Proteoliposomes as Tool for Assaying Membrane Transporter Functions and Interactions with Xenobiotics


Abstract
:1. Introduction
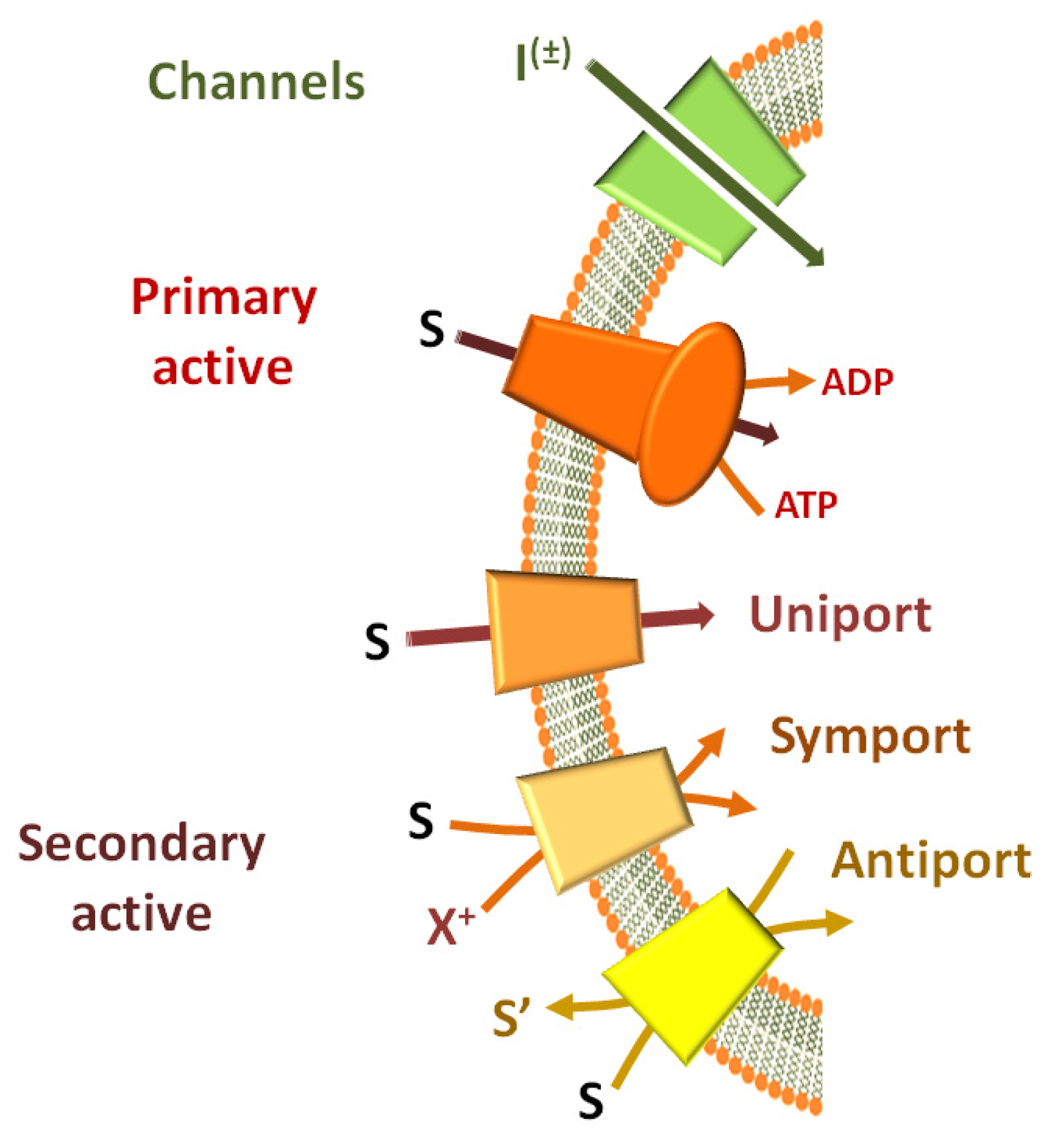
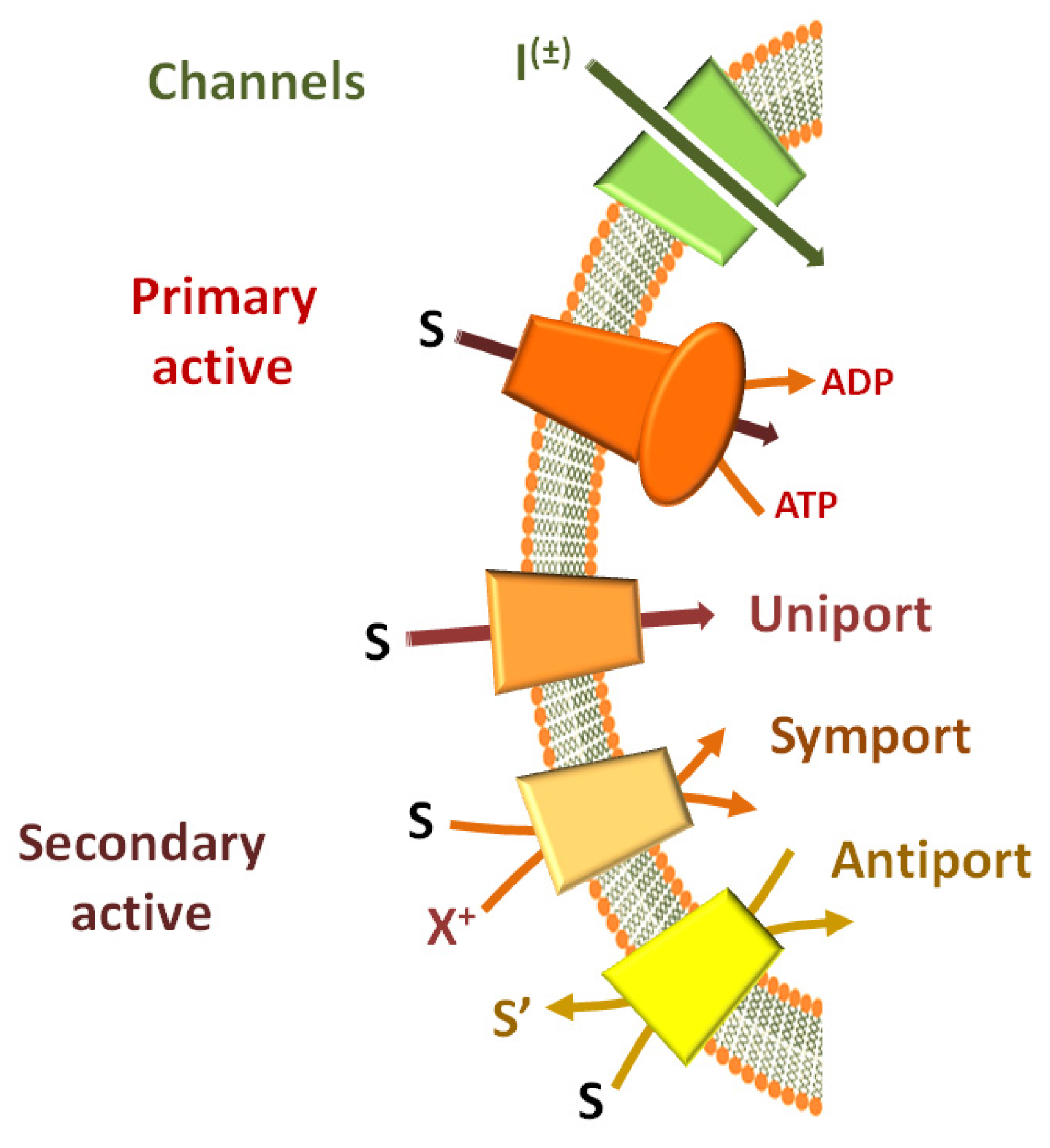
2. Experimental Tools for Studying Transport
2.1. Intact Cell Systems
2.2. Proteoliposomes
Methods of Proteoliposome Preparation
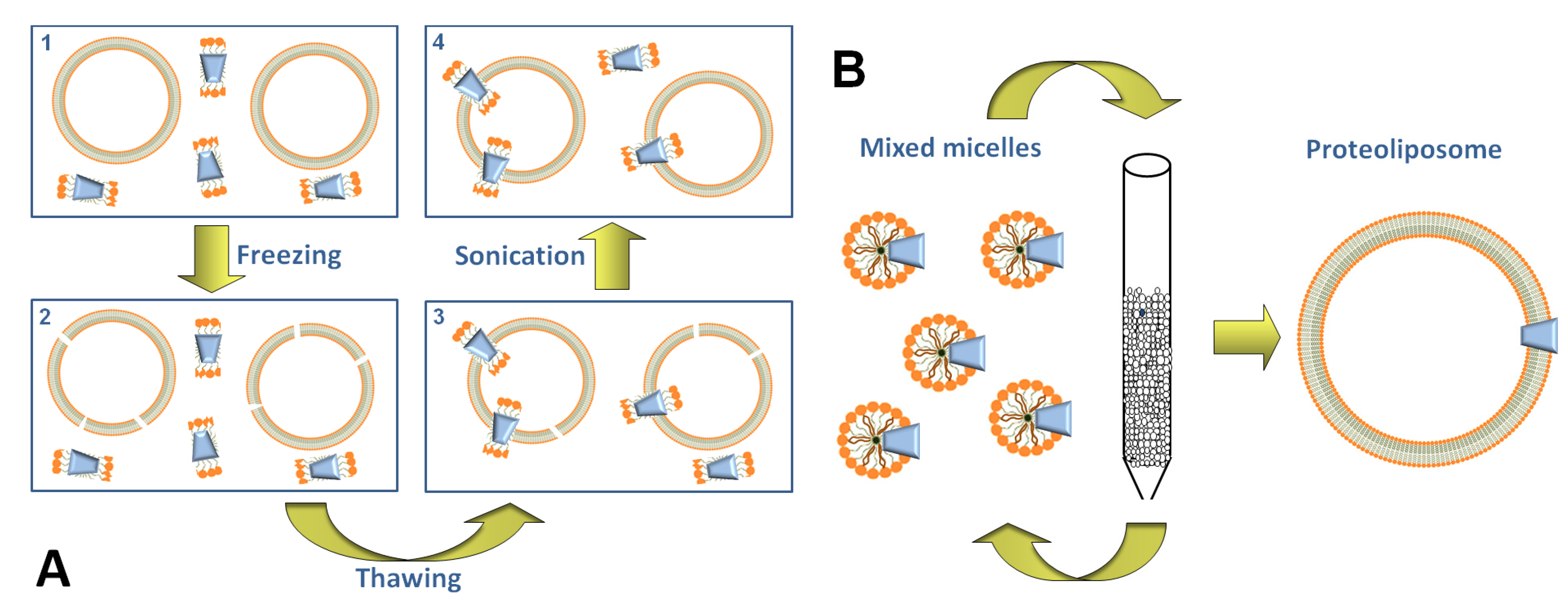
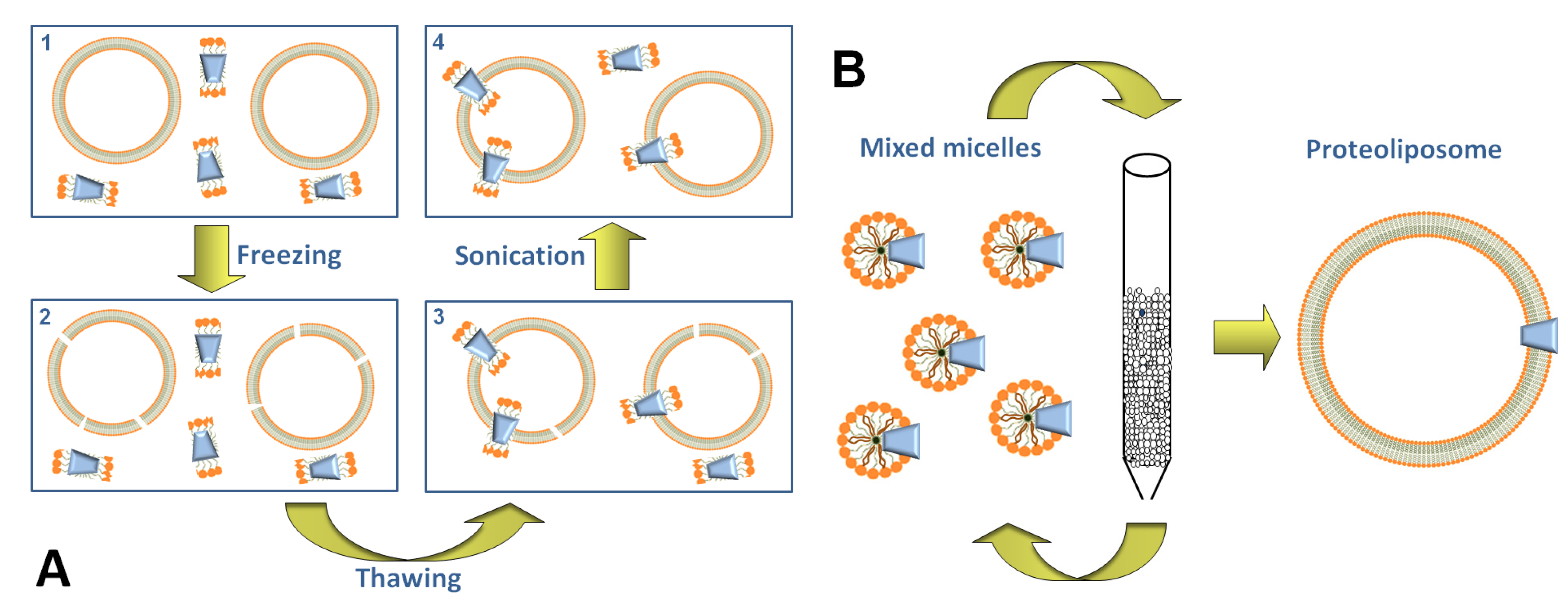
3. Transporter Function in Proteoliposomes
3.1. Using the Freeze-Thaw-Sonication Method
3.2. Using the Detergent-Removal Method
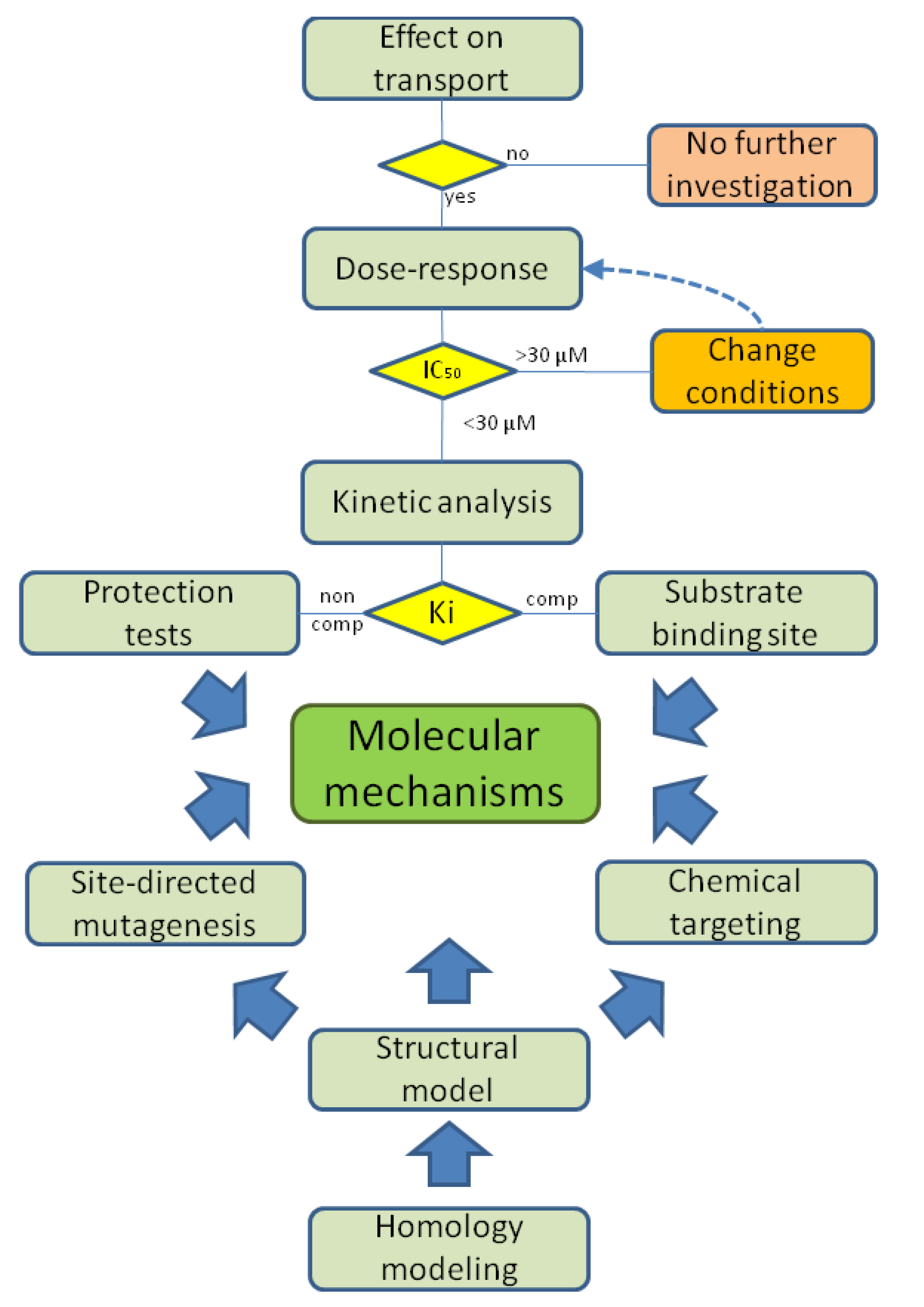
4. Using Proteoliposomes for Revealing Xenobiotic-Transporter Interaction Mechanisms


{kind=link}
{kind=link}
{kind=link}
| Molecules | CACT | ORCT | References | ||
|---|---|---|---|---|---|
| IC50 | Ki | IC50 | Ki | ||
| HgCl2 | 0.03 | 1.4 | [102,103] | ||
| Mersalyl | 0.05 | 1.1 | [102,103] | ||
| p-OHMB | 0.59 | [102] | |||
| p-CMBS | 0.61 | [102] | |||
| Cu2+ | 0.71 | [104] | |||
| Zn2+ | 40 | [104] | |||
| Cd2+ | 24 | [104] | |||
| Pb2+ | 50 | [104] | |||
| Co2+ | 210 | 94 | [104] | ||
| Mn2+ | 350 | 420 | [104] | ||
| Ni2+ | 83 | 53 | [104] | ||
| H2O2 | 170 | 710 | [105] | ||
| Cefonicid (*) | 6800 | 4900 | [106] | ||
| Cefonicid (**) | 120 | [106] | |||
| Ampicillin | 7600 | 9900 | [106] | ||
| Mildronate | 560 | 530 | [107] | ||
4.1. First Level Targets: Plasma Membrane Transporters
4.1.1. The Glutamine/Neutral Amino Acid Transporters B0AT1 and ASCT2
| Molecules | ASCT2 | B0AT1 | OCTN2 | References | |||
|---|---|---|---|---|---|---|---|
| IC50 | Ki | IC50 | Ki | IC50 | Ki | ||
| HgCl2 | 1.4 | 0.85 | 0.42 | 2.5 | 4.2 | [93,110,113] | |
| MethylHg | 2.4 | 1.1 | 0.89 | 0.33 | 7.4 | 13 | [93,110,113] |
| Mersalyl | 3.1 | 3.3 | 2 | [91,110] | |||
| Cu2+ | 20 | [110] | |||||
| 1,2,3,-dithiazoles | 3–30 | [49] | |||||
| Omeprazole (*) | 5.7 | 5.2 | [114] | ||||
| Omeprazole (**) | 20.4 | 14.6 | [114] | ||||
