SFN vs. Its Metabolites against H2O2
4. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Steward, W.P.; Brown, K. Cancer chemoprevention: A rapidly evolving field. Br. J. Cancer 2013, 109, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Tuorkey, M.J. Cancer Therapy with Phytochemicals: Present and Future Perspectives. Biomed. Environ. Sci. 2015, 28, 808–819. [Google Scholar] [CrossRef]
- Zubair, H.; Azim, S.; Ahmad, A.; Khan, M.A.; Patel, G.K.; Singh, S.; Singh, A.P. Cancer chemoprevention by phytochemicals: Nature’s healing touch. Molecules 2017, 22, 395. [Google Scholar] [CrossRef] [PubMed]
- Baena Ruiz, R.; Salinas Hernández, P. Cancer chemoprevention by dietary phytochemicals: Epidemiological evidence. Maturitas 2016, 94, 13–19. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Talalay, P.; Cho, C.G.; Posner, G.H. A major inducer of anticarcinogenic protective enzymes from broccoli: Isolation and elucidation of structure. Proc. Natl. Acad. Sci. USA 1992, 89, 2399–2403. [Google Scholar] [CrossRef] [PubMed]
- Lenzi, M.; Fimognari, C.; Hrelia, P. Sulforaphane as a promising molecule for fighting cancer. Cancer Treat. Res. 2014, 159, 207–223. [Google Scholar] [PubMed]
- Zhang, Y. Cancer-preventive isothiocyanates: Measurement of human exposure and mechanism of action. Mutat. Res. 2004, 555, 173–190. [Google Scholar] [CrossRef] [PubMed]
- Conaway, C.C.; Krzeminski, J.; Amin, S.; Chung, F.-L. Decomposition rates of isothiocyanate conjugates determine their activity as inhibitors of cytochrome p450. Chem. Res. Toxicol. 2001, 14, 1170–1176. [Google Scholar] [CrossRef] [PubMed]
- Al Janobi, A.A.; Mithen, R.F.; Gasper, A.V.; Shaw, P.N.; Middleton, R.J.; Ortori, C.A.; Barrett, D.A. Quantitative measurement of sulforaphane, iberin and their mercapturic acid pathway metabolites in human plasma and urine using liquid chromatography-tandem electrospray ionisation mass spectrometry. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2006, 844, 223–234. [Google Scholar] [CrossRef] [PubMed]
- Baille, T.A.; Slatter, J. Glutathione—A vehicle for the transport of chemically reactive metabolites in vivo. Acc. Chem. Res. 1991, 24, 264–270. [Google Scholar] [CrossRef]
- Hu, R.; Khor, T.O.; Shen, G.; Jeong, W.S.; Hebbar, V.; Chen, C.; Xu, C.; Reddy, B.; Chada, K.; Kong, A.N. Cancer chemoprevention of intestinal polyposis in ApcMin/+ mice by sulforaphane, a natural product derived from cruciferous vegetable. Carcinogenesis 2006, 27, 2038–2046. [Google Scholar] [CrossRef] [PubMed]
- Clarke, J.D.; Hsu, A.; Williams, D.E.; Dashwood, R.H.; Stevens, J.F.; Yamamoto, M.; Ho, E. Metabolism and tissue distribution of sulforaphane in Nrf2 knockout and wild-type mice. Pharm. Res. 2011, 28, 3171–3179. [Google Scholar] [CrossRef] [PubMed]
- Cornblatt, B.S.; Ye, L.; Dinkova-Kostova, A.T.; Erb, M.; Fahey, J.W.; Singh, N.K.; Chen, M.S.; Stierer, T.; Garrett-Mayer, E.; Argani, P.; et al. Preclinical and clinical evaluation of sulforaphane for chemoprevention in the breast. Carcinogenesis 2007, 28, 1485–1490. [Google Scholar] [CrossRef] [PubMed]
- Tang, L.; Li, G.; Song, L.; Zhang, Y. The principal urinary metabolites of dietary isothiocyanates, N-acetylcysteine conjugates, elicit the same anti-proliferative response as their parent compounds in human bladder cancer cells. Anticancer Drugs 2006, 17, 297–305. [Google Scholar] [CrossRef] [PubMed]
- Basten, G.P.; Bao, Y.; Williamson, G. Sulforaphane and its glutathione conjugate but not sulforaphane nitrile induce UDP-glucuronosyl transferase (UGT1A1) and glutathione transferase (GSTA1) in cultured cells. Carcinogenesis 2002, 23, 1399–1404. [Google Scholar] [CrossRef] [PubMed]
