Design, Synthesis, and Biological Evaluation of PKD Inhibitors

Abstract
: Protein kinase D (PKD) belongs to a family of serine/threonine kinases that play an important role in basic cellular processes and are implicated in the pathogenesis of several diseases. Progress in our understanding of the biological functions of PKD has been limited due to the lack of a PKD-specific inhibitor. The benzoxoloazepinolone CID755673 was recently reported as the first potent and kinase-selective inhibitor for this enzyme. For structure-activity analysis purposes, a series of analogs was prepared and their in vitro inhibitory potency evaluated.1. Introduction
Protein Kinase D (PKD) is a member of a novel family of serine/threonine kinases and diacylglycerol (DAG) receptors that have emerged as key regulators of many important cellular processes. The PKD family is now recognized as a subfamily of the Ca2+/calmodulin-dependent kinase superfamily. To date, three isoforms of PKD have been identified: PKD1 (formerly PKCμ) [1,2], PKD2 [3], and PKD3 (formerly PKCv) [4]. All three isoforms share a highly homologous sequence and a distinct structure that includes a catalytic domain, a pleckstrin homology (PH) domain, which mediates protein-protein interactions and PKD autoinhibition, and an N-terminal cysteine-rich DAG/phorbol ester binding domain (C1 domain). Both the activation of PKD and the regulatory mechanisms that control PKD activity have been well documented. PKD is activated by DAG-responsive protein kinase C (PKC) isoforms via transphosphorylation of the conserved Ser744 and Ser748 within the activation loop of PKD [5-7]. Subsequent autophosphorylation at multiple sites, including Ser916, confers full, sustained activity [8,9]. PKD is also subject to spatial regulation by DAG or phorbol esters and, as a result, is capable of shuttling between different subcellular compartments [10-12]. Accordingly, this canonical DAG/PKC/PKD pathway is crucial to fundamental PKD function in cells.
As a result of its central position in the signal transduction pathway, it is no surprise that PKD has been implicated in a variety of cellular processes, including cell proliferation, cell survival through oxidative stress-induced activation of nuclear factor-kappaB (NF-κB) signaling [13-15], gene expression by regulation of class IIa histone deacetylases (HDAC) [16-18], protein trafficking [19-21], cell motility [22,23], and immune responses [24-26]. These functions of PKD are known to impact many aspects of tumor biology, and extensive evidence indicates that PKD expression is dysregulated in multiple cancer types [27-30]. PKD also plays an active role in pathological processes such as cardiac hypertrophy [31,32], angiogenesis [33,34], and tumor cell proliferation and metastasis [35-37], making PKD an attractive therapeutic target for drug development [38].
Although extensive analysis of the role of PKD in biological processes has been hampered by the lack of both a three-dimensional structure and PKD specificity of early inhibitors (i.e. the isoquinoline sulfonamide H89 [39], staurosporine analogs [40,41], and resveratrol [42-44]), more potent and selective inhibitors have been reported recently [32,38,45-51]. The first breakthrough in this area came in 2008 with the identification and characterization of CID755673 (Figure 1), a potent and PKD-selective kinase inhibitor [52]. CID755673 was reported to inhibit all PKD isoforms with an IC50 of 200-300 nM, but showed specificity toward PKD over several related kinases [52]. Interestingly, CID755673 was not competitive with ATP for enzyme inhibition, suggesting an alternate binding site on the enzyme, which may account for the selectivity for PKD compared to other protein kinases. This compound was also shown to effectively block PKD-mediated cell functions as well as the tumor-promoting functions of PKD in prostate cancer cells [52,53]. Despite the high specificity of CID755673 and its potent in vitro inhibition of PKD, its cellular activity was relatively weak (EC50 = 11.8 μM) [52,53].
In addition to CID755673, promising ATP-competitive inhibitors continue to emerge in the literature [32,45,46,48-51]. The novel 2,6-naphthyridine 1a (Figure 1) was identified by a high throughput screen (HTS) as a dual PKC/PKD inhibitor. Modification of this chemotype led to the orally available naphthyridine inhibitors 1b and 1c (Figure 1) [50]. Both 1b and 1c were able to block PKD phosphorylation and nuclear export of HDAC in the target tissue in vivo; however, their moderate kinase selectivity complicated data interpretation. In an effort to reduce the off-target effects and improve PKD selectivity, a series of amidobipyridyl-based analogs was generated [51]. From this series, BPKDi (Figure 1) was identified as a potent and selective inhibitor of all three isoforms of PKD with single digit nanomolar IC50 values and improved selectivity relative to 1b [32,51]. Furthermore, BPKDi caused substantial inhibition of PKD1 signal-dependent phosphorylation and increased nuclear retention of class HDAC4 and HDAC5 in cardiomyocytes [32].
Another pan-PKD inhibitor, CRT0066101 (Figure 1), was also reported to inhibit all isoforms of PKD with single-digit nanomolar IC50s. CRT0066101 was shown to block cell proliferation, induce apoptosis, and reduce the viability of pancreatic cancer cells both in vitro and in vivo. As with the aforementioned ATP-competitive inhibitors, the most attractive features of CRT0066101 are its reported orally availability and efficacy in vivo [46,48]. Lastly, preliminary studies of the structure-activity relationships (SAR) of a novel 3,5-diarylazole 2a (Figure 1), which was identified in a HTS as a moderately potent kinase inhibitor, led to a series of promising benzamide analogs [49]. One analog in particular, 2b (Figure 1), was found to inhibit all PKD isoforms with low nanomolar IC50s, while showing a 9- and 3-fold preference for PKD1 versus PKD2 and PKD3, respectively. Notably, 2b shows high selectivity for PKD against a panel of other kinases, and pharmacokinetic studies in rats indicate that compound 2b is orally available [49].
While all of these ATP-competitive, orally available PKD inhibitors represent valuable tools for further study of PKD signaling, their substantial off-target activity may likely be due to the high sequence homology near the hinge-binding regions of PKCs and PKDs. Recent evidence suggests additional targets of CID755673 [54], but as this lead structure is not competitive with ATP for PKD inhibition, it can provide an orthogonal approach to gain further understanding of the structure and function of PKD. In an effort to enhance the selectivity and potency for potential in vivo applications, small molecule analogs of CID755673 were generated by modification of the core structure as well as the side chains. We describe herein the complete SAR conducted thus far, which led to the discovery of a novel benzothienothiazepinone series. The improved PKD1 inhibitory activity of some of these analogs has already been highlighted in previous communications [53,55].
2. Results and Discussion
2.1. First generation SAR
Our investigations began with the chromenopyridine-based CID797718, a by-product of the synthesis of the parental compound, CID755673 (Table 1). This compound was 10x less potent at PKD1 inhibition than CID755673. Efforts to improve the activity of CID797718 by substitution of the phenolic hydroxyl group (Table 1, entries 1-3), N-alkylation (Table 1, entry 4), and ortho-halogenation (Table 1, entry 5) resulted in a complete loss of PKD1 activity. Consequently, the chromenopyridine scaffold was abandoned.
Modifications to the benzofuroazepinone scaffold present in CID755673 proved to be more rewarding. The initial SAR analysis included modifications of the azepinone ring, substitution at the phenol and amide groups, and functionalization across the aryl moiety (Table 2). Changing the azepinone to either a 6- or 8-membered heterocycle resulted in a reduced inhibitory activity towards PKD1 (Table 2, entries 2–5). Substitutions of the phenolic hydroxyl group were not well tolerated either. Although both the methoxy and allyloxy substituents at that site showed moderate inhibitory activity towards PKD1 (Table 2, entries 6 and 7), the acetoxy and siloxy groups (Table 2, entries 8 and 9) showed poor inhibition and complete loss in activity, respectively. These results indicate that the aryl binding pocket of the protein may be restrictive in terms of both the size and the polarity of the aryl substituents. Similarly, alkylation/acylation of the amide or bioisosteric replacement with a dihydroimidazole resulted in weak or no PKD1 inhibition (Table 2, entries 10–14). These observations suggest that the amide functionality may provide crucial H-bonding interactions in the azepinone binding pocket that are required for optimal inhibitor-enzyme interactions.
Based on these results, subsequent synthetic analogs were biased to contain key features of the pharmacophore, i.e., the 7-membered ring azepine, the monosubstituted lactam, and a small R2 substituent (Table 3). Derivatives bearing either a Cl or F atom at the R1 position retained activity against PKD1, but did not show improved potency relative to CID755673 (Table 3, entries 1 and 2). In contrast, introduction of a Cl atom at either the R3 position, or both the R1 and R3 positions, provided inactive compounds (Table 3, entries 4 and 5). Interestingly, allyl and propenyl groups were well tolerated in this region, despite the aforementioned putative size restrictions of the aryl binding region (Table 3, entries 6 and 7). The inhibitory activity of these analogs also provides further support for a relatively hydrophobic aryl binding pocket. Finally, all modifications of the azepinone moiety at the 5 position (Z) yielded analogs with reduced inhibitory activity (Table 3, entries 8–12).
We next sought to investigate the replacement of the benzofuran core by a β-carboline skeleton (Table 4). Compound kb-NB123-57 (Table 4, entry 2), which retains all functionality of CID755673 except for the benzofuran oxygen being replaced with a nitrogen atom, exhibited comparable activity toward PKD1; however, there was no significant cellular activity. Variation of either the phenolic hydroxyl group or the lactam ring size in the β-carboline series did not provide any enhancement in activity (Table 4), and in cases where R2 was replaced with O-benzyl (Table 4, entries 3 and 4) or N-acyl (Table 4, entries 7 and 8), all inhibitory activity was lost. These modifications, accordingly, confirm that the 7-membered azepinone represents an optimal size and that the binding pocket of the protein is sterically demanding at the aryl binding region.
In the next SAR iteration, the benzofuran was replaced with a benzothiophene while the azepine was replaced by a thiazepine ring. Gratifyingly, both the benzothienothiazepinone kb-NB142-70 and the methoxy analog kb-NB165-09 showed a significance increase in potency toward PKD1, with IC50 values of 28.3 nM and 82.5 nM, respectively (Table 5, entries 2 and 3). This increased potency was further confirmed in cell-based assays, for which the cellular EC50 was lowered ca. 5-fold from 11.8 μM by CID755673 to 2.2 μM and 3.1 μM by kb-NB142-70 and kb-NB165-09, respectively. The remarkable activity of the methyl ether compared to the phenol indicates that a hydrogen bond donor at this position is not critical for activity. The lack of activity of the O-benzyl derivative for this series of analogs provides additional support for a sterically demanding aryl binding pocket.
2.2. Second generation SAR
The previous screening cycles led to the discovery of the benzothienothiazepinone kb-NB142-70 as the most potent analog with an IC50 of 28.3 nM, which was an almost 7-fold improvement in potency over the parent compound, CID755673. Furthermore, we had gained considerable understanding of the SAR of the tricyclic inhibitor scaffold. Accordingly, we updated our pharmacophore model based on the structure of kb-NB142-70, and devised 4 major structural zones (Figure 2): zone I (aryl moiety), zone II (thiophene), zone III (thiazepinone), and zone IV (amide function). Each of these zones was subjected to selected structural modifications in order to elucidate essential activity relationships.
In zone I, the phenolic substituent and the substitution across the aryl moiety were modified (Table 6). Most of the zone I analogs were less active than the lead, kb-NB142-70. Specifically, substitutions at R4 were detrimental to in vitro activity (Table 6, entries 7–9), at least when R2 = H. In contrast, halogenation at R1 (Table 6, entries 5 and 6) and replacement of the phenolic hydroxy group with amine variants (Table 6, entries 1–4) were surprisingly well tolerated. Notably, the azide analog, mcf292-08, maintained a high inhibitory activity both in vitro and in cells, with IC50 values of 74.9 nM and 2.2 μM, respectively, thereby providing further support for the limited significance of a hydrogen bond donor at this position. The ortho-iodinated analog, kb-NB165-31, was also a potent inhibitor with an IC50 value of 114 nM; however, its cellular activity was 4-fold lower than that of kb-NB142-70. Masking the hydroxyl group of the lead kb-NB142-70 was only slightly detrimental to the in vitro biological activity, as revealed by the methoxy analog kb-NB165-09. Moreover, preliminary results from an animal model suggest the glucuronidation of kb-NB142-70 at the phenolic position to be a major metabolic pathway.
Therefore, the methoxy analogs were systematically developed in addition to the corresponding phenols in order to potentially circumvent a rapid excretion scenario in vivo. As part of our zone II and III modifications, the oxidation state of the benzothiophene sulfur atom, the size of the thiazepinone ring, and the oxidation and substitution of the thiazepinone ring sulfur atom were explored (Table 7). Sulfur oxidations in zones II and III provided analogs with reduced activity (Table 7, entries 1–3), while increasing the thiazepinone ring size by the addition of a methylene group had only minor effects on PKD1 inhibition for both the hydroxy and methoxy analogs (Table 7, entries 5 and 6). Additionally, the replacement of the thiazepinone ring sulfur atom with an oxygen resulted in a loss of inhibitory activity (Table 7, entries 7 and 8), suggesting high hydrophobicity and polarizability to be preferred in zone III. Attempts to exchange the thiazepinone ring sulfur atom with a nitrogen atom were met with preparative difficulties. The desired diazepinone ring could not be formed, and, as a result, an acyclic precursor was submitted for testing (Table 7, entry 9). The lack of activity of this analog suggests that the zone III binding pocket may require the rigidity of a ring system for optimal binding interactions.
Furthermore, modifications to zone IV, which included functional group interconversions and replacement of the amide moiety, did not enhance the inhibitory activity (Table 8). These results suggest that the unique hydrogen bond donor-acceptor capability of the amide is critical for protein interactions within the zone IV binding pocket.
The results from both the initial and the second generation SAR firmly established that the benzothienothiazepinones displayed superior PKD1 inhibition compared to the benzofuroazepinones. The most potent analog, kb-NB142-70, inhibited PKD1 with an IC50 of 28 nM, which is nearly a 7-fold improvement in potency compared to the parental compound CID755673. This improved analog also demonstrated increased inhibition of PMA-induced autophosphorylation of endogenous PKD1 in LNCaP prostate cancer cells with an EC50 of 2.2 μM, which represents a ca. 5-fold improvement in activity over CID755673, which has an EC50 of 11.8 μM.
However, despite the improvement of in vitro and cellular activities, in vivo studies revealed a short plasma half-life for this compound. In order to enhance the resistance of kb-NB142-70 towards phase I and II metabolism, modifications to zone I were explored with the goal to install a more electron-deficient heteroarene ring (Table 9).
Gratifyingly, the methoxypyrimidine kmg-NB4-23 exhibited an IC50 of 124 nM, which represents only a slight decrease in activity compared to that of the parent compound, kb-NB165-09. This result not only validates our design, but also suggests that the zone I binding pocket is tolerant to a decrease in electron density in the aryl region. Surprisingly, the hydroxy analog kmg-NB4-69A showed a significant loss in activity relative to the parental compound, kb-NB142-70. This effect can be attributed to the susceptibility of the C-4 position of the pyrimidine towards nucleophilic attack [56]; the heterocycle spontaneously degrades in protic solvents, resulting in reduced inhibitory activity towards PKD1. More hydrolytically stable pyrimidine analogs containing a substituent at the R4 position also exhibited a dramatic loss in activity, which is consistent with the general SAR results and provides additional support for a sterically limited zone I binding pocket. Unfortunately, the low aqueous solubility (<0.4 mg/mL even in the presence of lipophilic solubilizing agents) of kmg-NB4-23 prevented the in vivo evaluation of this compound.
2.3. Synthetic chemistry
CID755673 was initially formed in conjunction with CID797718 in a synthetic route starting with commercially available ε-caprolactam, which was dibrominated at the α-position [57], and then treated with piperidine to afford the known α-oxolactam enamine 4 (Scheme 1) [58].
A Nenitzescu reaction between 4 and para-benzoquinone resulted in the formation of cyclic adduct 5, which was in turn subjected to heating in aqueous acid [59] to provide the desired CID755673. Although CID797718 was a byproduct of the synthesis of the target compound CID755673, it showed moderate activity and was therefore a desirable starting point for analog synthesis (Scheme 2). Functionalizations of this compound involved O- and N-alkylations, as well as chlorination across the aryl moiety. Allylation and TBS-protection of the phenolic hydroxy group of the parental compound, CID797718, provided the derivatives kb-NB77-83 and kb-NB77-78 in good yields. Treatment of kb-NB77-78 with CbzCl under basic conditions afforded the desilylated carbamate kb-NB77-91.
Introduction of a chlorine atom to the chromenopyridine scaffold was achieved by treating enamine 4 with 2-chloro-1,4-benzoquinone, followed by the same acid-promoted cis-trans isomerization-elimination as aforementioned in Scheme 1 (Scheme 3). The chlorinated analogs were isolated as a separable mixture of isomers. Unfortunately, all of the chromenopyridine-based analogs were inactive in the PKD1 inhibition assays, and therefore this scaffold was abandoned.
Our studies on the modification of CID755673 began with the azepinone ring (Scheme 4). The syntheses of 6- and 8-membered ring analogs were carried out in an analogous manner to the sequence shown in Scheme 1. The acid-mediated piperidine elimination reaction of the 8-membered ring derivative 7b proceeded with complete chemoselectivity toward the formation of the desired product kb-NB96-53. In contrast, treatment of 7a with acetic acid provided the N-acetylated analog of the desired compound and the corresponding chromenopyrrole as the major products. Attempts to cleave the N-acetyl functionality to obtain the desired analog kb-NB123-23A were unsuccessful. Gratifyingly, subjecting 7a to mCPBA at elevated temperatures afforded the desired 6-membered azepinone ring analog, kb-123-23A, in good yield. With the 6- and 8-membered ring analogs in hand, we next substituted the phenolic hydroxyl group and alkylated the azepinone amide nitrogen atom (Scheme 4).
Treatment of kb-NB123-23A with MeI and K2CO3 provided the O-methylated compound, kb-NB123-32, while use of a stronger base such as KOt-Bu gave the desired dimethylated analog, kb-NB123-37, in good yield. The 8-membered dimethylated product kb-NB96-59 was obtained in a similar manner to kb-NB123-37, although only in modest yield.
Due to the lack of potency improvements for the 6- and 8-membered azepinone analogs, further modifications to the aryl region of the parental compound CID755673 were explored. Functionalization of the phenol was achieved by treatment of CID755675 with base in the presence of an electrophile (Table 10, entries 1–4).
The dimethylated compound kb-NB96-04 and the diacylated compound kb-NB123-45-1 were obtained in a similar manner by the use of either a stronger base (KOt-Bu) or additional equivalents of the desired electrophile, respectively (Table 10, entries 5 and 6). Modification of the aryl region also included additions of halogens (Table 10, entries 7–9). These derivatives could be obtained by the treatment of CID755673 with either N-chlorosuccinimide in the case of the chlorinated analogs kb-NB77-88 and kb-NB96-43, or Selectfluor® in the case of the fluorinated analog kb-NB96-21.
Further modifications of these analogs were carried out in order to obtain a more complete SAR for the benzofuroazepinone series (Scheme 5). O-Allylation and N-methylation were performed on kb-NB77-88 and kb-NB77-77, respectively. Additionally, the microwave-mediated Claisen rearrangement of the O-allylated-benzoxoloazepinolone kb-NB77-84 provided the target compound kb-NB96-02 with the allyl functionality at the R1 position. Protection of the phenol of kb-NB96-02 with a silyl group provided a derivative which could be subjected to Ru-catalyzed olefin isomerization conditions. Subsequent TBS deprotection provided the isomerized product kb-NB96-30 in modest yield over the three steps.
In addition to alkylations and acetylations of the amide moiety, we also synthesized kb-NB165-15 to evaluate the effect of the isoelectronic replacement of the amide with an imidazoline ring (Scheme 6).
A TIPS protection of the parental compound CID755673 followed by N-alkylation provided 8. Debenzylation by transfer hydrogenation, mesylation of the primary alcohol, and displacement of the resulting mesylate with sodium azide afforded the desired alkyl azide 9 in good yield over the three steps. After protection of the phenol, the alkyl azide was reduced to the amine and protected, prior to treatment with Lawesson';s reagent to furnish thiolactam 10. Cyclodethionation of thiolactam 10 [60] and subsequent TIPS deprotection delivered the desired imidazole containing analog, kb-NB165-15.
At this time, all modifications to the azepinone ring including altering the ring size, N-alkylations/acetylations, and isoelectronic replacements of the amide moiety had resulted in inhibitors with either reduced potency toward PKD1 or no inhibitory effect at all. Thus, we explored the introduction of functionality on the azepinone methylene groups to determine if this region of the molecule could be optimized further (Scheme 7).
The synthesis of these analogs began with the oxidation of the O-acetylated derivative kb-NB123-36 with PDC-TBHP [61] in the presence of neutral alumina in order to install the desired ketone (11). The use of sonication proved to significantly improve both the yields and reproducibility of this oxidation. Subsequent acetyl deprotection in methanolic potassium carbonate solution provided kb-NB123-63. Access to further functionalized derivatives was achieved by acid-catalyzed condensation of kb-NB123-63 with O-benzylhydroxylamine or N-substituted hydrazines (Scheme 7). The newly installed keto functionality was reduced using sodium borohydride to furnish the hydroxy derivative kb-NB123-89.
We also investigated the replacement of the benzofuran core by a β-carboline scaffold (Scheme 8). β-Carbolines kb-NB123-57 and kb-NB123-59 were prepared from phenylhydrazine hydrochloride 14 [62,63] via a Fischer-like indole synthesis with the corresponding 6- and 7-membered ring α-keto lactams 12 and 13. Lactams 12 and 13 were obtained in situ upon acid-catalyzed hydrolysis of the corresponding enamines [64]. Debenzylation of the resulting β-carbolines by transfer hydrogenation furnished the final phenol derivatives [63]. The aniline analogs, kb-NB123-93 [65] and kb-NB123-94, were synthesized in a similar manner, starting from hydrazine hydrochloride 15 [66]. Subsequent cleavage of the acetyl group afforded the free amino β-carbolines (Scheme 8) [65].
We also synthesized a thio variant of the benzofuroazepinone scaffold to study the effects of sulfur incorporation (Scheme 9).
This analog was constructed by a thionyl chloride-mediated Higa cyclization [67] of benzyl protected hydroxycinnamate 16 to a benzo[b]thiophene derived acid chloride, which was subsequently converted to the methyl ester 17. Treatment of 17 with cysteamine hydrochloride in the presence of DBU furnished the benzyloxy-benzothienothiazepinone kb-NB123-66 [68]. Deprotection of the aryl benzyl ether kb-NB123-66 with boron tribromide provided kb-NB142-70 in good yields, and subsequent O-methylation gave kb-NB165-09 in excellent yield (Scheme 9). Gratifyingly, kb-NB142-70 was identified as the most potent analog with an IC50 of 28.3 nM for PKD1, which represents nearly 7-fold higher potency over the parental benzofuroazepinone CID755673. We therefore dissected this lead compound into four major structural zones in order to further probe the SAR, as shown in Figure 2.
In zone I, we modified the substituent on the phenolic hydroxyl group and the position of this group (Scheme 9). The positional isomer 18 could be isolated as a byproduct from the thionyl chloride-mediated cyclization of 16. Subsequent construction of the thiazepinone ring could be achieved via one-pot nucleophilic displacement-condensation reaction of 18 with cysteamine hydrochloride in a similar manner to the lead compound kb-NB142-70. Modification of this isomer by standard alkylation and protection-deprotection strategies provided the desired zone I analogs (Scheme 9). Zone I modifications also involved halogenations of the aryl moiety. Standard iodination conditions led to analog kb-NB165-31 ([Scheme 10 (a)]).
In contrast, the brominated analog kb-184-52 was synthesized from the corresponding sulfoxide kb-NB184-45 upon treatment with BBr3 [Scheme 10 (b)]. Additional zone I modifications, which included replacement of the phenol group by nitrogen, required the development of an alternative route (Scheme 11 and Table 11). Nucleophilic aromatic substitution of methyl 2-chloro-5-nitrobenzoate by methyl thioglycolate anion followed by immediate Dieckman cyclization [63,69] afforded the benzothiophene precursor 19. Cyclization of the corresponding triflate 20 with cysteamine hydrochloride provided the desired tricyclic core 21 in 50% yield (67% based on recovered starting material 19, Scheme 11). Subsequent reduction of the nitro group furnished aniline mcf292-03, which was further functionalized by treatment with t-butyl nitrite and TMS-azide using Moses'; method [70] to yield the aryl azide, mcf292-08 (Table 11, entry 1). The synthesis of the isothiocyanate mcf292-05 was realized by subjecting mcf292-03 to the modified Schotten-Baumann conditions reported by Nowick and co-workers [71] (Table 11, entry 2). Lastly, treatment of mcf292-03 with chlororacetyl chloride in the presence of 2,6-lutidine provided the desired chloroacetyl analog mcf292-09 in modest yield over two steps (Table 11, entry 3).
In our pursuit of zone II SAR, the sulfur ring atom of 17 was oxidized with trifluoroperacetic acid to the 3-chlorobenzo[b]thiophene-1-oxide 22 (Scheme 12). The vinylogous chloride 22 was then converted to the corresponding benzyloxybenzothienothiazepinone-6-oxide 23, which upon benzyl deprotection and methylation provided the desired sulfoxide analogs, kb-NB184-22 and kb-NB184-25, respectively (Scheme 12).
Zone III modifications included oxidations of the thiazepinone ring sulfur atom and variation of the thiazepinone ring size [Schemes 10 (b) and 13]. Selective oxidation of the thiazepinone ring sulfur atom could be achieved by treatment of kb-NB165-09 with peracid to furnish kb-NB184-45, as shown in Scheme 10 (b). Preparation of the 8-membered thiazepinone ring analog began with the synthesis of 25 via the ring opening of thiazinane-thione 24 [72] (Scheme 13). Aminothiol 25 was isolated as a thiol/disulfide mixture and used directly in the aforementioned cyclocondensation-deprotection sequence to provide the desired 8-membered thiazepinone analogs (Scheme 13).
In an effort to further assess the SAR effects of modifying zone III, we investigated the synthesis of benzothiophene analogs linked to a three-carbon chain by an ether or amine function, instead of the thioether present in the lead compound. Key to accomplishing the synthesis of the ether analog kb-NB184-36 was the use of the activated chloride 22, which was obtained via oxidation of the benzothiophene sulfur to the corresponding sulfoxide according to Scheme 12. A DMAP-catalyzed nucleophilic displacement of the chlorine atom with alcohol 26 afforded the cyclization precursor 27, which upon N-Boc deprotection and subsequent tandem cyclization-deoxygenation led to the benzyloxybenzothienooxazocinone analog kb-NB184-36 [Scheme 14 (a) ]. The methoxy analog kb-NB184-57 was synthesized in a similar manner [Scheme 14 (b) ].
In an analogous manner, the synthesis of diazepinone analog 31 was attempted; however, precursor 30 failed to undergo cyclization under both base- and Cu-mediated conditions [Scheme 15 (a) ] [73]. Therefore, we opted for a nucleophilic displacement at the activated chlorine atom in 29 with aminopropanol, followed by a TMSI-mediated deoxygenation of 32 to provide kb-NB184-80 [Scheme 15 (b) ]. Investigations regarding the use of these intermediates [Scheme 15 (b) ] as precursors for the desired analog 31 are still under investigation.
Finally, we explored structural modifications of zone IV by alkylation of the amide nitrogen and reduction of the amide (Schemes 16 and 17, respectively).
The N-methylated analog kb-NB165-17 was prepared from the benzyloxybenzothiazepinone kb-NB123-66 (Scheme 9) through an alkylation-deprotection sequence, while the dialkylated kb-NB165-16 was obtained in one step by treatment with NaH and MeI. The synthesis of kb-NB165-75 was accomplished via the N-alkylation of silyl protected kb-NB142-70 with the corresponding alkyl iodide, followed by functional group interconversions and a final deprotection to furnish the desired analog kb-NB165-75. The amide reduction of thiolactam 36 proceeded poorly upon treatment with Raney-Nickel in THF due to a competitive cleavage of the other C-S bonds present in this system (Scheme 17). Nonetheless, we were able to isolate kb-NB165-81 in low yields and debenzylate it to afford the desired phenol kb-NB165-83 (Scheme 17).
As mentioned in Section 2.2, preliminary in vivo studies revealed that the lead compound kb-NB142-70 and the methoxy analog kb-NB165-09 possessed a short plasma half-life (data not shown). Therefore, we further explored a zone I modification of kb-NB142-70 to install a more electron-deficient pyrimidine moiety in place of the phenol ether, a known site of active phase I and II metabolism. The synthetic route to arrive at this new thiazepinothiophenopyrimidinone scaffold is summarized in Scheme 18.
Starting with commercially available methyl 3-aminothiophene-2-carboxylate, formation of the pyrimidine moiety using potassium cyanate and chlorination with POCl3 provided dichloride 38 [74]. Regioselective palladium-catalyzed hydrogenolysis of 38 in the presence of Na2CO3 occurred exclusively at the C-4 position [75], and substitution of the remaining C-2 chloride with methoxide provided 40 in 79% yield over the two steps. Electrophilic bromination of 40 using bromine in acetic acid gave the desired C-7 bromo compound 41. Functionalization at C-6 was accomplished via selective metalation and trapping with Mander';s reagent to provide the required cyclization precursor 42 [76]. Formation of the thiazepinone moiety was achieved by a one-pot nucleophilic displacement-condensation of 42 with cysteamine hydrochloride to provide the desired methoxypyrimidine kmg-NB4-23 in good yield. The structure of kmg-NB4-23 was confirmed by x-ray analysis (Figure 3; this structure has been deposited at the Cambridge Crystallographic Data Centre and allocated the deposition number CCDC 822403). When kmg-NB4-23 was subjected to 4 M HCl in 1,4-dioxane, the desired hydroxypyrimidine kmg-NB4-69A was formed as the hydrochloride salt (Scheme 18).
