Advances in Lipid Nanoparticles for siRNA Delivery

{kind=link}
{kind=link}
Abstract
:1. Introduction
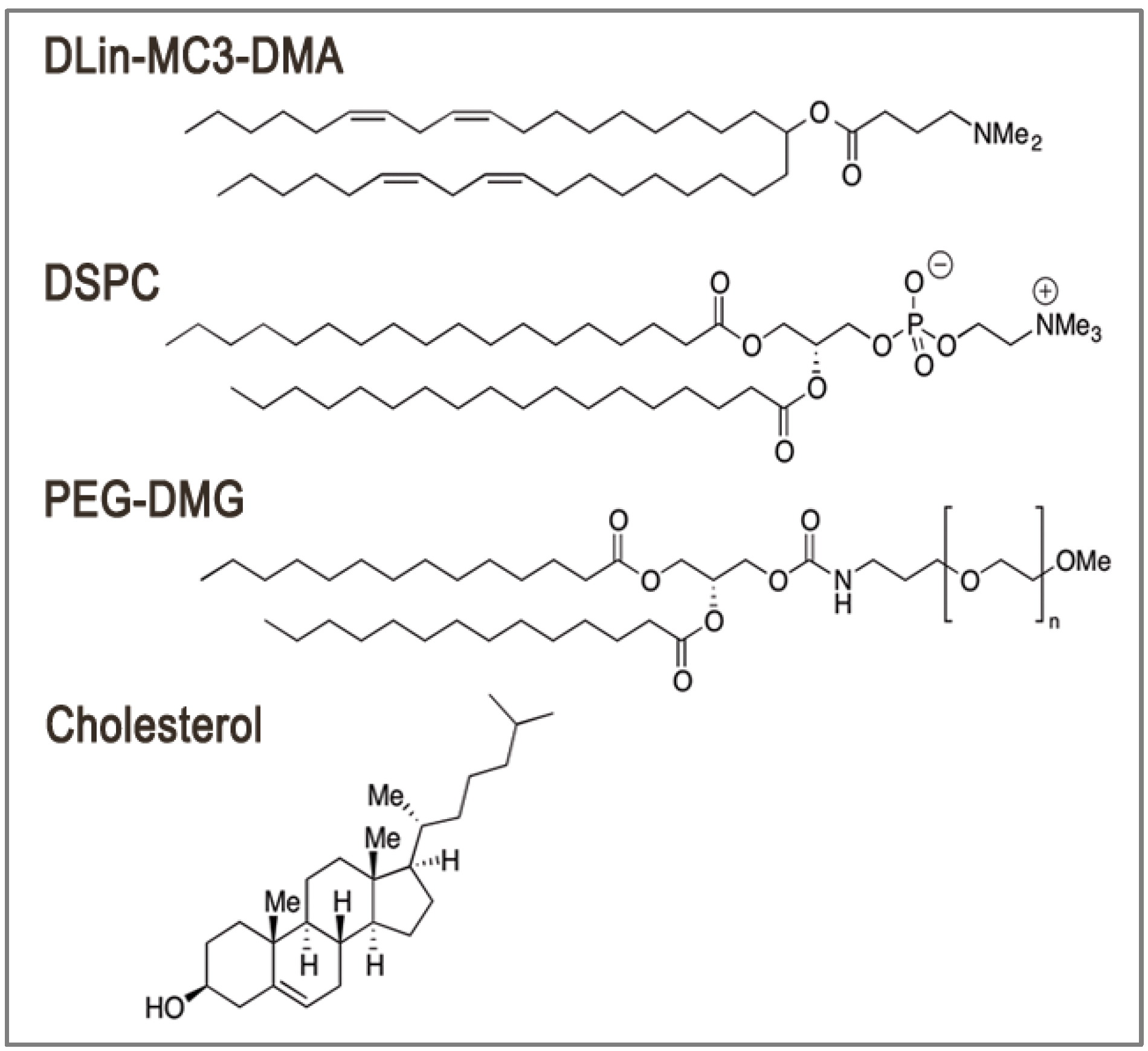
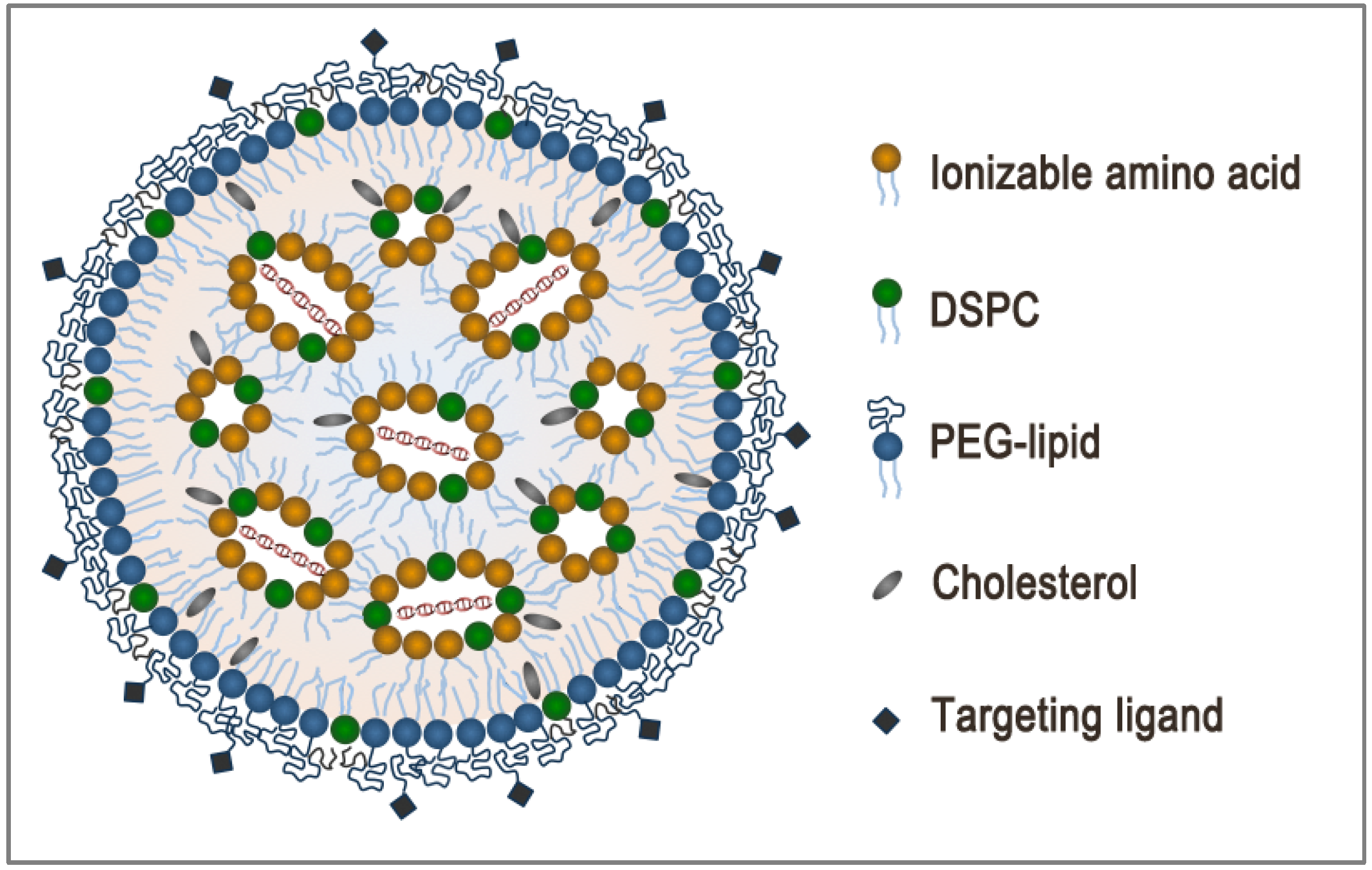
2. LNP Formulation: Composition, Structure and Size
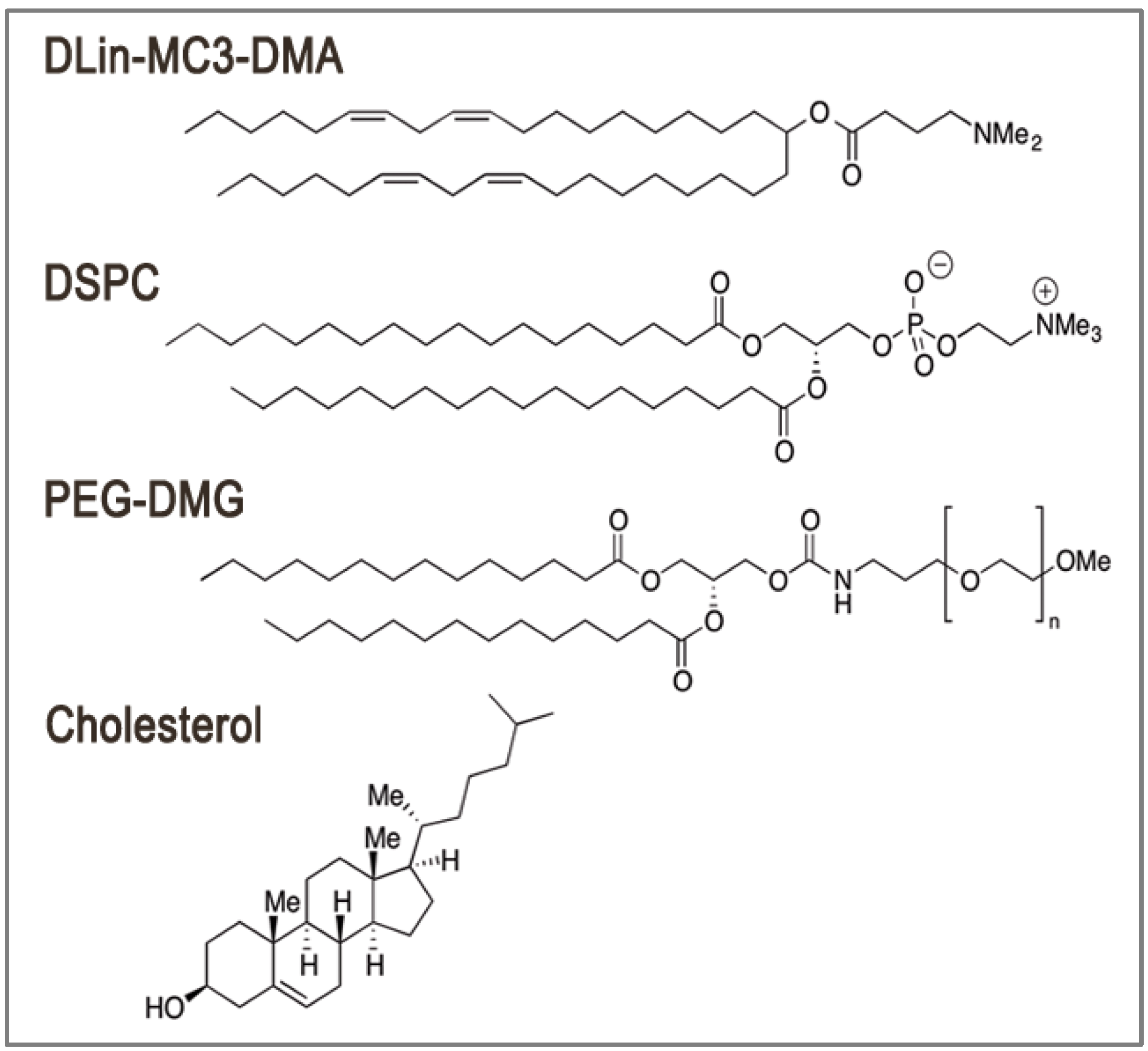
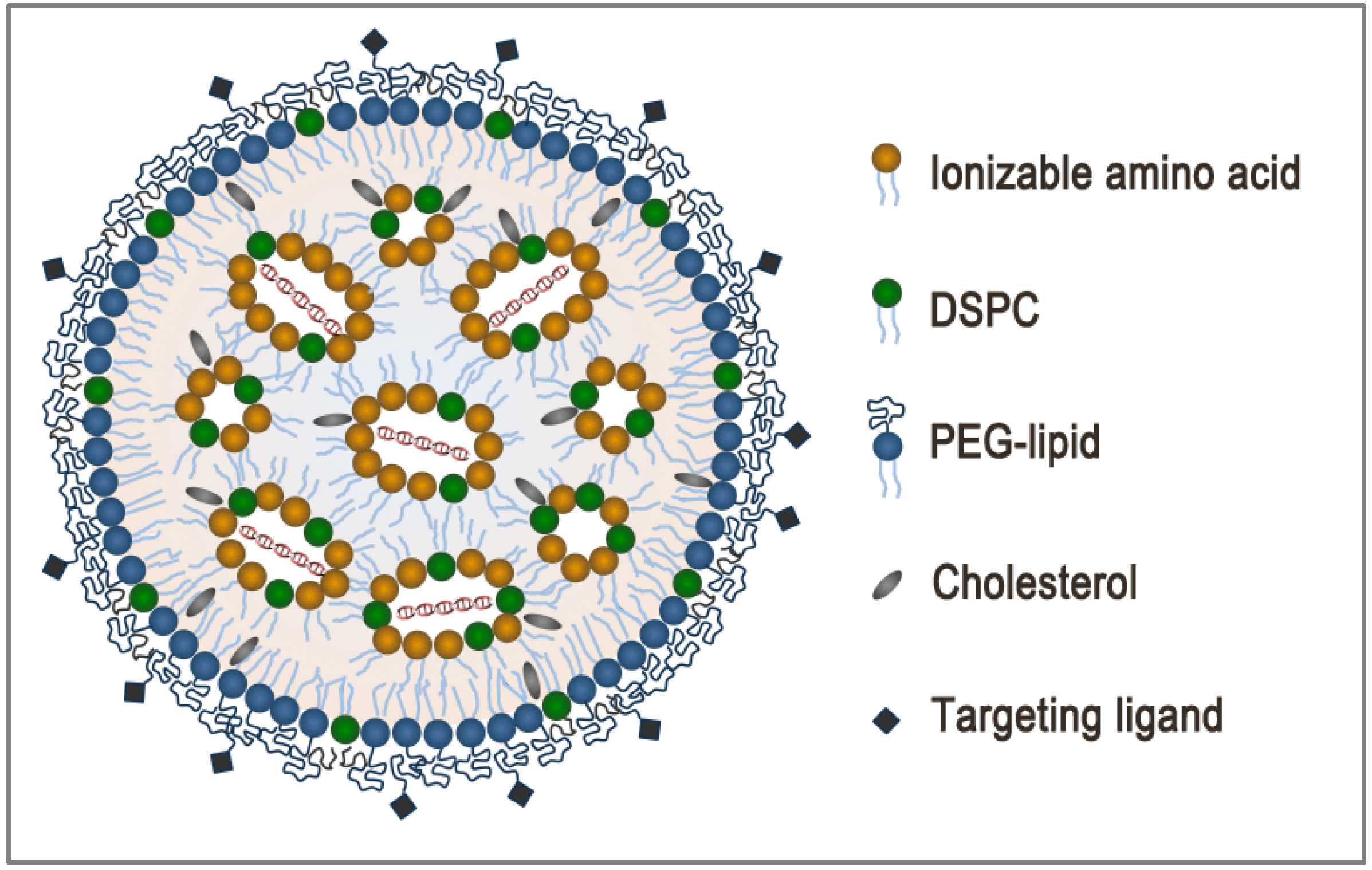
3. Encapsulation and Intracellular Release: Dual Function of Ionizable Amino Lipids
4. Steric Barrier: Stability and Circulation Lifetime
5. Cellular Uptake: Endogenous and Exogenous Targeting Ligands
6. Concluding Remarks
Acknowledgments
Conflicts of Interest
References
- Novina, C.D.; Sharp, P.A. The rnai revolution. Nature 2004, 430, 161–164. [Google Scholar] [CrossRef]
- Vaishnaw, A.K.; Gollob, J.; Gamba-Vitalo, C.; Hutabarat, R.; Sah, D.; Meyers, R.; de Fougerolles, T.; Maraganore, J. A status report on rnai therapeutics. Silence 2010, 1, 14. [Google Scholar] [CrossRef]
- McManus, M.T.; Sharp, P.A. Gene silencing in mammals by small interfering rnas. Nat. Rev. Genet. 2002, 3, 737–747. [Google Scholar] [CrossRef]
- Miele, E.; Spinelli, G.P.; di Fabrizio, E.; Ferretti, E.; Tomao, S.; Gulino, A. Nanoparticle-based delivery of small interfering RNA: Challenges for cancer therapy. Int. J. Nanomed. 2012, 7, 3637–3657. [Google Scholar]
- De Fougerolles, A.; Vornlocher, H.P.; Maraganore, J.; Lieberman, J. Interfering with disease: A progress report on siRNA-based therapeutics. Nat. Rev. Drug Discov. 2007, 6, 443–453. [Google Scholar] [CrossRef]
- Whitehead, K.A.; Langer, R.; Anderson, D.G. Knocking down barriers: Advances in sirna delivery. Nat. Rev. Drug Discov. 2009, 8, 129–138. [Google Scholar] [CrossRef]
- Manoharan, M.; Akinc, A.; Pandey, R.K.; Qin, J.; Hadwiger, P.; John, M.; Mills, K.; Charisse, K.; Maier, M.A.; Nechev, L.; et al. Unique gene-silencing and structural properties of 2'-fluoro-modified sirnas. Angew. Chem. Int. Ed. Engl. 2011, 50, 2284–2288. [Google Scholar]