- Lin, K.; Yang, R.; Zheng, Z.; Zhou, Y.; Geng, Y.; Hu, Y.; Wu, S.; Wu, W. Sulforaphane-cysteine-induced apoptosis via phosphorylated ERK1/2-mediated maspin pathway in human non-small cell lung cancer cells. Cell Death Discov. 2017, 3, 17025. [Google Scholar] [CrossRef] [PubMed]
- Tian, H.; Zhou, Y.; Yang, G.; Geng, Y.; Wu, S.; Hu, Y.; Lin, K.; Wu, W. Sulforaphane-cysteine suppresses invasion via downregulation of galectin-1 in human prostate cancer DU145 and PC3 cells. Oncol. Rep. 2016, 36, 1361–1368. [Google Scholar] [CrossRef] [PubMed]
- Chiao, J.W.; Chung, F.L.; Kancherla, R.; Ahmed, T.; Mittelman, A.; Conaway, C.C. Sulforaphane and its metabolite mediate growth arrest and apoptosis in human prostate cancer cells. Int. J. Oncol. 2002, 20, 631–636. [Google Scholar] [CrossRef] [PubMed]
- Conaway, C.C.; Wang, C.X.; Pittman, B.; Yang, Y.M.; Schwartz, J.E.; Tian, D.; McIntee, E.J.; Hecht, S.S.; Chung, F.L. Phenethyl isothiocyanate and sulforaphane and their N-acetylcysteine conjugates inhibit malignant progression of lung adenomas induced by tobacco carcinogens in A/J mice. Cancer Res. 2005, 65, 8548–8557. [Google Scholar] [CrossRef] [PubMed]
- Hwang, E.S.; Jeffery, E.H. Induction of quinone reductase by sulforaphane and sulforaphane N-acetylcysteine conjugate in murine hepatoma cells. J. Med. Food 2005, 8, 198–203. [Google Scholar] [CrossRef] [PubMed]
- Falkenberg, K.J.; Johnstone, R.W. Histone deacetylases and their inhibitors in cancer, neurological diseases and immune disorders. Nat. Rev. Drug Discov. 2014, 13, 673–691. [Google Scholar] [CrossRef] [PubMed]
- Myzak, M.C.; Karplus, P.A.; Chung, F.L.; Dashwood, R.H. A novel mechanism of chemoprotection by sulforaphane: Inhibition of histone deacetylase. Cancer Res. 2004, 64, 5767–5774. [Google Scholar] [CrossRef] [PubMed]
- Vanduchova, A.; Tomankova, V.; Anzenbacher, P.; Anzenbacherova, E. Influence of Sulforaphane Metabolites on Activities of Human Drug-Metabolizing Cytochrome P450 and Determination of Sulforaphane in Human Liver Cells. J. Med. Food 2016, 19, 1141–1146. [Google Scholar] [CrossRef] [PubMed]
- Chung, F.L.; Conaway, C.C.; Rao, C.V.; Reddy, B.S. Chemoprevention of colonic aberrant crypt foci in Fischer rats by sulforaphane and phenethyl isothiocyanate. Carcinogenesis 2000, 21, 2287–2291. [Google Scholar] [CrossRef] [PubMed]
- Kim, B.R.; Hu, R.; Keum, Y.S.; Hebbar, V.; Shen, G.; Nair, S.S.; Kong, A.N. Effects of Glutathione on Antioxidant Response Element-Mediated Gene Expression and Apoptosis Elicited by Sulforaphane. Cancer Res. 2003, 63, 7520–7525. [Google Scholar] [PubMed]
- Gasper, A.V.; Al-Janobi, A.; Smith, J.A.; Bacon, J.R.; Fortun, P.; Atherton, C.; Taylor, M.A.; Hawkey, C.J.; Barrett, D.A.; Mithen, R.F. Glutathione S-transferase M1 polymorphism and metabolism of sulforaphane from standard and high-glucosinolate broccoli. Am. J. Clin. Nutr. 2005, 82, 1283–1291. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Pausch, F.; Schlötzer-Schrehardt, U.; Brachvogel, B.; Pöschl, E. Induction of initial steps of angiogenic differentiation and maturation of endothelial cells by pericytes in vitro and the role of collagen IV. Histochem. Cell Biol. 2016, 145, 511–525. [Google Scholar] [CrossRef] [PubMed]
- Collins, A.R. The comet assay for DNA damage and repair: Principles, applications, and limitations. Mol. Biotechnol. 2004, 26, 249–261. [Google Scholar] [CrossRef]