Pyrimidine kmg-NB4-23 is a potent nanomolar inhibitor of PKD, thus confirming the validity of our design. In contrast, kmg-NB4-69A had only weak inhibitory effect against PKD. This lack of activity is attributed to the instability of the compound towards nucleophilic addition of H2O at the C-4 position [56]. Efforts to stabilize the C-4 position led to the design of compounds kmg-NB5-13 and kmg-NB5-15 (Scheme 19).
These analogs were successfully synthesized in a similar manner to kmg-NB4-23 and kmg-NB4-69A (Scheme 18). Unfortunately, preliminary biological data indicate an absence of inhibitory activity for both kmg-NB5-13 and kmg-NB5-15.
3. Experimental Section
3.1. General
Melting points were determined on a Meltemp capillary melting point apparatus fitted with a Fluke 51 II digital thermometer. Infrared spectra were recorded on a Smiths IdentifyIR ATR spectrometer or a Perkin Elmer Spectrum 100 FT-IR spectrometer using the Universal ATR Sampling Accessory for both oil and solid compounds. 1H NMR and 13C NMR spectra were obtained on a Bruker Avance 300, 400 or 600 instrument at 300/75 MHz, 400/100 MHz or 600/150 MHz, respectively. Chemical shifts were reported in parts per million (ppm) as referenced to residual solvent. 1H NMR spectra are tabulated as follows: chemical shift, multiplicity (app = apparent, b = broad, s = singlet, d = doublet, t = triplet, q = quartet, quint = quintuplet, sext, = sextuplet, m = multiplet), number of protons, coupling constant(s). 13C NMR were obtained using a proton-decoupled pulse sequence and are tabulated by observed peak. Mass spectra were obtained on a Waters Autospec double focusing mass spectrometer (EI) or a Waters Q-Tof mass spectrometer (ESI), at the University of Pittsburgh Mass Spectrometry facility.
3.2. Characterization data for all final synthetic compounds
9-Hydroxy-1,2,3,4-tetrahydro-chromeno[3,4-b]pyridin-5-one (CID797718)
Yield: 8%; m.p. (i-PrOH) 217–218 °C (lit. 213–216 °C) [59]; IR (ATR, neat) 3401, 3305 (br), 2937, 2879, 1662, 1583, 1449, 1342, 1219, 1184 cm−1; 1H-NMR (DMSO-d6, 600 MHz) δ 9.41 (s, 1 H), 7.09 (d, 1 H, J = 9.0 Hz), 6.74 (d, 1 H, J = 3.0 Hz), 6.66 (dd, 1 H, J = 9.0, 3.0 Hz), 5.91 (s, 1 H), 3.24–3.22 (m, 2 H), 2.59 (t, 2 H, J = 6.6 Hz), 1.90–1.85 (m, 2 H); 13C-NMR (DMSO-d6, 150 MHz) δ 158.1, 154.5, 140.9, 129.8, 122.9, 116.8, 114.8, 113.4, 106.4, 40.3, 21.6, 20.6; HRMS (ESI) m/z calcd for C12H11NO3 ([M+H]+) 218.0817, found 218.0802.
9-Allyloxy-1,2,3,4-tetrahydrochromeno[3,4-b]pyridin-5-one (kb-NB77-83)
Yield: 68%; m.p. 90–91 °C; IR (ATR, neat) 3419, 3394, 2917, 2864, 1702, 1599, 1510, 1189 cm−1; 1H-NMR (CDCl3, 600 MHz) δ 7.20 (d, 1 H, J = 9.0 Hz), 6.86 (d, 1 H, J = 3.0 Hz), 6.84 (dd, 1 H, J = 9.0, 3.0 Hz), 6.11–6.04 (m, 1 H), 5.44 (d, 1 H, J = 16.8 Hz), 5.32 (d, 1 H, J = 10.2 Hz), 4.74 (bs, 1 H), 4.57 (d, 2 H, J = 5.4 Hz), 3.41–3.37 (m, 2 H), 2.69 (t, 2 H, J = 6.6 Hz), 2.08–2.03 (m, 2 H); 13C-NMR (CDCl3, 150 MHz) δ 158.8, 155.4, 142.5, 133.3, 129.7, 122.6, 118.0, 117.1, 115.9, 113.0, 106.0, 69.5, 40.6, 21.6, 20.8; HRMS (ESI) m/z calcd for C15H15NO3 ([M+H]+) 258.1130, found 258.1138.
9-(tert-Butyldimethylsilyloxy)-1,2,3,4-tetrahydrochromeno[3,4-b]pyridin-5-one (kb-NB77-78)
Yield: 94%; m.p. 117–119 °C; IR (ATR, neat) 3418, 2929, 2856, 1721, 1699, 1602, 1508, 1430, 1251, 1172 cm−1; 1H-NMR (CDCl3, 600 MHz) δ 7.14 (d, 1 H, J = 9.0 Hz), 6.78 (d, 1 H, J = 3.0 Hz), 6.73 (dd, 1 H, J = 9.0, 3.0 Hz), 4.72 (bs, 1 H), 3.41–3.35 (m, 2 H), 2.67 (t, 2 H, J = 6.6 Hz), 2.09–2.02 (m, 2H), 1.00 (s, 9 H), 0.21 (s, 6 H); 13C-NMR (DMSO-d6, 150 MHz) δ 157.5, 151.7, 141.9, 129.5, 122.6, 117.1, 116.63, 116.57, 113.9, 111.0, 25.6 (3 C), 21.1, 20.0, 18.0, –4.5 (2 C); HRMS (ESI) m/z calcd for C18H25NO3Si ([M+H]+) 332.1682, found 332.1678.
Benzyl 9-hydroxy-5-oxo-1,2,3,4-tetrahydrochromeno[3,4-b]pyridine-4-carboxylate (kb-NB77-91)
Yield: 65%; m.p. 235–237 °C; IR (ATR, neat) 3887, 1724, 1679, 1404, 1308, 1261, 1160 cm−1; 1H-NMR (DMSO-d6, 600 MHz) δ 9.73 (s, 1 H), 7.38–7.33 (m, 4 H), 7.33–7.28 (m, 1 H), 7.23 (d, 1 H, J = 9.0 Hz), 6.98–6.95 (m, 2 H), 5.11 (s, 2 H), 3.30–3.50 (m, 2 H), 2.85 (t, 2 H, J = 6.6 Hz), 1.98–1.88 (m, 2 H); 13C-NMR (DMSO-d6, 150 MHz) δ 156.2, 154.03, 153.99, 144.8, 138.7, 136.1, 128.4 (2 C), 128.0, 127.7 (2 C), 125.0, 119.6, 118.4, 117.1, 108.5, 67.4, 44.2, 22.5, 22.1; HRMS (ESI) m/z calcd forC20H17NO5 ([M+Na]+) 374.1004, found 374.0991.
10-Chloro-9-hydroxy-1,2,3,4-tetrahydrochromeno[3,4-b]pyridin-5-one (kb-NB96-47-1)
Yield: 6%; m.p. 211–213 °C; IR (ATR, neat) 3434, 3225 (br), 2971, 2906, 1666, 1589, 1509, 1339, 1235 cm−1; 1H-NMR (DMSO-d6, 600 MHz) δ 10.02 (s, 1 H), 7.10 (d, 1 H, J = 9.0 Hz), 6.85 (d, 1 H, J = 9.0 Hz), 6.24 (bs, 1 H), 3.25–3.20 (m, 2 H), 3.15 (t, 2 H, J = 6.6 Hz), 1.80–1.74 (m, 2 H); 13C-NMR (DMSO-d6, 150 MHz) 156.8, 151.0, 140.9, 130.4, 120.7, 115.5, 113.4, 113.1, 112.4, 26.8, 20.9; HRMS (EI) m/z calcd for C12H10ClNO3 (M+) 251.0349, found 251.0349.
7-Hydroxy-2,3,4,5-tetrahydro-[1]benzoxolo[2,3-c]azepin-1-one (CID755673)
Yield: 83%; m.p. (i-PrOH) 245–247 °C (lit. 244–247 °C) [59]; IR (ATR, neat) 3187 (br), 3059, 2921, 1680, 1579, 1472, 1435, 1339, 1166 cm−1; 1H-NMR (DMSO-d6, 600 MHz) δ 9.36 (s, 1 H), 8.09 (t, 1 H, J = 4.8 Hz), 7.41 (d, 1 H, J = 9.0 Hz), 6.92 (d, 1 H, J = 2.4 Hz), 6.90 (dd, 1 H, J = 9.0, 2.4 Hz), 3.24 (dd, 2 H, J = 9, 4.8 Hz), 2.89 (t, 2 H, J = 6.6 Hz), 2.02–1.98 (m, 2 H); 13C-NMR (DMSO-d6, 150 MHz) δ 161.9, 153.9, 148.1, 144.3, 129.6, 123.5, 116.9, 112.4, 105.1, 41.2, 26.8, 24.3; HRMS (ESI) m/z calcd forC12H11NO3 ([M+H]+) 218.0817, found 218.0832.
6-Hydroxy-3,4-dihydrobenzoxolo[2,3-c]pyridin-1(2H)-one (kb-NB123-23A)
Yield: 77%; m.p. 265–268 °C; IR (ATR, neat) 3404, 3158 (bs), 1661, 1588, 1479, 1451, 1339, 1228, 1208, 1184 cm−1; 1H-NMR (DMSO-d6, 600 MHz) δ 9.43 (s, 1 H), 7.74 (bs, 1 H), 7.47 (d, 1 H, J = 9.0 Hz), 6.98 (d, 1 H, J = 2.4 Hz), 6.91 (dd, 1 H, J = 9.0, 2.4 Hz), 3.50 (td, 2 H, J = 7.2, 2.4 Hz), 2.88 (t, 2 H, J = 7.2 Hz); 13C-NMR (DMSO-d6, 150 MHz) δ 159.4, 153.8, 149.0, 144.5, 126.6, 124.6, 116.4, 112.6, 105.1, 40.3, 20.0; HRMS (ESI) m/z calcd for C11H9NO3 ([M+H]+) 203.0582, found 203.0588.
6-Methoxy-3,4-dihydrobenzoxolo[2,3-c]pyridin-1(2H)-one (kb-NB123-32) [77]
Yield: 94% m.p. 237–241 °C; IR (ATR, neat) 3197, 3088, 2895, 1672, 1585, 1480, 1432, 1329, 1214, 1192 cm−1; 1H-NMR (DMSO-d6, 600 MHz) δ 7.78 (s, 1 H), 7.58 (d, 1 H, J = 9.0 Hz), 7.26 (d, 1 H, J = 2.4 Hz), 7.06 (dd, 1 H, J = 9.0 Hz, 2.4 Hz), 3.81 (s, 3 H), 3.52 (td, 2 H, J = 7.2, 2.4 Hz), 2.94 (t, 2 H, J = 7.2 Hz); 13C-NMR (DMSO-d6, 150 MHz) δ 159.3, 156.0, 149.7, 144.7, 126.4, 125.0, 116.5, 112.9, 103.1, 55.7, 40.4, 20.1; HRMS (EI) m/z calcd for C12H11NO3 (M+) 217.0739, found 217.0743.
8-Hydroxy-3,4,5,6-tetrahydrobenzoxolo[2,3-c]azocin-1(2H)-one (kb-NB96-53)
Yield: 77%; m.p. 225–232 °C; IR (ATR, neat) 3348, 3166 (br), 2927, 1639, 1578, 1465, 1433, 1216, 1153 cm−1; 1H-NMR (DMSO-d6, 600 MHz) δ 9.33 (s, 1 H), 7.98 (t, 1 H, J = 6.7 Hz), 7.36 (d, 1 H, J = 8.8 Hz), 6.87 (d, 1 H, J = 2.2 Hz), 6.85 (dd, 1 H, J = 8.8, 2.4 Hz), 3.32–3.27 (m, 2 H), 2.79–2.74 (m, 2 H), 1.92–1.86 (m, 2 H), 1.67–1.62 (m, 2 H); 13C-NMR (DMSO-d6, 150 MHz) δ 162.9, 153.5, 147.4, 142.6, 129.1, 122.2, 115.4, 111.7, 104.5, 30.2, 22.9, 20.5; HRMS (ESI) m/z calcd for C13H13NO3 ([M+Na]+) 254.0793, found 254.0773.
8-Methoxy-2-methyl-3,4,5,6-tetrahydrobenzoxolo[2,3-c]azocin-1(2H)-one (kb-NB96-59)
Yield: 58%; m.p. 261–264 °C; IR (ATR, neat) 3168, 3038, 2928, 1651, 1581, 1478, 1435, 1210, 1152 cm−1; 1H-NMR (DMSO-d6, 600 MHz) δ 8.01 (s, 1 H), 7.47 (d, 1 H, J = 9.0 Hz), 7.12 (s, 1 H), 7.00 (d, 1 H, J = 9.0 Hz), 3.33–3.28 (m, 2 H), 3.80 (s, 3 H), 2.86–2.81 (m, 2 H), 1.93–1.86 (m, 2 H), 1.69–1.61 (m, 2 H); 13C-NMR (DMSO-d6, 150 MHz) δ 162.9, 155.8, 148.2, 142.9, 128.9, 122.7, 115.5, 112.1, 102.6, 55.7, 30.2, 23.0, 20.5; HRMS (ESI) m/z calcd for C14H15NO3 ([M+Na]+) 268.0950, found 268.0970.
7-Methoxy-2,3,4,5-tetrahydro-[1]benzoxolo[2,3-c]azepin-1-one (kb-NB77-56)
Yield: 74%; m.p. 261–263 °C; IR (ATR, neat) 3200, 3063, 2936, 1642, 1580, 1474, 1434, 1207, 1163 cm−1; 1H-NMR (DMSO-d6, 600 MHz) δ 8.13 (t, 1 H, J = 4.8 Hz), 7.53 (d, 1 H, J = 9.0 Hz), 7.18 (d, 1 H, J = 2.4 Hz), 7.05 (dd, 1 H, J = 9.0, 2.4 Hz), 3.81 (s, 3 H), 3.27 (dd, 2 H, J = 9.0, 5.4 Hz), 2.96 (t, 2 H, J = 6.6 Hz), 2.06–2.00 (m, 2 H); 13C-NMR (DMSO-d6, 150 MHz) δ 161.4, 155.7, 148.4, 144.1, 129.0, 123.5, 116.4, 112.3, 102.8, 55.7, 40.8, 26.3, 24.0; HRMS (ESI) m/z calcd for C13H14NO3 ([M+H]+) 232.0974,found 232.0966.
7-Allyloxy-2,3,4,5-tetrahydro-[1]benzoxolo[2,3-c]azepin-1-one (kb-NB77-84)
Yield: 69%; m.p. 198–200 °C; IR (ATR, neat) 3189, 3072, 2968, 2912, 1650, 1602, 1585, 1459, 1422, 1201, 1170 cm−1; 1H-NMR (DMSO-d6, 600 MHz) δ 8.13 (t, 1 H, J = 4.8 Hz), 7.53 (d, 1 H, J = 9.0 Hz), 7.20 (d, 1 H, J = 2.4 Hz), 7.08 (dd, 1 H, J = 9.0, 2.4 Hz), 6.11–6.04 (m, 1 H), 5.43 (dd, 1 H, J = 18, 1.8 Hz), 5.27 (dd, 1 H, J = 8.4, 1.8 Hz), 4.61 (d, 2 H, J = 5.22), 3.29–3.24 (m, 2 H), 2.94 (t, 2 H, J = 6.6 Hz), 2.06–1.98 (m, 2 H); 13C-NMR (DMSO-d6, 150 MHz) δ 161.4, 154.6, 148.4, 144.1, 133.8, 129.0, 123.5, 117.5, 116.8, 112.3, 104.1, 68.9, 40.8, 26.3, 23.9; HRMS (ESI) m/z calcd for C15H15NO3([M+Na]+) 280.0950, found 280.0959.
7-Acetoxy-2,3,4,5-tetrahydro-[1]benzoxolo[2,3-c]azepin-1-one (kb-NB123-36)
Yield: 91%; m.p. 196–197 °C; IR (ATR, neat) 3193, 3083, 2935, 1750, 1663, 1583, 1207, 1157, 1063 cm−1; 1H-NMR (DMSO-d6, 600 MHz) δ 8.21 (bs, 1 H), 7.66 (d, 1 H, J = 9.0 Hz), 7.47 (d, 1 H, J = 2.4 Hz), 7.22 (dd, 1 H, J = 9.0, 2.4 Hz), 3.27 (dd, 2 H, J = 8.4, 4.8 Hz), 2.94 (t, 2 H, J = 6.6 Hz), 2.29 (s, 3 H), 2.06–1.99 (m, 2 H); 13C-NMR (DMSO-d6, 150 MHz) δ 169.7, 161.2, 151.0, 146.3, 144.6, 129.0, 123.5, 122.6, 113.9, 112.3, 40.8, 26.3, 23.8, 20.9; HRMS (EI) m/z calcd for C14H13NO4 (M+) 259.0845, found 259.0850.
7-(tert-Butyldimethylsilyloxy)-2,3,4,5-tetrahydro-[1]benzoxolo[2,3-c]azepin-1-one (kb-NB77-77)
Yield: 91%; m.p. 209–212 °C; IR (ATR, neat) 3194, 3085, 2952, 2927, 1655, 1579, 1467, 1252, 1202, 1170 cm−1; 1H-NMR (DMSO-d6, 600 MHz) δ 8.13 (bs, 1 H), 7.50 (d, 1 H, J = 9.0 Hz), 7.06 (d, 1 H, J = 1.8 Hz), 6.96 (dd, 1 H, J = 8.4, 2.4 Hz), 3.28–3.23 (m, 2 H), 2.93 (t, 2 H, J = 6.6 Hz), 2.04–1.97 (m, 2 H), 0.97 (s, 9 H), 0.19 (s, 6 H); 13C-NMR (DMSO-d6, 150 MHz) δ 161.3, 151.0 148.8, 144.2, 129.3, 123.3, 120.4, 112.2, 110.3, 40.8, 26.p3, 25.6 (3 C), 23.9, 18.0, –4.6 (2 C); HRMS (ESI) m/z calcdfor C18H25NO3Si ([M+Na]+) 354.1501, found 354.1472.
6-Methoxy-2-methyl-3,4-dihydrobenzoxolo[2,3-c]pyridin-1(2H)-one (kb-NB123-37)
Yield: 83%; m.p. 168–172 °C; IR (ATR, neat) 2920, 1661, 1602, 1485, 1454, 1329, 1212, 1179 cm−1; 1H-NMR (DMSO-d6, 600 MHz) δ 7.57 (d, 1 H, J = 9.0 Hz), 7.25 (d, 1 H, J = 2.4 Hz), 7.05 (dd, 1 H, J = 9.0, 2.4 Hz), 3.81 (s, 3 H), 3.69 (t, 2 H, J = 7.2 Hz), 3.01 (t, 2 H, J = 7.2 Hz), 2.98 (s, 3 H); 13C-NMR (DMSO-d6, 150 MHz) δ 158.5, 156.0, 149.9, 144.5, 123.8, 116.4, 112.9, 103.0, 55.7, 48.6, 33.5, 19.3; HRMS (EI) m/z calcd for C13H13NO3 (M+) 231.0895, found 231.0899.
7-Hydroxy-2-methyl-2,3,4,5-tetrahydro-[1]benzoxolo[2,3-c]azepin-1-one (kb-NB142-25)
Yield: 36%; m.p. 282–284 °C; IR (ATR, neat) 3186, 1615, 1577, 1452, 1407, 1364, 1325, 1187 cm−1; 1H-NMR (DMSO-d6, 600 MHz) δ 9.36 (s, 1 H), 7.41 (d, 1 H, J = 9.0 Hz), 6.92 (d, 1 H, J = 2.4 Hz), 6.90 (dd, 1 H, J = 9.0, 2.4 Hz), 3.52–3.49 (m, 2 H), 3.06 (s, 3 H), 2.86 (t, 2 H, J = 6.6 Hz), 2.03–2.09 (m, 2 H); 13C-NMR (DMSO-d6, 150 MHz) δ 160.0, 153.5, 147.5, 144.0, 129.0, 122.4, 116.3, 111.9, 104.6, 49.3, 36.2, 25.5, 23.3; HRMS (EI) m/z calcd for C13H13NO3 (M+) 231.0895, found 231.0899.
7-Methoxy-2-methyl-2,3,4,5-tetrahydro-[1]benzoxolo[2,3-c]azepin-1-one (kb-NB96-04)
Yield: 34%; m.p. 150–151 °C; IR (ATR, neat) 3008, 2919, 1629, 1579, 1478, 1440, 1428, 1217, 1177 cm−1; 1H-NMR (DMSO-d6, 600 MHz) δ 7.53 (d, 1 H, J = 9.0 Hz), 7.16 (d, 1 H, J = 2.4 Hz), 7.05 (dd, 1 H, J = 9.0, 2.4 Hz), 3.81 (s, 3 H), 3.54–3.50 (m, 2 H), 3.07 (s, 3 H), 2.93 (t, 2 H, J = 6.6 Hz), 2.11–2.05 (m, 2 H); 13C-NMR (DMSO-d6, 150 MHz) δ 159.9, 155.7, 148.3, 144.3, 128.9, 122.9, 116.3, 112.3, 102.7, 55.7, 49.3, 36.2, 25.5, 23.4; HRMS (ESI) m/z calcd for C14H15NO3 ([M+Na]+) 268.0950, found 268.0935.
2-Acetyl-1-oxo-2,3,4,5-tetrahydro-[1]benzoxolo[2,3-c]azepin-7-yl acetate (kb-NB123-45-1)
Yield: 33%; m.p. 155–157 °C; IR (ATR, neat) 3069, 2949, 1753, 1699, 1669, 1570, 1402, 1363, 1213, 1168 cm−1; 1H-NMR (DMSO-d6, 600 MHz) δ 7.75 (d, 1 H, J = 9.0 Hz), 7.61 (s, 1 H, J = 2.4 Hz), 7.31 (dd, 1 H, J = 9.0, 2.4 Hz), 3.96–3.92 (m, 2 H), 3.03 (t, 2 H, J = 6.0 Hz), 2.45 (s, 3 H), 2.30 (s, 3 H), 2.08–2.03 (m, 2 H); 13C-NMR (DMSO-d6, 150 MHz) δ 171.3, 169.6, 162.3, 151.9, 146.6, 144.5, 128.4, 127.1, 123.1, 114.5, 112.7, 40.9, 26.2, 25.1, 21.6, 20.9; HRMS (EI) m/z calcd for C16H15NO5(M+) 301.0950, found 301.0964.
9-Hydroxy-3,5,6,7-tetrahydro-2H-benzoxolo[2,3-c]imidazo[1,2-a]azepine (kb-NB165-15)
Yield: 40%; m.p. 230–232 °C (dec., brown), 268–270 °C (dec., melts); IR (ATR, neat) 2918, 2871, 2528, 1624, 1590, 1550, 1445, 1392, 1285, 1195 cm−1; 1H-NMR (CD3OD, 600 MHz) δ 7.34 (d, 1 H, J = 9.6 Hz), 6.92–6.90 (m, 2 H), 3.81 (t, 2 H, J = 10.2 Hz), 3.63 (t, 2 H, J = 10.2 Hz), 3.44–3.41 (t, 2 H, J = 4.8 Hz), 2.95 (t, 2 H, J = 6.0 Hz), 2.22–2.17 (m, 2 H); 13C-NMR (CD3OD, 150 MHz) δ 159.8, 155.6, 150.3, 142.4, 130.6, 125.8, 117.9, 112.8, 105.6, 55.0, 52.0, 49.4, 26.5, 25.1; HRMS (EI) m/z calcd for C14H14N2O2 (M+) 242.1055, found 242.1052.
6-Chloro-7-hydroxy-2,3,4,5-tetrahydro-[1]benzoxolo[2,3-c]azepin-1-one (kb-NB77-88)
Yield: 86%; m.p. >300 °C; IR (ATR, neat) 3886, 3036 (br), 2928, 1629, 1566, 1428, 1340, 1174 cm−1; 1H-NMR (DMSO-d6, 600 MHz) δ 10.0 (s, 1 H), 8.22 (t, 1 H, J = 4.8 Hz), 7.43 (d, 1 H, J = 9.0 Hz), 7.10 (d, 1 H, J = 9.0 Hz), 3.26–3.20 (m, 4 H), 2.04–1.98 (m, 2 H); 13C-NMR (DMSO-d6, 150 MHz) δ 161.1, 149.5, 147.9, 144.6, 126.0, 123.4, 116.8, 111.4, 110.8, 40.2, 27.0, 26.3; HRMS (ESI) m/z calcd for C12H10ClNO3 ([M+Na]+), 274.0247, found 274.0226.
6-Fluoro-7-hydroxy-2,3,4,5-tetrahydro-[1]benzoxolo[2,3-c]azepin-1-one (kb-NB96-21)
Yield: 29%; m.p. >300 °C; IR (ATR, neat) 3197 (br), 2925, 2385, 1625, 1577, 1477, 1437, 1345, 1032 cm−1; 1H-NMR (DMSO-d6, 600 MHz) δ 9.67 (bs, 1 H), 8.19 (t, 1 H, J = 4.2 Hz), 7.26 (d, 1 H, J = 9.0 Hz), 7.08 (dd, 1 H, J = 8.6 Hz, JHF = 8.6 Hz), 3.25 (dd, 2 H, J = 8.4, 4.8 Hz), 3.09 (t, 2 H, J = 6.6 Hz), 2.05–1.97 (m, 2 H); 13C-NMR (DMSO-d6, 150 MHz) δ 161.1, 147.8 (d, JCF = 7.5 Hz), 144.3 (d, JCF = 244 Hz), 143.9, 139.8 (d, JCF = 10.3 Hz), 121.8 (d, JCF = 3.5 Hz), 118.3 (d, JCF = 4.5 Hz), 118.1, 107.4 (d, JCF = 4.2 Hz), 40.5, 26.7, 25.4; 19F NMR (DMSO-d6, 400 MHz) δ –149.09 (d, JFH = 8.4 Hz); HRMS (ESI) m/z calcd for C12H10FNO3 ([M+Na]+) 258.0542, found 258.0566.
7-Allyloxy-6-chloro-2,3,4,5-tetrahydro-[1]benzoxolo[2,3-c]azepin-1-one (kb-NB96-50)
Yield: 83%; representative experimental data: m.p. 192–194 °C; IR (ATR, neat) 3200, 3075, 2928, 1674, 1650, 1573, 1464, 1422, 1259, 1173, 1065 cm−1; 1H-NMR (DMSO-d6, 600 MHz) δ 8.28 (t, 1 H, J = 4.2 Hz), 7.58 (d, 1 H, J = 9.0 Hz), 7.33 (d, 1 H, J = 9.0 Hz), 6.10–6.02 (m, 1 H), 5.44 (dd, 1 H, J = 17.4, 1.8 Hz), 5.28 (dd, 1 H, J = 9, 1.8 Hz), 4.69 (s, 2 H), 3.28–3.21 (m, 4 H), 2.05–1.97 (m, 2 H); 13C-NMR (DMSO-d6, 150 MHz) δ 160.9, 150.1, 148.9, 145.1, 133.5, 126.2, 123.4, 117.7, 115.2, 114.6, 110.8, 70.4, 30.7, 26.9, 26.3; HRMS (ESI) m/z calcd for C15H14ClNO3 ([M+Na]+) 314.0560, found 314.0540.
8-Chloro-7-hydroxy-2,3,4,5-tetrahydro-[1]benzoxolo[2,3-c]azepin-1-one (kb-NB96-47-5)
Yield: 5%; representative experimental data: m.p. 327–332 °C; IR (ATR, neat) 3293, 3196 (br), 2938, 1652, 1578, 1464, 1437, 1233, 1141 cm−1; 1H-NMR (DMSO-d6, 600 MHz) δ 10.12 (s, 1 H), 8.14 (bs, 1 H), 7.73 (s, 1 H), 7.11 (s, 1 H), 3.28–3.22 (m, 2 H), 2.89 (t, J = 6.6 Hz, 2 H), 2.03–1.98 (m, 2 H); 13C-NMR (DMSO-d6, 150 MHz) δ161.2, 149.4, 147.0, 144.3, 128.0, 123.0, 120.9, 112.7, 105.7, 40.7, 26.3, 23.7; HRMS (ESI) m/z calcd for C12H10ClNO3 ([M+Na]+) 274.0247, found 274.0224.
6,8-Dichloro-7-hydroxy-2,3,4,5-tetrahydro-[1]benzoxolo[2,3-c]azepin-1-one (kb-NB96-43)
Yield: 73%; m.p. 298–301 °C; 1H-NMR (DMSO-d6, 600 MHz) δ 9.91 (s, 1 H), 8.29 (bs, 1 H), 7.83 (s, 1 H), 3.25–3.20 (m, 4 H), 2.04–1.99 (m, 2 H); 13C-NMR (DMSO-d6, 150 MHz) δ 160.8, 147.2, 145.7, 145.1, 125.1, 123.4, 123.1, 114.8, 111.7, 40.1, 26.9, 26.1; IR (ATR, neat) 3377, 3077 (br), 2969, 1643, 1568, 1428, 1327, 1233, 1171 cm–1; HRMS (ESI) m/z calcd for C12H9Cl2NO3 ([M+Na]+), 307.9857, found 307.9882.
6-Allyl-7-hydroxy-2,3,4,5-tetrahydro-[1]benzoxolo[2,3-c]azepin-1-one (kb-NB96-02)
Yield: 87%; m.p. 255–257 °C; IR (ATR, neat) 3174 (br), 3056, 2920, 1641, 1571, 1476, 1426, 1358, 1343, 1272, 1118 cm−1; 1H-NMR (DMSO-d6, 600 MHz) δ 9.19 (s, 1 H), 8.12 (t, 1 H, J = 4.8 Hz), 7.28 (d, 1 H, J = 9.0 Hz), 6.98 (d, 1 H, J = 9.0 Hz), 6.00 (ddt, 1 H, J = 17.4, 10.2, 5.4 Hz,), 4.97 (dd, 1 H, J = 10.2, 1.2 Hz), 4.79 (dd, 1 H, J = 17.4, 1.2 Hz), 3.64 (d, 2 H, J = 5.4 Hz), 3.20 (dd, 2 H, J = 9.0, 5.4 Hz), 3.14 (t, 2 H, J = 6.6 Hz), 2.02–1.95 (m, 2 H); 13C-NMR (DMSO-d6, 150 MHz) δ 161.6, 150.9, 148.1, 143.8, 137.5, 127.5, 124.0, 118.4, 116.1, 114.8, 109.7, 40.3, 29.1, 27.3, 25.9; HRMS (ESI) m/z calcd forC15H15NO3 ([M+Na]+), 280.0950, found 280.0960.