- Manoharan, M. RNA interference and chemically modified small interfering RNAs. Curr. Opin. Chem. Biol. 2004, 8, 570–579. [Google Scholar] [CrossRef]
- Engels, J.W. Gene silencing by chemically modified sirnas. New Biotechnol. 2013, 30, 302–307. [Google Scholar] [CrossRef]
- Soutschek, J.; Akinc, A.; Bramlage, B.; Charisse, K.; Constien, R.; Donoghue, M.; Elbashir, S.; Geick, A.; Hadwiger, P.; Harborth, J.; et al. Therapeutic silencing of an endogenous gene by systemic administration of modified sirnas. Nature 2004, 432, 173–178. [Google Scholar] [CrossRef]
- Allen, T.M.; Cullis, P.R. Liposomal drug delivery systems: From concept to clinical applications. Adv. Drug Deliv. Rev. 2012, 65, 36–48. [Google Scholar] [CrossRef]
- Daka, A.; Peer, D. RNAi-based nanomedicines for targeted personalized therapy. Adv. Drug Deliv. Rev. 2012, 64, 1508–1521. [Google Scholar] [CrossRef]
- Semple, S.C.; Akinc, A.; Chen, J.; Sandhu, A.P.; Mui, B.L.; Cho, C.K.; Sah, D.W.; Stebbing, D.; Crosley, E.J.; Yaworski, E.; et al. Rational design of cationic lipids for siRNA delivery. Nat. Biotechnol. 2010, 28, 172–176. [Google Scholar] [CrossRef]
- Jayaraman, M.; Ansell, S.M.; Mui, B.L.; Tam, Y.K.; Chen, J.; Du, X.; Butler, D.; Eltepu, L.; Matsuda, S.; Narayanannair, J.K.; et al. Maximizing the potency of sirna lipid nanoparticles for hepatic gene silencing in vivo. Angew. Chem. Int. Ed. Engl. 2012, 51, 8529–8533. [Google Scholar] [CrossRef]
- Huang, L.; Liu, Y. In vivo delivery of rnai with lipid-based nanoparticles. Annu. Rev. Biomed. Eng. 2011, 13, 507–530. [Google Scholar] [CrossRef]
- Burnett, J.C.; Rossi, J.J.; Tiemann, K. Current progress of siRNA/shRNA therapeutics in clinical trials. Biotechnol. J. 2011, 6, 1130–1146. [Google Scholar] [CrossRef]
- Barros, S.A.; Gollob, J.A. Safety profile of RNAi nanomedicines. Adv. Drug Deliv. Rev. 2012, 64, 1730–1737. [Google Scholar] [CrossRef]
