
Highly Sensitive Bacteriophage-Based Detection of Brucella abortus in Mixed Culture and Spiked Blood




Abstract
:1. Introduction
2. Results
2.1. Brucellaphage Propagation and DNA Extraction
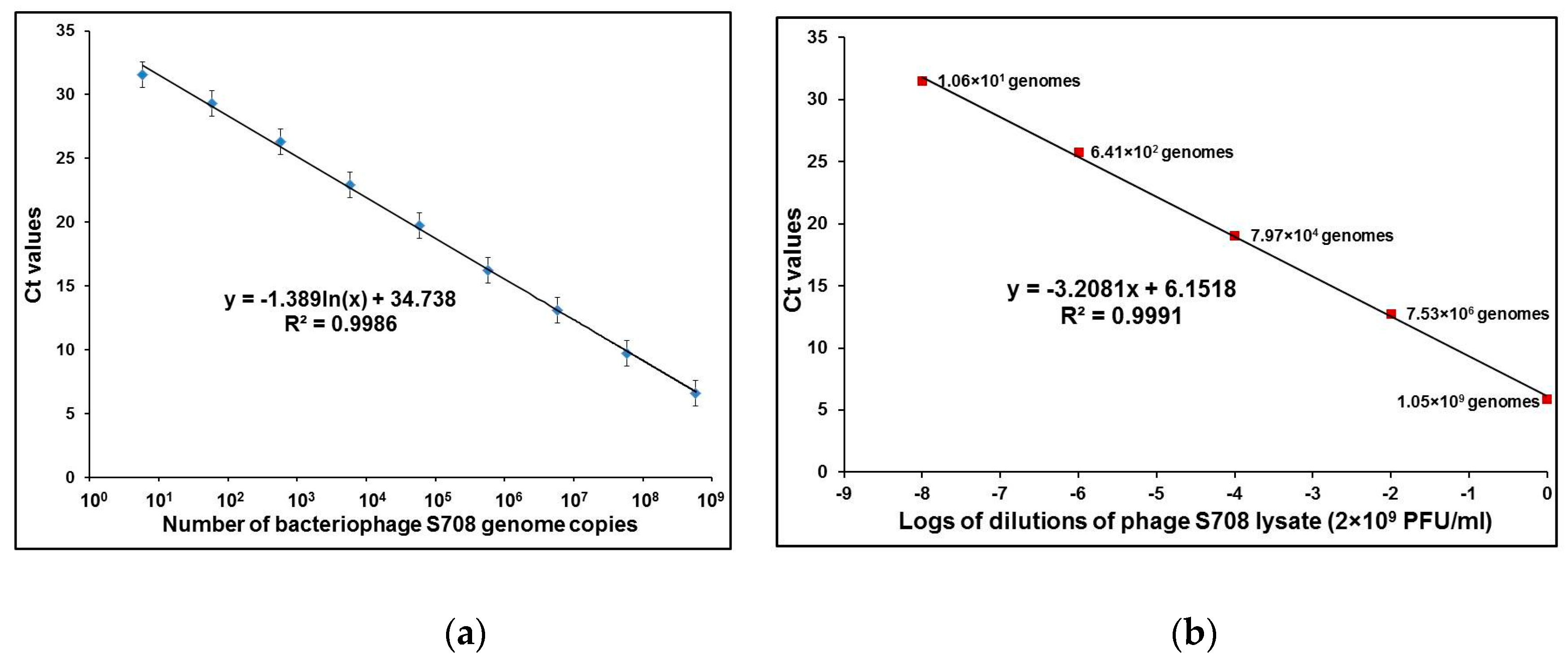
2.2. Performance Testing of qPCR with Phage DNA and Intact Phage Particles
2.3. Sensitivity of Phage-Based qPCR Detection of Brucella abortus in Broth Culture
2.4. Specificity of qPCR Tests with the Bk Bacteriophage
2.5. Assay Performance with Simulated Clinical Samples
3. Discussion
4. Materials and Methods
4.1. Bacterial Strains, Bacteriophages and Growth Media
4.2. Phage DNA Isolation
4.3. Phage S708 Amplification on Different Culture Dilutions of B. abortus
4.4. Testing Different Brucellaphages for qPCR Limits of Detection
4.5. Testing Phage Specificity on Non-Brucella Bacteria
4.6. Phage Amplification in Simulated Clinical Samples Containing B. abortus
4.7. Primer Design
4.8. qPCR Assays
4.9. Statistical Analysis
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Dean, A.S.; Crump, L.; Greter, H.; Schelling, E.; Zinsstag, J. Global burden of human brucellosis: A systematic review of disease frequency. PLoS Negl. Trop. Dis. 2012, 6, e1865. [Google Scholar] [CrossRef] [PubMed]
- Moreno, E. Retrospective and prospective perspectives on zoonotic brucellosis. Front. Microbiol. 2014, 5, 213. [Google Scholar] [CrossRef] [PubMed]
- Moreno, E.; Moriyón, I. The genus Brucella. In Prokaryotes—A Handbook on the Biology of Bacteria, 3rd ed.; Dworkin, M., Falkow, S., Rosenberg, E., Schleifer, K.H., Stackebrandt, E., Eds.; Springer-Verlag: New York, NY, USA, 2006; Volume 5, pp. 315–456. [Google Scholar]
- Olsen, S.C.; Palmer, M.V. Advancement of knowledge of Brucella over the past 50 years. Vet. Pathol. 2014, 51, 1076–1089. [Google Scholar] [CrossRef] [PubMed]
- Doganay, G.D.; Doganay, M. Brucella as a potential agent of bioterrorism. Recent Pat. Antiinfect. Drug. Discov. 2013, 8, 27–33. [Google Scholar] [CrossRef] [PubMed]
- CDC/USDA Federal Select Agent Program, Select Agents and Toxins List. Available online: https://www.selectagents.gov/selectagentsandtoxinslist.htmL (accessed on 23 January 2017).
- Al Dahouk, S.; Nöckler, K. Implications of laboratory diagnosis on brucellosis therapy. Expert Rev. Anti-Infect. Ther. 2011, 9, 833–845. [Google Scholar] [CrossRef] [PubMed]