6-[(E)-2-(Prop-1-enyl) ]-7-hydroxy-2,3,4,5-tetrahydro-[1]benzoxolo[2,3-c]azepin-1-one (kb-NB96-30)
Yield: 37%; m.p. >300 °C; IR (ATR, neat) 3147 (br), 2908, 1644, 1568, 1477, 1417, 1245, 1165 cm−1; 1H-NMR (CD3OD, 600 MHz) δ 7.23 (d, 1 H, J = 9.0 Hz), 6.97 (d, 1 H, J = 9.0 Hz), 6.69 (dd, 1 H, J = 15.6, 1.2 Hz), 6.22 (dq, 1 H, J = 15.6, 6.6 Hz), 3.40–3.37 (m, 2 H), 3.15 (t, 2 H, J = 6.6 Hz), 2.15–2.10 (m, 2 H), 1.95 (dd, 3 H, J = 6.6, 1.8 Hz); 13C-NMR (CD3OD, 150 MHz) δ 165.2, 152.0, 150.5, 144.4, 133.8, 128.0, 127.9, 124.8, 120.7, 118.2, 111.0, 42.4, 29.5, 28.3, 19.3; HRMS (ESI) m/z calcd for C15H15NO3 ([M+Na]+) 280.0950, found 280.0950.
7-Hydroxy-3,4-dihydro-[1]benzoxolo[2,3-c]azepine-1,5(2H)-dione (kb-NB123-63)
Yield: 38%; m.p. >300 °C; IR (ATR, neat) 3189 (br), 3072, 2919, 1674, 1645, 1544, 1461, 1349, 1262, 1223 cm−1; 1H-NMR (DMSO-d6, 600 MHz) δ 8.87 (t, 1 H, J = 4.8 Hz), 7.58 (d, 1 H, J = 9.0 Hz), 7.57 (d, 1 H, J = 2.4 Hz), 6.97 (dd, 1 H, J = 9.0, 2.4 Hz), 3.51–3.46 (m, 2 H), 2.93–2.87 (m, 2 H); 13C-NMR (DMSO-d6, 150 MHz) δ 195.7, 159.8, 155.4, 151.3, 147.9, 125.5, 119.1, 117.1, 112.5, 107.2, 44.1, 36.0; HRMS (EI) m/z calcd for C12H9NO4 (M+) 231.0532, found 231.0530.
5,7-Dihydroxy-2,3,4,5-tetrahydro-[1]benzoxolo[2,3-c]azepin-1-one (kb-NB123-89)
Yield: 79%; m.p. 280–282 °C; IR (ATR, neat) 3189 (br), 3074, 2918, 1646, 1577, 1455, 1436, 1337, 1178 cm−1; 1H-NMR (CD3OD, 600 MHz) δ 7.39 (d, 1 H, J = 9.0 Hz), 7.23 (d, 1 H, J = 2.4 Hz), 6.97 (dd, 1 H, J = 9.0, 2.4 Hz), 5.14 (d, 1 H, J = 4.2 Hz), 3.58 (dd, 1 H, J = 15.0, 10.2 Hz), 3.37–3.32 (m, 1 H), 2.32–2.26 (m, 1 H), 2.21–2.14 (m, 1 H); 13C-NMR (CD3OD, 150 MHz) δ 164.4, 155.0, 150.4, 144.2, 130.1, 127.5, 118.2, 113.0, 107.3, 64.6, 37.3, 37.1; HRMS (EI) m/z calcd for C12H11NO4 (M+) 233.0688, found 233.0691.
7-Hydroxy-5-(2-phenylhydrazono)-2,3,4,5-tetrahydro-[1]benzoxolo[2,3-c]azepin-1-one (kb-NB142-05)
Yield: 13%; m.p. 234–236 °C; IR (ATR, neat) 3218 (br), 2924, 1643, 1600, 1553, 1447, 1341, 1250, 1144 cm−1; 1H-NMR (CD3OD, 600 MHz) δ 7.93 (d, 1 H, J = 2.4 Hz), 7.41 (d, 1 H, J = 8.4 Hz), 7.34–7.27 (m, 4 H), 7.01 (dd, 1 H, J = 9, 2.4 Hz), 6.87 (tt, 1 H, J = 6.0, 1.2 Hz), 3.60–3.57 (m, 2 H), 3.00–2.97 (m, 2 H); 13C-NMR (CD3OD, 150 MHz) δ 165.7, 155.4, 150.8, 146.8, 144.6, 139.2, 130.2 (2 C), 127.5, 124.8, 121.3, 118.1, 114.5 (2 C), 112.8, 110.6, 39.0, 32.4; HRMS (EI) m/z calcd for C18H15N3O3 (M+) 321.1113, found 321.1110.
7-Hydroxy-5-[2-{(4-methylphenyl)sulfony}hydrazono]-2,3,4,5-tetrahydro-[1]benzoxolo[2,3-c]azepin-1-one (kb-NB142-11)
Yield: 46%; m.p. 220–224 °C; IR (ATR, neat) 3212 (br), 1648, 1559, 1448, 1334, 1161 cm−1; 1H-NMR (DMSO-d6, 600 MHz) δ 10.69 (s, 1 H), 9.53 (s, 1 H), 8.48 (bs, 1 H), 7.91 (d, 2 H, J = 7.2 Hz), 7.55 (s, 1 H), 7.45 (d, 1 H, J = 9.0 Hz), 7.35 (d, 2 H, J = 7.80 Hz),6.94 (d, 1 H, J = 8.4 Hz), 3.33–3.28 (m, 2 H), 2.76–2.83 (m, 2 H), 2.33 (s, 3 H); 13C-NMR (CD3OD, 150 MHz) δ 164.6, 155.8, 151.2, 150.6, 147.0, 145.6, 137.3, 130.6 (2 C), 129.2 (2 C), 127.0, 122.3, 118.3, 112.7,110.8, 38.6, 32.9, 21.5; HRMS (EI) m/z calcd for C19H17N3O5S (M+), 422.0787, found 422.0817.
5-(Benzyloxyimino)-7-hydroxy-2,3,4,5-tetrahydro-[1]benzoxolo[2,3-c]azepin-1-one (kb-NB142-10)
Yield: 74%; m.p. 103–110 °C; IR (ATR, neat) 3217 (br), 2925, 1648, 1552, 1467, 1448, 1351, 1334, 1209, 1185 cm−1; 1H-NMR (CD3OD, 600 MHz) δ 7.66 (t, 1 H, J = 1.8 Hz), 7.47 (dd, 2 H, J = 7.8, 0.6 Hz), 7.41 (dd, 1 H, J = 9.0, 1.2 Hz), 7.37 (dt, 2 H, J = 7.8, 7.2 Hz), 7.31 (td, 1 H, J = 7.2, 0.6 Hz), 6.98 (ddd, 1 H, J = 9, 2.4, 1.8 Hz), 5.33 (s, 2 H), 3.45–3.42 (m, 2 H), 3.11–3.07 (m, 2 H); 13C-NMR (CD3OD, 150 MHz) δ 164.7, 155.7, 153.7, 150.7, 146.9, 139.0, 129.5 (2 C), 129.5 (2 C), 129.1, 126.8, 120.7, 118.3, 112.9, 110.7, 78.1, 38.7, 31.2.
2,3,4,9-Tetrahydro-6-hydroxy-1H-pyrido[3,4-b]indol-1-one (kb-NB123-59)
[63]. Yield: 95%; m.p. 244–247 °C; IR (ATR, neat) 3391, 3267, 1650, 1618, 1498, 1335, 1207 cm−1; 1H-NMR (DMSO-d6, 600 MHz) δ 11.26 (s, 1 H), 8.85 (d, 1 H, J = 2.4 Hz), 7.47 (s, 1 H), 7.18 (dd, 1 H, J = 8.4, 3.0 Hz), 6.83 (d, 1 H, J = 2.4 Hz), 6.74 (dd, 1 H, J = 8.4, 2.4 Hz), 3.50–3.43 (m, 2 H), 2.84–2.79 (m, 2 H); 13C-NMR (DMSO-d6, 150 MHz) δ 162.0, 150.9, 131.8, 127.5, 125.5, 117.1, 115.1, 113.0, 103.0, 41.2, 20.4; HRMS (EI) m/z calcd for C11H10N2O2 (M+) 202.0742, found 202.0752.
3,4,5,10-Tetrahydro-7-hydroxy-azepino[3,4-b]indol-1(2H)-one (kb-NB123-57)
Yield: 89%; IR (ATR, neat) 3362, 3276, 1600, 1545, 1484, 1362 cm−1; 1H-NMR (DMSO-d6, 300 MHz) δ 10.82 (s, 1 H), 8.78 (s, 1 H), 7.86 (t, 1 H, J = 4.5 Hz), 7.19 (d, 1 H, J = 8.7 Hz), 6.79 (d, 1 H, J = 1.8 Hz), 6.73 (dd, 1 H, J = 8.7, 2.3 Hz), 3.30–3.22 (m, 2 H), 2.91 (t, 2 H, J = 6.3 Hz), 2.07–1.94 (m, 2 H); 13C-NMR (DMSO-d6, 150 MHz) δ 164.2, 150.6, 130.5, 128.1, 127.6, 115.7, 115.2, 112.7, 102.8, 41.6, 26.9, 25.4; HRMS (EI) m/z calcd for C12H12N2O2 (M+) 216.0899, found 216.0898.
2,3,4,9-Tetrahydro-6-benzyloxy-1H-pyrido[3,4-b]indol-1-one (kb-NB123-52)
[63]. Yield: 52%; m.p. 208–210 °C; IR (ATR, neat) 3233, 2905, 2691, 1567, 1508, 1379, 1242 cm−1; 1H-NMR (DMSO-d6, 600 MHz) δ 11.45 (s, 1 H), 7.53 (s, 1 H), 7.47 (d, 2 H, J = 7.2 Hz), 7.39 (t, 2 H, J = 7.8 Hz), 7.32 (t, 1 H, J = 7.2 Hz), 7.29 (d, 1 H, J = 9.0 Hz), 7.17 (d, 1 H, J = 1.8 Hz), 6.94 (dd, 1 H, J = 9.0, 1.8 Hz),5.10 (s, 2 H), 3.48 (td, 2 H, J = 6.6, 1.8 Hz), 2.87 (t, 2 H, J = 6.6 Hz); 13C-NMR (DMSO-d6, 150 MHz)δ 161.9, 152.6, 137.6, 132.5, 128.4 (2 C), 127.8, 127.7 (2 C), 127.7, 125.1, 117.7, 115.6, 113.4, 102.4,69.7, 41.2, 20.5; HRMS (EI) m/z calcd for C18H16N2O2 (M+) 292.1212, found 292.1223.
3,4,5,10-Tetrahydro-7-benzyloxy-azepino[3,4-b]indol-1(2H)-one (kb-NB123-53)
Yield: 53%; representative experimental data: IR (ATR, neat) 3227, 3194, 3033, 2920, 1623, 1543, 1478, 1453, 1276, 1197 cm−1; 1H-NMR (DMSO-d6, 600 MHz) δ 11.03 (s, 1 H), 7.95 (bs, 1 H), 7.50–7.42 (d, 2 H), 7.41–7.33 (m, 2 H), 7.32–7.29 (m, 2 H), 7.11 (bs, 1 H), 6.97–6.91 (m, 1 H), 5.10 (s, 2 H), 3.38–3.34 (m, 2 H), 2.96 (bs, 2 H), 2.02 (bs, 2 H); 13C-NMR (DMSO-d6, 150 MHz) δ 164.1, 152.3, 137.6, 131.3, 128.4, 127.8, 127.7, 127.6, 116.4, 115.6, 113.0, 102.2, 69.7, 41.6, 26.8, 25.4; HRMS (EI) m/z calcd for C19H18N2O2 (M+) 306.1368, found 306.1366.
6-Amino-2,3,4,9-tetrahydro-1H-pyrido[3,4-b]indol-1-one (kb-NB142-08)
Yield: 69%; m.p. 280–282 °C (lit. 280–282 °C) [65]; IR (ATR, neat) 3356, 3230, 1653, 1501, 1327, 1225 cm−1; 1H-NMR (CD3OD, 600 MHz) δ 7.25 (d, 1 H, J = 8.4 Hz), 6.93 (s, 1 H), 6.86 (d, 1 H, J = 8.4 Hz), 3.62 (t, 2 H, J = 7.2 Hz), 2.94 (t, 2 H, J = 7.2 Hz); 13C-NMR (CD3OD, 150 MHz) δ 165.2, 141.1, 134.4, 127.6, 127.2, 120.0, 118.5, 113.9, 105.9, 42.8, 21.6; HRMS (EI) m/z calcd for C11H11N3O (M+) 201.0902,found 201.0901.
6-Amino-3,4,5,10-tetrahydro-1H-azepino[3,4-b]indol-1(2H)-one (kb-NB142-01)
Yield: 68% m.p. 202–207 °C; IR (ATR, neat) 3208, 2921, 1618, 1542, 1479, 1450, 1335, 1296 cm−1; 1H-NMR (DMSO-d6, 600 MHz) δ 10.85 (s, 1 H), 7.93–7.85 (m, 1 H), 7.22–7.17 (m, 1 H), 6.86–6.83 (m, 1 H), 6.78–6.74 (m, 1 H), 6.39 (bs, 2 H), 3.30–3.24 (m, 2 H), 2.94–2.88 (m, 2 H), 2.04–1.98 (m, 2 H); 13C-NMR (DMSO-d6, 150 MHz) δ 164.3, 140.7, 130.0, 128.3, 127.0, 115.8, 115.1, 112.4, 101.9, 41.6, 27.0, 25.5; HRMS (EI) m/z calcd for C12H13N3O (M+) 215.1059, found 215.1062.
N-(2,3,4,9-Tetrahydro-1-oxo-1H-pyrido[3,4-b]indol-6-yl)-acetamide (kb-NB123-93)
Yield: 24%; m.p. >300 °C (lit > 320 °C) [65]; IR (ATR, neat) 3206, 1642, 1588, 1542, 1484, 1437, 1270 cm−1; 1H-NMR (DMSO-d6, 600 MHz) δ 11.50 (s, 1 H), 9.80 (s, 1 H), 7.91 (s, 1 H), 7.54 (s, 1 H), 7.32–7.22 (m, 2 H), 3.52–3.45 (m, 2 H), 2.89–2.83 (m, 2 H), 2.03 (s, 3 H); 13C-NMR (DMSO-d6, 150 MHz) δ 167.8, 161.8, 133.8, 131.9, 127.8, 124.6, 118.0, 117.9, 112.4, 109.8, 41.1, 23.9, 20.4; HRMS (EI) m/z calcd for C13H13N3O2 (M+) 243.1008, found 243.1009.
N-(3,4,5,10)-Tetrahydro-1-oxo-1H-azepino[3,4-b]indol-7-yl)-(2H)-acetamide (kb-NB123-94)
Yield: 75%; m.p. > 300 °C; IR (ATR, neat) 3270, 1618, 1547, 1477, 1453, 1267 cm−1; 1H-NMR (DMSO-d6, 600 MHz) δ 11.06 (s, 1 H), 9.80 (s, 1 H), 7.95 (t, 1 H, J = 4.5 Hz), 7.88 (s, 1 H), 7.32–7.25 (m, 2 H), 3.30–3.26 (m, 2 H), 2.95 (t, J = 6.6 Hz, 2 H), 2.00–2.06 (m, 5 H); 13C-NMR (DMSO-d6, 150 MHz) δ 167.7, 164.1, 132.6, 131.5, 127.8, 127.2, 118.1, 116.6, 112.0, 109.7, 41.5, 26.9, 25.4, 23.9; HRMS (EI) m/z calcd for C14H15N3O2 (M+) 257.1164, found 257.1166.
3,4-Dihydro-9-benzyloxy-[1]benzothieno[2,3-f]-1,4-thiazepin-5(2H)-one (kb-NB123-66)
Yield: 87%; representative experimental data: m.p. 247–249 °C; IR (ATR, neat) 3165, 3037, 1650, 1500, 1282 cm−1; 1H-NMR (DMSO-d6, 600 MHz) δ 8.47 (t, 1 H, J = 5.6 Hz), 7.90 (d, 1 H, J = 8.8 Hz), 7.49 (d, 2 H, J = 7.5 Hz), 7.40 (t, 2 H, J = 7.7 Hz), 7.36–7.32 (m, 1 H), 7.29–7.27 (m, 1 H), 7.26–7.23 (m, 1 H), 5.20 (s, 2 H), 3.64–3.60 (m, 2 H), 3.41–3.37 (m, 2 H); 13C-NMR (DMSO-d6, 150 MHz) δ 165.1, 156.4, 139.4, 136.9, 133.3, 131.2, 128.5, 127.9, 127.8, 123.8, 118.0, 105.8, 69.6, 42.4, 33.4; HRMS (EI) m/z calcd for C18H15NO2S2 (M+) 341.0544, found 341.0543.
3,4-Dihydro-9-hydroxy-[1]benzothieno[2,3-f]-1,4-thiazepin-5(2H)-one (kb-NB142-70)
Yield: 93%; representative experimental data: m.p. 218–220 °C (dec., dark brown), 235–238 °C (dec.); IR (ATR, neat) 3269, 1633, 1597, 1496, 1432, 1197 cm−1; 1H-NMR (DMSO-d6, 600 MHz) δ 9.73 (s, 1 H), 8.42 (t, 1 H, J = 5.5 Hz), 7.77 (d, 1 H, J = 8.7 Hz), 7.11 (d, 1 H, J = 1.9 Hz), 7.01 (dd, 1 H, J = 8.7, 1.6 Hz), 3.64–3.59 (m, 2 H), 3.40–3.36 (m, 2 H); 13C-NMR (DMSO-d6, 150 MHz) δ 165.2, 155.3, 139.6, 132.8, 129.3, 127.l, 123.6, 117.8, 107.0, 42.5, 33.3; HRMS (EI) m/z calcd for C11H9NO2S2 (M+) 251.0075,found 251.0080.
3,4-Dihydro-9-methoxy-[1]benzothieno[2,3-f]-1,4-thiazepin-5(2H)-one (kb-NB165-09)
[68]. Yield: 92%; m.p. 202–204 °C (lit. 209–209.5 °C) [78]; IR (ATR, neat) 3156, 3018, 2916, 1633, 1499, 1403, 1284 cm−1; 1H-NMR (DMSO-d6, 600 MHz) δ 8.47 (t, 1 H, J = 5.3 Hz), 7.89 (d, 1 H, J = 8.3 Hz), 7.19–7.15 (m, 2 H), 3.84 (s, 3 H), 3.65–3.60 (m, 2 H), 3.42–3.38 (m, 2 H); 13C-NMR (DMSO-d6, 150 MHz) δ 165.2, 157.4, 139.4, 133.4, 130.9, 127.7, 123.8, 117.5, 104.4, 55.4, 42.4, 33.4; HRMS (EI) m/z calcd for C12H11NO2S2 (M+) 265.0231, found 265.0232.
3,4-Dihydro-9-amino-[1]benzothieno[2,3-f]-1,4-thiazepin-5(2H)-one (mcf292-03)
Yield: 71%; m.p. 198.6–199.0 °C; IR (ATR, neat) 3370, 3254, 3146, 3008, 2915, 1623, 1599, 1556, 1491, 1454, 1430, 1403, 1346, 1333, 1312, 1286, 1243, 1204, 1184, 1129, 1083, 975, 887, 837, 798, 766, 749, 719, 691, 677, 663 cm−1; 1H-NMR (CD3OD, 400 MHz) δ 7.58 (dd, 1 H, J = 8.8, 0.4 Hz), 7.15 (dd, 1 H, J = 2.4, 0.4 Hz), 6.98 (dd, 1 H, J = 8.8, 2.0 Hz), 3.77–3.71 (m, 2 H), 3.42–3.37 (m, 2 H); 13C-NMR (CD3OD, 100 MHz) δ 169.3, 147.0, 141.5, 132.4, 130.8, 130.7, 124.0, 119.7, 108.4, 44.3, 35.1; MS (EI) m/z 250 (M+, 100); HRMS (EI) m/z calcd for C11H10N2OS2 (M+) 250.0235, found 250.0225.
3,4-Dihydro-9-azido-[1]benzothieno[2,3-f]-1,4-thiazepin-5(2H)-one (mcf292-08)
Yield: 53%, 96% purity estimated by 1H-NMR; m.p. 193 °C (dec.); IR (ATR, neat) 3260, 3154, 3016, 2922, 2115, 1631, 1592, 1495, 1467, 1441, 1422, 1400, 1340, 1284, 1252, 1234, 1215, 1198, 1144, 1113, 975, 889, 835, 809, 792, 751, 721 cm−1; 1H-NMR (DMSO-d6, 400 MHz) δ 8.53 (bt, 1 H, J = 5.6 Hz), 8.06 (d, 1 H, J = 8.8 Hz), 7.39 (d, 1 H, J = 1.6 Hz), 7.31 (dd, 1 H, J = 8.6, 2.2 Hz), 3.68–3.60 (m, 2 H), 3.45–3.38 (m, 2 H); 13C-NMR (DMSO-d6, 100 MHz) δ 164.9, 139.4, 136.9, 135.2, 134.2, 127.7, 124.6, 119.2, 112.0, 42.4, 33.4; MS (EI) m/z 276 (M+, 14), 248 ([M–N2]+, 50), 68 (100); HRMS (EI) m/z calcd for C11H8N4OS2 (M+) 276.0140, found 276.0137.
3,4-Dihydro-9-isothiocyanato-[1]benzothieno[2,3-f]-1,4-thiazepin-5(2H)-one (mcf292-05)
Yield: 58%, 98% purity estimated by 1H-NMR; m.p. 253.0–253.3 °C (softening point: 251.6 °C); IR (ATR, neat) 3260, 3155, 3025, 2922, 2067 (broad), 1633, 1590, 1497, 1469, 1457, 1441, 1420, 1400, 1340, 1321, 1282, 1254, 1241, 1141, 941, 900, 852, 807, 792, 744 cm−1; 1H-NMR (DMSO-d6, 400 MHz) δ 8.56 (bt, 1 H, J = 5.8 Hz), 8.11 (dd, 1 H, J = 8.8, 0.4 Hz), 7.80 (dd, 1 H, J = 2.0, 0.4 Hz), 7.58 (dd, 1 H, J = 8.8, 2.0 Hz), 3.68–3.61 (m, 2 H), 3.46–3.39 (m, 2 H); 13C-NMR (DMSO-d6, 100 MHz) δ 164.7, 139.0, 137.7, 134.4, 128.2, 127.2, 124.9, 124.5, 119.7, 42.4, 33.3; MS (EI) m/z 292 (M+, 47), 113 (57), 95 (86), 83 (96), 81 (100); HRMS (EI) m/z calcd for C12H8N2OS3 (M+) 291.9799, found 291.9795.
3,4-Dihydro-9-(2-chloroacetamido)-[1]benzothieno[2,3-f]-1,4-thiazepin-5(2H)-one (mcf292-09)
Yield: 65%; m.p. 229–230 °C (dec.); IR (ATR, neat) 3381, 3262, 3155, 3010, 2928, 1668, 1649, 1635, 1571, 1523, 1495, 1467, 1446, 1403, 1338, 1277, 1266, 1243, 1187, 1144, 986, 891, 876, 816, 790, 773, 734, 729 cm−1; 1H-NMR (DMSO-d6, 300 MHz) δ 8.48 (bt, 1 H, J = 5.8 Hz), 8.25 (d, 1 H, J = 1.5 Hz), 7.95 (d, 1 H, J = 8.7 Hz), 7.63 (dd, 1 H, J = 8.7, 2.1 Hz), 4.29 (s, 2 H), 3.70–3.58 (m, 2 H), 3.45–3.38 (m, 2 H); 13C-NMR (DMSO-d6, 100 MHz) δ 165.0, 164.8, 138.7, 135.9, 133.7, 132.9, 128.2, 123.2, 119.8, 112.4, 43.6, 42.5, 33.2; MS (EI) m/z 328 (40), 326 (M+, 100); HRMS (EI) m/z calcd for C13H11ClN2O2S2 (M+) 325.9950, found 325.9952.
3,4-Dihydro-9-hydroxy-10-iodo-[1]benzothieno[2,3-f]-1,4-thiazepin-5(2H)-one (kb-NB165-31)
Yield: 66%; m.p. 128 °C (dec.); IR (ATR, neat) 3335, 3071 (br), 1595, 1485, 1387, 1292 cm−1; 1H-NMR (DMSO-d6, 600 MHz) δ 10.52 (s, 1 H), 8.54 (t, 1 H, J = 5.4 Hz), 7.80 (d, 1 H, J = 8.4 Hz), 7.11 (d, 1 H, J = 8.4 Hz), 3.47 (q, 2 H, J = 5.4 Hz), 3.30–3.26 (m, 2 H); 13C-NMR (DMSO-d6, 150 MHz) δ 165.1, 155.2, 138.6, 138.3, 132.0, 128.7, 123.9, 115.4, 78.7, 42.2, 35.5; HRMS (EI) m/z calcd for C11H8INO2S2 (M+) 376.9041, found 376.9045.
3,4-Dihydro-9-hydroxy-10-bromo-[1]benzothieno[2,3-f]-1,4-thiazepin-5(2H)-one (kb-NB184-52)
Yield: 25%; m.p. 235–238 °C (dec., bprown); IR (ATR, neat) 3350, 3083, 2918, 1595 1492, 1396, 1303 cm−1; 1H-NMR (DMSO-d6, 600 MHz) δ 10.38 (s, 1 H), 8.52 (t, 1 H, J = 5.4 Hz), 7.78 (d, 1 H, J = 8.4 Hz), 7.16 (d, 1 H, J = 8.4 Hz), 3.53 (q, 2 H, J = 5.4 Hz), 3.31–3.27 (m, 2 H); 13C-NMR (DMSO-d6, 150 MHz) δ 164, 152.6, 136.5, 136.4, 132.0, 129.1, 122.8, 117.1, 103.3, 42.6, 34.8; HRMS (EI) m/z calcd for C11H8BrNO2S2 (M+) 328.9149 found 328.9164.
3,4-Dihydro-7-benzyloxy-[1]benzothieno[2,3-f]-1,4-thiazepin-5(2H)-one (kb-NB184-38)
Yield: 53%; m.p. 213–215 °C; IR (ATR, neat) 3159, 3036, 2923, 1647, 1462, 1257 cm−1; 1H-NMR (DMSO-d6, 600 MHz) δ 8.47 (t, 1 H, J = 5.4 Hz), 7.49 (d, 2 H, J = 7.2 Hz), 7.22–7.31 (m, 5 H), 7.19 (d, 1 H, J = 3.0 Hz), 5.35 (s, 2 H), 3.68–3.62 (m, 2 H), 3.42–3.38 (m, 2 H); 13C-NMR (DMSO-d6, 150 MHz) δ 165.0, 152.7, 140.0, 136.6, 132.1, 129.0, 128.6 (2 C), 128.0, 127.9, 127.4 (2 C), 126.6, 115.4, 108.7, 69.7, 42.6, 33.1; HRMS (EI) m/z calcd for C18H15NO2S2 (M+) 341.0544, found 341.0543.
3,4-Dihydro-7-hydroxy-[1]benzothieno[2,3-f]-1,4-thiazepin-5(2H)-one (kb-NB184-40)
Yield: 73%; m.p. 269–272 °C (dec., dark brown), 278–281 °C (dec.); IR (ATR, neat) 3255, 3155, 3015, 1620, 1439, 1285 cm−1; 1H-NMR (DMSO-d6, 600 MHz) δ 10.55 (s, 1 H), 8.45 (t, 1 H, J = 5.4 Hz), 7.29 (t, 1 H, J = 7.8 Hz), 7.24 (d, 1 H, J = 7.9 Hz), 6.89 (d, 1 H, J = 7.5 Hz), 3.65–3.60 (m, 2 H), 3.40–3.36 (m, 2 H); 13C-NMR (DMSO-d6, 150 MHz) δ 165.2, 152.1, 140.3, 131.8, 128.8, 126.65, 126.61, 113.7, 110.8, 42.5, 33.2; HRMS (EI) m/z calcd for C11H9NO2S2 (M+) 251.0075, found 251.0066.
3,4-Dihydro-7-methoxy-[1]benzothieno[2,3-f]-1,4-thiazepin-5(2H)-one (kb-NB184-44)
Yield: 99%; m.p. 220–224 °C; IR (ATR, neat) 3163, 3032, 2932, 1634, 1467, 1261 cm−1; 1H-NMR (DMSO-d6, 600 MHz) δ 8.48 (t, 1 H, J = 4.8 Hz), 7.44 (t, 1 H, J = 7.8 Hz), 7.37 (d, 1 H, J = 7.8 Hz), 7.09 (d, 1 H, J = 7.8 Hz), 3.96 (s, 3 H), 3.66–3.61 (m, 2 H), 3.42–3.37 (m, 2 H); 13C-NMR (DMSO-d6, 150 MHz) δ 165.0, 153.8, 139.9, 132.0, 128.9, 127.4, 126.7, 115.1, 107.2, 55.9, 42.5, 33.1; HRMS (ESI) m/z calcd for C12H11NO2S2 ([M+Na]+) 288.0129, found 288.0102.
3,4-Dihydro-9-hydroxy-6-oxide-[1]benzothieno[2,3-f]-1,4-thiazepin-5(2H)-one (kb-NB184-22)
Yield: 80%; m.p. 160–163 °C (dec., brown) 280–282 °C (dec., melts); IR (ATR, neat) 3369 (br), 1648, 1608, 1577, 1452, 1335, 1245, 998 cm−1; 1H-NMR (DMSO-d6, 600 MHz) δ 9.90 (bs, 1 H), 7.80 (bs, 1 H), 7.47 (s, 1 H), 7.33 (d, 1 H, J = 9.0 Hz), 7.04 (d, 1 H, J = 9.0 Hz), 3.73–3.63 (m, 2 H), 3.00–2.90 (m, 2 H); 13C-NMR (DMSO-d6, 150 MHz) δ 175.8, 156.2, 146.6, 127.3, 123.5, 121.7, 118.1, 109.5, 101.7, 42.6, 22.7; MS (ESI) m/z 268 ([M+H]+).