- Tabernero, J.; Shapiro, G.I.; LoRusso, P.M.; Cervantes, A.; Schwartz, G.K.; Weiss, G.J.; Paz-Ares, L.; Cho, D.C.; Infante, J.R.; Alsina, M.; et al. First-in-humans trial of an RNA interference therapeutic targeting vegf and ksp in cancer patients with liver involvement. Cancer Discov. 2013, 3, 406–417. [Google Scholar] [CrossRef]
- Alabi, C.; Vegas, A.; Anderson, D. Attacking the genome: Emerging sirna nanocarriers from concept to clinic. Curr. Opin. Pharmacol. 2012, 12, 427–433. [Google Scholar] [CrossRef]
- Crawford, R.; Dogdas, B.; Keough, E.; Haas, R.M.; Wepukhulu, W.; Krotzer, S.; Burke, P.A.; Sepp-Lorenzino, L.; Bagchi, A.; Howell, B.J. Analysis of lipid nanoparticles by cryo-em for characterizing siRNA delivery vehicles. Int. J. Pharm. 2011, 403, 237–244. [Google Scholar] [CrossRef]
- Leung, A.K.; Hafez, I.M.; Baoukina, S.; Belliveau, N.M.; Zhigaltsev, I.V.; Afshinmanesh, E.; Tieleman, D.P.; Hansen, C.L.; Hope, M.J.; Cullis, P.R. Lipid nanoparticles containing sirna synthesized by microfluidic mixing exhibit an electron-dense nanostructured core. J. Phys. Chem. C Nanomater. Interfaces 2012, 116, 18440–18450. [Google Scholar] [CrossRef]
- Belliveau, N.M.; Huft, J.; Lin, P.J.C.; Chen, S.; Leung, A.K.K.; Leaver, T.J.; Wild, A.W.; Lee, J.B.; Taylor, R.J.; Tam, Y.K.; et al. Microfluidic synthesis of highly potent limit-size lipid nanoparticles for in vivo delivery of sirna. Mol. Ther. Nucleic Acids 2012, 1, e37. [Google Scholar] [CrossRef]
- Zhigaltsev, I.V.; Maurer, N.; Edwards, K.; Karlsson, G.; Cullis, P.R. Formation of drug-arylsulfonate complexes inside liposomes: A novel approach to improve drug retention. J. Control. Release 2006, 110, 378–386. [Google Scholar] [CrossRef]
