Improving Encapsulation of Hydrophilic Chloroquine Diphosphate into Biodegradable Nanoparticles: A Promising Approach against Herpes Virus Simplex-1 Infection

 , , , , ,
, , , , ,
Abstract
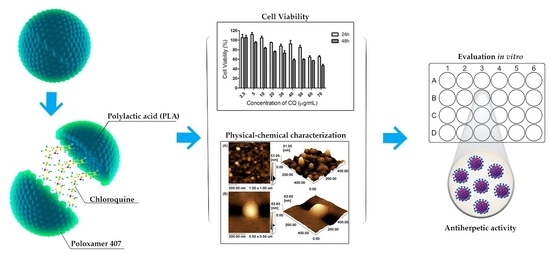
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Nanoprecipitation
2.2.1. Effect of Drug/Polymer Ratio
2.2.2. pH Changes in the Aqueous Phase
2.3. Emulsification with Solvent Evaporation
2.4. Particle Size and Zeta Potential Measurements
2.5. Drug-Loading Efficiency
2.6. Atomic Force Microscopy (AFM)
2.7. In Vitro Drug Release
2.8. Cells and Viruses
2.9. Cell Viability Studies
2.10. Cell Infection
2.11. Antiviral Activity
2.12. Statistical
3. Results
3.1. Preparation of Drug-Loaded Nanoparticles by the Nanoprecipitation Method
3.2. Preparation of Drug-Loaded Nanoparticles by the Emulsification-Solvent Evaporation Method
3.3. Morphology
3.4. In Vitro Drug Release
3.5. Cell Viability Studies
3.6. Antiviral Activity
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Li, T.; Liu, L.; Wu, H.; Chen, S.; Zhu, Q.; Gao, H.; Yu, X.; Wang, Y.; Su, W.; Yao, X.; et al. Anti-herpes simplex virus type 1 activity of Houttuynoid A, a flavonoid from Houttuynia cordata Thunb. Antivir. Res. 2017, 144, 273–280. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Liu, Y.; Wei, F.; Shen, M.; Zhong, Y.; Li, S.; Chen, L.; Ma, N.; Liu, B.; Mao, Y.; et al. The antiviral activity of arbidol hydrochloride against herpes simplex virus type i in vitro and in vivo. Int. J. Antimicrob. Agents 2017, 51, 96–106. [Google Scholar] [CrossRef]
- de Oliveira, A.; Prince, D.; Lo, C.-Y.; Lee, L.H.; Chu, T.-C. Antiviral activity of theaflavin digallate against herpes simplex virus type 1. Antivir. Res. 2015, 118, 56–67. [Google Scholar] [CrossRef] [PubMed]
- Johnson, K.E.; Chikoti, L.; Chandran, B. Herpes Simplex Virus 1 Infection Induces Activation and Subsequent Inhibition of the IFI16 and NLRP3 Inflammasomes. J. Virol. 2013, 87, 5005–5018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Terlizzi, M.E.; Occhipinti, A.L.; Maffei, A.; Massimo, E.; Gribaudo, G. Inhibition of herpes simplex type 1 and type 2 infections by Oximacro®, a cranberry extract with a high content of A-type proanthocyanidins (PACs-A). Antivir. Res. 2016, 132, 154–164. [Google Scholar] [CrossRef] [PubMed]
- Strand, M.; Islam, K.; Edlund, K.; Oberg, C.T.; Allard, A.; Bergström, T.; Mei, Y.-F.; Elofsson, M.; Wadell, G. 2-[4,5-Difluoro-2-(2-fluorobenzoylamino)-benzoylamino] benzoic acid, an antiviral compound with activity against acyclovir-resistant isolates of herpes simplex virus types 1 and 2. Antimicrob. Agents Chemother. 2012, 56, 5735–5743. [Google Scholar] [CrossRef] [PubMed]
- Gavanji, S.; Sayedipour, S.S.; Larki, B.; Bakhtari, A. Antiviral activity of some plant oils against herpes simplex virus type 1 in Vero cell culture. J. Acute Med. 2015, 5, 62–68. [Google Scholar] [CrossRef]