3,4-Dihydro-9-methoxy-6-oxide-[1]benzothieno[2,3-f]-1,4-thiazepin-5(2H)-one (kb-NB184-25)
Yield: 58%; m.p. 284–287 °C; IR (ATR, neat) 3340, 1506, 1477, 1337, 1248 cm−1; 1H-NMR (DMSO-d6, 600 MHz) δ 7.93 (bs, 1 H), 7.60 (d, 1 H, J = 2.4 Hz), 7.46 (d, 1 H, J = 9.0 Hz), 7.18 (dd, 1 H, J = 9.0, 2.4 Hz), 3.86 (s, 3 H), 3.75–3.69 (m, 2 H), 3.00–2.95 (m, 2 H); 13C-NMR (DMSO-d6, 150 MHz) δ 175.7, 158.1, 146.5, 127.5, 123.7, 123.2, 117.3, 107.4, 102.0, 55.8, 42.6, 22.7; MS (ESI) m/z 282 ([M+H]+); HRMS (EI) m/z calcd for C12H11NO3S2 (M+) 281.0180, found 281.0180.
3,4-Dihydro-9-methoxy-1-oxide-[1]benzothieno[2,3-f]-1,4-thiazepin-5(2H)-one (kb-NB184-45)
Yield: 54%; m.p. 215–217 °C; IR (ATR, neat) 3156, 3020, 2915, 1638, 1507 cm−1; 1H-NMR (DMSO-d6, 600 MHz) δ 8.99 (bs, 1 H), 8.03 (d, 1 H, J = 9.0 Hz), 7.59 (d, 1 H, J = 2.4 Hz), 7.25 (dd, 1 H, J = 8.4, 2.4 Hz), 3.94–3.87 (m, 1 H), 3.87 (s, 3 H), 3.59–3.51 (m, 3 H); 13C-NMR (DMSO-d6, 150 MHz) δ 162.4, 157.8, 141.9, 140.1, 133.6, 131.5, 124.0, 117.9, 104.6, 55.5, 50.4, 32.6; HRMS (EI) m/z calcd for C12H11NO3S2 (M+) 281.0180, found 281.0177.
2,3,4,5-Tetrahydro-10-benzyloxybenzo[b]thieno[2,3-f]-1,5-thiazocin-6-one (kb-NB165-89)
Yield: 42%; m.p. 198–199 °C; IR (ATR, neat) 3162, 3033, 2937, 1644, 1619, 1600, 1497, 1384, 1274, 1193 cm−1; 1H-NMR (DMSO-d6, 600 MHz) δ 8.02 (bs, 1 H), 7.90–7.84 (m, 1 H), 7.51–7.45 (m, 2 H), 7.43–7.37 (m, 2 H), 7.36–7.31 (m, 1 H), 7.26–7.19 (m, 2 H), 5.20 (s, 2 H), 3.50–3.43 (m, 2 H), 3.30–3.24 (m, 2 H), 1.92–1.89 (m, 2 H); 13C-NMR (DMSO-d6, 150 MHz) δ 164.8, 156.4, 138.7, 136.9, 130.3, 128.5, 128.5, 127.9, 127.7, 127.4, 123.6, 117.3, 105.8, 69.6, 30.5, 27.4; MS (EI) m/z 356 (23), 355 (M+, 100), 357 (12); HRMS (EI) m/z calcd for C19H17NO2S2 (M+) 355.0701, found 355.0689.
2,3,4,5-Tetrahydro-10-hydroxybenzo[b]thieno[2,3-f]-1,5-thiazocin-6-one (kb-NB165-92)
Yield: 92%; m.p. 139–142 °C; IR (ATR, neat) 3256 (br), 3169 (br), 1615, 1492, 1444, 1182 cm−1; 1H-NMR (DMSO-d6, 600 MHz) δ 9.71 (s, 1 H), 7.97 (t, 1 H, J = 7.8 Hz), 7.74 (d, 1 H, J = 8.6 Hz), 7.09 (s, 1 H), 6.98 (d, 1 H, J = 8.6 Hz), 3.50–3.43 (m, 2 H), 3.30–3.23 (m, 2 H), 1.92–1.85 (m, 2 H); 13C-NMR (DMSO-d6, 150 MHz) δ 164.9, 155.3, 139.0, 128.4, 127.0, 123.4, 117.1, 106.8, 30.5, 27.3; MS (EI) m/z 266 (15), 265 (M+, 100), 267 (11); HRMS (EI) m/z calcd for C12H11NO2S2 (M+) 265.0231, found 265.0230.
2,3,4,5-Tetrahydro-10-methoxybenzo[b]thieno[2,3-f]-1,5-thiazocin-6-one (kb-NB184-02)
Yield: 77%; m.p. 185–188 °C; IR (ATR, neat) 3152, 3026, 2939, 1636, 1498, 1395, 1209 cm−1; 1H-NMR (DMSO-d6, 600 MHz) δ 8.02 (t, 1 H, J = 7.1 Hz), 7.87 (d, 1 H, J = 9.4 Hz), 7.16–7.13 (m, 2 H), 3.84 (s, 3 H), 3.50–3.44 (m, 2 H), 3.30–3.26 (m, 2 H), 1.92–1.87 (m, 2 H); 13C-NMR (DMSO-d6, 150 MHz) δ 164.8, 157.4, 138.7, 130.1, 127.4, 123.6, 116.8, 104.3, 55.4, 30.5, 27.4; MS (EI) m/z 280 (16), 279 (M+, 100); HRMS (EI) m/z calcd for C13H13NO2S2 (M+) 279.0388, found 279.0379.
2,3,4,5-Tetrahydro-10-benzyloxybenzo[b]thieno[2,3-f]-1,5-oxazocin-6-one (kb-NB184-36)
Yield: 51%; m.p. 195–200 °C; IR (ATR, neat) 2912 (br), 2298, 1637, 1607, 1528, 1455, 1422 1222 cm−1; 1H-NMR (DMSO-d6, 600 MHz) δ 8.00 (t, 1 H, J = 7.2 Hz), 7.75 (d, 1 H, J = 8.4 Hz), 7.47 (d, 2 H, J = 7.8 Hz), 7.40 (t, 2 H, J = 7.8 Hz), 7.37–7.32 (m, 1 H), 7.26 (d, 1 H, J = 2.4 Hz), 7.18 (dd, 1 H, J = 8.4, 2.4 Hz), 5.17 (s, 2 H), 4.49 (t, 2 H, J = 5.4 Hz), 3.40–3.35 (m, 2 H), 1.87 (quint, 2 H, J = 5.4 Hz); 13C-NMR (DMSO-d6, 150 MHz) δ 165.1, 156.2, 149.2, 137.0, 133.5, 129.2, 128.5 (2 C), 127.9, 127.6 (2 C), 123.8, 118.3, 112.0, 104.7, 69.5, 68.6, 37.8, 29.2; HRMS (ESI) m/z calcd for C19H17NO3S ([M+Na]+) 362.0827, found 362.0809.
2,3,4,5-Tetrahydro-10-methoxybenzo[b]thieno[2,3-f]-1,5-oxazocin-6-one (kb-NB184-57)
Yield: 35%; m.p. 233–237 °C; IR (ATR, neat) 3154, 3027, 2919, 1631, 1467, 1422, 1220 cm−1; 1H-NMR (DMSO-d6, 600 MHz) δ 8.00 (t, 1 H, J = 6.6 Hz), 7.73 (d, 1 H, J = 9.0 Hz), 7.15 (d, 1 H, J = 2.4 Hz), 7.09 (dd, 1 H, J = 9.0, 2.4 Hz), 4.50 (t, 2 H, J = 5.4 Hz), 3.81 (s, 3 H), 3.42–3.32 (m, 2 H), 1.92–1.84 (m, 2 H); 13C-NMR (DMSO-d6, 150 MHz) δ 165.1, 157.2, 149.2, 133.5, 128.9, 123.7, 117.9, 112.0, 103.3, 68.6, 55.3, 37.8, 29.2; HRMS (ESI) m/z calcd for C13H13NO3S ([M+Na]+) 286.0514, found 286.0510.
Methyl 3-(3-hydroxypropylamino)-5-methoxybenzo[b]thiophene-2-carboxylate (kb-NB184-80)
Yield: 71%; m.p. 116–120 °C; IR (ATR, neat) 3481, 2921, 1627, 1578, 1440, 1224 cm−1; 1H-NMR (CDCl3, 600 MHz) δ 7.60 (d, 1 H, J = 8.4 Hz), 7.57 (d, 1 H, J = 2.4 Hz), 7.11 (dd, 1 H, J = 8.4, 2.4 Hz), 3.87 (s, 3 H), 3.87 (s, 3 H), 3.90–3.87 (m, 2 H), 3.84 (t, 1 H, J = 6.6 Hz), 1.99 (quint, 2 H, J = 6.6 Hz), 1.25 (s, 2 H); 13C-NMR (CDCl3, 150 MHz) δ 166.4, 157.0, 151.8, 133.3, 133.0, 124.4, 118.1, 107.5, 101.9, 60.5, 55.8, 51.7, 43.7, 33.6; HRMS (EI) m/z calcd for C14H17NO4S (M+) 295.0878, found 295.0879.
2,3-Dihydro-4-methyl-9-methoxy-[1]benzothieno[2,3-f]-1,4-thiazepin-5(2H)-one (kb-NB165-16)
Yield: 71%; m.p. 160–161 °C; IR (ATR, neat) 2928, 1625, 1598, 1497, 1397, 1207 cm−1; 1H-NMR (CD3OD, 600 MHz) δ 7.77 (d, 1 H, J = 9.0 Hz), 7.33 (d, 1 H, J = 2.4 Hz), 7.14 (dd, 1 H, J = 9.0, 2.4 Hz), 3.88 (s, 3 H), 3.86–3.83 (m, 2 H), 3.53–3.50 (m, 2 H), 3.22 (s, 3 H); 13C-NMR (CD3OD, 150 MHz) δ 167.5, 159.6, 141.3, 136.0, 133.2, 129.2, 124.5, 118.9, 105.7, 56.0, 51.3, 35.8, 35.0; HRMS (EI) m/z calcd for C13H13NO2S2 (M+) 279.0388, found 279.0386.
2,3-Dihydro-4-methyl-9-hydroxy-[1]benzothieno[2,3-f]-1,4-thiazepin-5(2H)-one (kb-NB165-17)
Yield: 56%; m.p. 265–268 °C; IR (ATR, neat) 3193 (br), 2384, 1610, 1587, 1494, 1401 cm−1; 1H-NMR (CD3OD, 600 MHz) δ 7.69 (d, 1 H, J = 9.0 Hz), 7.26 (d, 1 H, J = 2.4 Hz), 7.03 (dd, 1 H, J = 9.0, 2.4 Hz), 3.85 (t, 2 H, J = 5.4 Hz), 3.50 (t, 2 H, J = 5.4 Hz), 3.21 (s, 3 H); 13C-NMR (CD3OD, 150 MHz) δ 167.6, 156.9, 141.5, 135.5, 131.9, 128.9, 124.4, 118.8, 108.5, 51.4, 35.8, 34.9; HRMS (EI) m/z calcd for C12H11NO2S2 (M+) 265.0231, found 265.0235.
2,3-Dihydro-4-(2-aminoethyl)-9-hydroxy-[1]benzothieno[2,3-f]-1,4-thiazepin-5(2H)-one (kb-NB165-75)
Yield: quant.; m.p. 250–254 °C; IR (ATR, neat) 3251 (br), 2918 (br), 1579, 1500, 1427, 1178 cm−1; 1H-NMR (DMSO-d6, 600 MHz) δ 9.82 (s, 1 H), 7.99 (bs, 2 H), 7.79 (d, 1 H, J = 9.0 Hz), 7.18 (d, 1 H, J = 2.4 Hz), 7.04 (dd, 1 H, J = 9.0, 2.4 Hz), 3.85 (t, 2 H, J = 4.8 Hz), 3.73 (t, 2 H, J = 6.0 Hz), 3.48 (t, 2 H, J = 4.8 Hz), 3.10–3.03 (m, 2 H); 13C-NMR (DMSO-d6, 150 MHz) δ 165.2, 155.5, 139.5, 133.6, 129.3, 127.0, 123.7, 117.9, 107.1, 48.3, 45.8, 37.3, 32.8; HRMS (EI) m/z calcd for C13H14N2O2S2 (M+) 294.0497, found 294.0492.
2,3,4,5-Tetrahydro-9-benzyloxy-[1]benzothieno[2,3-f]-1,4-thiazepine (kb-NB165-81)
Yield: 15%; m.p. 144–145 °C; IR (ATR, neat) 2915, 1596, 1443, 1270, 1192 cm−1; 1H-NMR (CD3OD, 600 MHz) δ 7.67 (d, J = 9.0 Hz, 1 H), 7.48 (d, 2 H, J = 7.2 Hz), 7.38 (t, 2 H, J = 7.2 Hz), 7.37–7.30 (m, 2 H), 7.06 (dd, 1 H, J = 9.0, 2.4 Hz), 5.14 (s, 2 H), 4.14 (s, 2 H), 3.44–3.41 (m, 2 H), 2.82–2.79 (m, 2 H); 13C-NMR (CD3OD, 150 MHz) δ 158.4, 145.0, 142.7, 138.7, 131.3, 129.5 (2 C), 128.9, 128.7 (2 C), 128.0, 124.2, 116.5, 107.2, 71.3, 55.3, 35.5; MS (EI) m/z 327 (M+); HRMS (EI) m/z calcd for C18H17NOS2 (M+) 327.0752, found 327.0749.
2,3,4,5-Tetrahydro-9-hydroxy-[1]benzothieno[2,3-f]-1,4-thiazepine (kb-NB165-83)
Yield: 70%; m.p. 182–184 °C (dec., dark brown); IR (ATR, neat) 2947 (br), 2920, 1598, 1436, 1183 cm−1; 1H-NMR (CD3OD, 600 MHz) δ 7.59 (d, 1 H, J = 9.0 Hz), 7.16 (d, 1 H, J = 2.4 Hz,), 6.88 (dd, 1 H, J = 9.0, 2.4 Hz), 4.18 (s, 2 H), 3.49–3.45 (m, 2 H), 2.86–2.81 (m, 2 H); 13C-NMR (CD3OD, 150 MHz) δ 156.5, 143.9, 142.8, 130.1, 127.9, 124.1, 116.1, 108.4, 55.2, 35.1; MS (EI) m/z 238 (14), 237 (M+, 100), 239 (10); HRMS (EI) m/z calcd for C11H11NOS2 (M+) 237.0282, found 237.0289.
2-Methoxy-7H,8H,9H-1,4-thiazepino [7′,6′-5,4]thiopheno[3,2-d]pyrimidin-6-one (kmg-NB4-23)
Yield: 68%; m.p. 308 °C (dec.); IR (ATR, neat) 3260, 3153, 3015, 1636, 1554, 1495, 1467, 1374, 1269, 1353, 1323 cm−1; 1H-NMR (DMSO-d6, 300 MHz) δ 9.36 (s, 1 H), 8.70 (t, 1 H, J = 5.4 Hz), 3.98 (s, 1 H), 3.68 (app dd, 2 H, J = 6.0 Hz), 3.40–3.36 (m, 2 H); 13C-NMR (DMSO-d6, 75 MHz) δ 164.2, 163.1, 159.6, 155.9, 138.5, 129.7, 124.1, 54.8, 42.7, 31.9; HRMS (ESI) m/z calcd for C10H10N3O2S2 ([M+H]+) 268.0214, found 268.0237.
2-Hydroxy-7H,8H,9H-1,4-thiazepino[7′,6′-5,4]thiopheno[3,2-d]pyrimidin-6-one (hydrochloride salt, kmg-NB4-69A)
Yield: 85%; m.p. 335.9 °C (dec); IR (ATR) cm−1 3452, 3267, 3176, 2591, 2032, 1912, 1700, 1623, 1463, 1240; 1H-NMR (DMSO-d6, 400 MHz) δ 9.20 (s, 1 H), 8.74 (t, 1 H, J = 6.2 Hz), 7.79 (bs, 1 H), 3.68–3.60 (m, 2 H), 3.39–3.32 (m, 2 H); 13C-NMR (DMSO-d6, 100 MHz) δ 164.0, 161.0, 159.7, 153.6, 140.9, 128.4, 119.6, 42.6, 32.2; HRMS (ESI) m/z calcd for C9H8N3O2S2 ([M+H]+) 254.0058, found 254.0041.
2,4-Dimethoxy-7H,8H,9H-1,4-thiazepino[7′,6′-5,4]thiopheno[3,2-d]pyrimidin-6-one (kmg-NB5-13)
Yield: 77%; m.p. 288.0 °C (dec); IR (ATR, neat) 3321, 1642, 1579, 1545, 1491, 1476, 1458, 1346, 1331, 1206 cm−1; 1H-NMR (DMSO-d6, 500 MHz) δ 8.65 (t, 1 H, J = 5.7 Hz), 4.09 (s, 3 H), 3.96 (s, 3 H), 3.69–3.64 (m, 2 H), 3.38–3.34 (m, 2 H); 13C-NMR (DMSO-d6, 125 MHz) δ 165.5, 164.1, 163.5, 159.9, 135.9, 130.3, 111.5, 54.74, 54.72, 42.8, 31.8.
4-Hydroxy-2-methoxy-7H,8H,9H-1,4-thiazepino[7′,6′-5,4]thiopheno[3,2-d]pyrimidin-6-one (kmg-NB5-15)
Yield: 77%; m.p. 295.0 °C (dec); IR (ATR, neat) 3266, 3170, 2740, 1674, 1646, 1603, 1465, 1407, 1316 cm−1; 1H-NMR (DMSO-d6, 400 MHz) δ 12.72 (s, 1 H), 8.56 (t, 1 H, J = 5.2 Hz), 3.94 (s, 3 H), 3.66–3.60 (m, 2 H), 3.32–3.28 (m, 2 H); 13C-NMR (DMSO-d6, 125 MHz) δ 164.0, 158.4, 156.8, 153.8, 135.7, 130.6, 118.6, 54.9, 42.7, 31.9; MS (EI) m/z 283 (M+, 100). HRMS (ESI) m/z calcd for C10H9N3O3S2Na ([M+Na]+) 305.9983, found 305.9995.
3.2. IMAP-based kinase counterscreening assay
An automated, HTS formatted IMAP-based PKD Fluorescence Polarization (FP) assay was used to assess the specificity of the PKD analogs as previously described [52]. Briefly, PKD1 kinase reactions were assembled by the addition of 3× concentrations of substrate/ATP (300 nm/60 μm), experimental compound, and PKD1 enzyme (0.18 milliunits/mL) in a miniaturized reaction volume (i.e., 6 μL). Kinase reactions were incubated for 90 min at room temperature and stopped by the addition of IMAP binding reagent. Assay plates were then incubated for 2 h and IMAP-based FP data were captured on a Molecular Devices Spectra-Max M5 (Sunnyvale, CA, USA). The IC50 determinations for each compound in the PKD1 IMAP FP assay were conducted within the linear range of the captured signal readout (n = 3).
3.3. In vitro radiometric PKD kinase assays
In vitro radiometric PKD kinase assays were conducted as previously described [52]. Briefly, 50 ng recombinant human PKD1 (Biomol International, Plymouth Meeting, PA, USA) was incubated at 30 °C for 10 min with a reaction mixture containing 2.5 μg syntide-2 (Sigma), 70 μM ATP, and 1 μCi γ-32 P-ATP (PerkinElmer, Boston, MA, USA) in kinase buffer (50 mM Tris-HCl pH 7.5, 4 μM MgCl2, and 10 mM P-mercaptoethanol). 25 μL of the reaction mixture was then spotted on Whatman P81 filter paper (Whatman Inc., Clifton, NJ, USA), and filter papers were washed three times with 0.5% phosphoric acid, dried, then counted using a Beckman LS6500 multi-purpose scintillation counter.
3.4. Determination of cellular IC50 for PKD 1 inhibition
Inhibition of PKD 1 in cells was determined by densitometry analysis of Western blotting data for PKD1 autophosphorylation at Ser916 in LNCaP cells as previously described [52]. LNCaP cells were pre-treated with PKD inhibitors for 45 min at various concentrations, and then stimulated with 10 nM phorbol 12-myristate 13-acetate (PMA) for 20 min. Cells were then collected and lysed in lysis buffer containing 200 mM Tris-HCl, pH 7.4, 100 μM 4-(2-aminoethyl) benzenesulfonyl fluoride, 1 mM EGTA, and 1% Triton X-100. Cell lysates were probed by Western blot analysis using primary antibodies targeting p-S916-PKD1 (Millipore), PKD1 (Cell Signaling Technology), or GAPDH. Densitometry analysis of visualized bands was used to determine the cellular IC50 values for PKD1 inhibition.
3.5. Statistical analysis
GraphPad Prism V software was used to determine statistical significance. Each assay was repeated two or three times with triplicate determination at each point. A p value of < 0.05 was considered significant.
4. Conclusions
An extensive SAR study starting with the HTS-based confirmed hit CID755673 led to the discovery of the benzothienothiazepinone kb-NB142-70 and its methoxy analog kb-NB165-09, which were ca. 7-fold more potent at PKD1 inhibition. A 4-zone pharmacophore model was developed based on these new lead compounds, and systematic modifications of each zone led to a novel series of benzothienothiazepinones with equal or greater potencies compared to CID755673. Five of these analogs were further investigated, and each of them inhibited PMA-induced autophosphorylation of PKD1 [53]. These analogs also caused dramatic arrest in cell proliferation and inhibited cell migration and invasion [53]. Furthermore, counterscreens on a panel of other kinases clearly demonstrated a high selectivity profile in this series for PKD inhibition [53]. In order to decrease the susceptibility of these compounds to metabolic pathways, a pyrimidine moiety was installed in zone I, leading to a promising new lead structure, kmg-NB4-23. Because of the inadequate solubility properties of the pyrimidine-based inhibitors, additional modifications to zone I are currently being explored in our laboratories. Overall, the results of this extensive SAR study suggest that the zone I binding region is restrictive in both size and polarity of the aryl substituents. Additionally, it appears that the hydrogen bond donor-acceptor abilities of the amide moiety are critical for optimal interactions with the zone IV binding site. The synthesis of the azide analog mcf292-08 and the electrophilic reactive probe molecules mcf292-05 and mcf292-09 as tools for photoaffinity/affinity labeling studies was undertaken to gain key structural insights into the possible protein binding site. The latter information is currently pending and will be utilized for future SAR studies.