- Semple, S.C.; Klimuk, S.K.; Harasym, T.O.; Dos Santos, N.; Ansell, S.M.; Wong, K.F.; Maurer, N.; Stark, H.; Cullis, P.R.; Hope, M.J.; et al. Efficient encapsulation of antisense oligonucleotides in lipid vesicles using ionizable aminolipids: Formation of novel small multilamellar vesicle structures. Biochim. Biophys. Acta 2001, 1510, 152–166. [Google Scholar] [CrossRef]
- Maurer, N.; Wong, K.F.; Stark, H.; Louie, L.; McIntosh, D.; Wong, T.; Scherrer, P.; Semple, S.C.; Cullis, P.R. Spontaneous entrapment of polynucleotides upon electrostatic interaction with ethanol-destabilized cationic liposomes. Biophys. J. 2001, 80, 2310–2326. [Google Scholar] [CrossRef]
- Hafez, I.M.; Maurer, N.; Cullis, P.R. On the mechanism whereby cationic lipids promote intracellular delivery of polynucleic acids. Gene Ther. 2001, 8, 1188–1196. [Google Scholar] [CrossRef]
- Xu, Y.; Szoka, F.C., Jr. Mechanism of DNA release from cationic liposome/DNA complexes used in cell transfection. Biochemistry 1996, 35, 5616–5623. [Google Scholar] [CrossRef]
- Bailey, A.L.; Cullis, P.R. Modulation of membrane fusion by asymmetric transbilayer distributions of amino lipids. Biochemistry 1994, 33, 12573–12580. [Google Scholar] [CrossRef]
- Heyes, J.; Palmer, L.; Bremner, K.; MacLachlan, I. Cationic lipid saturation influences intracellular delivery of encapsulated nucleic acids. J. Control. Release 2005, 107, 276–287. [Google Scholar] [CrossRef]
- Maier, M.A.; Jayaraman, M.; Matsuda, S.; Liu, J.; Barros, S.; Querbes, W.; Tam, Y.K.; Ansell, S.M.; Kumar, V.; Qin, J.; et al. Biodegradable lipids enabling rapidly eliminated lipid nanoparticles for systemic delivery of RNAi therapeutics. Mol. Ther. 2013, 21, 1570–1578. [Google Scholar] [CrossRef]
- Allen, T.M. The use of glycolipids and hydrophilic polymers in avoiding rapid uptake of liposomes by the mononuclear phagocyte system. Adv. Drug Deliv. Rev. 1994, 13, 285–309. [Google Scholar] [CrossRef]
- Ishida, T.; Harashima, H.; Kiwada, H. Liposome clearance. Biosci. Rep. 2002, 22, 197–224. [Google Scholar] [CrossRef]