- Lopes, N.; Ray, S.; Espada, S.F.; Bomfim, W.A.; Ray, B.; Faccin-Galhardi, L.C.; Linhares, R.E.C.; Nozawa, C. Green seaweed Enteromorpha compressa (Chlorophyta, Ulvaceae) derived sulphated polysaccharides inhibit herpes simplex virus. Int. J. Biol. Macromol. 2017, 102, 605–612. [Google Scholar] [CrossRef] [PubMed]
- Priengprom, T.; Ekalaksananan, T.; Kongyingyoes, B.; Suebsasana, S.; Aromdee, C.; Pientong, C. Synergistic effects of acyclovir and 3, 19-isopropylideneandrographolide on herpes simplex virus wild types and drug-resistant strains. BMC Complement. Altern. Med. 2015, 15, 56. [Google Scholar] [CrossRef] [PubMed]
- Bhalekar, M.R.; Upadhaya, P.G.; Madgulkar, A.R. European Journal of Pharmaceutical Sciences Fabrication and ef fi cacy evaluation of chloroquine nanoparticles in CFA-induced arthritic rats using TNF- α ELISA. PHASCI 2016, 84, 1–8. [Google Scholar] [CrossRef]
- Rainsford, K.D.; Parke, A.L.; Clifford-Rashotte, M.; Kean, W.F. Therapy and pharmacological properties of hydroxychloroquine and chloroquine in treatment of systemic lupus erythematosus, rheumatoid arthritis and related diseases. Inflammopharmacology 2015, 23, 231–269. [Google Scholar] [CrossRef] [PubMed]
- An, N.; Chen, Y.; Wang, C.; Yang, C.; Wu, Z.H.; Xue, J.; Ye, L.; Wang, S.; Liu, H.F.; Pan, Q. Chloroquine Autophagic Inhibition Rebalances Th17/Treg-Mediated Immunity and Ameliorates Systemic Lupus Erythematosus. Cell. Physiol. Biochem. 2017, 44, 412–422. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Zeng, X.; Liang, X.; Yang, Y.; Li, X. The chemotherapeutic potential of PEG- b -PLGA copolymer micelles that combine chloroquine as autophagy inhibitor and docetaxel as an anti-cancer drug. Biomaterials 2014, 35, 9144–9154. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Dong, Y.; Zeng, X.; Liang, X.; Li, X.; Tao, W.; Chen, H.; Jiang, Y.; Mei, L.; Feng, S.S. The effect of autophagy inhibitors on drug delivery using biodegradable polymer nanoparticles in cancer treatment. Biomaterials 2014, 35, 1932–1943. [Google Scholar] [CrossRef] [PubMed]
- Jia, L.; Wang, J.; Wu, T.; Wu, J.; Ling, J.; Cheng, B. In vitro and in vivo antitumor effects of chloroquine on oral squamous cell carcinoma. Mol. Med. Rep. 2017, 16, 5779–5786. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, M.L.; Xu, Y.Z.; Lu, W.J.; Li, Y.H.; Tan, S.S.; Lin, H.J.; Wu, T.M.; Li, Y.; Wang, S.Y.; Zhao, Y.L. Chloroquine potentiates the anticancer effect of sunitinib on renal cell carcinoma by inhibiting autophagy and inducing apoptosis. Oncol. Lett. 2018, 15, 2839–2846. [Google Scholar] [CrossRef] [PubMed]
- Fu, Z.; Xi, C.; Kuang, J.; Feng, H.; Chen, L.; Liang, J.; Shen, X.; Yuen, S.; Chenghong, P.; Baiyong, S.; et al. CQ sensitizes human pancreatic cancer cells to gemcitabine through the lysosomal apoptotic pathway via reactive oxygen species. Mol. Oncol. 2018, 12, 529–544. [Google Scholar] [CrossRef] [PubMed]