- Liu, H.; Smith, A.J.; Lott, M.C.; Bao, Y.; Bowater, R.P.; Reddan, J.R.; Wormstone, I.M. Sulforaphane can protect lens cells against oxidative stress: Implications for cataract prevention. Investig. Ophthalmol. Vis. Sci. 2013, 54, 5236–5248. [Google Scholar] [CrossRef] [PubMed]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef] [PubMed]
- Cooley, L.S.; Handsley, M.M.; Zhou, Z.; Lafleur, M.A.; Pennington, C.J.; Thompson, E.W.; Pöschl, E.; Edwards, D.R. Reversible transdifferentiation of blood vascular endothelial cells to a lymphatic-like phenotype in vitro. J. Cell Sci. 2010, 123, 3808–3816. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Behray, M.; Wang, Q.; Wang, W.; Zhou, Z.; Chao, Y.; Bao, Y. Anti-cancer activities of allyl isothiocyanate and its conjugated silicon quantum dots. Sci. Rep. 2018, 8, 1084. [Google Scholar] [CrossRef] [PubMed]
- Cotgreave, I.A.; Moldéus, P. Methodologies for the application of monobromobimane to the simultaneous analysis of soluble and protein thiol components of biological systems. J. Biochem. Biophys. Methods 1986, 13, 231–249. [Google Scholar] [CrossRef]
- Wang, W.; He, Y.; Yu, G.; Li, B.; Sexton, D.W.; Wileman, T.; Roberts, A.A.; Hamilton, C.J.; Liu, R.; Chao, Y.; et al. Sulforaphane protects the liver against CdSe quantum dot-induced cytotoxicity. PLoS ONE 2015, 10, e0138771. [Google Scholar] [CrossRef] [PubMed]
- Friedl, P.; Wolf, K. Tumour-cell invasion and migration: Diversity and escape mechanisms. Nat. Rev. Cancer 2003, 3, 362–374. [Google Scholar] [CrossRef] [PubMed]
- Weis, S.M.; Cheresh, D.A. Tumor angiogenesis: Molecular pathways and therapeutic targets. Nat. Med. 2011, 17, 1359–1370. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Atkinson, S.J.; Akbareian, S.E.; Zhou, Z.; Munsterberg, A.; Robinson, S.D.; Bao, Y. Sulforaphane exerts anti-angiogenesis effects against hepatocellular carcinoma through inhibition of STAT3/HIF-1α/VEGF signalling. Sci. Rep. 2017, 7, 12651. [Google Scholar] [CrossRef] [PubMed]
- Townsend, D.M.; Tew, K.D.; Tapiero, H. The importance of glutathione in human disease. Biomed. Pharmacother. 2003, 57, 145–155. [Google Scholar] [CrossRef]
- Sestili, P.; Paolillo, M.; Lenzi, M.; Colombo, E.; Vallorani, L.; Casadei, L.; Martinelli, C.; Fimognari, C. Sulforaphane induces DNA single strand breaks in cultured human cells. Mutat. Res. 2010, 689, 65–73. [Google Scholar] [CrossRef] [PubMed]
- Rajendran, P.; Kidane, A.I.; Yu, T.W.; Dashwood, W.M.; Bisson, W.H.; Löhr, C.V.; Ho, E.; Williams, D.E.; Dashwood, R.H. HDAC turnover, CtIP acetylation and dysregulated DNA damage signaling in colon cancer cells treated with sulforaphane and related dietary isothiocyanates. Epigenetics 2013, 8, 612–623. [Google Scholar] [CrossRef] [PubMed]
- Piberger, A.L.; Köberle, B.; Hartwig, A. The broccoli-born isothiocyanate sulforaphane impairs nucleotide excision repair: XPA as one potential target. Arch. Toxicol. 2014, 88, 647–658. [Google Scholar] [CrossRef] [PubMed]
- Naumann, P.; Liermann, J.; Fortunato, F.; Schmid, T.E.; Weber, K.; Debus, J.; Combs, S.E. Sulforaphane enhances irradiation effects in terms of perturbed cell cycle progression and increased DNA damage in pancreatic cancer cells. PLoS ONE 2017, 12, e018940. [Google Scholar] [CrossRef] [PubMed]
- Bao, C.; Kim, M.C.; Chen, J.; Song, J.; Ko, H.W.; Lee, H.J. Sulforaphene Interferes with Human Breast Cancer Cell Migration and Invasion through Inhibition of Hedgehog Signaling. J. Agric. Food Chem. 2016, 64, 5515–5524. [Google Scholar] [CrossRef] [PubMed]