- Klibanov, A.L.; Maruyama, K.; Torchilin, V.P.; Huang, L. Amphipathic polyethyleneglycols effectively prolong the circulation time of liposomes. FEBS Lett. 1990, 268, 235–237. [Google Scholar] [CrossRef]
- Webb, M.S.; Saxon, D.; Wong, F.M.; Lim, H.J.; Wang, Z.; Bally, M.B.; Choi, L.S.; Cullis, P.R.; Mayer, L.D. Comparison of different hydrophobic anchors conjugated to poly(ethylene glycol): Effects on the pharmacokinetics of liposomal vincristine. Biochim. Biophys. Acta 1998, 1372, 272–282. [Google Scholar] [CrossRef]
- Woodle, M.C.; Lasic, D.D. Sterically stabilized liposomes. Biochim. Biophys. Acta 1992, 1113, 171–199. [Google Scholar] [CrossRef]
- Wheeler, J.J.; Palmer, L.; Ossanlou, M.; MacLachlan, I.; Graham, R.W.; Zhang, Y.P.; Hope, M.J.; Scherrer, P.; Cullis, P.R. Stabilized plasmid-lipid particles: Construction and characterization. Gene Ther. 1999, 6, 271–281. [Google Scholar] [CrossRef]
- Mok, K.W.; Lam, A.M.; Cullis, P.R. Stabilized plasmid-lipid particles: Factors influencing plasmid entrapment and transfection properties. Biochim. Biophys. Acta 1999, 1419, 137–150. [Google Scholar] [CrossRef]
- Song, L.Y.; Ahkong, Q.F.; Rong, Q.; Wang, Z.; Ansell, S.; Hope, M.J.; Mui, B. Characterization of the inhibitory effect of PEG-lipid conjugates on the intracellular delivery of plasmid and antisense DNA mediated by cationic lipid liposomes. Biochim. Biophys. Acta 2002, 1558, 1–13. [Google Scholar] [CrossRef]
- Monck, M.A.; Mori, A.; Lee, D.; Tam, P.; Wheeler, J.J.; Cullis, P.R.; Scherrer, P. Stabilized plasmid-lipid particles: Pharmacokinetics and plasmid delivery to distal tumors following intravenous injection. J. Drug Target. 2000, 7, 439–452. [Google Scholar] [CrossRef]
- Tam, P.; Monck, M.; Lee, D.; Ludkovski, O.; Leng, E.C.; Clow, K.; Stark, H.; Scherrer, P.; Graham, R.W.; Cullis, P.R. Stabilized plasmid-lipid particles for systemic gene therapy. Gene Ther. 2000, 7, 1867–1874. [Google Scholar] [CrossRef]