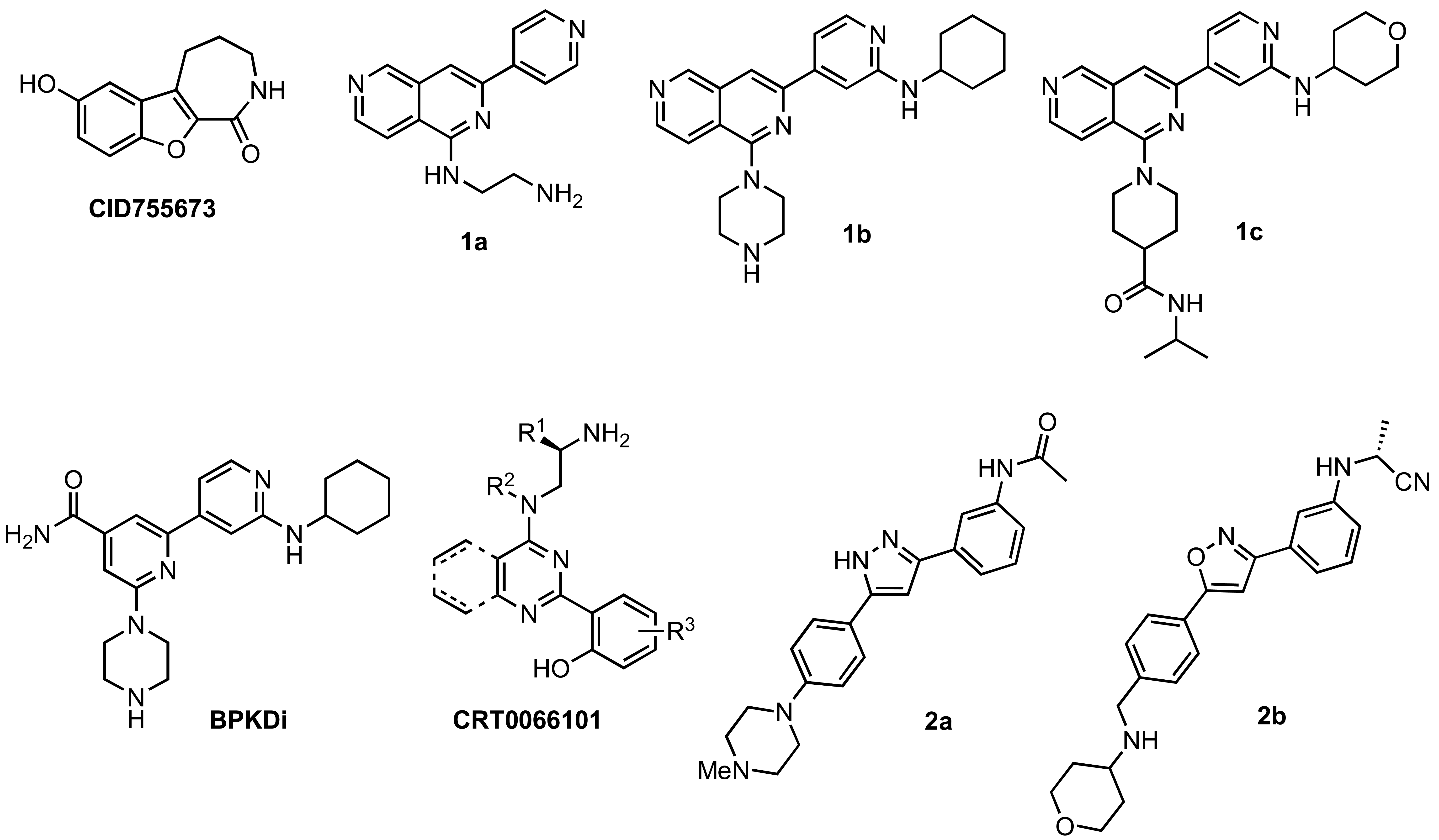
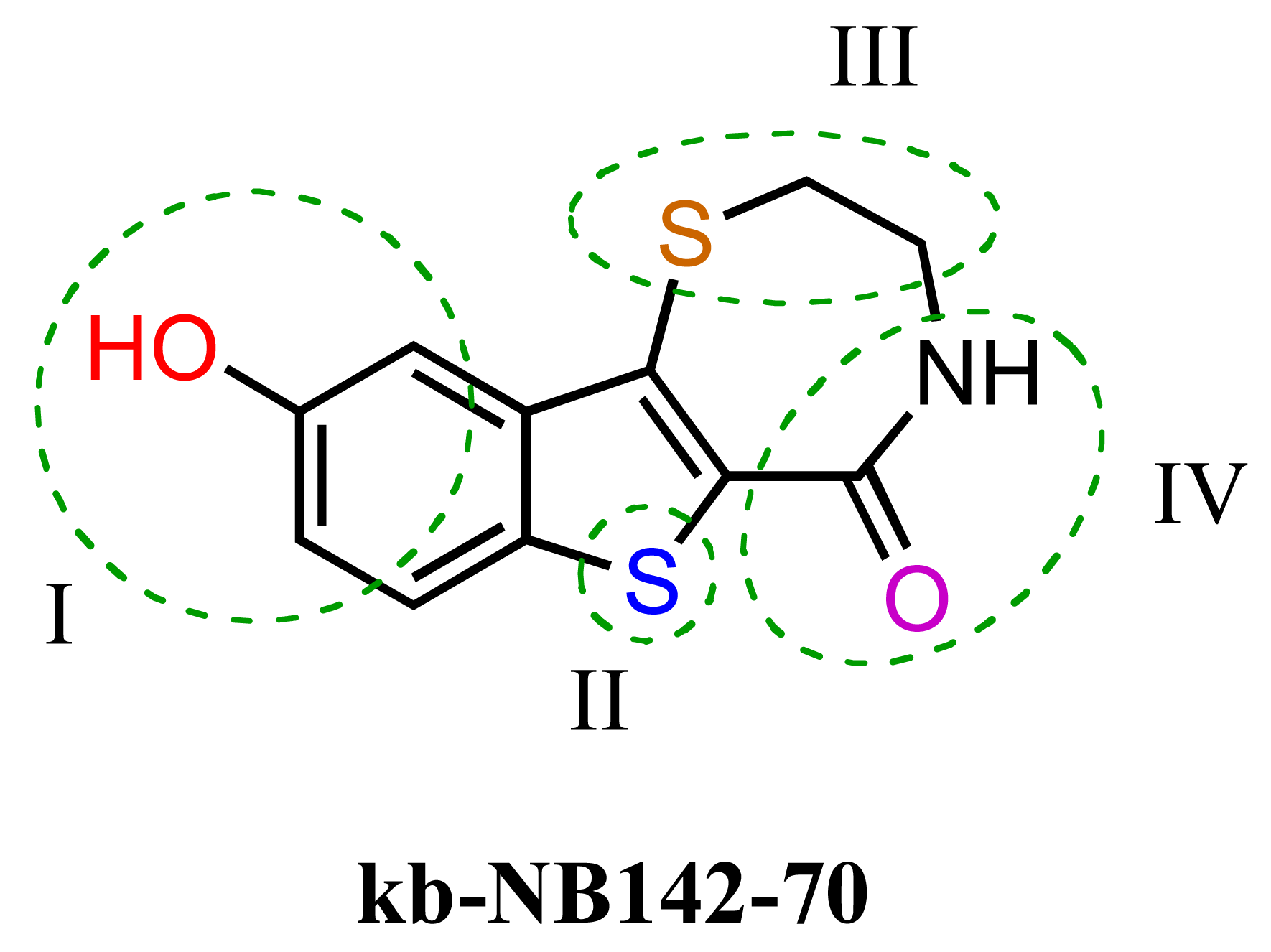
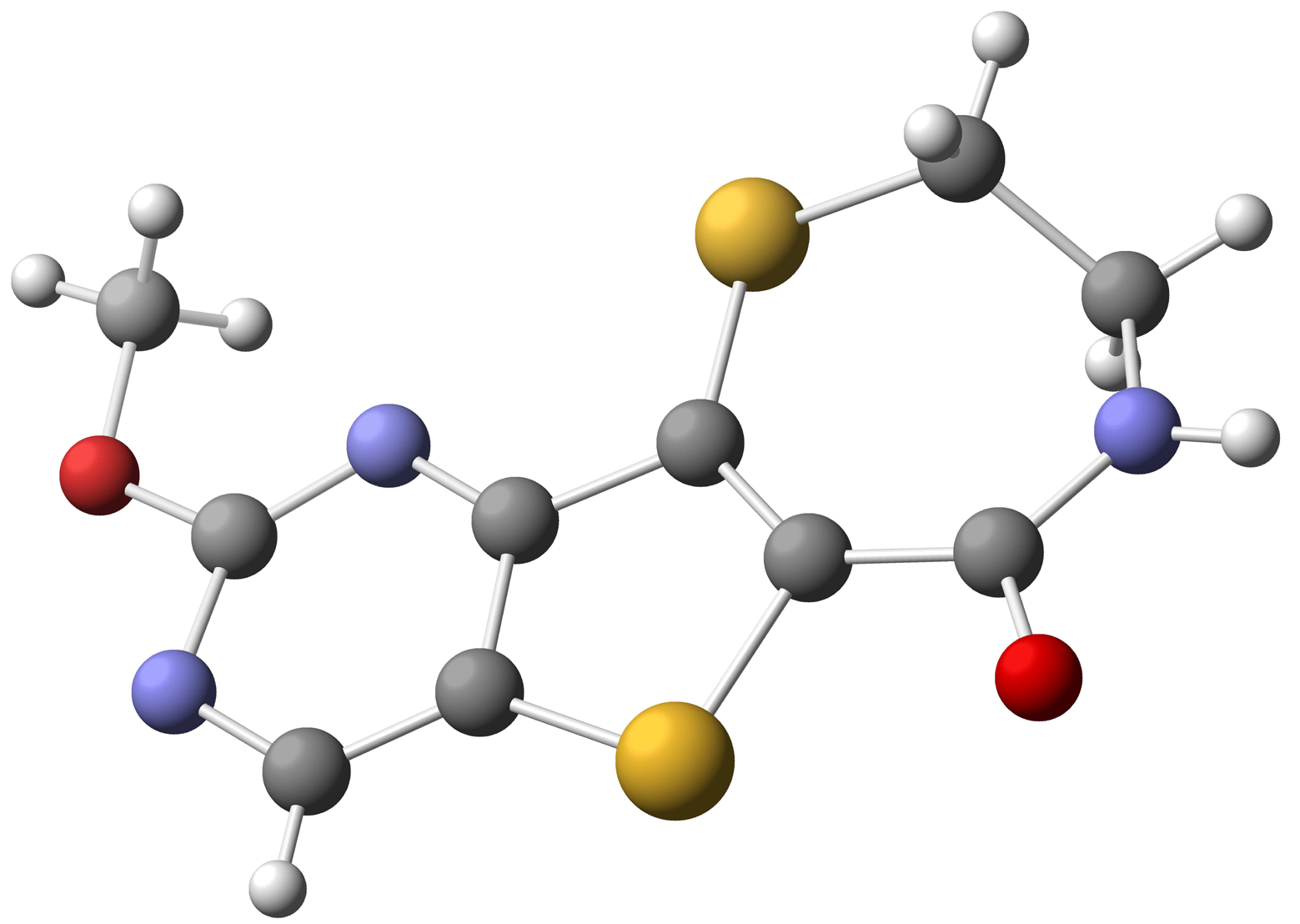
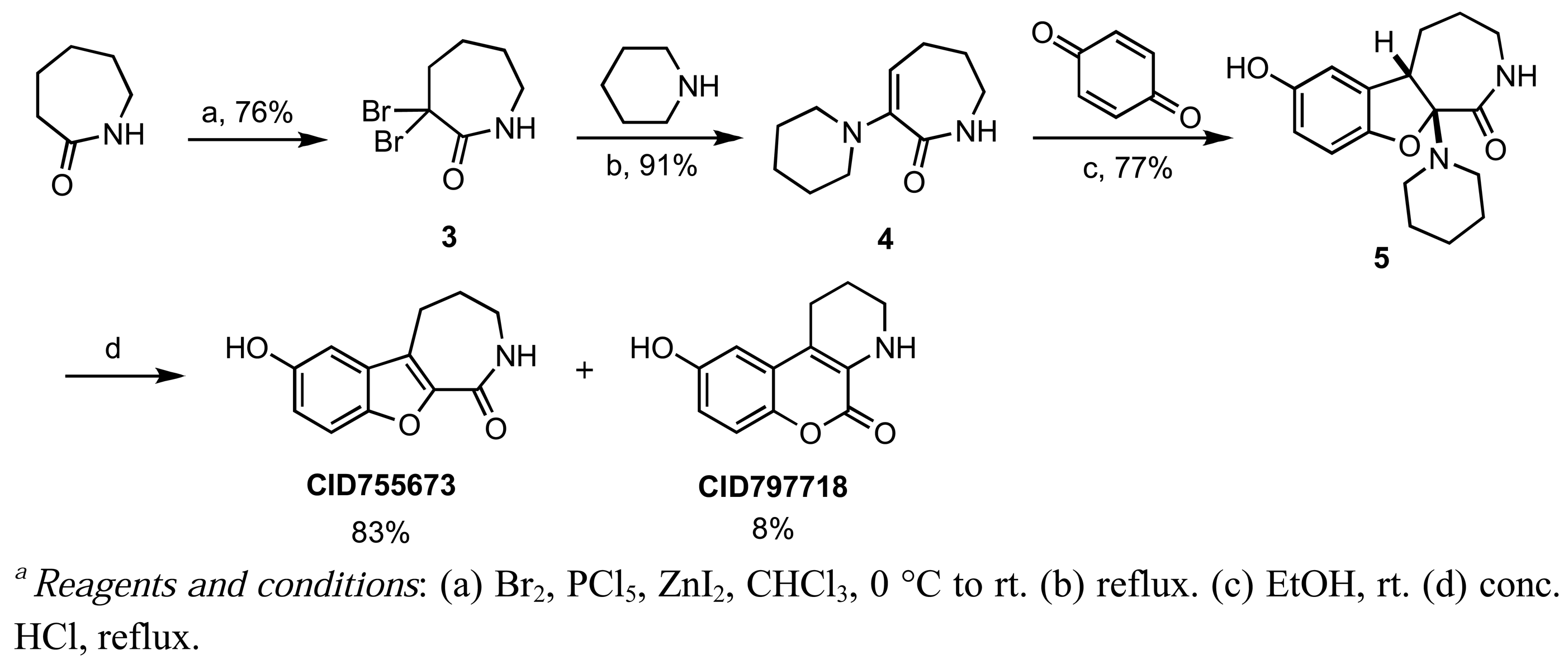
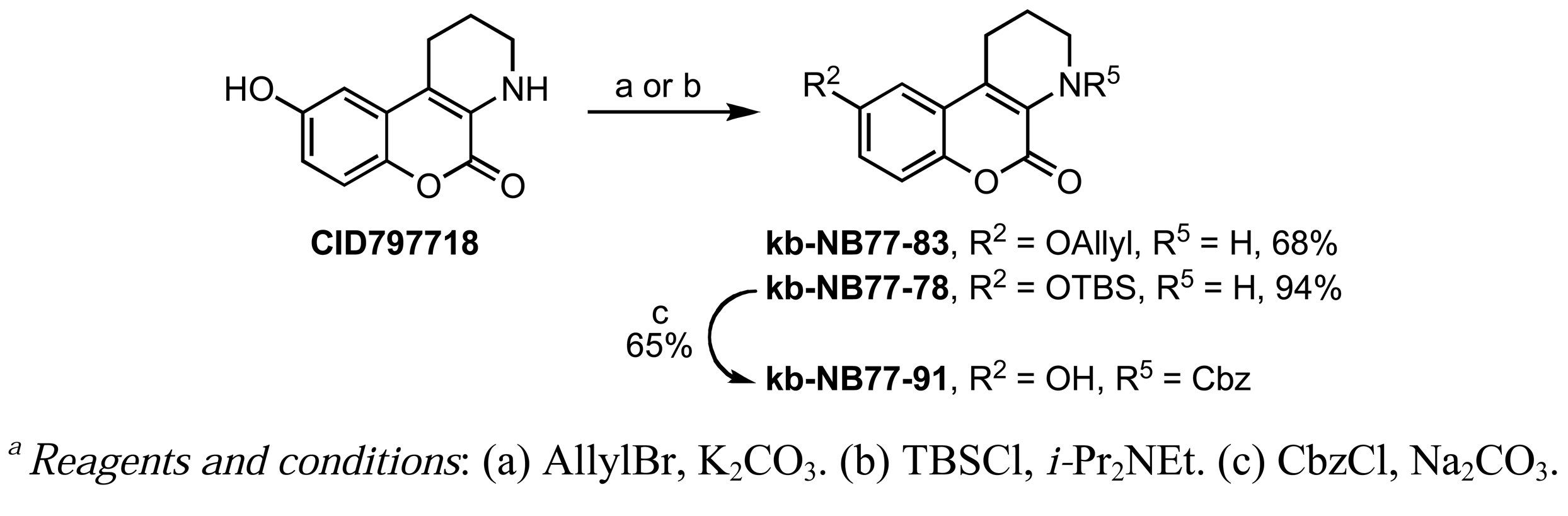

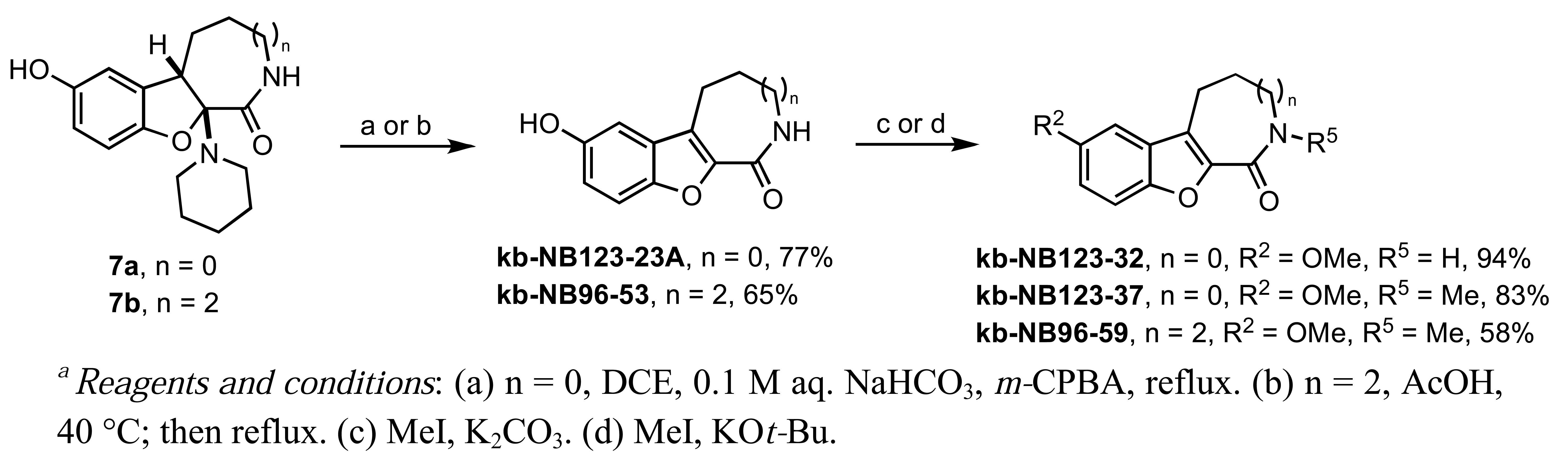
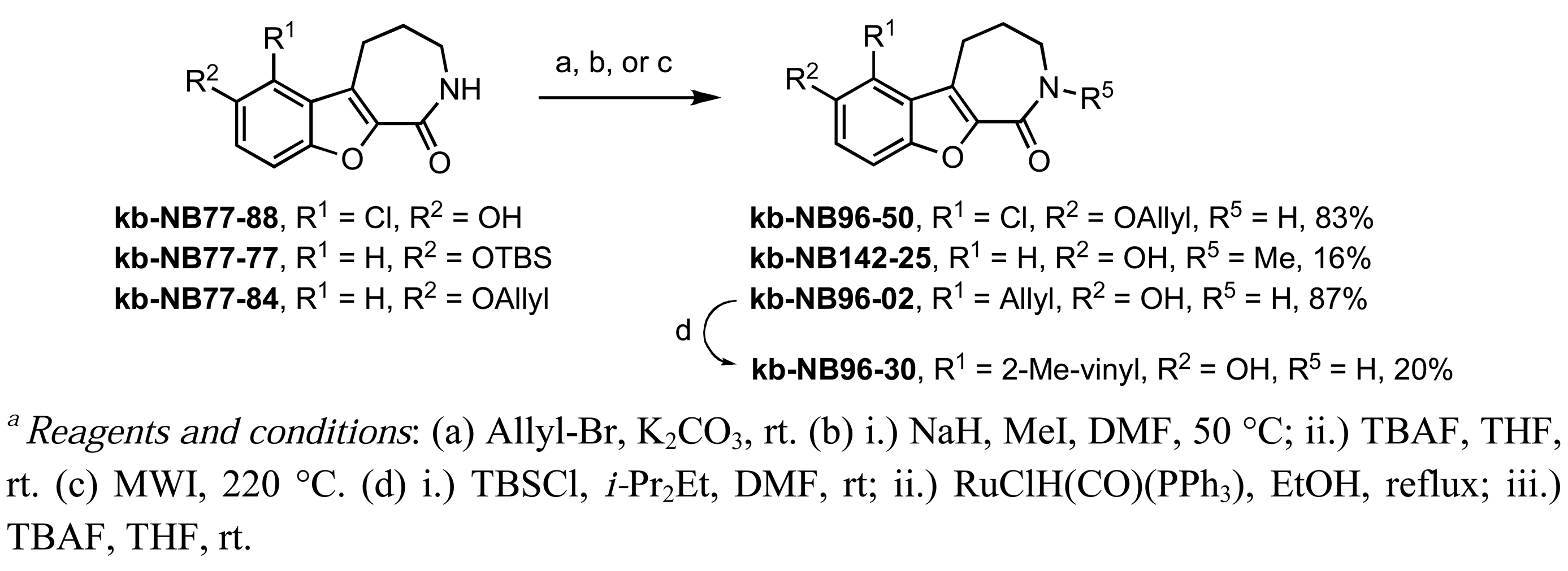
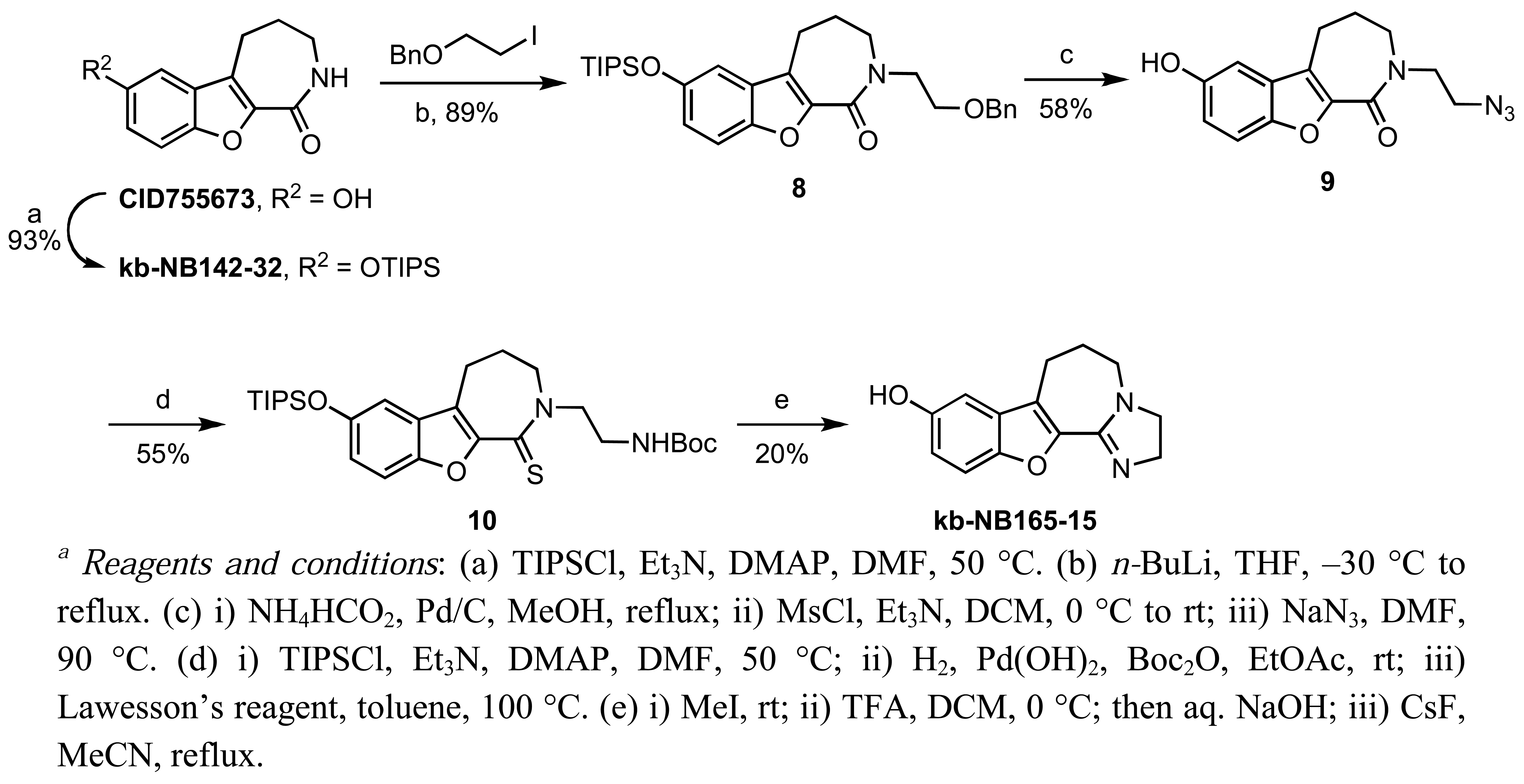

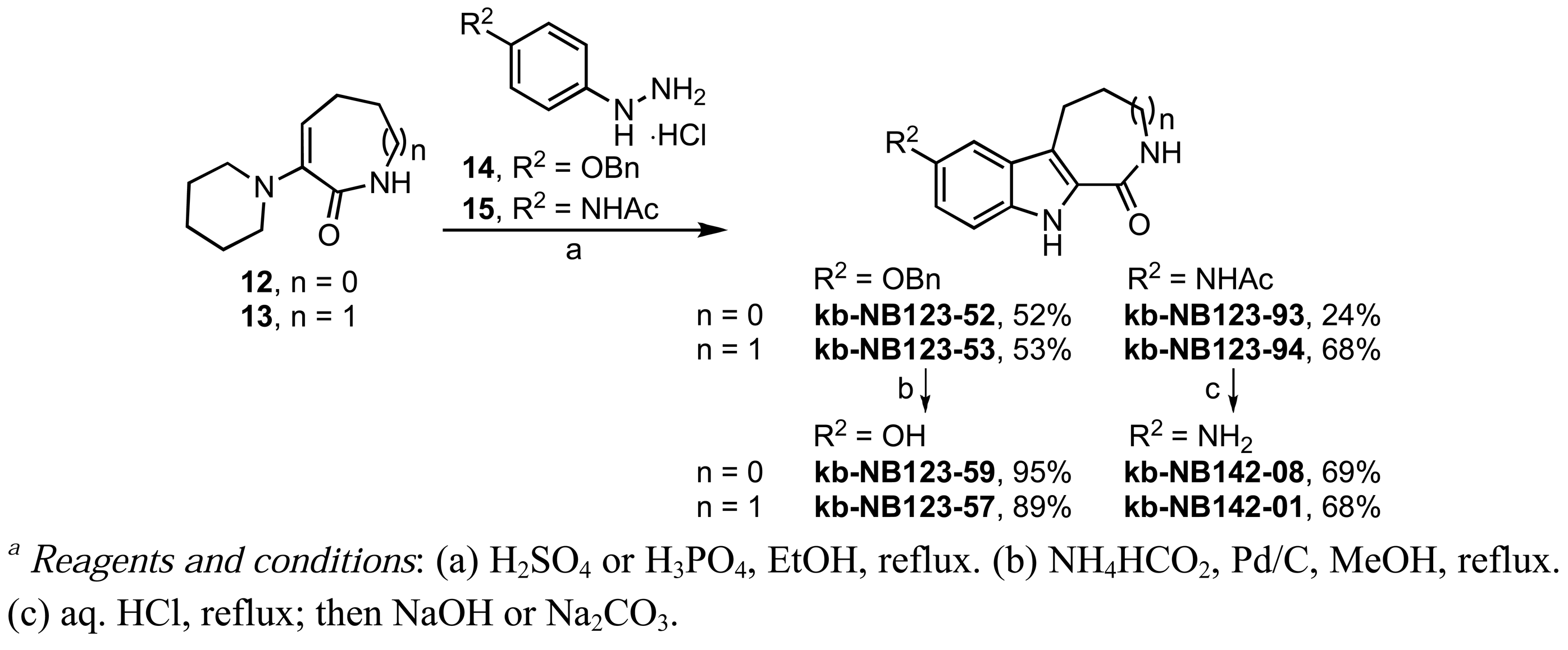
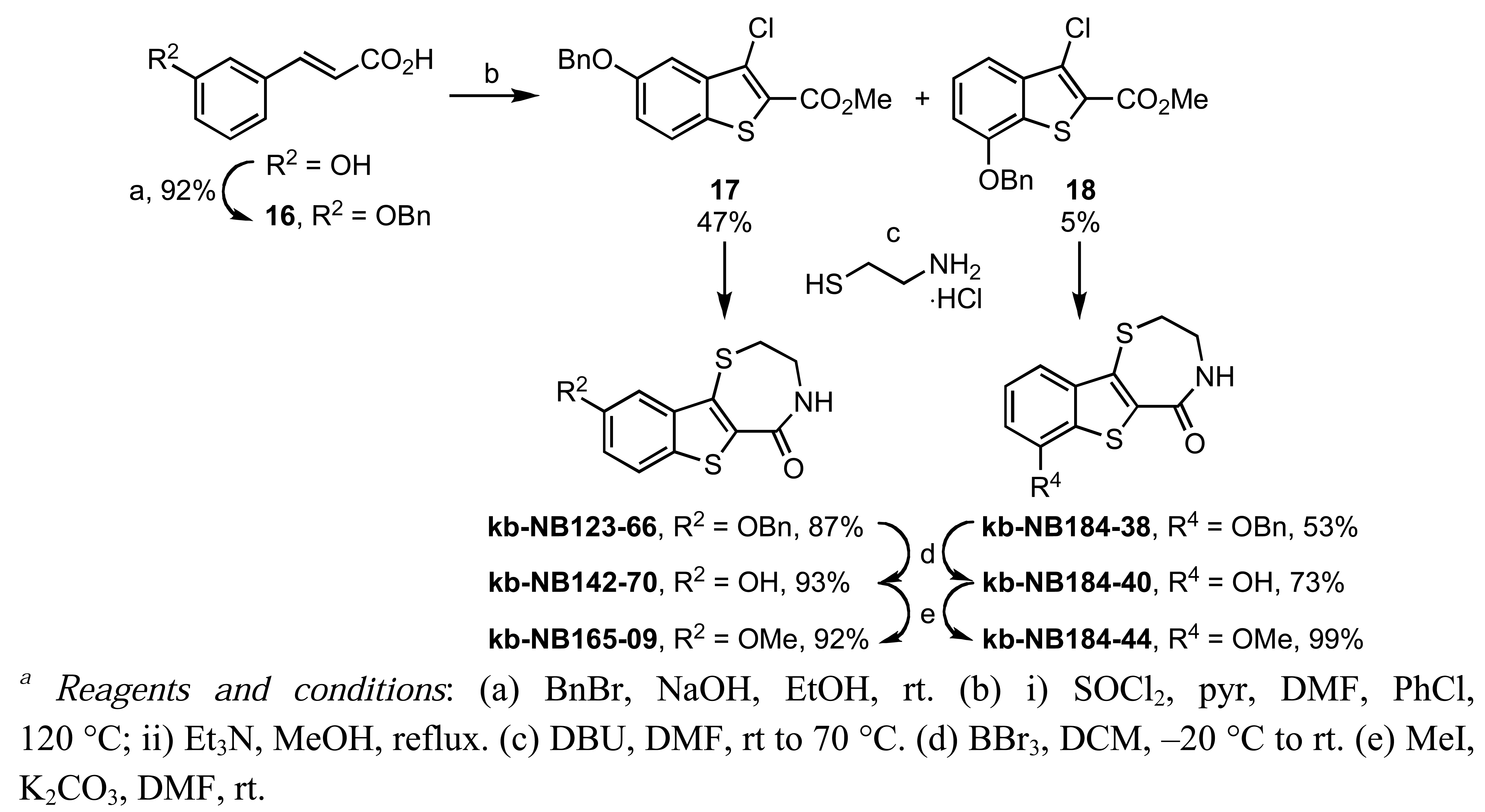
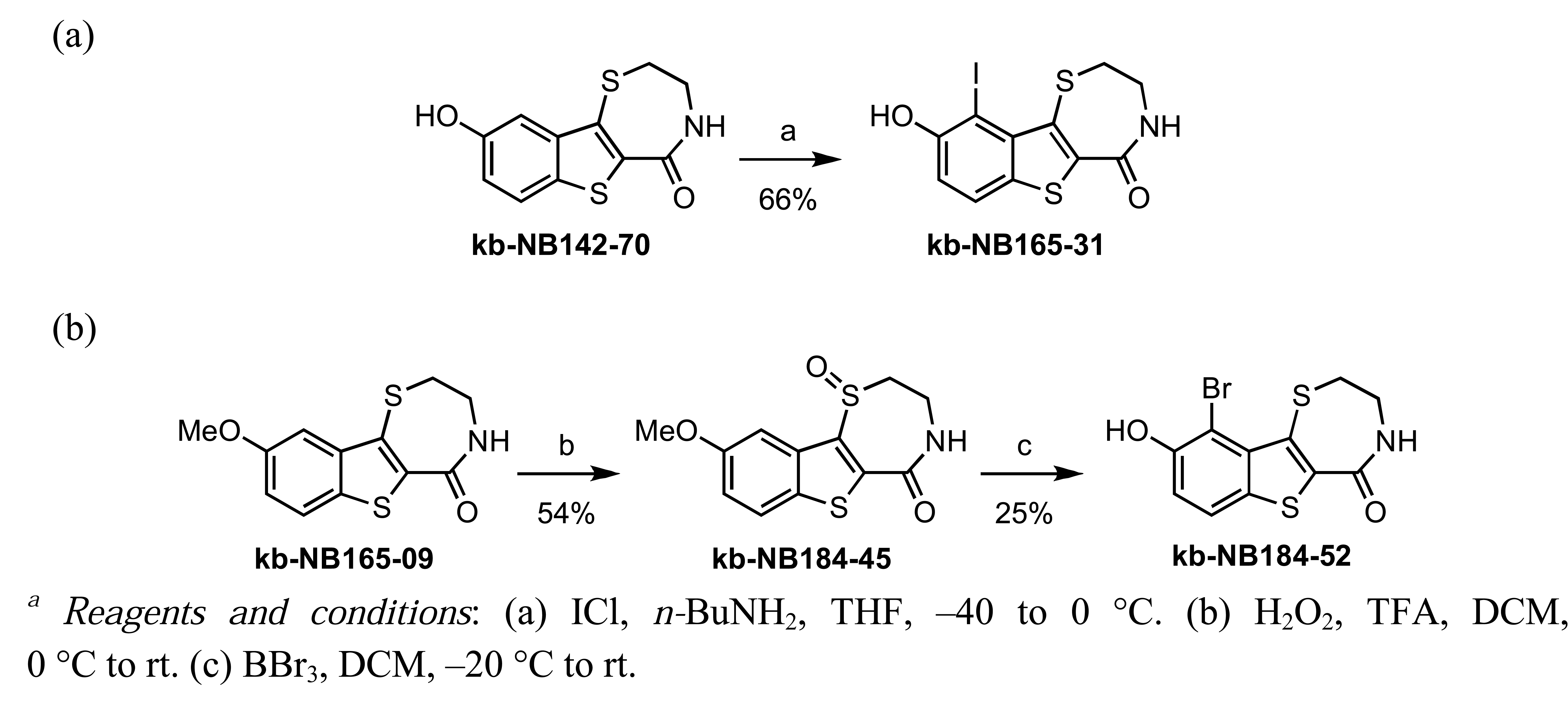
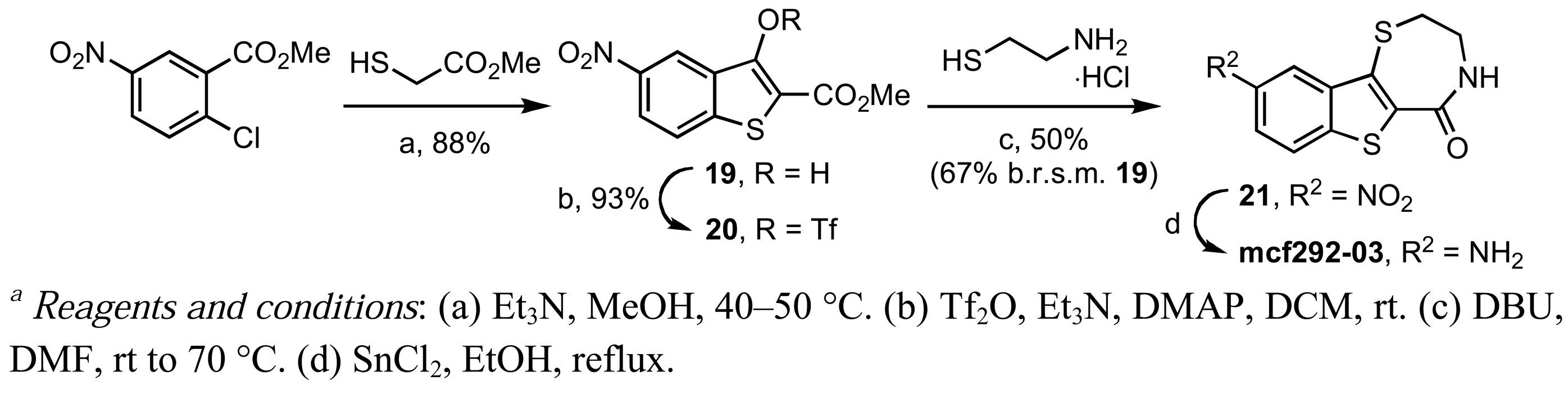
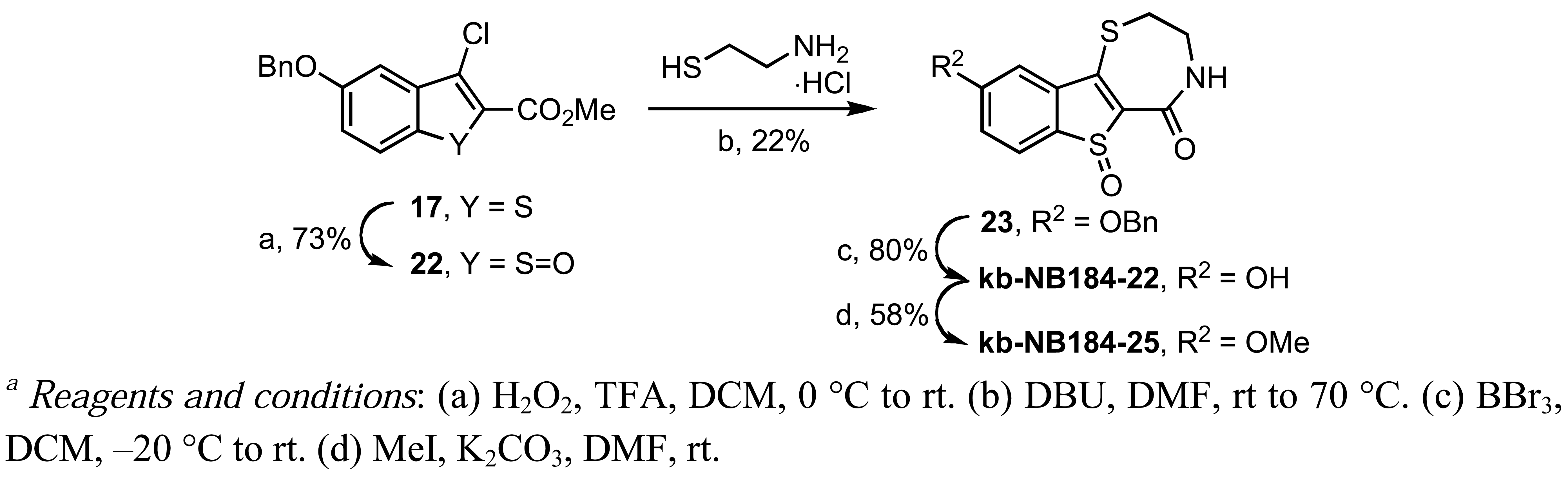
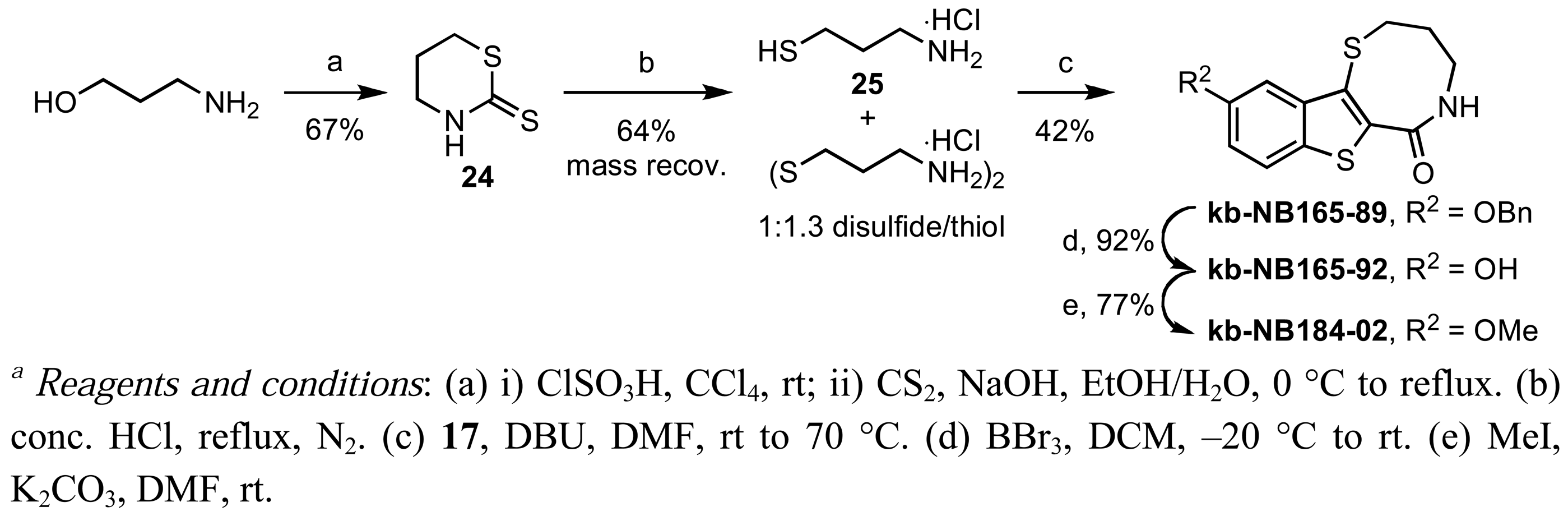


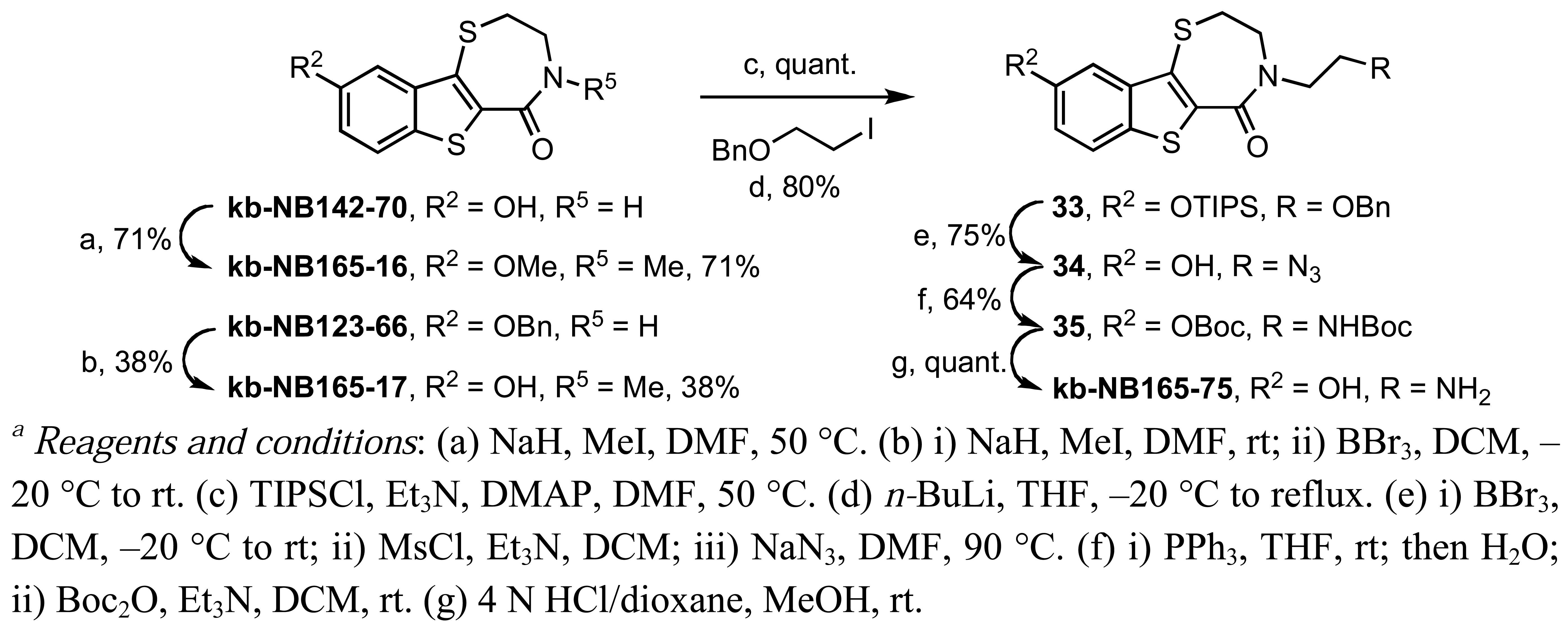


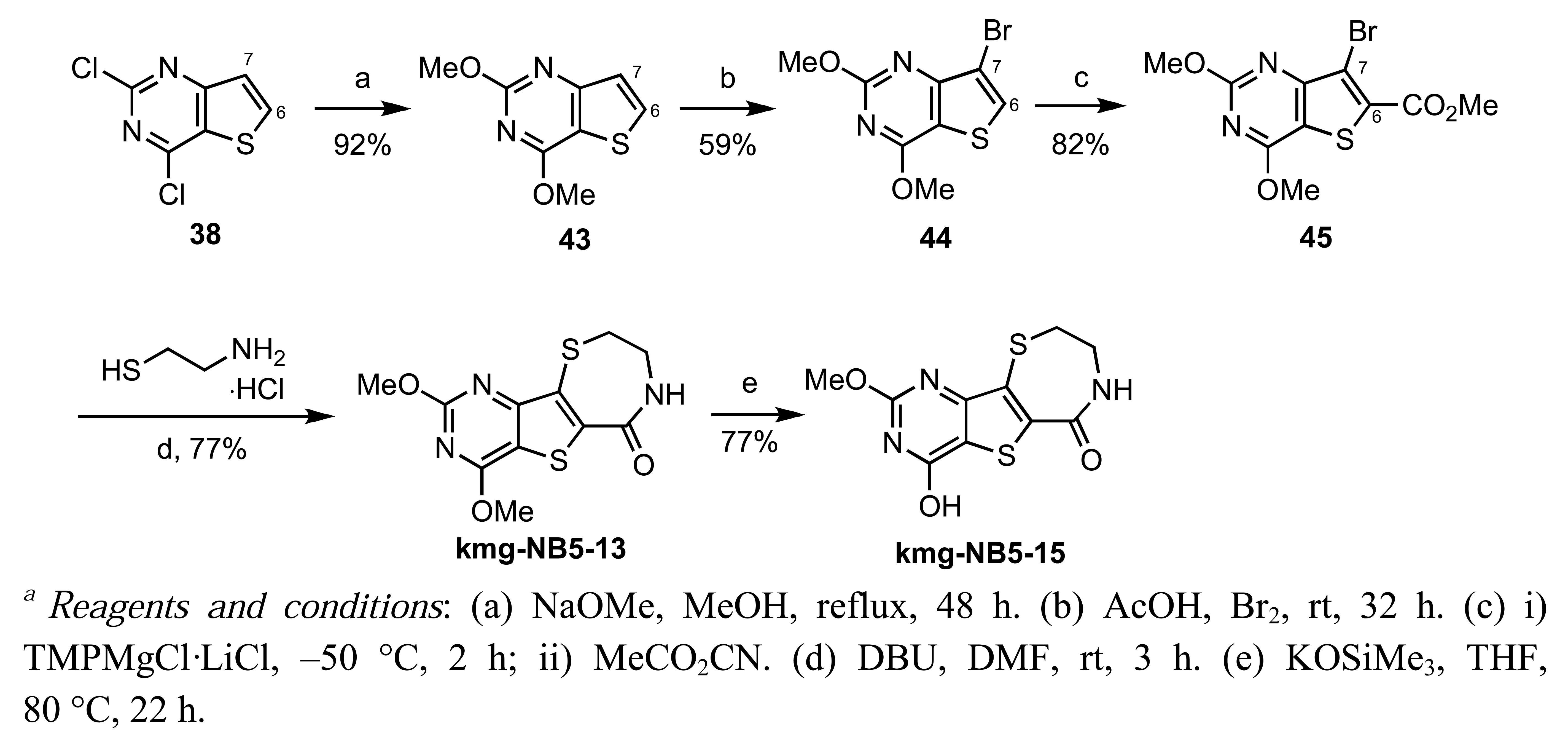

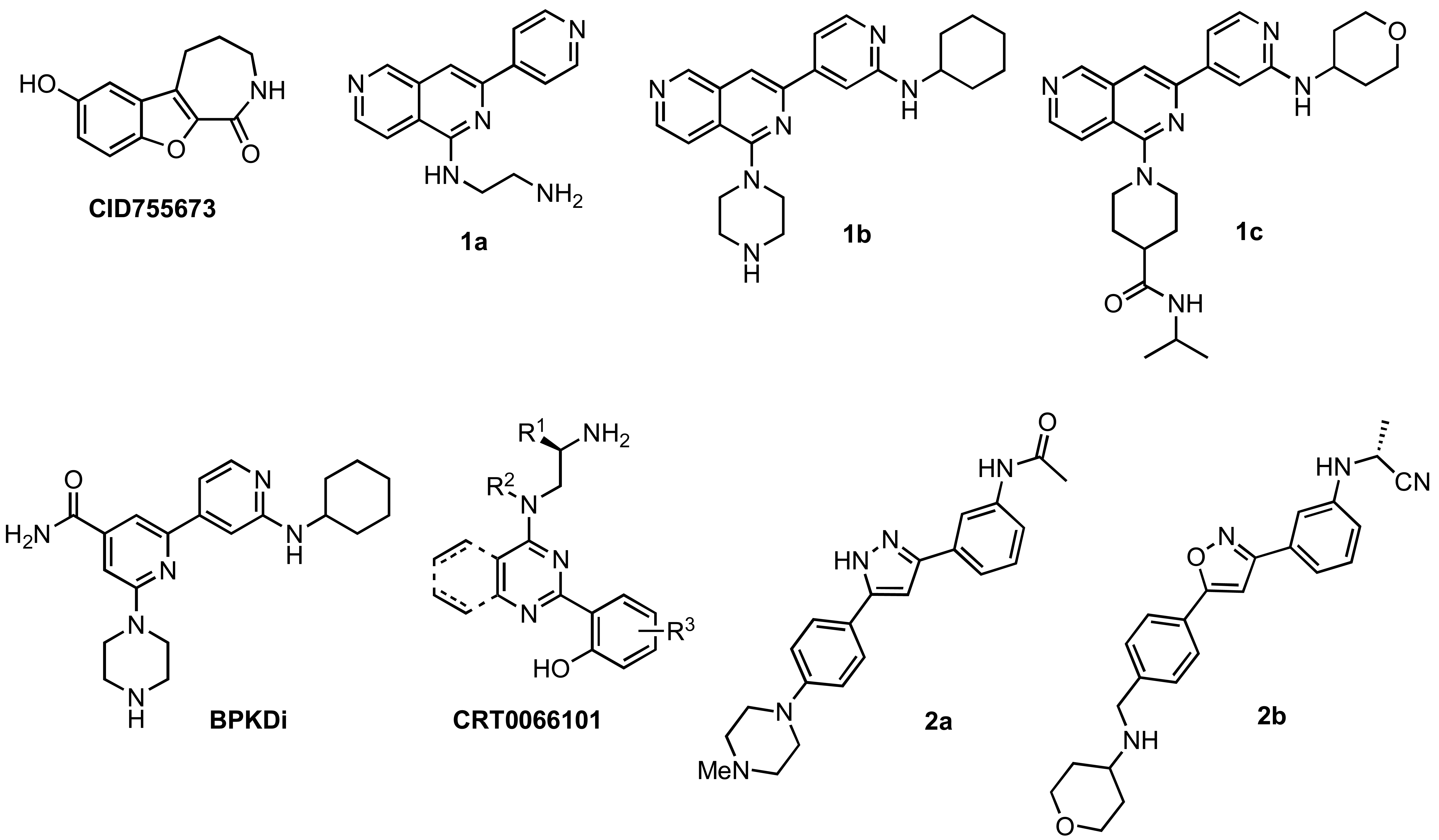{kind=link}
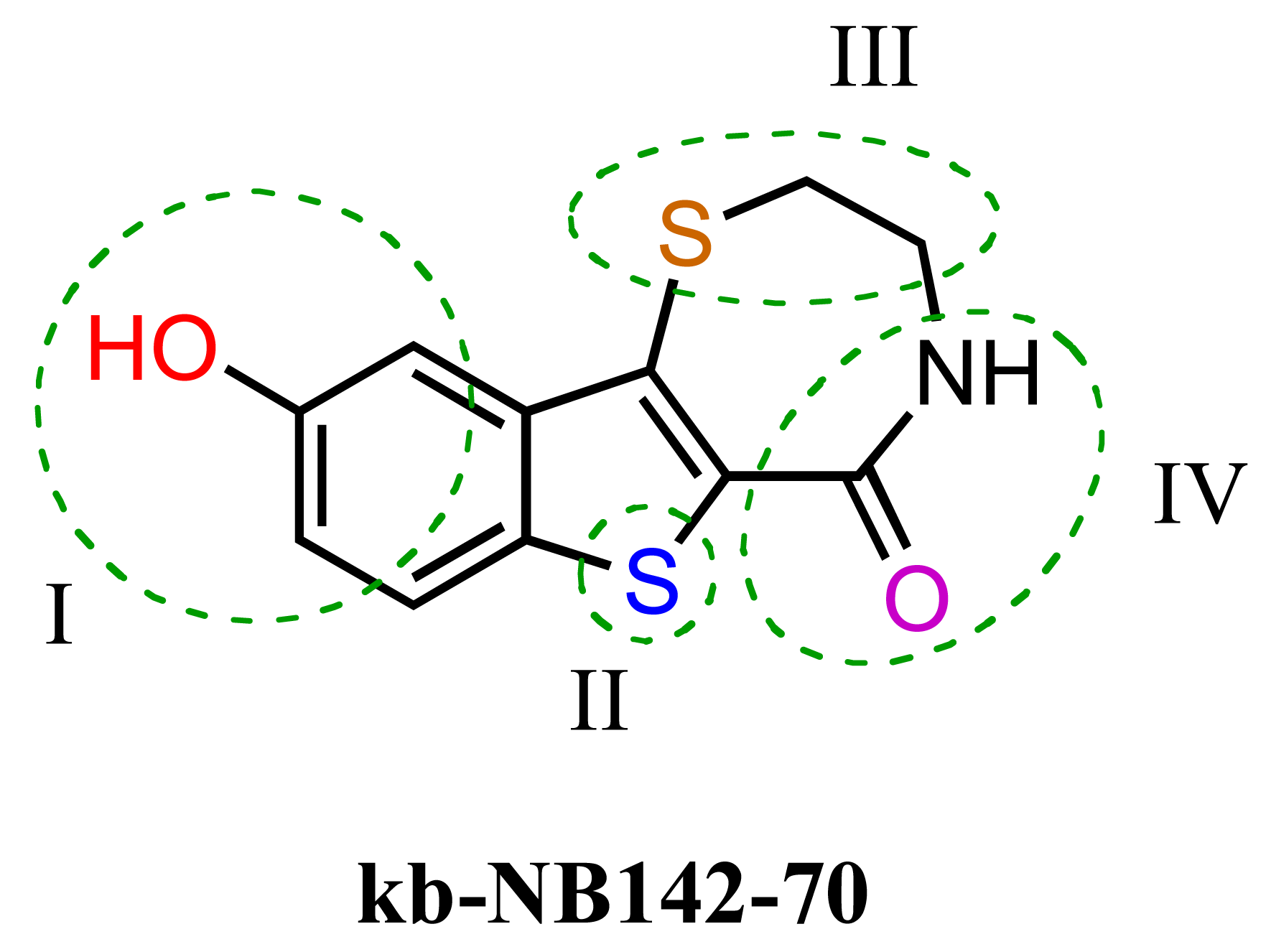{kind=link}
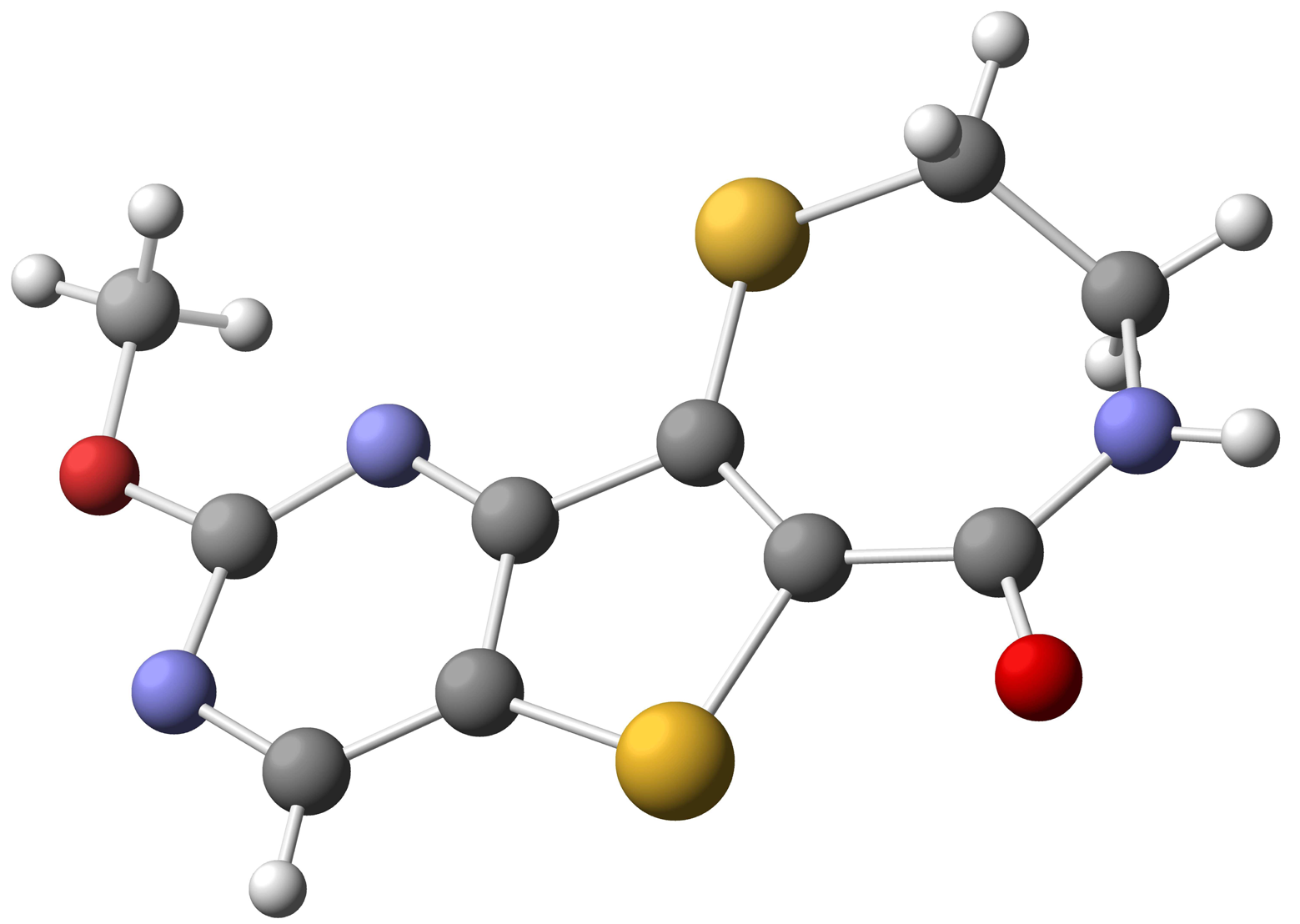{kind=link}
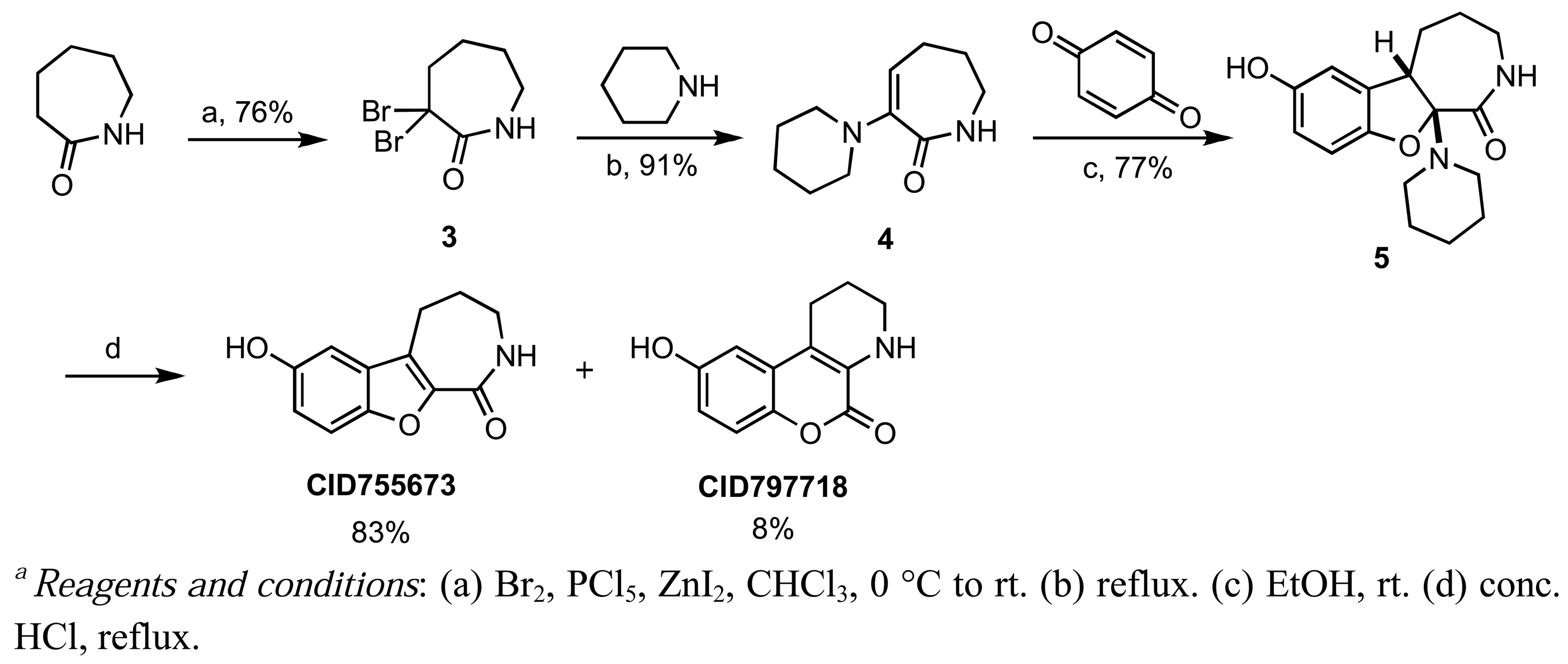{kind=link}
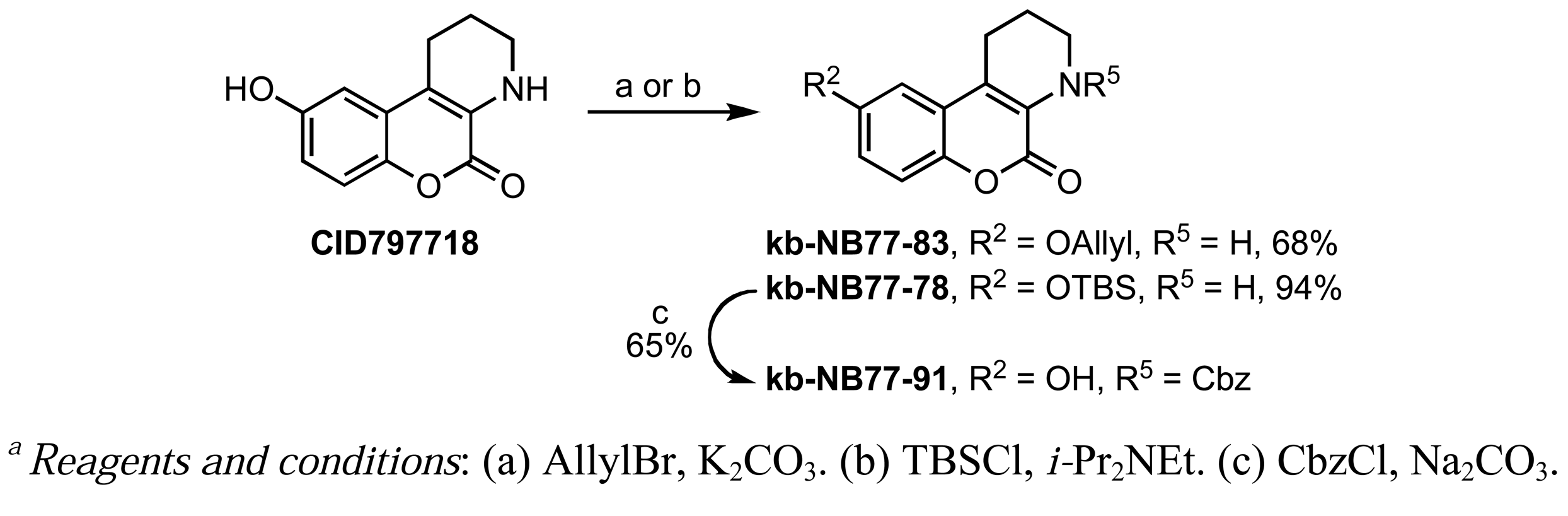{kind=link}
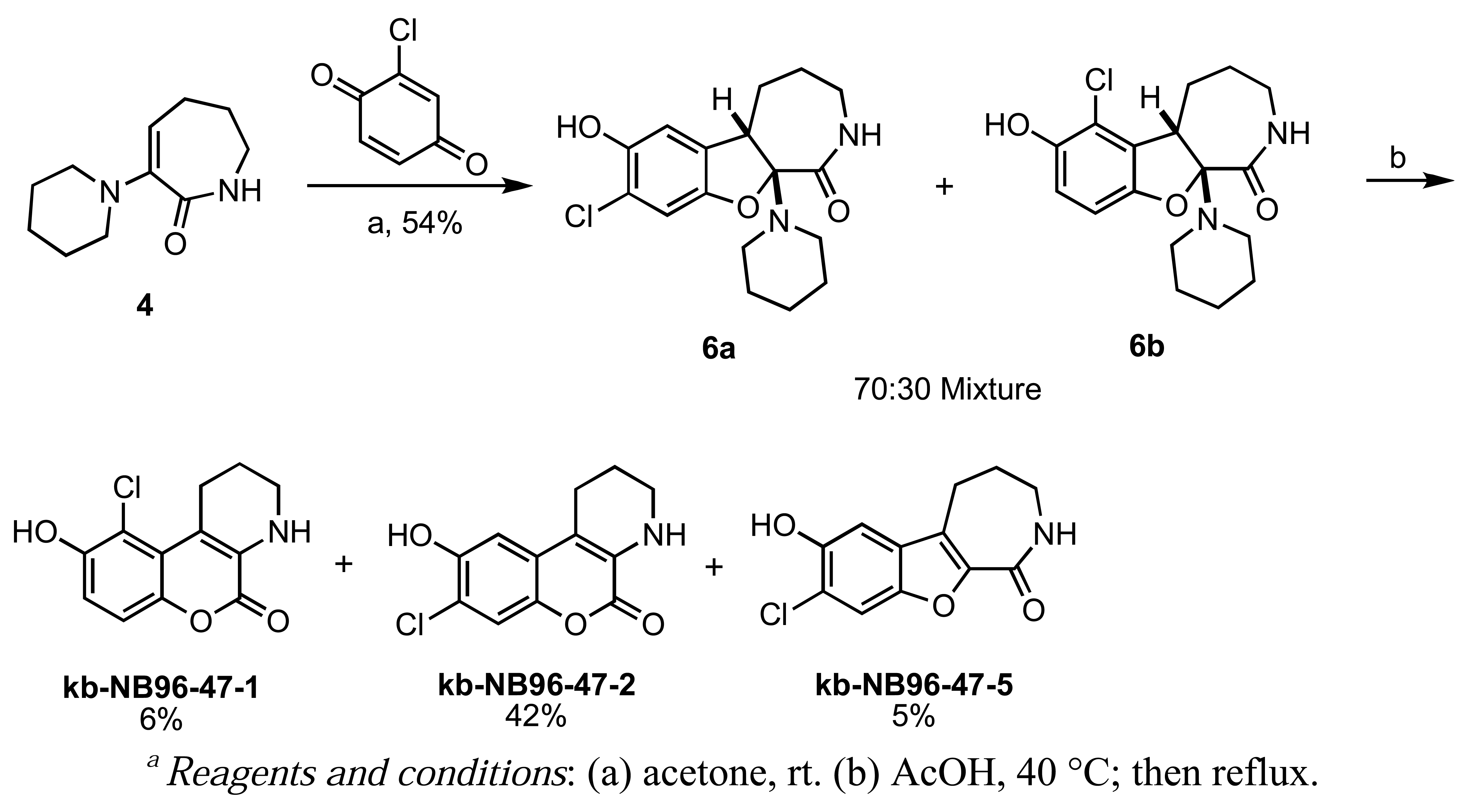{kind=link}
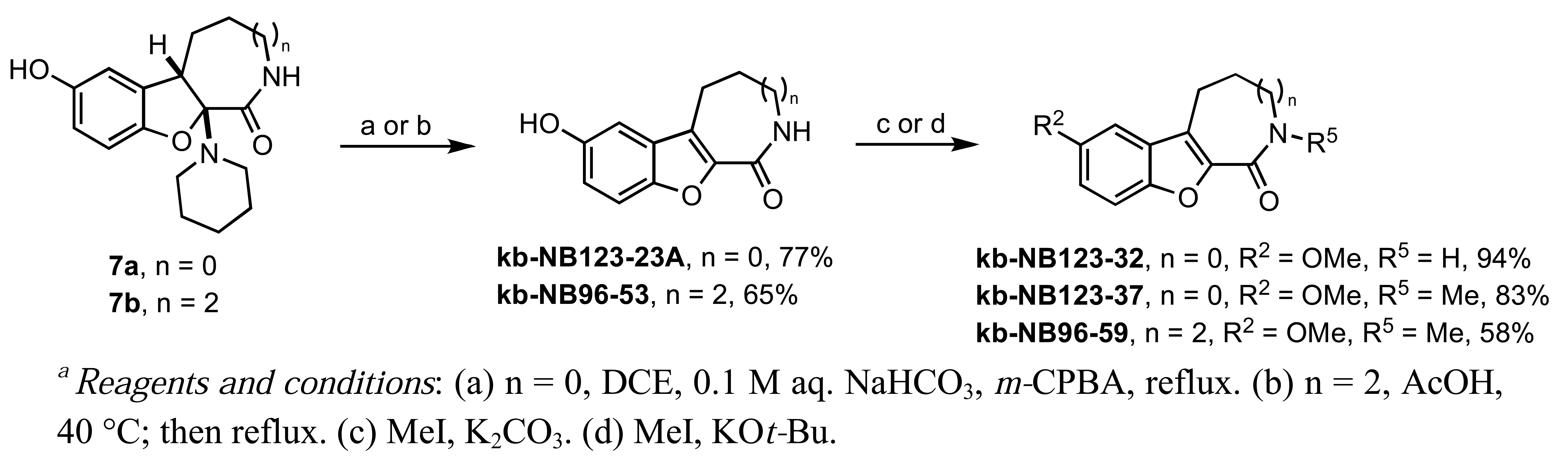{kind=link}
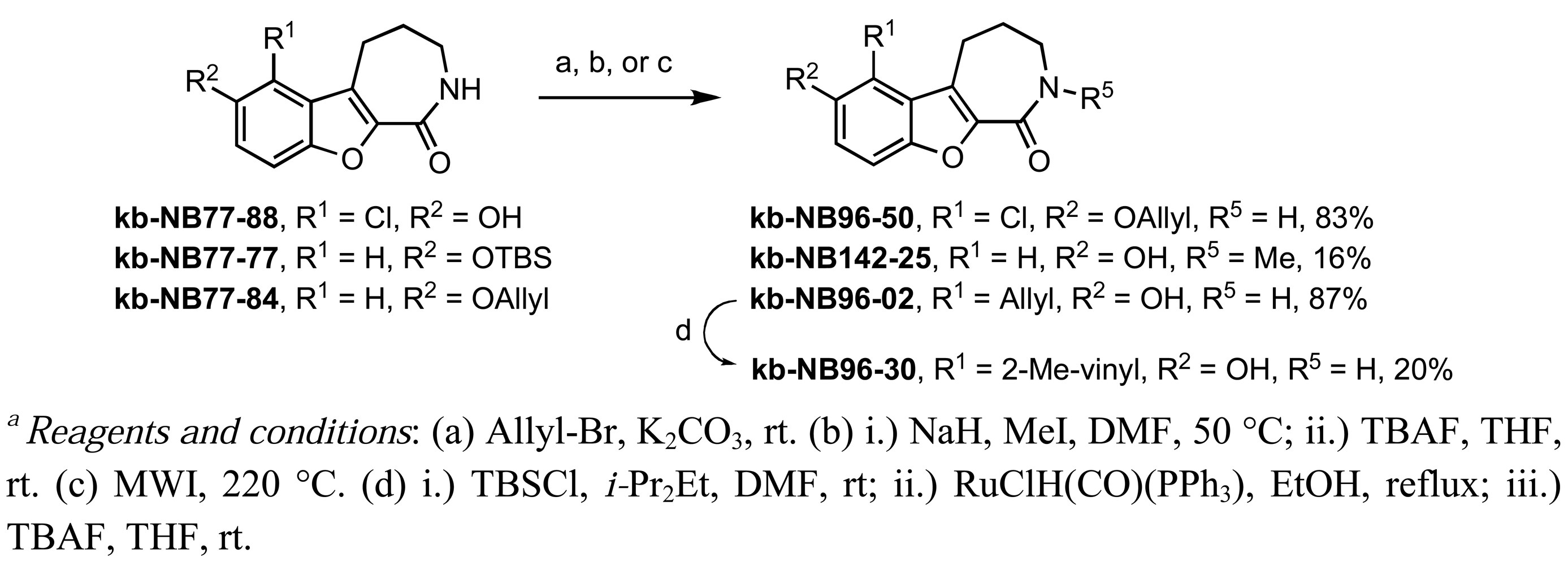{kind=link}
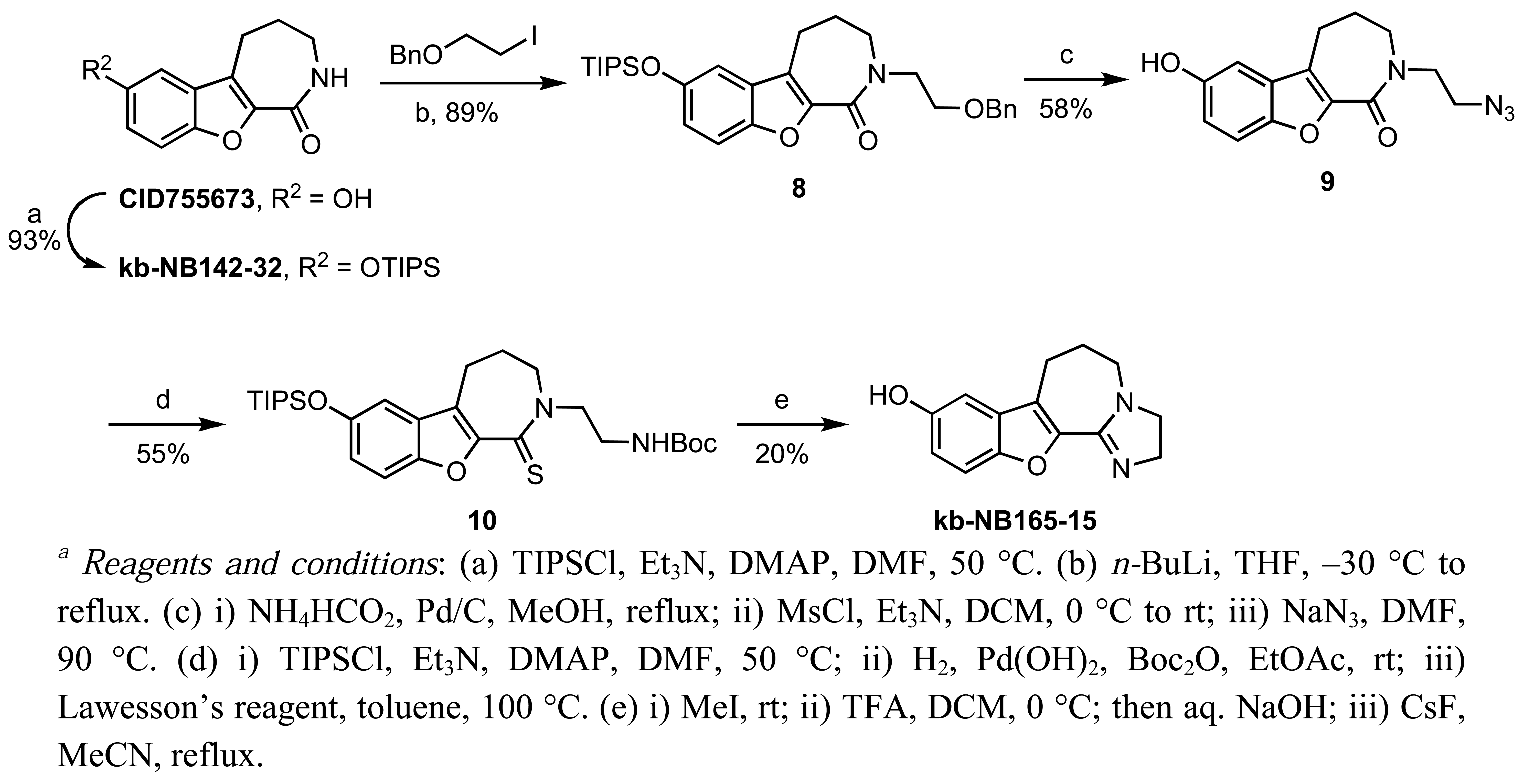{kind=link}
 | ||||||
|---|---|---|---|---|---|---|
| Entry | Compound | Structure | IC50 | |||
| R1 | R2 | R5 | IMAP-FP PKD1 (μM)a | Radiometric PKD1 (μM)b | ||
| 1 | CID797718 | H | OH | H | 13.7 ± 0.42 (n = 3) | 2.34 ± 0.16 (n = 3) |
| 2 | kb-NB77-83 | H | OAllyl | H | not inhibitory | n.d. |
| 3 | kb-NB77-78 | H | OTBS | H | not inhibitory | n.d. |
| 4 | kb-NB77-91 | H | OH | Cbz | not inhibitory | n.d. |
| 5 | kb-NB96-47-1 | Cl | OH | H | not inhibitory | n.d. |
aPKD1 IC50 was determined using an automated, HTS formatted IMAP-based PKD Fluorescence Polarization (FP) assay as previously described [52]. Each IC50 was calculated as the mean ± SEM of at least three independent experiments with triplicate determinations at each concentration in each experiment; n = number of independent experiments.bPKD1 IC50 was determined using a radiometric kinase activity assay as previously described [52]. Each IC50 was calculated as the mean ± SEM of at least three independent experiments with triplicate determinations at each concentration in each experiment; n = number of independent experiments.
 | |||||||
|---|---|---|---|---|---|---|---|
| Entry | Compound | Structure | IC50 | ||||
| R2 | R5 | n | IMAP-FP PKD1 (μM)a | Radiometric PKD1 (μM)b | Cellular PKD1 (μM)c | ||
| 1 | CID755673 | OH | H | 1 | 0.64 ± 0.03 (n = 3) | 0.18 ± 0.02 (n = 5) | 11.8 ± 4.0 (n = 3) |
| 2 | kb-NB123-23A | OH | H | 0 | 12.6 ± 1.3 (n = 3) | 1.41 (n = 1) | n.d. |
| 3 | kb-NB123-32 | OMe | H | 0 | >100 | n.d. | n.d. |
| 4 | kb-NB96-53 | OH | H | 2 | 8.3 ± 0.6 (n = 3) | 1.03 (n = 1) | n.d. |
| 5 | kb-NB96-59 | OMe | H | 2 | >100 | n.d. | n.d. |
| 6 | kb-NB77-56 | OMe | H | 1 | 2.40 ± 0.14 (n = 3) | 2.39 (n = 1) | n.d. |
| 7 | kb-NB77-84 | OAllyl | H | 1 | 2.6 ± 0.2 (n = 3) | 1.23 (n = 1) | n.d. |
| 8 | kb-NB123-36 | OAc | H | 1 | 84.89 ± 3.21 (n = 3) | n.d. | n.d. |
| 9 | kb-NB77-77 | OTBS | H | 1 | not inhibitory | n.d. | n.d. |
| 10 | kb-NB123-37 | OMe | Me | 0 | not inhibitory | n.d. | n.d. |
| 11 | kb-NB142-25 | OH | Me | 1 | n.d. | 4.0 ± 1.1 (n = 2) | n.d. |
| 12 | kb-NB96-04 | OMe | Me | 1 | >100 | n.d. | n.d. |
| 13 | kb-NB123-45-1 | OAc | Ac | 1 | not inhibitory | n.d. | n.d. |
| 14 | kb-NB165-15 | 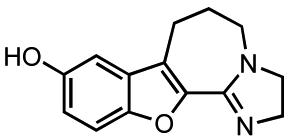 | n.d. | not inhibitory | n.d. | ||
aPKD1 IC50 was determined using an automated, HTS formatted IMAP-based PKD Fluorescence Polarization (FP) assay as previously described [52]. Each IC50 was calculated as the mean ± SEM of at least three independent experiments with triplicate determinations at each concentration in each experiment; n = number of independent experiments.bPKD1 IC50 was determined using a radiometric kinase activity assay as previously described [52]. Each IC50 was calculated as the mean ± SEM with triplicate determinations at each concentration in each experiment; n = number of independent experiments.cCellular IC50 was determined by densitometry analysis of Western blotting data for PKD1 autophosphorylation at S916 in LNCaP cells as previously described [53]. Each IC50 was calculated as the mean ± SEM of at least two independent experiments; n = number of independent experiments.
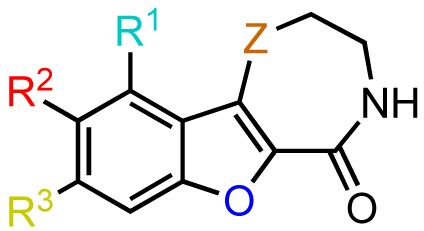 | |||||||
|---|---|---|---|---|---|---|---|
| Entry | Compound | Structure | IC50 | ||||
| Z | R1 | R2 | R3 | IMAP-FP PKD1 (μM)a | Radiometric PKD1 (μM)b | ||
| 1 | kb-NB77-88 | CH2 | Cl | OH | H | 1.4 ± 0.1 (n = 3) | 0.89 (n = 1) |