- Keyaerts, E.; Li, S.; Vijgen, L.; Rysman, E.; Verbeeck, J.; Van Ranst, M.; Maes, P. Antiviral Activity of Chloroquine against Human Coronavirus OC43 Infection in Newborn Mice. Antimicrob. Agents Chemother. 2009, 53, 3416–3421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Juvenal, K.; Farias, S.; Renata, P.; Machado, L.; Antônio, B. Chloroquine Inhibits Dengue Virus Type 2 Replication in Vero Cells but Not in C6/36 Cells. 2013, 2013, 282734. [Google Scholar]
- Juvenal, K.; Farias, S.; Renata, P.; Machado, L.; Pereira, A.; Muniz, C. Antiviral Activity of Chloroquine Against Dengue Virus Type 2 Replication in Aotus Monkeys. 2015, 28, 161–169. [Google Scholar] [CrossRef] [Green Version]
- Marco Isaac, B.-L.; María del Carmen, M.-G.; José Guillermo, V.-R.; Mario Enrique, R.-M.; Sergio, F.-H.; César, R.-B.; Margarito, S.-G.; Blanca Ariadna, C.-A. Effect of Tenofovir/Emtricitabine/Efavirenz with and without Chloroquine in Patients with HIV/AIDS C3: Double Blinded Randomized Clinical Trial. J. Pharmacovigil. 2015, 3. [Google Scholar] [CrossRef]
- Long, J.; Wright, E.; Molesti, E.; Temperton, N.; Barclay, W. Antiviral therapies against Ebola and other emerging viral diseases using existing medicines that block virus entry. F1000Research 2015, 4. [Google Scholar] [CrossRef] [PubMed]
- Delvecchio, R.; Higa, L.; Pezzuto, P.; Valadão, A.; Garcez, P.; Monteiro, F.; Loiola, E.; Dias, A.; Silva, F.; Aliota, M.; et al. Chloroquine, an Endocytosis Blocking Agent, Inhibits Zika Virus Infection in Different Cell Models. Viruses 2016, 8, 322. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.; Santhosh, S.R.; Tiwari, M.; Lakshmana Rao, P.V.; Parida, M. Assessment of in vitro prophylactic and therapeutic efficacy of chloroquine against chikungunya virus in vero cells. J. Med. Virol. 2010, 82, 817–824. [Google Scholar] [CrossRef] [PubMed]
- Santos-Magalhães, N.S.; Mosqueira, V.C.F. Nanotechnology applied to the treatment of malaria. Adv. Drug Deliv. Rev. 2010, 62, 560–575. [Google Scholar] [CrossRef] [PubMed]
- Pushpalatha, R.; Selvamuthukumar, S.; Kilimozhi, D. Nanocarrier mediated combination drug delivery for chemotherapy—A review. J. Drug Deliv. Sci. Technol. 2017, 39, 362–371. [Google Scholar] [CrossRef]
- Crucho, C.I.C.; Teresa, M. Polymeric nanoparticles: A study on the preparation variables and characterization methods. Mater. Sci. Eng. C. 2017, 80, 771–784. [Google Scholar] [CrossRef] [PubMed]
- Baruah, U.K.; Gowthamarajan, K.; Vanka, R.; Karri, V.V.S.R.; Selvaraj, K.; Jojo, G.M. Malaria treatment using novel nano-based drug delivery systems. J. Drug Target. 2017, 25, 567–581. [Google Scholar] [CrossRef] [PubMed]
- Mishra, B.; Patel, B.B.; Tiwari, S. Colloidal nanocarriers: A review on formulation technology, types and applications toward targeted drug delivery. Nanomedicine 2010, 6, 9–24. [Google Scholar] [CrossRef] [PubMed]
- Vrignaud, S.; Benoit, J.P.; Saulnier, P. Strategies for the nanoencapsulation of hydrophilic molecules in polymer-based nanoparticles. Biomaterials 2011, 32, 8593–8604. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dalpiaz, A.; Sacchetti, F.; Baldisserotto, A.; Pavan, B.; Maretti, E.; Iannuccelli, V.; Leo, E. Application of the “in-oil nanoprecipitation” method in the encapsulation of hydrophilic drugs in PLGA nanoparticles. J. Drug Deliv. Sci. Technol. 2016, 32, 283–290. [Google Scholar] [CrossRef]