- Ambegia, E.; Ansell, S.; Cullis, P.; Heyes, J.; Palmer, L.; MacLachlan, I. Stabilized plasmid-lipid particles containing PEG-diacylglycerols exhibit extended circulation lifetimes and tumor selective gene expression. Biochim. Biophys. Acta 2005, 1669, 155–163. [Google Scholar] [CrossRef]
- Judge, A.; McClintock, K.; Phelps, J.R.; Maclachlan, I. Hypersensitivity and loss of disease site targeting caused by antibody responses to pegylated liposomes. Mol. Ther. 2006, 13, 328–337. [Google Scholar] [CrossRef]
- Semple, S.C.; Harasym, T.O.; Clow, K.A.; Ansell, S.M.; Klimuk, S.K.; Hope, M.J. Immunogenicity and rapid blood clearance of liposomes containing polyethylene glycol-lipid conjugates and nucleic acid. J. Pharmacol. Exp. Ther. 2005, 312, 1020–1026. [Google Scholar]
- Heyes, J.; Hall, K.; Tailor, V.; Lenz, R.; MacLachlan, I. Synthesis and characterization of novel poly(ethylene glycol)-lipid conjugates suitable for use in drug delivery. J. Control. Release 2006, 112, 280–290. [Google Scholar] [CrossRef]
- Chonn, A.; Semple, S.C.; Cullis, P.R. Association of blood proteins with large unilamellar liposomes in vivo. Relation to circulation lifetimes. J. Biol. Chem. 1992, 267, 18759–18765. [Google Scholar]
- Cullis, P.R.; Chonn, A.; Semple, S.C. Interactions of liposomes and lipid-based carrier systems with blood proteins: Relation to clearance behaviour in vivo. Adv. Drug Deliv. Rev. 1998, 32, 3–17. [Google Scholar] [CrossRef]
- Mendez, A.J.; He, J.L.; Huang, H.S.; Wen, S.R.; Hsia, S.L. Interaction of rabbit lipoproteins and red blood cells with liposomes of egg yolk phospholipids. Lipids 1988, 23, 961–967. [Google Scholar] [CrossRef]
- Rensen, P.C.; Schiffelers, R.M.; Versluis, A.J.; Bijsterbosch, M.K.; van Kuijk-Meuwissen, M.E.; van Berkel, T.J. Human recombinant apolipoprotein e-enriched liposomes can mimic low-density lipoproteins as carriers for the site-specific delivery of antitumor agents. Mol. Pharmacol. 1997, 52, 445–455. [Google Scholar]