4.1.2. The Carnitine Transporter OCTN2
4.2. Second Level Targets: Mitochondrial Transporters
4.2.1. The Carnitine/Acylcarnitine Transporter, CACT
4.2.2. The Ornithine/Citrulline Transporter, ORCT
5. Conclusions
Abbreviations
| OCTN | Organic Cation Transporter Novel |
| ASCT2 | Alanine, Serine, Cysteine, Transporter 2 |
| B0AT1 | Broad specificity Amino acid Transporter 1 |
| TEA | tetraethylamonium |
| CACT | Carnitine/Acyl-Carnitine Transporter |
| ORCT | Ornithine/Citrulline transporter |
| LeuT | Leucine Transporter |
| GltPh | Glutamate transporter from Pyrococcus horikoshii |
| methyl-Hg | methyl mercury |
| NAC | N-Acetyl Cysteine |
Acknowledgements
Conflicts of interest
References
- Lahjouji, K.; Mitchell, G.A.; Qureshi, I.A. Carnitine transport by organic cation transporters and systemic carnitine deficiency. Mol. Genet. Metab. 2001, 73, 287–297. [Google Scholar] [CrossRef]
- Chou, J.Y.; Matern, D.; Mansfield, B.C.; Chen, Y.T. Type I glycogen storage diseases: Disorders of the glucose-6-phosphatase complex. Curr. Mol. Med. 2002, 2, 121–143. [Google Scholar] [CrossRef]
- Daniel, H.; Kottra, G. The proton oligopeptide cotransporter family SLC15 in physiology and pharmacology. Pflug. Arch. 2004, 447, 610–618. [Google Scholar] [CrossRef]
- Palmieri, F. Diseases caused by defects of mitochondrial carriers: A review. Biochim. Biophys. Acta 2008, 1777, 564–578. [Google Scholar] [CrossRef]
- Broer, S.; Palacin, M. The role of amino acid transporters in inherited and acquired diseases. Biochem. J. 2011, 436, 193–211. [Google Scholar] [CrossRef]
- Law, C.J.; Maloney, P.C.; Wang, D.N. Ins and outs of major facilitator superfamily antiporters. Annu. Rev. Microbiol. 2008, 62, 289–305. [Google Scholar] [CrossRef]
- Herrera, M.; Garvin, J.L. Aquaporins as gas channels. Pflug. Arch. 2011, 462, 623–630. [Google Scholar] [CrossRef]
- Forrest, L.R.; Kramer, R.; Ziegler, C. The structural basis of secondary active transport mechanisms. Biochim. Biophys. Acta 2011, 1807, 167–188. [Google Scholar] [CrossRef]