- Miladi, K.; Sfar, S.; Fessi, H.; Elaissari, A. Encapsulation of alendronate sodium by nanoprecipitation and double emulsion: From preparation to in vitro studies. Ind. Crops Prod. 2015, 72, 24–33. [Google Scholar] [CrossRef]
- Peltonen, L.; Aitta, J.; Hyvönen, S.; Karjalainen, M.; Hirvonen, J. Improved entrapment efficiency of hydrophilic drug substance for nanoprecipitation of poly(l)lactide nanoparticles. AAPS PharmSciTech 2004, 5, 16. [Google Scholar] [CrossRef]
- Koyama, A.H.; Uchida, T. Inhibition of multiplication of herpes simplex virus type 1 by ammonium chloride and chloroquine. Virology 1984, 138, 332–335. [Google Scholar] [CrossRef]
- Lebon, P. Inhibition of Herpes Simplex Virus Type 1-induced Interferon Synthesis by Monoclonal Antibodies against Viral Glycoprotein D and by Lysosomotropic Drugs. J. Gen. Virol. 1985, 66, 2781–2786. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harley, C.A.; Dasgupta, A.; Wilson, D.W. Characterization of herpes simplex virus-containing organelles by subcellular fractionation: role for organelle acidification in assembly of infectious particles. J. Virol. 2001, 75, 1236–1251. [Google Scholar] [CrossRef] [PubMed]
- McClain, L.; Zhi, Y.; Cheng, H.; Ghosh, A.; Piazza, P.; Yee, M.B.; Kumar, S.; Milosevic, J.; Bloom, D.C.; Arav-Boger, R.; et al. Broad-spectrum non-nucleoside inhibitors of human herpesviruses. Antivir. Res. 2015, 121, 16–23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, A.K.; Sidhu, G.S.; Friedman, R.M.; Maheshwari, R.K. Mechanism of Enhancement of the Antiviral Action of Interferon Against Herpes Simplex Virus-1 by Chloroquine. J. Interf. Cytokine Res. 1996, 16, 725–731. [Google Scholar] [CrossRef] [PubMed]
- Fessi, H.; Puisieux, F.; Devissaguet, J.P.; Ammoury, N.; Benita, S. Nanocapsule formation by interfacial polymer deposition following solvent displacement. Int. J. Pharm. 1989, 55, 1–4. [Google Scholar] [CrossRef]
- Souto, E.B.; Severino, P.; Santana, M.H.A. Preparation of Polymeric Nanoparticles from Pre-Formed Polymers—Part II. Polímeros 2012, 22, 101–106. [Google Scholar] [CrossRef]
- Caldas dos Santos, T.; Rescignano, N.; Boff, L.; Reginatto, F.H.; Simões, C.M.O.; de Campos, A.M.; Mijangos, C. In vitro antiherpes effect of C-glycosyl flavonoid enriched fraction of Cecropia glaziovii encapsulated in PLGA nanoparticles. Mater. Sci. Eng. C 2017, 75, 1214–1220. [Google Scholar] [CrossRef] [PubMed]
- Baram-Pinto, D.; Shukla, S.; Perkas, N.; Gedanken, A.; Sarid, R. Inhibition of Herpes Simplex Virus Type 1 Infection by Silver Nanoparticles Capped with Mercaptoethane Sulfonate. Bioconjug. Chem. 2009, 20, 1497–1502. [Google Scholar] [CrossRef] [PubMed]
- dos Santos-Silva, A.M.; de Caland, L.B.; de SL Oliveira, A.L.C.; de Araújo-Júnior, R.F.; Fernandes-Pedrosa, M.F.; Cornélio, A.M.; da Silva-Júnior, A.A. Designing structural features of novel benznidazole-loaded cationic nanoparticles for inducing slow drug release and improvement of biological efficacy. Mater. Sci. Eng. C 2017, 78, 978–987. [Google Scholar] [CrossRef] [PubMed]