| 2 | kb-NB96-21 | CH2 | F | OH | H | 1.3 ± 0.05 (n = 3) | 0.24 (n = 1) |
| 3 | kb-NB96-50 | CH2 | Cl | OAllyl | H | not inhibitory | n.d. |
| 4 | kb-NB96-47-5 | CH2 | H | OH | Cl | >100 | n.d. |
| 5 | kb-NB96-43 | CH2 | Cl | OH | Cl | not inhibitory | n.d. |
| 6 | kb-NB96-02 | CH2 | Allyl | OH | H | 2.4 ± 0.3 (n = 3) | 1.58 (n = 1) |
| 7 | kb-NB96-30 | CH2 | Propenyl | OH | H | 1.0 ± 0.1 (n = 3) | 0.24 (n = 1) |
| 8 | kb-NB123-63 | C=O | H | OH | H | 14.9 ± 1.2 (n = 3) | 0.85 ± 0.11 (n = 2) |
| 9 | kb-NB123-89 | CHOH | H | OH | H | 24.09 ± 0.71 (n = 3) | 1.23 ± 0.21 (n = 2) |
| 10 | kb-NB142-05 | C=NNHPh | H | OH | H | 21.70 ± 0.52 (n = 3) | 1.13 (n = 1) |
| 11 | kb-NB142-11 | C=NNHTs | H | OH | H | 38.21 ± 1.17 (n = 3) | n.d. |
| 12 | kb-NB142-10 | C=NOBn | H | OH | H | not inhibitory | n.d. |
aPKD1 IC50 was determined using an automated, HTS formatted IMAP-based PKD Fluorescence Polarization (FP) assay as previously described [52]. Each IC50 was calculated as the mean ± SEM of at least three independent experiments with triplicate determinations at each concentration in each experiment; n = number of independent experiments.bPKD1 IC50 was determined using a radiometric kinase activity assay as previously described [52]. Each IC50 was calculated as the mean ± SEM with triplicate determinations at each concentration in each experiment; n = number of independent experiments.
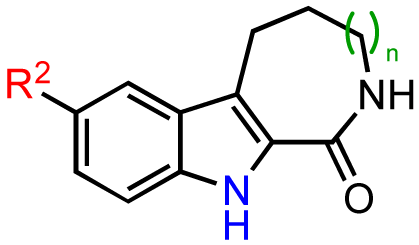 | ||||||
|---|---|---|---|---|---|---|
| Entry | Compound | Structure | IC50 | |||
| R2 | n | IMAP-FP PKD1 (μM)a | Radiometric PKD1 (μM)b | Cellular PKD1 (μM)c | ||
| 1 | kb-NB123-59 | OH | 0 | 19.4 ± 1.4 (n = 3) | 1.57 ± 0.20 (n = 2) | n.d. |
| 2 | kb-NB123-57 | OH | 1 | 2.14 ± 0.12 (n = 3) | 0.13 ± 0.01 (n = 3) | >50 (n = 3) |
| 3 | kb-NB123-52 | OBn | 0 | not inhibitory | n.d. | n.d. |
| 4 | kb-NB123-53 | OBn | 1 | not inhibitory | n.d. | n.d. |
| 5 | kb-NB142-08 | NH2 | 0 | 74.4 ± 2.2 (n = 3) | 15.74 ± 0.19 (n = 2) | n.d. |
| 6 | kb-NB142-01 | NH2 | 1 | 47.1 ± 2.5 (n = 3) | 9.68 ± 1.01 (n = 2) | n.d. |
| 7 | kb-NB123-93 | NHAc | 0 | not inhibitory | n.d. | n.d. |
| 8 | kb-NB123-94 | NHAc | 1 | not inhibitory | n.d. | n.d. |
aPKD1 IC50 was determined using an automated, HTS formatted IMAP-based PKD Fluorescence Polarization (FP) assay as previously described [52]. Each IC50 was calculated as the mean ± SEM of at least three independent experiments with triplicate determinations at each concentration in each experiment; n = number of independent experiments.bPKD1 IC50 was determined using a radiometric kinase activity assay as previously described [52]. Each IC50 was calculated as the mean ± SEM of at least two independent experiments with triplicate determinations at each concentration in each experiment; n = number of independent experiments.cCellular IC50 was determined by densitometry analysis of Western blotting data for PKD1 autophosphorylation at S916 in LNCaP cells as previously described [53]. Each IC50 was calculated as the mean ± SEM of at least three independent experiments; n = number of independent experiments.
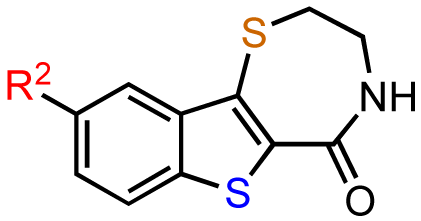 | |||||||
|---|---|---|---|---|---|---|---|
| Entry | Compound | Structure | IC50 | ||||
| R2 | IMAP-FP PKD1 (μM)a | Radiometric PKD1 (μM)b | cellular PKD1 (μM)c | ||||
| 1 | kb-NB123-66 | OBn | not inhibitory | n.d. | n.d. | ||
| 2 | kb-NB142-70 | OH | 0.71 ± 0.02 (n = 3) | 0.028 ± 0.002 (n = 3) | 2.22 ± 0.59 (n = 3) | ||
| 3 | kb-NB165-09 | OMe | n.d. | 0.08 ± 0.01 (n = 4) | 3.13 ± 0.54 (n = 3) | ||
aPKD1 IC50 was determined using an automated, HTS formatted IMAP-based PKD Fluorescence Polarization (FP) assay as previously described [52]. Each IC50 was calculated as the mean ± SEM of at least three independent experiments with triplicate determinations at each concentration in each experiment; n = number of independent experiments.bPKD1 IC50 was determined using a radiometric kinase activity assay as previously described [52]. Each IC50 was calculated as the mean ± SEM of at least three independent experiments with triplicate determinations at each concentration in each experiment; n = number of independent experiments.cCellular IC50 was determined by densitometry analysis of Western blotting data for PKD1 autophosphorylation at S916 in LNCaP cells as previously described [53]. Each IC50 was calculated as the mean ± SEM of at least three independent experiments; n = number of independent experiments.
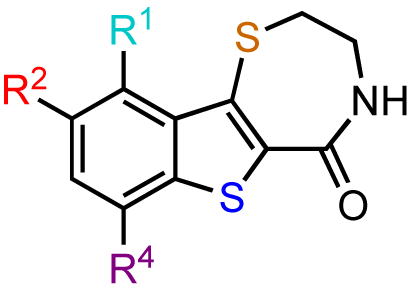 | |||||||