- Saier, M.H., Jr.; Yen, M.R.; Noto, K.; Tamang, D.G.; Elkan, C. The transporter classification database: Recent advances. Nucleic Acids Res. 2009, 37, D274–D278. [Google Scholar] [CrossRef]
- Hediger, M.A.; Clemencon, B.; Burrier, R.E.; Bruford, E.A. The ABCs of membrane transporters in health and disease (SLC series): Introduction. Mol. Asp. Med. 2013, 34, 95–107. [Google Scholar] [CrossRef]
- Nyblom, M.; Oberg, F.; Lindkvist-Petersson, K.; Hallgren, K.; Findlay, H.; Wikstrom, J.; Karlsson, A.; Hansson, O.; Booth, P.J.; Bill, R.M.; et al. Exceptional overproduction of a functional human membrane protein. Protein Expr. Purif. 2007, 56, 110–120. [Google Scholar] [CrossRef]
- Wright, E.M.; Loo, D.D.; Hirayama, B.A. Biology of human sodium glucose transporters. Physiol. Rev. 2011, 91, 733–794. [Google Scholar] [CrossRef]
- Wright, E.M. Glucose transport families SLC5 and SLC50. Mol. Asp. Med. 2013, 34, 183–196. [Google Scholar] [CrossRef]
- Bodoy, S.; Fotiadis, D.; Stoeger, C.; Kanai, Y.; Palacin, M. The small SLC43 family: Facilitator system l amino acid transporters and the orphan EEG1. Mol. Asp. Med. 2013, 34, 638–645. [Google Scholar] [CrossRef]
- Fotiadis, D.; Kanai, Y.; Palacin, M. The SLC3 and SLC7 families of amino acid transporters. Mol. Asp. Med. 2013, 34, 139–158. [Google Scholar] [CrossRef]
- Kanai, Y.; Clemencon, B.; Simonin, A.; Leuenberger, M.; Lochner, M.; Weisstanner, M.; Hediger, M.A. The SLC1 high-affinity glutamate and neutral amino acid transporter family. Mol. Asp. Med. 2013, 34, 108–120. [Google Scholar] [CrossRef]
- Anderson, C.M.; Stahl, A. SLC27 fatty acid transport proteins. Mol. Asp. Med. 2013, 34, 516–528. [Google Scholar] [CrossRef]
- Indiveri, C.; Pochini, L.; Oppedisano, F.; Tonazzi, A. The carnitine transporter network: Interactions with drugs. Curr. Chem. Biol. 2010, 4, 108–123. [Google Scholar]