- de Oliveira, A.R.; Mesquita, P.C.; Machado, P.R.L.; Farias, K.J.S.; de Almeida, Y.M.B.; Fernandes-Pedrosa, M.F.; Cornélio, A.M.; do Egito, E.S.T.; da Silva-Júnior, A.A. Monitoring structural features, biocompatibility and biological efficacy of gamma-irradiated methotrexate-loaded spray-dried microparticles. Mater. Sci. Eng. C 2017, 80, 438–448. [Google Scholar] [CrossRef] [PubMed]
- Pujol, C.A.; Sepúlveda, C.S.; Richmond, V.; Maier, M.S.; Damonte, E.B. Polyhydroxylated sulfated steroids derived from 5α-cholestanes as antiviral agents against herpes simplex virus. Arch. Virol. 2016, 161, 1993–1999. [Google Scholar] [CrossRef] [PubMed]
- Bisignano, C.; Mandalari, G.; Smeriglio, A.; Trombetta, D.; Pizzo, M.; Pennisi, R.; Sciortino, M. Almond Skin Extracts Abrogate HSV-1 Replication by Blocking Virus Binding to the Cell. Viruses 2017, 9, 178. [Google Scholar] [CrossRef] [PubMed]
- Biswal, I.; Dinda, A.; Mohanty, S.; Dhara, M.; Das, D.; Chowdary, K.A.; Si, S. Influence of drug/polymer ratio on the encapsulation efficiency of highly hydrophilic drug. Asian J. Chem. 2011, 23, 1973–1978. [Google Scholar]
- Halayqa, M.; Domańska, U. PLGA biodegradable nanoparticles containing perphenazine or chlorpromazine hydrochloride: Effect of formulation and release. Int. J. Mol. Sci. 2014, 15, 23909–23923. [Google Scholar] [CrossRef] [PubMed]
- Cheow, W.S.; Hadinoto, K. Colloids and Surfaces A: Physicochemical and Engineering Aspects Enhancing encapsulation efficiency of highly water-soluble antibiotic in poly(lactic-co-glycolic acid) nanoparticles: Modifications of standard nanoparticle preparation methods. Colloids Surfaces A Physicochem. Eng. Asp. 2010, 370, 79–86. [Google Scholar] [CrossRef]
- Rivas-Granizo, P.; Santos, S.R.C.J.; Ferraz, H.G. Development of a Stability-Indicating LC Assay Method for Determination of Chloroquine. Chromatogr. Suppl. 2009, 69, 137–141. [Google Scholar] [CrossRef]
- Maruyama, A.; Ishihara, T.; Kim, J.S.; Kim, S.W.; Akaike, T. Nanoparticle DNA carrier with poly(l-lysine) grafted polysaccharide copolymer and poly(d,l-lactic acid). Bioconjug. Chem. 1997, 8, 735–742. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Zhang, J.; Watanabe, W. Physical and chemical stability of drug nanoparticles. Adv. Drug Deliv. Rev. 2011, 63, 456–469. [Google Scholar] [CrossRef] [PubMed]
- Hoo, C.M.; Starostin, N.; West, P.; Mecartney, M.L. A comparison of atomic force microscopy (AFM) and dynamic light scattering (DLS) methods to characterize nanoparticle size distributions. J. Nanopart. Res. 2008, 10, 89–96. [Google Scholar] [CrossRef]
- Roopkishora; Singh, A.; Singh, C.L. Development and Characterization of Hydroxyl Chloroquine Sulphate. Int. J. Pharm. Sci. Drug Res. 2015, 7, 22–26. [Google Scholar]