- Bisgaier, C.L.; Siebenkas, M.V.; Williams, K.J. Effects of apolipoproteins A-IV and A-I on the uptake of phospholipid liposomes by hepatocytes. J. Biol. Chem. 1989, 264, 862–866. [Google Scholar]
- Yan, X.; Kuipers, F.; Havekes, L.M.; Havinga, R.; Dontje, B.; Poelstra, K.; Scherphof, G.L.; Kamps, J.A. The role of apolipoprotein e in the elimination of liposomes from blood by hepatocytes in the mouse. Biochem. Biophys. Res. Commun. 2005, 328, 57–62. [Google Scholar] [CrossRef]
- Akinc, A.; Querbes, W.; De, S.; Qin, J.; Frank-Kamenetsky, M.; Jayaprakash, K.N.; Jayaraman, M.; Rajeev, K.G.; Cantley, W.L.; Dorkin, J.R.; et al. Targeted delivery of rnai therapeutics with endogenous and exogenous ligand-based mechanisms. Mol. Ther. 2010, 18, 1357–1364. [Google Scholar] [CrossRef]
- Sapra, P.; Allen, T.M. Ligand-targeted liposomal anticancer drugs. Progr. Lipid Res. 2003, 42, 439–462. [Google Scholar] [CrossRef]
- Cressman, S.; Dobson, I.; Lee, J.B.; Tam, Y.Y.; Cullis, P.R. Synthesis of a labeled RGD-lipid, its incorporation into liposomal nanoparticles, and their trafficking in cultured endothelial cells. Bioconjug. Chem. 2009, 20, 1404–1411. [Google Scholar] [CrossRef]
- Allen, T.M.; Sapra, P.; Moase, E.; Moreira, J.; Iden, D. Adventures in targeting. J. Liposome Res. 2002, 12, 5–12. [Google Scholar] [CrossRef]
- Di Paolo, D.; Ambrogio, C.; Pastorino, F.; Brignole, C.; Martinengo, C.; Carosio, R.; Loi, M.; Pagnan, G.; Emionite, L.; Cilli, M.; et al. Selective therapeutic targeting of the anaplastic lymphoma kinase with liposomal sirna induces apoptosis and inhibits angiogenesis in neuroblastoma. Mol. Ther. 2011, 19, 2201–2212. [Google Scholar] [CrossRef]