- Yonezawa, A.; Inui, K. Novel riboflavin transporter family RFVT/SLC52: Identification, nomenclature, functional characterization and genetic diseases of RFVT/SLC52. Mol. Asp. Med. 2013, 34, 693–701. [Google Scholar] [CrossRef]
- Zhao, R.; Goldman, I.D. Folate and thiamine transporters mediated by facilitative carriers (SLC19A1-3 and SLC46A1) and folate receptors. Mol. Asp. Med. 2013, 34, 373–385. [Google Scholar] [CrossRef]
- Palmieri, F. The mitochondrial transporter family SLC25: Identification, properties and physiopathology. Mol. Asp. Med. 2013, 34, 465–484. [Google Scholar] [CrossRef]
- Hirabayashi, Y.; Nomura, K.H.; Nomura, K. The acetyl-CoA transporter family SLC33. Mol. Asp. Med. 2013, 34, 586–589. [Google Scholar] [CrossRef]
- Csala, M.; Marcolongo, P.; Lizak, B.; Senesi, S.; Margittai, E.; Fulceri, R.; Magyar, J.E.; Benedetti, A.; Banhegyi, G. Transport and transporters in the endoplasmic reticulum. Biochim. Biophys. Acta 2007, 1768, 1325–1341. [Google Scholar] [CrossRef]
- Lawal, H.O.; Krantz, D.E. SLC18: Vesicular neurotransmitter transporters for monoamines and acetylcholine. Mol. Asp. Med. 2013, 34, 360–372. [Google Scholar] [CrossRef]
- Bobulescu, I.A.; Moe, O.W. Luminal Na(+)/H (+) exchange in the proximal tubule. Pflug. Arch. 2009, 458, 5–21. [Google Scholar] [CrossRef]
- Forster, I.C.; Hernando, N.; Biber, J.; Murer, H. Phosphate transporters of the SLC20 and SLC34 families. Mol. Asp. Med. 2013, 34, 386–395. [Google Scholar] [CrossRef]
- Verrey, F.; Ristic, Z.; Romeo, E.; Ramadan, T.; Makrides, V.; Dave, M.H.; Wagner, C.A.; Camargo, S.M. Novel renal amino acid transporters. Annu. Rev. Physiol. 2005, 67, 557–572. [Google Scholar] [CrossRef]
- Broer, S. Amino acid transport across mammalian intestinal and renal epithelia. Physiol. Rev. 2008, 88, 249–286. [Google Scholar] [CrossRef]
- TransportDB. Available online: http://www.membranetransport.org (accessed on 12 September 2013).