- Mondal, A.; Biswas, R.; Rhee, Y.H.; Kim, J.; Ahn, J.C. Sulforaphene promotes Bax/Bcl2, MAPK-dependent human gastric cancer AGS cells apoptosis and inhibits migration via EGFR, p-ERK1/2 down-regulation. Gen. Physiol. Biophys. 2016, 35, 25–34. [Google Scholar] [PubMed]
- Nishikawa, T.; Tsuno, N.H.; Okaji, Y.; Sunami, E.; Shuno, Y.; Sasaki, K.; Hongo, K.; Kaneko, M.; Hiyoshi, M.; Kawai, K.; et al. The inhibition of autophagy potentiates anti-angiogenic effects of sulforaphane by inducing apoptosis. Angiogenesis 2010, 13, 227–238. [Google Scholar] [CrossRef] [PubMed]
- Jackson, S.J.T.; Singletary, K.W.; Venema, R.C. Sulforaphane suppresses angiogenesis and disrupts endothelial mitotic progression and microtubule polymerization. Vasc. Pharmacol. 2007, 46, 77–84. [Google Scholar] [CrossRef] [PubMed]
- Davis, R.; Singh, K.P.; Kurzrock, R.; Shankar, S. Sulforaphane inhibits angiogenesis through activation of FOXO transcription factors. Oncol. Rep. 2009, 22, 1473–1478. [Google Scholar] [PubMed]
- Hynes, R.O. Cell-matrix adhesion in vascular development. J. Thromb. Haemost. 2007, 5, 32–40. [Google Scholar] [CrossRef] [PubMed]
- Popova, S.N.; Lundgren-Åkerlund, E.; Wiig, H.; Gullberg, D. Physiology and pathology of collagen receptors. Acta Physiol. 2007, 190, 179–187. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Wang, W.; Shan, Y.; Barrera, L.N.; Howie, A.F.; Beckett, G.J.; Wu, K.; Bao, Y. Synergy between sulforaphane and selenium in the up-regulation of thioredoxin reductase and protection against hydrogen peroxide-induced cell death in human hepatocytes. Food Chem. 2012, 133, 300–307. [Google Scholar] [CrossRef] [PubMed]
- Bacon, J.R.; Plumb, G.W.; Howie, A.F.; Beckett, G.J.; Wang, W.; Bao, Y. Dual action of sulforaphane in the regulation of thioredoxin reductase and thioredoxin in human HepG2 and caco-2 cells. J. Agric. Food Chem. 2007, 55, 1170–1176. [Google Scholar] [CrossRef] [PubMed]
- Taguchi, K.; Motohashi, H.; Yamamoto, M. Molecular mechanisms of the Keap1-Nrf2 pathway in stress response and cancer evolution. Genes Cells 2011, 16, 123–140. [Google Scholar] [CrossRef] [PubMed]
- Menegon, S.; Columbano, A.; Giordano, S. The Dual Roles of NRF2 in Cancer. Trends Mol. Med. 2016, 22, 578–593. [Google Scholar] [CrossRef] [PubMed]
- Kansanen, E.; Kuosmanen, S.M.; Leinonen, H.; Levonenn, A.L. The Keap1-Nrf2 pathway: Mechanisms of activation and dysregulation in cancer. Redox Biol. 2013, 1, 45–49. [Google Scholar] [CrossRef] [PubMed]
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, P.; Wang, W.; Zhou, Z.; Smith, A.J.O.; Bowater, R.P.; Wormstone, I.M.; Chen, Y.; Bao, Y. Chemopreventive Activities of Sulforaphane and Its Metabolites in Human Hepatoma HepG2 Cells. Nutrients 2018, 10, 585. https://doi.org/10.3390/nu10050585
Liu P, Wang W, Zhou Z, Smith AJO, Bowater RP, Wormstone IM, Chen Y, Bao Y. Chemopreventive Activities of Sulforaphane and Its Metabolites in Human Hepatoma HepG2 Cells. Nutrients. 2018; 10(5):585. https://doi.org/10.3390/nu10050585
Chicago/Turabian StyleLiu, Peng, Wei Wang, Zhigang Zhou, Andrew J. O. Smith, Richard P. Bowater, Ian Michael Wormstone, Yuqiong Chen, and Yongping Bao. 2018. "Chemopreventive Activities of Sulforaphane and Its Metabolites in Human Hepatoma HepG2 Cells" Nutrients 10, no. 5: 585. https://doi.org/10.3390/nu10050585
APA StyleLiu, P., Wang, W., Zhou, Z., Smith, A. J. O., Bowater, R. P., Wormstone, I. M., Chen, Y., & Bao, Y. (2018). Chemopreventive Activities of Sulforaphane and Its Metabolites in Human Hepatoma HepG2 Cells. Nutrients, 10(5), 585. https://doi.org/10.3390/nu10050585