- Li, S.D.; Huang, L. Targeted delivery of antisense oligodeoxynucleotide and small interference RNA into lung cancer cells. Mol. Pharm. 2006, 3, 579–588. [Google Scholar] [CrossRef]
- Tam, Y.Y.C.; Chen, S.; Zaifman, J.; Tam, Y.K.; Lin, P.J.C.; Ansell, S.; Roberge, M.; Ciufolini, M.A.; Cullis, P.R. Small molecule ligands for enhanced intracellular delivery of lipid nanoparticle formulations of siRNA. Nanomed. Nanotechnol. Biol. Med. 2013, 9, 665–674. [Google Scholar] [CrossRef]
- Li, S.D.; Chono, S.; Huang, L. Efficient oncogene silencing and metastasis inhibition via systemic delivery of siRNA. Mol. Ther. 2008, 16, 942–946. [Google Scholar] [CrossRef]
- Chen, Y.; Sen, J.; Bathula, S.R.; Yang, Q.; Fittipaldi, R.; Huang, L. Novel cationic lipid that delivers siRNA and enhances therapeutic effect in lung cancer cells. Mol. Pharm. 2009, 6, 696–705. [Google Scholar] [CrossRef]
- Cabral, H.; Matsumoto, Y.; Mizuno, K.; Chen, Q.; Murakami, M.; Kimura, M.; Terada, Y.; Kano, M.R.; Miyazono, K.; Uesaka, M.; et al. Accumulation of sub-100 nm polymeric micelles in poorly permeable tumours depends on size. Nat. Nanotechnol. 2011, 6, 815–823. [Google Scholar] [CrossRef]
- Huo, S.; Ma, H.; Huang, K.; Liu, J.; Wei, T.; Jin, S.; Zhang, J.; He, S.; Liang, X.J. Superior penetration and retention behavior of 50 nm gold nanoparticles in tumors. Cancer Res. 2013, 73, 319–330. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Tam, Y.Y.C.; Chen, S.; Cullis, P.R. Advances in Lipid Nanoparticles for siRNA Delivery. Pharmaceutics 2013, 5, 498-507. https://doi.org/10.3390/pharmaceutics5030498
Tam YYC, Chen S, Cullis PR. Advances in Lipid Nanoparticles for siRNA Delivery. Pharmaceutics. 2013; 5(3):498-507. https://doi.org/10.3390/pharmaceutics5030498
Chicago/Turabian StyleTam, Yuen Yi C., Sam Chen, and Pieter R. Cullis. 2013. "Advances in Lipid Nanoparticles for siRNA Delivery" Pharmaceutics 5, no. 3: 498-507. https://doi.org/10.3390/pharmaceutics5030498