- Indiveri, C.; Galluccio, M.; Scalise, M.; Pochini, L. Strategies of bacterial over expression of membrane transporters relevant in human health: The successful case of the three members of OCTN subfamily. Mol. Biotechnol. 2013, 54, 724–736. [Google Scholar] [CrossRef]
- Transporter Classification Database. Available online: http://www.tcdb.org/pdb_structure.php (accessed on 12 September 2013).
- Mizuno, N.; Niwa, T.; Yotsumoto, Y.; Sugiyama, Y. Impact of drug transporter studies on drug discovery and development. Pharmacol. Rev. 2003, 55, 425–461. [Google Scholar] [CrossRef]
- Sugiura, T.; Kato, Y.; Tsuji, A. Role of SLC xenobiotic transporters and their regulatory mechanisms PDZ proteins in drug delivery and disposition. J. Control. Release 2006, 116, 238–246. [Google Scholar] [CrossRef]
- Giacomini, K.M.; Huang, S.M.; Tweedie, D.J.; Benet, L.Z.; Brouwer, K.L.; Chu, X.; Dahlin, A.; Evers, R.; Fischer, V.; Hillgren, K.M.; et al. Membrane transporters in drug development. Nat. Rev. 2010, 9, 215–236. [Google Scholar] [CrossRef]
- Han, H.K. Role of transporters in drug interactions. Arch. Pharm. Res. 2011, 34, 1865–1877. [Google Scholar] [CrossRef]
- DeGorter, M.K.; Xia, C.Q.; Yang, J.J.; Kim, R.B. Drug transporters in drug efficacy and toxicity. Annu. Rev. Pharmacol. Toxicol. 2012, 52, 249–273. [Google Scholar] [CrossRef]
- Mandery, K.; Glaeser, H.; Fromm, M.F. Interaction of innovative small molecule drugs used for cancer therapy with drug transporters. Br. J. Pharmacol. 2012, 165, 345–362. [Google Scholar] [CrossRef]
- Rask-Andersen, M.; Masuram, S.; Fredriksson, R.; Schioth, H.B. Solute carriers as drug targets: Current use, clinical trials and prospective. Mol. Asp. Med. 2013, 34, 702–710. [Google Scholar] [CrossRef]
- Giacomini, K.M.; Huang, S.M. Transporters in drug development and clinical pharmacology. Clin. Pharmacol. Ther. 2013, 94, 3–9. [Google Scholar] [CrossRef]
- Lounkine, E.; Keiser, M.J.; Whitebread, S.; Mikhailov, D.; Hamon, J.; Jenkins, J.L.; Lavan, P.; Weber, E.; Doak, A.K.; Cote, S.; et al. Large-scale prediction and testing of drug activity on side-effect targets. Nature 2012, 486, 361–367. [Google Scholar]
- Tsuji, A. Transporter-mediated drug interactions. Drug Metab. Pharmacokinet. 2002, 17, 253–274. [Google Scholar] [CrossRef]
- Nakanishi, T.; Tamai, I. Solute carrier transporters as targets for drug delivery and pharmacological intervention for chemotherapy. J. Pharm. Sci. 2011, 100, 3731–3750. [Google Scholar] [CrossRef]
- Sharom, F.J. The P-glycoprotein multidrug transporter. Essays Biochem. 2011, 50, 161–178. [Google Scholar] [CrossRef]
- Macheda, M.L.; Rogers, S.; Best, J.D. Molecular and cellular regulation of glucose transporter (GLUT) proteins in cancer. J. Cell. Physiol. 2005, 202, 654–662. [Google Scholar] [CrossRef]
- Minchinton, A.I.; Tannock, I.F. Drug penetration in solid tumours. Nat. Rev. Cancer 2006, 6, 583–592. [Google Scholar] [CrossRef]
- Okabe, M.; Szakacs, G.; Reimers, M.A.; Suzuki, T.; Hall, M.D.; Abe, T.; Weinstein, J.N.; Gottesman, M.M. Profiling SLCO and SLC22 genes in the NCI-60 cancer cell lines to identify drug uptake transporters. Mol. Cancer Ther. 2008, 7, 3081–3091. [Google Scholar] [CrossRef]
- Ganapathy, V.; Thangaraju, M.; Prasad, P.D. Nutrient transporters in cancer: Relevance to Warburg hypothesis and beyond. Pharmacol. Ther. 2009, 121, 29–40. [Google Scholar] [CrossRef]
- Falasca, M.; Linton, K.J. Investigational ABC transporter inhibitors. Expert Opin. Investig. Drugs 2012, 21, 657–666. [Google Scholar] [CrossRef]
- Oppedisano, F.; Catto, M.; Koutentis, P.A.; Nicolotti, O.; Pochini, L.; Koyioni, M.; Introcaso, A.; Michaelidou, S.S.; Carotti, A.; Indiveri, C. Inactivation of the glutamine/amino acid transporter ASCT2 by 1,2,3-dithiazoles: Proteoliposomes as a tool to gain insights in the molecular mechanism of action and of antitumor activity. Toxicol. Appl. Pharmacol. 2012, 265, 93–102. [Google Scholar] [CrossRef]
- Zamek-Gliszczynski, M.J.; Lee, C.A.; Poirier, A.; Bentz, J.; Chu, X.; Ellens, H.; Ishikawa, T.; Jamei, M.; Kalvass, J.C.; Nagar, S.; et al. ITC recommendations for transporter kinetic parameter estimation and translational modeling of transport-mediated PK and DDIs in humans. Clin. Pharmacol. Ther. 2013, 94, 64–79. [Google Scholar] [CrossRef]
- Hillgren, K.M.; Keppler, D.; Zur, A.A.; Giacomini, K.M.; Stieger, B.; Cass, C.E.; Zhang, L. Emerging transporters of clinical importance: An update from the international transporter consortium. Clin. Pharmacol. Ther. 2013, 94, 52–63. [Google Scholar] [CrossRef]
- Huang, S.M.; Strong, J.M.; Zhang, L.; Reynolds, K.S.; Nallani, S.; Temple, R.; Abraham, S.; Habet, S.A.; Baweja, R.K.; Burckart, G.J.; et al. New era in drug interaction evaluation: US Food and Drug Administration update on CYP enzymes, transporters, and the guidance process. J. Clin. Pharmacol. 2008, 48, 662–670. [Google Scholar] [CrossRef]
- Zhang, L.; Zhang, Y.; Huang, S.M. Scientific and regulatory perspectives on metabolizing enzyme-transporter interplay and its role in drug interactions: Challenges in predicting drug interactions. Mol. Pharm. 2009, 6, 1766–1774. [Google Scholar] [CrossRef]
- Agarwal, S.; Chinn, L.; Zhang, L. An overview of transporter information in package inserts of recently approved new molecular entities. Pharm. Res. 2013, 30, 899–910. [Google Scholar] [CrossRef]
- Guidotti, G.G.; Luneburg, B.; Borghetti, A.F. Amino acid uptake in isolated chick embryo heart cells. Biochem. J. 1969, 114, 97–105. [Google Scholar]
- Palmieri, F.; Klingenberg, M. Direct methods for measuring metabolite transport and distribution in mitochondria. Methods Enzymol. 1979, 56, 279–301. [Google Scholar] [CrossRef]
- Burchell, A. Endoplasmic reticulum phosphate transport. Kidney Int. 1996, 49, 953–958. [Google Scholar] [CrossRef]
- U.S. Department of Health and Human Services Food and Drug Administration Center for Drug Evaluation and Research (CDER). Drug Interaction Studies—Study Design, Data Analysis, Implications for Dosing, and Labeling Recommendations. Clin. Pharmacol. 2012, 1–79.