- Al Dahouk, S.; Sprague, L.D.; Neubauer, H. New developments in the diagnostic procedures for zoonotic brucellosis in humans. Rev. Sci. Tech. 2013, 32, 177–188. [Google Scholar] [CrossRef] [PubMed]
- Public health consequences of a false-positive laboratory test result for Brucella—Florida, Georgia, and Michigan, 2005. MMWR 2008, 57, 603–605.
- Farrell, I.D. The development of a new selective medium for the isolation of Brucella abortus from contaminated sources. Res. Vet. Sci. 1974, 16, 280–286. [Google Scholar] [PubMed]
- Marín, C.M.; Jiménez-de-Bagüés, M.P.; Barberán, M.; Blasco, J.M. Comparison of two selective media for the isolation of Brucella melitensis from naturally infected sheep and goats. Vet. Rec. 1996, 138, 409–411. [Google Scholar] [CrossRef] [PubMed]
- De Miguel, M.J.; Marín, C.M.; Muñoz, P.M.; Dieste, L.; Grilló, M.J.; Blasco, J.M. Development of a selective culture medium for primary isolation of the main Brucella species. J. Clin. Microbiol. 2011, 49, 1458–1463. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, A.C.; Almendra, C.; Cardoso, R.; Pereira, M.S.; Beja-Pereira, A.; Luikart, G.; Corrêa de Sá, M.I. Development and evaluation of a selective medium for Brucella suis. Res. Vet. Sci. 2012, 93, 565–567. [Google Scholar] [CrossRef] [PubMed]
- Yagupsky, P. Detection of brucellae in blood cultures. J. Clin. Microbiol. 1999, 37, 3437–3442. [Google Scholar] [PubMed]
- Yagupsky, P.; Peled, N.; Press, J. Use of BACTEC 9240 blood culture system for detection of Brucella melitensis in synovial fluid. J. Clin. Microbiol. 2001, 39, 738–739. [Google Scholar] [CrossRef] [PubMed]
- Traxler, R.M.; Lehman, M.W.; Bosserman, E.A.; Guerra, M.A.; Smith, T.L. A literature review of laboratory-acquired brucellosis. J. Clin. Microbiol. 2013, 51, 3055–3062. [Google Scholar] [CrossRef] [PubMed]
- Bricker, B.J.; Halling, S.M. Differentiation of Brucella abortus bv. 1, 2, and 4, Brucella melitensis, Brucella ovis, and Brucella suis bv. 1 by PCR. J. Clin. Microbiol. 1994, 32, 2660–2666. [Google Scholar] [PubMed]
- Navarro, E.; Casao, M.A.; Solera, J. Diagnosis of human brucellosis using PCR. Expert Rev. Mol. Diagn. 2004, 4, 115–123. [Google Scholar] [CrossRef] [PubMed]