|---|---|---|---|---|---|---|---|
| Entry | Compound | Structure | IC50 | ||||
| R1 | R2 | R4 | % PKD1 activity at 1 μM | Radiometric PKD1 (μM)a | Cellular PKD1 (μM)b | ||
| 1 | mcf292-03 | H | NH | H | 74.4 ± 1.1 (n = 2) | 3.17 (n = 1) | n.d. |
| 2 | mcf292-08 | H | N3 | H | n.d. | 0.08 ± 0.01 (n = 5) | 2.17 ± 0.22 (n = 3) |
| 3 | mcf292-05 | H | N=C=S | H | n.d. | 2.77 (n = 1) | n.d. |
| 4 | mcf292-09 | H | NHCOCH2Cl | H | n.d. | 1.50 (n = 1) | n.d. |
| 5 | kb-NB165-31 | I | OH | H | 13.6 (n = 1) | 0.11 ± 0.02 (n = 3) | 8.6 ± 2.0 (n = 3) |
| 6 | kb-NB184-52 | Br | OH | H | 12.7 ± 0.2 (n = 2) | 0.048 (n = 1) | n.d. |
| 7 | kb-NB184-38 | H | H | OBn | 98.6 ± 4.1 (n = 2) | n.d. | n.d. |
| 8 | kb-NB184-40 | H | H | OH | 99 ± 11 (n = 2) | n.d. | n.d. |
| 9 | kb-NB184-44 | H | H | OMe | 77.5 ± 3.6 (n = 2) | n.d. | n.d. |
aPKD1 IC50 was determined using a radiometric kinase activity assay as previously described [52]. Each IC50 was calculated as the mean ± SEM with triplicate determinations at each concentration in each experiment; n = number of independent experiments.bCellular IC50 was determined by densitometry analysis of Western blotting data for PKD1 autophosphorylation at S916 in LNCaP cells as previously described [53]. Each IC50 was calculated as the mean ± SEM of at least three independent experiments; n = number of independent experiments.
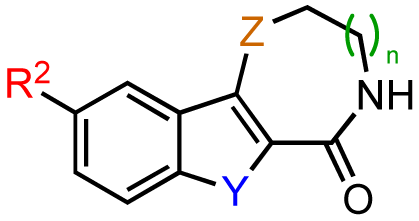 | ||||||||
|---|---|---|---|---|---|---|---|---|
| Entry | Compound | Structure | IC50 | |||||
| Y | Z | R2 | n | % PKD1 activity at 1 μM | Radiometric PKD1 (μM)a | Cellular PKD1 (μM)b | ||
| 1 | kb-NB184-22 | S=O | S | OH | 1 | 66.5 ± 6.1 (n = 2) | n.d. | n.d. |
| 2 | kb-NB184-25 | S=O | S | OMe | 1 | 50.4 ± 2.3 (n = 2) | 1.08 (n = 1) | n.d. |
| 3 | kb-NB184-45 | S | S=O | OMe | 1 | 97 ± 16 (n = 2) | n.d. | n.d. |
| 4 | kb-NB165-89 | S | S | OBn | 2 | 84.3 (n = 1) | 21.7 (n = 1) | n.d. |
| 5 | kb-NB165-92 | S | S | OH | 2 | 16.7 (n = 1) | 0.11 ± 0.01 (n = 3) | 2.56 ± 0.66 (n = 2) |
| 6 | kb-NB184-02 | S | S | OMe | 2 | 29.5 (n = 1) | 0.19 ± 0.03 (n = 3) | 18.6 ± 2.0 (n = 3) |
| 7 | kb-NB184-36 | S | O | OBn | 2 | 83.3 ± 3.8 (n = 2) | n.d. | n.d. |
| 8 | kb-NB184-57 | S | O | OMe | 2 | 62.0 ± 3.5 (n = 2) | n.d. | n.d. |
| 9 | kb-NB184-80 |  | 91.3 ± 1.5 (n = 2) | not inhibitory | n.d. | |||
aPKD1 IC50 was determined using a radiometric kinase activity assay as previously described [52]. Each IC50 was calculated as the mean ± SEM with triplicate determinations at each concentration in each experiment; n = number of independent experiments.bCellular IC50 was determined by densitometry analysis of Western blotting data for PKD1 autophosphorylation at S916 in LNCaP cells as previously described [53]. Each IC50 was calculated as the mean ± SEM of at least two independent experiments; n = number of independent experiments.
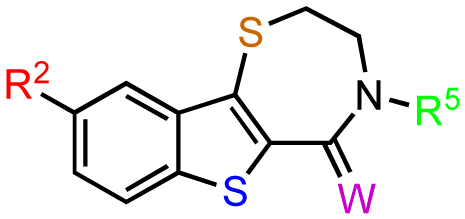 | |||||||
|---|---|---|---|---|---|---|---|
| Entry | Compound | Structure | IC50 | ||||
| W | R2 | R5 | % PKD1 activity at 1 μM | Radiometric PKD1 (μ M)a | |||
| 1 | kb-NB165-16 | O | OMe | Me | 4.57 ± 0.78 (n = 2) | ||
| 2 | kb-NB165-17 | O | OH | Me | 0.45 ± 0.05 (n = 2) | ||
| 3 | kb-NB165-75 | O | OH | (CH2)2NH2 | 55.6 (n = 1) | 0.757 (n = 1) | |
| 4 | kb-NB165-81 | -- | OBn | H | 78.3 (n = 1) | 39.6 (n = 1) | |
| 5 | kb-NB165-83 | -- | OH | H | 92.4 (n = 1) | 16.4 (n = 1) | |
aPKD1 IC50 was determined using a radiometric kinase activity assay as previously described [52]. Each IC50 was calculated as the mean ± SEM with triplicate determinations at each concentration in each experiment; n = number of independent experiments.
 | |||||
|---|---|---|---|---|---|
| Entry | Compound | Structure | IC50 | ||
| R2 | R4 | Radiometric PKD1 (μM)a | Cellular PKD1 (μ M)b | ||
| 1 | kmg-NB4-23 | OMe | H | 0.12 ± 0.03 (n = 4) | 6.8 ± 1.3 (n = 3) |
| 2 | kmg-NB4-69A | OH | H | 25.3 (n = 1) | n.d. |
| 3 | kmg-NB5-13C | OMe | OMe | >30.0 (n = 2) | n.d. |
| 4 | kmg-NB5-15A | OMe | OH | >30.0 (n = 2) | n.d. |
aPKD1 IC50 was determined using a radiometric kinase activity assay as previously described [52]. Each IC50 was calculated as the mean ± SEM with triplicate determinations at each concentration in each experiment; n = number of independent experiments.bCellular IC50 was determined by densitometry analysis of Western blotting data for PKD1 autophosphorylation at S916 in LNCaP cells as previously described [53]. Each IC50 was calculated as the mean ± SEM of at least three independent experiments; n = number of independent experiments.