- Pochini, L.; Scalise, M.; Galluccio, M.; Indiveri, C. OCTN cation transporters in health and disease: Role as drug targets and assay development. J. Biomol. Screen. 2013. [Google Scholar] [CrossRef]
- Whorton, M.R.; MacKinnon, R. Crystal structure of the mammalian GIRK2 K+ channel and gating regulation by G proteins, PIP2, and sodium. Cell 2011, 147, 199–208. [Google Scholar] [CrossRef]
- Brohawn, S.G.; del Marmol, J.; MacKinnon, R. Crystal structure of the human K2P TRAAK, a lipid- and mechano-sensitive K+ ion channel. Science 2012, 335, 436–441. [Google Scholar] [CrossRef]
- Miller, A.N.; Long, S.B. Crystal structure of the human two-pore domain potassium channel K2P1. Science 2012, 335, 432–436. [Google Scholar] [CrossRef]
- Song, C.; Weichbrodt, C.; Salnikov, E.S.; Dynowski, M.; Forsberg, B.O.; Bechinger, B.; Steinem, C.; de Groot, B.L.; Zachariae, U.; Zeth, K. Crystal structure and functional mechanism of a human antimicrobial membrane channel. Proc. Natl. Acad. Sci. USA 2013, 110, 4586–4591. [Google Scholar] [CrossRef]
- Gregoriadis, G.; Ryman, B.E. Liposomes as carriers of enzymes or drugs: A new approach to the treatment of storage diseases. Biochem. J. 1971, 124, 58P. [Google Scholar]
- Gregoriadis, G. The carrier potential of liposomes in biology and medicine (first of two parts). N. Engl. J. Med. 1976, 295, 704–710. [Google Scholar] [CrossRef]
- Gregoriadis, G. The carrier potential of liposomes in biology and medicine (second of two parts). N. Engl. J. Med. 1976, 295, 765–770. [Google Scholar] [CrossRef]
- Boddapati, S.V.; D’Souza, G.G.; Weissig, V. Liposomes for drug delivery to mitochondria. Methods Mol. Biol. 2010, 605, 295–303. [Google Scholar] [CrossRef]
- Kasahara, M.; Hinkle, P.C. Reconstitution of D-glucose transport catalyzed by a protein fraction from human erythrocytes in sonicated liposomes. Proc. Nat. Acad. Sci. USA 1976, 73, 396–400. [Google Scholar] [CrossRef]
- Kasahara, M.; Hinkle, P.C. Reconstitution and purification of the D-glucose transporter from human erythrocytes. J. Biol. Chem. 1977, 252, 7384–7390. [Google Scholar]
- Newman, M.J.; Foster, D.L.; Wilson, T.H.; Kaback, H.R. Purification and reconstitution of functional lactose carrier from Escherichia coli. J. Biol. Chem. 1981, 256, 11804–11808. [Google Scholar]
- Viitanen, P.; Garcia, M.L.; Kaback, H.R. Purified reconstituted lac carrier protein from Escherichia coli is fully functional. Proc. Natl. Acad. Sci. USA 1984, 81, 1629–1633. [Google Scholar] [CrossRef]
- Kramer, R.; Klingenberg, M. Reconstitution of adenine nucleotide transport from beef heart mitochondria. Biochemistry 1979, 18, 4209–4215. [Google Scholar] [CrossRef]
- Ueno, M.; Tanford, C.; Reynolds, J.A. Phospholipid vesicle formation using nonionic detergents with low monomer solubility. Kinetic factors determine vesicle size and permeability. Biochemistry 1984, 23, 3070–3076. [Google Scholar] [CrossRef]
- Kramer, R.; Heberger, C. Functional reconstitution of carrier proteins by removal of detergent with a hydrophobic ion exchange column. Biochim. Biophys. Acta 1986, 863, 289–296. [Google Scholar] [CrossRef]
- Palmieri, F.; Indiveri, C.; Bisaccia, F.; Iacobazzi, V. Mitochondrial metabolite carrier proteins: Purification, reconstitution, and transport studies. Methods Enzymol. 1995, 260, 349–369. [Google Scholar] [CrossRef]
- Klingenberg, M.; Winkler, E. The reconstituted isolated uncoupling protein is a membrane potential driven H+ translocator. EMBO J. 1985, 4, 3087–3092. [Google Scholar]
- Spagnoletta, A.; de Palma, A.; Prezioso, G.; Scalera, V. A micro-batchwise technique method for rapid reconstitution of functionally active mitochondrial ADP/ATP carrier from Jerusalem artichoke (Helianthus tuberosus L.) tubers. J. Biochem. Biophys. Methods 2008, 70, 954–957. [Google Scholar] [CrossRef]
- Pochini, L.; Scalise, M.; Galluccio, M.; Amelio, L.; Indiveri, C. Reconstitution in liposomes of the functionally active human OCTN1 (SLC22A4) transporter overexpressed in Escherichia coli. Biochem. J. 2011, 439, 227–233. [Google Scholar] [CrossRef]
- Indiveri, C. Studying amino acid transport using liposomes. Methods Mol. Biol. 2010, 606, 55–68. [Google Scholar] [CrossRef]
- Rigaud, J.L.; Levy, D. Reconstitution of membrane proteins into liposomes. Methods Enzymol. 2003, 372, 65–86. [Google Scholar] [CrossRef]
- Klingenberg, M. Molecular aspects of the adenine nucleotide carrier from mitochondria. Arch. Biochem. Biophys. 1989, 270, 1–14. [Google Scholar] [CrossRef]
- Palmieri, F.; Indiveri, C.; Bisaccia, F.; Kramer, R. Functional properties of purified and reconstituted mitochondrial metabolite carriers. J. Bioenerg. Biomembr. 1993, 25, 525–535. [Google Scholar] [CrossRef]
- Palmieri, F. Mitochondrial carrier proteins. FEBS Lett. 1994, 346, 48–54. [Google Scholar] [CrossRef]
- Vickers, M.F.; Mani, R.S.; Sundaram, M.; Hogue, D.L.; Young, J.D.; Baldwin, S.A.; Cass, C.E. Functional production and reconstitution of the human equilibrative nucleoside transporter (hENT1) in Saccharomyces cerevisiae. Interaction of inhibitors of nucleoside transport with recombinant hENT1 and a glycosylation-defective derivative (hENT1/N48Q). Biochem. J. 1999, 339, 21–32. [Google Scholar] [CrossRef]
- Quick, M.; Wright, E.M. Employing Escherichia coli to functionally express, purify, and characterize a human transporter. Proc. Natl. Acad. Sci. USA 2002, 99, 8597–8601. [Google Scholar] [CrossRef]
- Komatsu, T.; Hiasa, M.; Miyaji, T.; Kanamoto, T.; Matsumoto, T.; Otsuka, M.; Moriyama, Y.; Omote, H. Characterization of the human MATE2 proton-coupled polyspecific organic cation exporter. Int. J. Biochem. Cell Biol. 2011, 43, 913–918. [Google Scholar] [CrossRef]