- Magalhães, G.A., Jr.; Moura Neto, E.; Sombra, V.G.; Richter, A.R.; Abreu, C.M.W.S.; Feitosa, J.P.A.; Paula, H.C.B.; Goycoolea, F.M.; de Paula, R.C.M. Chitosan/Sterculia striata polysaccharides nanocomplex as a potential chloroquine drug release device. Int. J. Biol. Macromol. 2016, 88, 244–253. [Google Scholar] [CrossRef] [PubMed]
- Kang, T.; Zhu, Q.; Jiang, D.; Feng, X.; Feng, J.; Jiang, T.; Yao, J.; Jing, Y.; Song, Q.; Jiang, X.; et al. Synergistic targeting tenascin C and neuropilin-1 for specific penetration of nanoparticles for anti-glioblastoma treatment. Biomaterials 2016, 101, 60–75. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.; Zhang, H.; Chen, Z.; Zheng, Y. Penetration of lipid membranes by gold nanoparticles: Insights into cellular uptake, cytotoxicity, and their relationship. ACS Nano 2010, 4, 5421–5429. [Google Scholar] [CrossRef] [PubMed]
- James, S.H.; Prichard, M.N. Current and future therapies for herpes simplex virus infections: mechanism of action and drug resistance. Curr. Opin. Virol. 2014, 8, 54–61. [Google Scholar] [CrossRef] [PubMed]
- Burrel, S.; Bonnafous, P.; Hubacek, P.; Agut, H.; Boutolleau, D. Impact of novel mutations of herpes simplex virus 1 and 2 thymidine kinases on acyclovir phosphorylation activity. Antivir. Res. 2012, 96, 386–390. [Google Scholar] [CrossRef] [PubMed]
- Piret, J.; Boivin, G. Antiviral drug resistance in herpesviruses other than cytomegalovirus. Rev. Med. Virol. 2014, 24, 186–218. [Google Scholar] [CrossRef] [PubMed]
- Piret, J.; Boivin, G. Resistance of Herpes Simplex Viruses to Nucleoside Analogues: Mechanisms, Prevalence, and Management. Antimicrob. Agents Chemother. 2011, 55, 459–472. [Google Scholar] [CrossRef] [PubMed]
- Andrei, G.; Snoeck, R. Herpes simplex virus drug-resistance. Curr. Opin. Infect. Dis. 2013, 26, 551–560. [Google Scholar] [CrossRef] [PubMed]
- Savarino, A.; Di Trani, L.; Donatelli, I.; Cauda, R.; Cassone, A. New insights into the antiviral effects of chloroquine. Lancet Infect. Dis. 2006, 6, 67–69. [Google Scholar] [CrossRef]
- Inglot, A.D. Comparison of the Antiviral Activity in vitro of some Non-steroidal Anti-inflammatory Drugs. J. Gen. Virol. 1969, 4, 203–214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cavalli, R.; Donalisio, M.; Civra, A.; Ferruti, P.; Ranucci, E.; Trotta, F.; Lembo, D. Enhanced antiviral activity of Acyclovir loaded into β-cyclodextrin-poly(4-acryloylmorpholine) conjugate nanoparticles. J. Control. Release 2009, 137, 116–122. [Google Scholar] [CrossRef] [PubMed]
- Mesquita, P.C.; Silva, E.S.; Streck, L.; Damasceno, I.Z.; Maia, A.M.S.; Fernandes-Pedrosa, M.F.; Silva-Júnio, A.A. Cationic functionalized biocompatible polylactide nanoparticles for slow release of proteins. Colloids Surfaces A Physicochem. Eng. Asp. 2017, 513, 442–451. [Google Scholar] [CrossRef]
- Wurm, M.C.; Möst, T.; Bergauer, B.; Rietzel, D.; Neukam, F.W.; Cifuentes, S.C.; von Wilmowsky, C. In Vitro evaluation of Polylactic acid (PLA) manufactured by fused deposition modeling. J. Biol. Eng. 2017, 11, 29. [Google Scholar] [CrossRef] [PubMed]