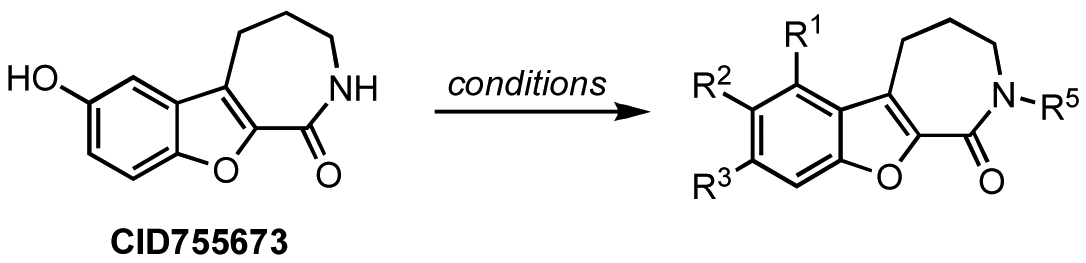 | |||||||
|---|---|---|---|---|---|---|---|
| Entry | Conditions | Structure | Yield, % | Product | |||
| R1 | R2 | R3 | R5 | ||||
| 1 | MeI, K2CO3 | H | OMe | H | H | 74 | kb-NB77-56 |
| 2 | AllylBr, K2CO3 | H | OAllyl | H | H | 69 | kb-NB77-84 |
| 3 | TBSCl, i-Pr2NEt | H | OTBS | H | H | 91 | kb-NB77-77 |
| 4 | AcCl (2 equiv), DMAP | H | OAc | H | H | 91 | kb-NB123-36 |
| 5 | MeI, KOt-Bu | H | OMe | H | Me | 34 | kb-NB96-04 |
| 6 | AcCl (3 equiv), DMAP | H | OAc | H | Ac | 33 | kb-NB123-45-1 |
| 7 | N-Chlorosuccinimide (1.05 equiv) | Cl | OH | H | H | 86 | kb-NB77-88 |
| 8 | Selectflour® | F | OH | H | H | 29 | kb-NB96-21 |
| 9 | N-Chlorosuccinimide (2 equiv) | Cl | OH | Cl | H | 73 | kb-NB96-43 |
 | ||||
|---|---|---|---|---|
| Entry | Condition | Structure | Yield, % (from 21) | Product |
| R2 | ||||
| 1 | tBuONO, TMSN3, MeCN, rt | N3 | 38% | mcf292-08 |
| 2 | CSCl2, NaHCO3, CHCl3/H2O, rt | NCS | 41% | mcf292-05 |
| 3 | ClCH2COCl, 2,6-lutidine, DCM, rt | NHCOCH2Cl | 46% | mcf292-09 |
Acknowledgements
This study was supported in part by the National Institutes of Health (Grants R03MH082038-01, R01CA129127, R01CA142580), the NIH Roadmap Program (Grants U54MH074411, GM067082), the Fiske Drug Discovery Fund, and the Elsa U. Pardee Foundation. The authors thank Steven Geib (University of Pittsburgh) for the x-ray structure determination of kmg-NB4-23, Donna Huryn (University of Pittsburgh) for advice on the design of analogs, Ms. Rachel Byerly (University of Pittsburgh) for technical assistance, and Julie Eiseman (University of Pittsburgh Hillman Cancer Center) for her suggestions on metabolism and in vivo studies.
References and Notes
- Johannes, F.J.; Prestle, J.; Eis, S.; Oberhagemann, P.; Pfizenmaier, K. PKCμ is a novel, atypical member of the protein kinase C family. J. Biol. Chem. 1994, 269, 6140–6148. [Google Scholar]
- Valverde, A.M.; Sinnett-Smith, J.; Van Lint, J.; Rozengurt, E. Molecular cloning and characterization of protein kinase D: A target for diacylglycerol and phorbol esters with a distinctive catalytic domain. Proc. Natl. Acad. Sci. USA 1994, 91, 8572–8576. [Google Scholar]
- Sturany, S.; Van Lint, J.; Muller, F.; Wilda, M.; Hameister, H.; Hocker, M.; Brey, A.; Gern, U.; Vandenheede, J.; Gress, T.; Adler, G.; Seufferlein, T. Molecular cloning and characterization of the human protein kinase D2. A novel member of the protein kinase D family of serine threonine kinases. J. Biol. Chem. 2001, 276, 3310–3318. [Google Scholar]
- Hayashi, A.; Seki, N.; Hattori, A.; Kozuma, S.; Saito, T. PKCν, a new member of the protein kinase C family, composes a fourth subfamily with PKCμ. Biochim. Biophys. Acta 1999, 1450, 99–106. [Google Scholar]
- Waldron, R.T.; Iglesias, T.; Rozengurt, E. Phosphorylation-dependent protein kinase D activation. Electrophoresis 1999, 20, 382–390. [Google Scholar]
- Zugaza, J.L.; Sinnett-Smith, J.; Van Lint, J.; Rozengurt, E. Protein kinase D (PKD) activation in intact cells through a protein kinase C-dependent signal transduction pathway. EMBO J. 1996, 15, 6220–6230. [Google Scholar]
- Waldron, R.T.; Rozengurt, E. Protein kinase C phosphorylates protein kinase D activation loop Ser744 and Ser748 and releases autoinhibition by the pleckstrin homology domain. J. Biol. Chem. 2003, 278, 154–163. [Google Scholar]
- Jacamo, R.; Sinnett-Smith, J.; Rey, O.; Waldron, R.T.; Rozengurt, E. Sequential protein kinase C (PKC)-dependent and PKC-independent protein kinase D catalytic activation via Gq-coupled receptors: Differential regulation of activation loop Ser(744) and Ser(748) phosphorylation. J. Biol. Chem. 2008, 283, 12877–12887. [Google Scholar]
- Matthews, S.A.; Rozengurt, E.; Cantrell, D. Characterization of serine 916 as an in vivo autophosphorylation site for protein kinase D/Protein kinase Cμ. J. Biol. Chem. 1999, 274, 26543–26549. [Google Scholar]
- Rozengurt, E.; Rey, O.; Waldron, R.T. Protein kinase D signaling. J. Biol. Chem. 2005, 280, 13205–13208. [Google Scholar]
- Sinnett-Smith, J.; Jacamo, R.; Kui, R.; Wang, Y.M.; Young, S.H.; Rey, O.; Waldron, R.T.; Rozengurt, E. Protein kinase D mediates mitogenic signaling by Gq-coupled receptors through protein kinase C-independent regulation of activation loop Ser744 Ser748 phosphorylation. J. Biol. Chem. 2009, 284, 13434–13445. [Google Scholar]
- Wang, Q.J. PKD at the crossroads of DAG and PKC signaling. Trends Pharmacol. Sci 2006, 27, 317–323. [Google Scholar]
- Storz, P.; Doppler, H.; Johannes, F.J.; Toker, A. Tyrosine phosphorylation of protein kinase D in the pleckstrin homology domain leads to activation. J. Biol. Chem. 2003, 278, 17969–17976. [Google Scholar]
- Storz, P.; Doppler, H.; Toker, A. Protein kinase D mediates mitochondrion-to-nucleus signaling and detoxification from mitochondrial reactive oxygen species. Mol. Cell. Biol. 2005, 25, 8520–8530. [Google Scholar]
- Waldron, R.T.; Rozengurt, E. Oxidative stress induces protein kinase D activation in intact cells. Involvement of Src and dependence on protein kinase C. J. Biol. Chem. 2000, 275, 17114–17121. [Google Scholar]
- Ha, C.H.; Jhun, B.S.; Kao, H.Y.; Jin, Z.G. VEGF stimulates HDAC7 phosphorylation and cytoplasmic accumulation modulating matrix metalloproteinase expression and angiogenesis. Arterioscler. Thromb. Vasc. Biol. 2008, 28, 1782–1788. [Google Scholar]
- Ha, C.H.; Wang, W.; Jhun, B.S.; Wong, C.; Hausser, A.; Pfizenmaier, K.; McKinsey, T.A.; Olson, E.N.; Jin, Z.G. Protein kinase D-dependent phosphorylation and nuclear export of histone deacetylase 5 mediates vascular endothelial growth factor-induced gene expression and angiogenesis. J. Biol. Chem. 2008, 283, 14590–14599. [Google Scholar]
- Wang, S.; Li, X.; Parra, M.; Verdin, E.; Bassel-Duby, R.; Olson, E.N. Control of endothelial cell proliferation and migration by VEGF signaling to histone deacetylase 7. Proc. Natl. Acad. Sci. USA 2008, 105, 7738–7743. [Google Scholar]
- Bankaitis, V.A. Cell biology. Slick recruitment to the Golgi. Science 2002, 295, 290–291. [Google Scholar]
- Cuenda, A.; Nebreda, A.R. p38δ and PKD1: Kinase switches for insulin secretion. Cell 2009, 136, 209–210. [Google Scholar]
- Ghanekar, Y.; Lowe, M. Protein kinase D: Activation for Golgi carrier formation. Trends Cell Biol. 2005, 15, 511–514. [Google Scholar]
- Prigozhina, N.L.; Waterman-Storer, CM. Protein kinase D-mediated anterograde membrane trafficking is required for fibroblast motility. Curr. Biol. 2004, 14, 88–98. [Google Scholar]
- Woods, A.J.; White, D.P.; Caswell, P.T.; Norman, J.C. PKD1/PKCμ promotes ανβ3integrin recycling and delivery to nascent focal adhesions. EMBO J. 2004, 23, 2531–2543. [Google Scholar]
- Matthews, S.A.; Liu, P.; Spitaler, M.; Olson, E.N.; McKinsey, T.A.; Cantrell, D.A.; Scharenberg, A.M. Essential role for protein kinase D family kinases in the regulation of class II histone deacetylases in B lymphocytes. Mol. Cell. Biol. 2006, 26, 1569–1577. [Google Scholar]
- Marklund, U.; Lightfoot, K.; Cantrell, D. Intracellular location and cell context-dependent function of protein kinase D. Immunity 2003, 19, 491–501. [Google Scholar]
- Medeiros, R.B.; Dickey, D.M.; Chung, H.; Quale, A.C.; Nagarajan, L.R.; Billadeau, D.D.; Shimizu, Y. Protein kinase D1 and the β1 integrin cytoplasmic domain control β1 integrin function via regulation of Rap1 activation. Immunity 2005, 23, 213–226. [Google Scholar]
- Jaggi, M.; Rao, P.S.; Smith, D.J.; Hemstreet, G.P.; Balaji, K.C. Protein kinase Cμ is down-regulated in androgen-independent prostate cancer. Biochem. Biophys. Res. Commun. 2003, 307, 254–260. [Google Scholar]
- Chen, J.; Deng, F.; Singh, S.V.; Wang, Q.J. Protein kinase D3 (PKD3) contributes to prostate cancer cell growth and survival through a PKCε/PKD3 pathway downstream of Akt and ERK 1/2. Cancer Res. 2008, 68, 3844–3853. [Google Scholar]
- Eiseler, T.; Doppler, H.; Yan, I.K.; Goodison, S.; Storz, P. Protein kinase D1 regulates matrix metalloproteinase expression and inhibits breast cancer cell invasion. Breast Cancer Res. 2009, 11, R13. [Google Scholar]
- Kim, M.; Jang, H.R.; Kim, J.H.; Noh, S.M.; Song, K.S.; Cho, J.S.; Jeong, H.Y.; Norman, J.C.; Caswell, P.T.; Kang, G.H.; Kim, S.Y.; Yoo, H.S.; Kim, Y.S. Epigenetic inactivation of protein kinase D1 in gastric cancer and its role in gastric cancer cell migration and invasion. Carcinogenesis 2008, 29, 629–637. [Google Scholar]
- Fielitz, J.; Kim, M.S.; Shelton, J.M.; Qi, X.; Hill, J.A.; Richardson, J.A.; Bassel-Duby, R.; Olson, E.N. Requirement of protein kinase D1 for pathological cardiac remodeling. Proc. Natl. Acad. Sci. USA 2008, 105, 3059–3063. [Google Scholar]
- Monovich, L.; Vega, R.B.; Meredith, E.; Miranda, K.; Rao, C.; Capparelli, M.; Lemon, D.D.; Phan, D.; Koch, K.A.; Chapo, J.A.; Hood, D.B.; McKinsey, T.A. A novel kinase inhibitor establishes a predominant role for protein kinase D as a cardiac class IIa histone deacetylase kinase. FEBS Lett. 2010, 584, 631–637. [Google Scholar]
- Altschmied, J.; Haendeler, J. A new kid on the block: PKD1: A promising target for antiangiogenic therapy? Arterioscler. Thromb. Vasc. Biol. 2008, 28, 1689–1690. [Google Scholar]
- Ha, C.H.; Jin, Z.G. Protein kinase D1, a new molecular player in VEGF signaling and angiogenesis. Mol. Cells 2009, 28, 1–5. [Google Scholar]
- Jaggi, M.; Rao, P.S.; Smith, D.J.; Wheelock, M.J.; Johnson, K.R.; Hemstreet, G.P.; Balaji, K.C. E-cadherin phosphorylation by protein kinase D1/protein kinase Cμ is associated with altered cellular aggregation and motility in prostate cancer. Cancer Res. 2005, 65, 483–492. [Google Scholar]
- Sinnett-Smith, J.; Zhukova, E.; Hsieh, N.; Jiang, X.; Rozengurt, E. Protein kinase D potentiates DNA synthesis induced by Gq-coupled receptors by increasing the duration of ERK signaling in swiss 3T3 cells. J. Biol. Chem. 2004, 279, 16883–16893. [Google Scholar]
- Wong, C.; Jin, Z.G. Protein kinase C-dependent protein kinase D activation modulates ERK signal pathway and endothelial cell proliferation by vascular endothelial growth factor. J. Biol. Chem. 2005, 280, 33262–33269. [Google Scholar]
- LaValle, C.R.; George, K.M.; Sharlow, E.R.; Lazo, J.S.; Wipf, P.; Wang, Q.J. Protein kinase D as a potential new target for cancer therapy. Biochim. Biophys. Acta (BBA) Rev. Cancer 2010, 1806, 183–192. [Google Scholar]
- Johannes, F.-J.; Prestle, J.; Dieterich, S.; Oberhagemann, P.; Link, G.; Pfizenmaier, K. Characterization of activators and inhibitors of protein kinase Cμ. Eur. J. Biochem. 1995, 227, 303–307. [Google Scholar]
- Martiny-Baron, G.; Kazanietz, M.G.; Mischak, H.; Blumberg, P.M.; Kochs, G.; Hug, H.; Marme, D.; Schachtele, C. Selective inhibition of protein kinase C isozymes by the indolocarbazole Go 6976. J. Biol. Chem. 1993, 268, 9194–9197. [Google Scholar]
- Gschwendt, M.; Dieterich, S.; Rennecke, J.; Kittstein, W.; Mueller, H.J.; Johannes, F.J. Inhibition of protein kinase Cμ by various inhibitors. Differentiation from protein kinase C isoenzymes. FEBS Lett. 1996, 392, 77–80. [Google Scholar]
- Haworth, R.S.; Avkiran, M. Inhibition of protein kinase D by resveratrol. Biochem. Pharmacol. 2001, 62, 1647–1651. [Google Scholar]
- Aggarwal, B.B.; Bhardwaj, A.; Aggarwal, R.S.; Seeram, N.P.; Shishodia, S.; Takada, Y. Role of resveratrol in prevention and therapy of cancer: Preclinical and clinical studies. Anticancer Res. 2004, 24, 2783–2840. [Google Scholar]
- Stewart, J.R.; Christman, K.L.; O′Brian, C.A. Effects of resveratrol on the autophosphorylation of phorbol ester-responsive protein kinases: Inhibition of protein kinase D but not protein kinase C isozyme autophosphorylation. Biochem. Pharmacol. 2000, 60, 1355–1359. [Google Scholar]
- Raynham, T.M.; Hammonds, T.R.; Charles, M.D.; Pave, G.A.; Foxton, C.H.; Blackaby, W.P.; Stevens, A.P.; Ekwuru, C.T. Pyridine benzamides and pyrazine benzamides used as PKD inhibitors. WO 2008/074997 A1 2008. [Google Scholar]
- Raynham, T.M.; Hammonds, T.R.; Gillatt, J.H.; Charles, M.D.; Pave, G.A.; Foxton, C.H.; Carr, J.L.; Mistry, N.S. Amino-ethyl-amino-aryl (AEAA) compounds and their use. WO 2007/125331 A2 2007. [Google Scholar]
- Singh, R.; Li, H.; Zhao, H.; Payan, D.G.; Kolluri, R.; Tso, K.; Ramphal, J.; Gu, S. Cyclic amine substituted pyrimidinediamines as PKC inhibitors. WO 2009/012421 A1 2009. [Google Scholar]
- Harikumar, K.B.; Kunnumakkara, A.B.; Ochi, N.; Tong, Z.; Deorukhkar, A.; Sung, B.; Kelland, L.; Jamieson, S.; Sutherland, R.; Raynham, T.; Charles, M.; Bagherzadeh, A.; Foxton, C.; Boakes, A.; Farooq, M.; Maru, D.; Diagaradjane, P.; Matsuo, Y.; Sinnett-Smith, J.; Gelovani, J.; Krishnan, S.; Aggarwal, B.B.; Rozengurt, E.; Ireson, C.R.; Guha, S. A novel small-molecule inhibitor of protein kinase D blocks pancreatic cancer growth in vitro and in vivo. Mol. Cancer Ther. 2010, 9, 1136–1146. [Google Scholar]
- Gamber, G.G.; Meredith, E.; Zhu, Q.; Yan, W.; Rao, C.; Capparelli, M.; Burgis, R.; Enyedy, I.; Zhang, J.-H.; Soldermann, N.; Beattie, K.; Rozhitskaya, O.; Koch, K.A.; Pagratis, N.; Hosagrahara, V.; Vega, R.B.; McKinsey, T.A.; Monovich, L. 3,5-Diarylazoles as novel and selective inhibitors of protein kinase D. Bioorg. Med. Chem. Lett. 2011, 21, 1447–1451. [Google Scholar]
- Meredith, E.L.; Ardayfio, O.; Beattie, K.; Dobler, M.R.; Enyedy, I.; Gaul, C.; Hosagrahara, V.; Jewell, C.; Koch, K.; Lee, W.; Lehmann, H.; McKinsey, T.A.; Miranda, K.; Pagratis, N.; Pancost, M.; Patnaik, A.; Phan, D.; Plato, C.; Qian, M.; Rajaraman, V.; Rao, C.; Rozhitskaya, O.; Ruppen, T.; Shi, J.; Siska, S.J.; Springer, C.; van Eis, M.; Vega, R.B.; von Matt, A.; Yang, L.; Yoon, T.; Zhang, J.-H.; Zhu, N.; Monovich, L.G. Identification of orally available naphthyridine protein kinase D inhibitors. J. Med. Chem. 2010, 53, 5400–5421. [Google Scholar]
- Meredith, E.L.; Beattie, K.; Burgis, R.; Capparelli, M.; Chapo, J.; DiPietro, L.; Gamber, G.; Enyedy, I.; Hood, D.B.; Hosagrahara, V.; Jewell, C.; Koch, K.A.; Lee, W.; Lemon, D.D.; McKinsey, T.A.; Miranda, K.; Pagratis, N.; Phan, D.; Plato, C.; Rao, C.; Rozhitskaya, O.; Soldermann, N.; Springer, C.; van Eis, M.; Vega, R.B.; Yan, W.; Zhu, Q.; Monovich, L.G. Identification of potent and selective amidobipyridyl inhibitors of protein kinase D. J. Med. Chem. 2010, 53, 5422–5438. [Google Scholar]
- Sharlow, E.R.; Giridhar, K.V.; LaValle, C.R.; Chen, J.; Leimgruber, S.; Barrett, R.; Bravo-Altamirano, K.; Wipf, P.; Lazo, J.S.; Wang, Q.J. Potent and selective disruption of protein kinase D functionality by a benzoxoloazepinolone. J. Biol. Chem. 2008, 283, 33516–33526. [Google Scholar]
- LaValle, C.R.; Bravo-Altamirano, K.; Giridhar, K.V.; Chen, J.; Sharlow, E.R.; Lazo, J.S.; Wipf, P.; Wang, Q.J. Novel protein kinase D inhibitors cause potent arrest in prostate cancer cell growth and motility. BMC Chem. Biol. 2010, 10, 1–16. [Google Scholar]
- Torres-Marquez, E.; Sinnett-Smith, J.; Guha, S.; Kui, R.; Waldron, R.T.; Rey, O.; Rozengurt, E. CID755673 enhances mitogenic signaling by phorbol esters, bombesin and EGF through a protein kinase D-independent pathway. Biochem. Biophys. Res. Commun. 2010, 391, 63–68. [Google Scholar]
- Bravo-Altamirano, K.; George, K.M.; Frantz, M.-C.; LaValle, C.R.; Tandon, M.; Leimgruber, S.; Sharlow, E.R.; Lazo, J.S.; Wang, Q.J.; Wipf, P. Synthesis and structure-activity relationships of benzothienothiazepinone inhibitors of protein kinase D. ACS Med. Chem. Lett. 2011, 2, 154–159. [Google Scholar]
- Gacek, M.; Ongstad, L.; Undheim, K. C-Alkylation of pyrimidines by an oxoalkyl reactant. Acta Chem. Scand. 1979, B33, 150–151. [Google Scholar]
- Wineman, R.J.; Hsu, E.-P.T.; Anagnostopoulos, C.E. α-Halogenation products of s-caprolactam and their transformation to DL-Lysine. J. Am. Chem. Soc. 1958, 80, 6233–6237. [Google Scholar]
- Grimm, J.B.; Stables, J.P.; Brown, M.L. Design, synthesis, and development of novel caprolactam anticonvulsants. Bioorg. Med. Chem. 2003, 11, 4133–4141. [Google Scholar]
- Lyubchanskaya, V.M.; Alekseeva, L.M.; Savina, S.A.; Shashkov, A.S.; Granik, V.G. α-Oxolactam enamines as new synthons in the Nenitzescu reaction. Russ. Chem. Bull. 2002, 51, 1886–1893. [Google Scholar]
- Kotsuki, H.; Sugino, A.; Sakai, H.; Yasuoka, H. A novel synthesis of chiral DBU/DBN-related molecules for use in asymmetric base catalysis. Heterocycles 2000, 53, 2561–2567. [Google Scholar]
- Muzart, J. Chromium-catalyzed oxidations in organic synthesis. Chem. Rev. 1992, 92, 113–140. [Google Scholar]
- Iwata, Y.; Arisawa, M.; Hamada, R.; Kita, Y.; Mizutani, M.Y.; Tomioka, N.; Itai, A.; Miyamoto, S. Discovery of novel aldose reductase inhibitors using a protein structure-based approach: 3D-Database search followed by design and synthesis. J. Med. Chem. 2001, 44, 1718–1728. [Google Scholar]
- Di Fabio, R.; Micheli, F.; Alvaro, G.; Cavanni, P.; Donati, D.; Gagliardi, T.; Fontana, G.; Giovannini, R.; Maffeis, M.; Mingardi, A.; Tranquillini, M.E.; Vitulli, G. From pyrroles to 1-oxo-2,3,4,9-tetrahydro-1 H-[β]-carbolines: A new class of orally bioavailable mGluR1 antagonists. Bioorg. Med. Chem. Lett. 2007, 17, 2254–2259. [Google Scholar]
- Glushkov, R.G.; Volskova, V.A.; Smirnova, V.G.; Magidson, O.Y. Lactam enamines and the synthesis of condensed indole derivatives. Dokl. Akad. Nauk SSSR 1969, 187, 327–329. [Google Scholar]
- Haro-Castellanos, J.A.; Gutiérrez-Carrillo, A.; Romero-Martínez, A.; James-Molina, G.; Ramírez-Chavarín, L.; Pérez-Yescas, W. Síntesis de 6-acetamido- y 6-amino-1,2,3,4-tetrahidro-1-oxo-β-carbolinas. Rev. Soc. Quim. Mex. 2002, 46, 79–82. [Google Scholar]
- Pégurier, C.; Collart, P.; Danhaive, P.; Defays, S.; Gillard, M.; Gilson, F.; Kogej, T.; Pasau, P.; Van Houtvin, N.; Van Thuyne, M.; van Keulen, B. Pyrazolone methylamino piperidine derivatives as novel CCR3 antagonists. Bioorg. Med. Chem. Lett. 2007, 17, 4228–4231. [Google Scholar]
- Higa, T.; Krubsack, A.J. Oxidations by thionyl chloride. VI. Mechanism of the reaction with cinnamic acids. J. Org. Chem. 1975, 40, 3037–3045. [Google Scholar]
- Khatana, S.S.; Boschelli, D.H.; Kramer, J.B.; Connor, D.T.; Barth, H.; Stoss, P. Preparation of benzothieno[2,3-f]-1,4-oxazepin- and -thiazepin-5(2H)-ones and of benzothieno[3,2-e]-1,4-diazepin-5-ones. J. Org. Chem. 1996, 61, 6060–6062. [Google Scholar]
- Connor, D.T.; Cetenko, W.A.; Mullican, M.D.; Sorenson, R.J.; Unangst, P.C.; Weikert, R.J.; Adolphson, R.L.; Kennedy, J.A.; Thueson, D.O. Novel benzothiophene-, benzofuran-, and naphthalenecarboxamidotetrazoles as potential antiallergy agents. J. Med. Chem. 1992, 35, 958–965. [Google Scholar]
- Barral, K.; Moorhouse, A.D.; Moses, J.E. Efficient conversion of aromatic amines into azides: A one-pot synthesis of triazole linkages. Org. Lett. 2007, 9, 1809–1811. [Google Scholar]
- Nowick, J.S.; Holmes, D.L.; Noronha, G.; Smith, E.M.; Nguyen, T.M.; Huang, S.-L. Synthesis of peptide isocyanates and isothiocyanates. J. Org. Chem. 1996, 61, 3929–3934. [Google Scholar]
- Amir, N.; Motonishi, M.; Fujita, M.; Miyashita, Y.; Fujisawa, K.; Okamoto, K.-I. Synthesis of novel S-bridged heterotrinuclear complexes containing six-membered chelate rings: Structural, spectroscopic, and electrochemical properties of [Co{Rh(apt)3}2]3+ (apt = 3-aminopropane-thiolate). Eur. J. Inorg. Chem. 2006, 2006, 1041–1049. [Google Scholar]
- Boschelli, D.; Connor, D.T.; Kramer, J.B.; Unangst, P.C. Method for treating inflammatory disease in humans. US 1996/5489586 1996. [Google Scholar]
- Babu, S.; Cheng, Z.; Reynolds, M.E.; Savage, S.J.; Tian, Q.; Yajima, H. Process for making thienopyrimidine compounds. WO 2009/055730 2009. [Google Scholar]
- Zhang, Y.-M.; Gu, M.; Ma, H.; Tang, J.; Lu, W.; Nan, F.-J. An efficient synthesis of 2-chloropyrimidines via Pd-catalyzed regioselective dechlorination of 2,4-dichloropyridines in the presence of NaHCO3. Chin. J. Chem. 2008, 26, 962–964. [Google Scholar]
- Piller, F.M.; Knochel, P. Regio- and chemoselective synthesis of fully substituted thiophenes. Org. Lett. 2009, 11, 445–448. [Google Scholar]
- Brouillette, W.J.; Einspahr, H.M. Bicyclic imides with bridgehead nitrogen. Synthesis and x-ray crystal structure of a bicyclic 2,4-oxazolidinedione. J. Org. Chem. 1984, 49, 5113–5116. [Google Scholar]
- Elberling, J.A.; Nagasawa, H.T. The twelve to fifteen-membered ring homologs of proline. J. Heterocycl. Chem. 1972, 9, 411–414. [Google Scholar]
© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
George, K.M.; Frantz, M.-C.; Bravo-Altamirano, K.; LaValle, C.R.; Tandon, M.; Leimgruber, S.; Sharlow, E.R.; Lazo, J.S.; Wang, Q.J.; Wipf, P. Design, Synthesis, and Biological Evaluation of PKD Inhibitors. Pharmaceutics 2011, 3, 186-228. https://doi.org/10.3390/pharmaceutics3020186
George KM, Frantz M-C, Bravo-Altamirano K, LaValle CR, Tandon M, Leimgruber S, Sharlow ER, Lazo JS, Wang QJ, Wipf P. Design, Synthesis, and Biological Evaluation of PKD Inhibitors. Pharmaceutics. 2011; 3(2):186-228. https://doi.org/10.3390/pharmaceutics3020186
Chicago/Turabian StyleGeorge, Kara M., Marie-Céline Frantz, Karla Bravo-Altamirano, Courtney R. LaValle, Manuj Tandon, Stephanie Leimgruber, Elizabeth R. Sharlow, John S. Lazo, Q. Jane Wang, and Peter Wipf. 2011. "Design, Synthesis, and Biological Evaluation of PKD Inhibitors" Pharmaceutics 3, no. 2: 186-228. https://doi.org/10.3390/pharmaceutics3020186