- Filippo, C.A.; Ardon, O.; Longo, N. Glycosylation of the OCTN2 carnitine transporter: Study of natural mutations identified in patients with primary carnitine deficiency. Biochim. Biophys. Acta 2011, 1812, 312–320. [Google Scholar] [CrossRef]
- Keller, T.; Schwarz, D.; Bernhard, F.; Dotsch, V.; Hunte, C.; Gorboulev, V.; Koepsell, H. Cell free expression and functional reconstitution of eukaryotic drug transporters. Biochemistry 2008, 47, 4552–4564. [Google Scholar] [CrossRef]
- Pochini, L.; Oppedisano, F.; Indiveri, C. Reconstitution into liposomes and functional characterization of the carnitine transporter from renal cell plasma membrane. Biochim. Biophys. Acta 2004, 1661, 78–86. [Google Scholar] [CrossRef]
- Oppedisano, F.; Pochini, L.; Galluccio, M.; Cavarelli, M.; Indiveri, C. Reconstitution into liposomes of the glutamine/amino acid transporter from renal cell plasma membrane: Functional characterization, kinetics and activation by nucleotides. Biochim. Biophys. Acta 2004, 1667, 122–131. [Google Scholar] [CrossRef]
- Oppedisano, F.; Indiveri, C. Reconstitution into liposomes of the B degrees -like glutamine-neutral amino acid transporter from renal cell plasma membrane. Biochim. Biophys. Acta 2008, 1778, 2258–2265. [Google Scholar] [CrossRef]
- Oppedisano, F.; Pochini, L.; Galluccio, M.; Indiveri, C. The glutamine/amino acid transporter (ASCT2) reconstituted in liposomes: Electrical nature of the glutamine/glutamate antiport. Ital. J. Biochem. 2007, 56, 275–278. [Google Scholar]
- Oppedisano, F.; Pochini, L.; Broer, S.; Indiveri, C. The B0AT1 amino acid transporter from rat kidney reconstituted in liposomes: Kinetics and inactivation by methylmercury. Biochim. Biophys. Acta 2011, 1808, 2551–2558. [Google Scholar] [CrossRef]
- Mouro-Chanteloup, I.; Cochet, S.; Chami, M.; Genetet, S.; Zidi-Yahiaoui, N.; Engel, A.; Colin, Y.; Bertrand, O.; Ripoche, P. Functional reconstitution into liposomes of purified human RhCG ammonia channel. PLoS One 2010, 5, e8921. [Google Scholar] [CrossRef]
- Scalise, M.; Galluccio, M.; Pochini, L.; Indiveri, C. Over-expression in Escherichia coli, purification and reconstitution in liposomes of the third member of the OCTN sub-family: The mouse carnitine transporter OCTN3. Biochem. Biophys. Res. Commun. 2012, 422, 59–63. [Google Scholar] [CrossRef]
- Pingitore, P.; Pochini, L.; Scalise, M.; Galluccio, M.; Hedfalk, K.; Indiveri, C. Large scale production of the active human ASCT2 (SLC1A5) transporter in Pichia pastoris—Functional and kinetic asymmetry revealed in proteoliposomes. Biochim. Biophys. Acta 2013, 1828, 2238–2246. [Google Scholar] [CrossRef]
- Song, P.; Rekow, S.S.; Singleton, C.A.; Sekhon, H.S.; Dissen, G.A.; Zhou, M.; Campling, B.; Lindstrom, J.; Spindel, E.R. Choline transporter-like protein 4 (CTL4) links to non-neuronal acetylcholine synthesis. J. Neurochem. 2013, 126, 451–461. [Google Scholar] [CrossRef]
- Pochini, L.; Scalise, M.; Galluccio, M.; Pani, G.; Siminovitch, K.A.; Indiveri, C. The human OCTN1 (SLC22A4) reconstituted in liposomes catalyzes acetylcholine transport which is defective in the mutant L503F associated to the Crohn’s disease. Biochim. Biophys. Acta 2012, 1818, 559–565. [Google Scholar] [CrossRef]
- Verchere, A.; Broutin, I.; Picard, M. Photo-induced proton gradients for the in vitro investigation of bacterial efflux pumps. Sci. Rep. 2012, 2, 306. [Google Scholar]
- Raunser, S.; Haase, W.; Bostina, M.; Parcej, D.N.; Kuhlbrandt, W. High-yield expression, reconstitution and structure of the recombinant, fully functional glutamate transporter GLT-1 from Rattus norvegicus. J. Mol. Biol. 2005, 351, 598–613. [Google Scholar] [CrossRef]
- Adam, Y.; Edwards, R.H.; Schuldiner, S. Expression and function of the rat vesicular monoamine transporter 2. Am. J. Physiol. Cell Physiol. 2008, 294, C1004–C1011. [Google Scholar] [CrossRef]
- Tonazzi, A.; Indiveri, C. Chemical modification of the mitochondrial ornithine/citrulline carrier by SH reagents: Effects on the transport activity and transition from carrier to pore-like function. Biochim. Biophys. Acta 2003, 1611, 123–130. [Google Scholar] [CrossRef]
- Indiveri, C.; Tonazzi, A.; Dierks, T.; Kramer, R.; Palmieri, F. The mitochondrial carnitine carrier: Characterization of SH-groups relevant for its transport function. Biochim. Biophys. Acta. 1992, 1140, 53–58. [Google Scholar] [CrossRef]
- Tonazzi, A.; Indiveri, C. Effects of heavy metal cations on the mitochondrial ornithine/citrulline transporter reconstituted in liposomes. Biometals 2011, 24, 1205–1215. [Google Scholar] [CrossRef]
- Tonazzi, A.; Console, L.; Indiveri, C. Inhibition of mitochondrial carnitine/acylcarnitine transporter by H2O2: Molecular mechanism and possible implication in pathophysiology. Chem. Biol. Interact. 2013, 203, 423–439. [Google Scholar] [CrossRef]
- Pochini, L.; Galluccio, M.; Scumaci, D.; Giangregorio, N.; Tonazzi, A.; Palmieri, F.; Indiveri, C. Interaction of beta-lactam antibiotics with the mitochondrial carnitine/acylcarnitine transporter. Chem. Biol. Interact. 2008, 173, 187–194. [Google Scholar] [CrossRef]
- Oppedisano, F.; Fanello, D.; Calvani, M.; Indiveri, C. Interaction of mildronate with the mitochondrial carnitine/acylcarnitine transport protein. J. Biochem. Mol. Toxicol. 2008, 22, 8–14. [Google Scholar] [CrossRef]
- Agency for Toxic Substance and Disease Registry (ATSDR), Toxicological Profile for Mercury; Department of Health and Humans Services, Public Health Services, Centers for Disease Control: Atlanta, GA, USA, 1999.