- Shah, A.; Farooq, A.V.; Tiwari, V.; Kim, M.-J.; Shukla, D. HSV-1 infection of human corneal epithelial cells: receptor-mediated entry and trends of re-infection. Mol. Vis. 2010, 16, 2476–2486. [Google Scholar] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Formulation | CQ 1:PLA 2 Ratio | AP 3 pH | Size (nm) | PdI 4 | ZP 5 (mV) | EE 6 (%) |
|---|---|---|---|---|---|---|
| B-NP | 0 | 6.4 | 106.2 ± 2.5 | 0.157 ± 0.03 | −7.95 ± 3.2 | |
| 1 | 1:5 | 6.4 | 173.5 ± 8.5 | 0.113 ± 0.04 | −3.13 ± 3.2 | 10.6 ± 1.3 |
| 2 | 1:10 | 6.4 | 189.1 ± 6.5 | 0.080 ± 0.03 | −6.85 ± 5.3 | 8.4 ± 2.6 |
| 3 | 1:15 | 6.4 | 226.4 ± 9.2 | 0.073 ± 0.03 | −14.42 ± 2.1 | 3.4 ± 1.4 |
| B-NP | 0 | 11.0 | 114.5 ± 6.0 | 0.089 ± 0.02 | −1.91 ± 0.8 | |
| 4 | 1:10 | 11.0 | 200.6 ± 11.4 | 0.069 ± 0.01 | −18.15 ± 3.3 | 11.4 ± 2.0 |
| B-NP | 0 | 8.4 | 118.6 ± 6.0 | 0.046 ± 0.01 | −11.63 ± 3.0 | |
| 5 | 1:10 | 8.4 | 231.4 ± 11.5 | 0.096 ± 0.02 | −5.68 ± 4.1 | 25.0 ± 1.6 |
| Formulation | Physicochemical Properties | Kinetic Models [k (R)] | ||||||
|---|---|---|---|---|---|---|---|---|
| Size (nm) | PdI 1 | ZP 2 (mV) | EE 3 (%) | First Order | Bhaskar | Freundlich | Parabolic | |
| B-NP | 283.9 ± 53.2 | 0.27 ± 0.05 | −25.4 ± 11.6 | - | - | - | - | - |
| 6 | 297.3 ± 26.1 | 0.30 ± 0.03 | −20.0 ± 12.0 | 64.1 ± 5.0 | 0.004 h−1 (0.88) | 0.26 h0.65 (0.94) | 63.99 (0.94) | 2.25 h−0.5 (0.95) |
| Samples (Treatment 48 h) | CC50 1 (µg mL−1) | IC50 2 (µg mL−1) | SI 3 |
|---|---|---|---|
| B-NP | >500 | 4 N.A | - |
| CQ | 222.6 ± 5.4 | 6.7 ± 0.6 | 33.0 |
| CQ-NP | 67.9 ± 2.1 | 4.3 ± 1.4 | 15.6 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lima, T.L.C.; Feitosa, R.d.C.; Dos Santos-Silva, E.; Dos Santos-Silva, A.M.; Siqueira, E.M.d.S.; Machado, P.R.L.; Cornélio, A.M.; Do Egito, E.S.T.; Fernandes-Pedrosa, M.d.F.; Farias, K.J.S.; et al. Improving Encapsulation of Hydrophilic Chloroquine Diphosphate into Biodegradable Nanoparticles: A Promising Approach against Herpes Virus Simplex-1 Infection. Pharmaceutics 2018, 10, 255. https://doi.org/10.3390/pharmaceutics10040255
Lima TLC, Feitosa RdC, Dos Santos-Silva E, Dos Santos-Silva AM, Siqueira EMdS, Machado PRL, Cornélio AM, Do Egito EST, Fernandes-Pedrosa MdF, Farias KJS, et al. Improving Encapsulation of Hydrophilic Chloroquine Diphosphate into Biodegradable Nanoparticles: A Promising Approach against Herpes Virus Simplex-1 Infection. Pharmaceutics. 2018; 10(4):255. https://doi.org/10.3390/pharmaceutics10040255
Chicago/Turabian StyleLima, Tábata Loíse Cunha, Renata de Carvalho Feitosa, Emanuell Dos Santos-Silva, Alaine Maria Dos Santos-Silva, Emerson Michell da Silva Siqueira, Paula Renata Lima Machado, Alianda Maira Cornélio, Eryvaldo Sócrates Tabosa Do Egito, Matheus de Freitas Fernandes-Pedrosa, Kleber Juvenal Silva Farias, and et al. 2018. "Improving Encapsulation of Hydrophilic Chloroquine Diphosphate into Biodegradable Nanoparticles: A Promising Approach against Herpes Virus Simplex-1 Infection" Pharmaceutics 10, no. 4: 255. https://doi.org/10.3390/pharmaceutics10040255