- Van Iwaarden, P.R.; Driessen, A.J.; Konings, W.N. What we can learn from the effects of thiol reagents on transport proteins. Biochim. Biophys. Acta 1992, 1113, 161–170. [Google Scholar] [CrossRef]
- Oppedisano, F.; Galluccio, M.; Indiveri, C. Inactivation by Hg2+ and methylmercury of the glutamine/amino acid transporter (ASCT2) reconstituted in liposomes: Prediction of the involvement of a CXXC motif by homology modelling. Biochem. Pharmacol. 2010, 80, 1266–1273. [Google Scholar] [CrossRef]
- Jordan, I.K.; Kondrashov, F.A.; Adzhubei, I.A.; Wolf, Y.I.; Koonin, E.V.; Kondrashov, A.S.; Sunyaev, S. A universal trend of amino acid gain and loss in protein evolution. Nature 2005, 433, 633–638. [Google Scholar] [CrossRef]
- Abajian, C.; Rosenzweig, A.C. Crystal structure of yeast Sco1. J. Biol. Inorg. Chem. 2006, 11, 459–466. [Google Scholar] [CrossRef]
- Pochini, L.; Peta, V.; Indiveri, C. Inhibition of the OCTN2 carnitine transporter by HgCl2 and methylmercury in the proteoliposome experimental model: Insights in the mechanism of toxicity. Toxicol. Mech. Methods 2013, 23, 68–76. [Google Scholar] [CrossRef]
- Pochini, L.; Scalise, M.; Indiveri, C. Inactivation by omeprazole of the carnitine transporter (OCTN2) reconstituted in liposomes. Chem. Biol. Interact. 2009, 179, 394–401. [Google Scholar] [CrossRef]
- Koepsell, H.; Lips, K.; Volk, C. Polyspecific organic cation transporters: Structure, function, physiological roles, and biopharmaceutical implications. Pharm. Res. 2007, 24, 1227–1251. [Google Scholar] [CrossRef]
- Hopf, T.A.; Colwell, L.J.; Sheridan, R.; Rost, B.; Sander, C.; Marks, D.S. Three-dimensional structures of membrane proteins from genomic sequencing. Cell 2012, 149, 1607–1621. [Google Scholar] [CrossRef]
- Shin, J.M.; Cho, Y.M.; Sachs, G. Chemistry of covalent inhibition of the gastric (H+, K+)-ATPase by proton pump inhibitors. J. Am. Chem. Soc. 2004, 126, 7800–7811. [Google Scholar] [CrossRef]
- Piermatti, O.; Fringuelli, F.; Pochini, L.; Indiveri, C.; Palmerini, C.A. Synthesis and characterization of carnitine nitro-derivatives. Bioorg. Med. Chem. 2008, 16, 1444–1451. [Google Scholar] [CrossRef]
- Indiveri, C.; Tonazzi, A.; Palmieri, F. Identification and purification of the ornithine/citrulline carrier from rat liver mitochondria. Eur. J. Biochem. 1992, 207, 449–454. [Google Scholar] [CrossRef]
- Tonazzi, A.; Giangregorio, N.; Indiveri, C.; Palmieri, F. Identification by site-directed mutagenesis and chemical modification of three vicinal cysteine residues in rat mitochondrial carnitine/acylcarnitine transporter. J. Biol. Chem. 2005, 280, 19607–19612. [Google Scholar] [CrossRef]
- Indiveri, C.; Iacobazzi, V.; Tonazzi, A.; Giangregorio, N.; Infantino, V.; Convertini, P.; Console, L.; Palmieri, F. The mitochondrial carnitine/acylcarnitine carrier: Function, structure and physiopathology. Mol. Asp. Med. 2011, 32, 223–233. [Google Scholar] [CrossRef]
- Camacho, J.A.; Obie, C.; Biery, B.; Goodman, B.K.; Hu, C.A.; Almashanu, S.; Steel, G.; Casey, R.; Lambert, M.; Mitchell, G.A.; et al. Hyperornithinaemia-hyperammonaemia-homocitrullinuria syndrome is caused by mutations in a gene encoding a mitochondrial ornithine transporter. Nat. Genet. 1999, 22, 151–158. [Google Scholar] [CrossRef]
- Schroder, E.; Eaton, P. Hydrogen peroxide as an endogenous mediator and exogenous tool in cardiovascular research: Issues and considerations. Curr. Opin. Pharmacol. 2008, 8, 153–159. [Google Scholar] [CrossRef]
- Bae, Y.S.; Oh, H.; Rhee, S.G.; Yoo, Y.D. Regulation of reactive oxygen species generation in cell signaling. Mol. Cells 2011, 32, 491–509. [Google Scholar] [CrossRef]
- Indiveri, C.; Tonazzi, A.; Stipani, I.; Palmieri, F. The purified and reconstituted ornithine/citrulline carrier from rat liver mitochondria: Electrical nature and coupling of the exchange reaction with H+ translocation. Biochem. J. 1997, 15, 349–355. [Google Scholar]
- Indiveri, C.; Tonazzi, A.; Stipani, I.; Palmieri, F. The purified and reconstituted ornithine/citrulline carrier from rat liver mitochondria catalyses a second transport mode: Ornithine+/H+ exchange. Biochem. J. 1999, 341, 705–711. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Scalise, M.; Pochini, L.; Giangregorio, N.; Tonazzi, A.; Indiveri, C. Proteoliposomes as Tool for Assaying Membrane Transporter Functions and Interactions with Xenobiotics. Pharmaceutics 2013, 5, 472-497. https://doi.org/10.3390/pharmaceutics5030472
Scalise M, Pochini L, Giangregorio N, Tonazzi A, Indiveri C. Proteoliposomes as Tool for Assaying Membrane Transporter Functions and Interactions with Xenobiotics. Pharmaceutics. 2013; 5(3):472-497. https://doi.org/10.3390/pharmaceutics5030472
Chicago/Turabian StyleScalise, Mariafrancesca, Lorena Pochini, Nicola Giangregorio, Annamaria Tonazzi, and Cesare Indiveri. 2013. "Proteoliposomes as Tool for Assaying Membrane Transporter Functions and Interactions with Xenobiotics" Pharmaceutics 5, no. 3: 472-497. https://doi.org/10.3390/pharmaceutics5030472