- López-Goñi, I.; García-Yoldi, D.; Marín, C.M.; de Miguel, M.J.; Muñoz, P.M.; Blasco, J.M.; Jacques, I.; Grayon, M.; Cloeckaert, A.; Ferreira, A.C.; et al. Evaluation of a multiplex PCR assay (Bruce-ladder) for molecular typing of all Brucella species, including the vaccine strains. J. Clin. Microbiol. 2008, 6, 3484–3487. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wang, Z.; Zhang, Y.; Bai, L.; Zhao, Y.; Liu, C.; Ma, A.; Yu, H. Polymerase chain reaction-based assays for the diagnosis of human brucellosis. Ann. Clin. Microbiol. Antimicrob. 2014, 13, 31. [Google Scholar] [CrossRef] [PubMed]
- Wareth, G.; Melzer, F.; Tomaso, H.; Roesler, U.; Neubauer, H. Detection of Brucella abortus DNA in aborted goats and sheep in Egypt by real-time PCR. BMC Res. Notes 2015, 8, 212. [Google Scholar] [CrossRef] [PubMed]
- Vrioni, G.; Pappas, G.; Priavali, E.; Gartzonika, C.; Levidiotou, S. An eternal microbe: Brucella DNA load persists for years after clinical cure. Clin. Infect. Dis. 2008, 46, 131–136. [Google Scholar] [CrossRef] [PubMed]
- Castaño, M.J.; Solera, J. Chronic brucellosis and persistence of Brucella melitensis DNA. J. Clin. Microbiol. 2009, 47, 2084–2089. [Google Scholar] [CrossRef] [PubMed]
- Dauphin, L.A.; Hutchins, R.J.; Bost, L.A.; Bowen, M.D. Evaluation of automated and manual commercial DNA extraction methods for recovery of Brucella DNA from suspensions and spiked swabs. J. Clin. Microbiol. 2009, 47, 3920–3926. [Google Scholar] [CrossRef] [PubMed]
- Droževkina, M.S. The present position in Brucella phage research. Bull. World Health Organ. 1963, 29, 43–57. [Google Scholar] [PubMed]
- Corbel, M.J.; Thomas, E.L. The Brucella-phages: Their Properties, Characterization and Applications, Booklet 2266; Ministry of Agriculture, Fisheries and Food: Pinner, Middlesex, UK, 1980. [Google Scholar]
- Segondy, M.; Allardet-Servent, A.; Caravano, R.; Ramuz, M. Common physical map of four Brucella bacteriophage genomes. FEMS Microbiol. Lett. 1988, 56, 177–181. [Google Scholar] [CrossRef]
- Rigby, C.E.; Cerqueira-Campos, M.L.; Kelly, H.A.; Surujballi, O.P. Properties and partial genetic characterization of Nepean phage and other lytic phages of Brucella species. Can. J. Vet. Res. 1989, 53, 319–325. [Google Scholar] [PubMed]
- Flores, V.; López-Merino, A.; Mendoza-Hernandez, G.; Guarneros, G. Comparative genomic analysis of two brucellaphages of distant origins. Genomics 2012, 99, 233–240. [Google Scholar] [CrossRef] [PubMed]
- Farlow, J.; Filippov, A.A.; Sergueev, K.V.; Hang, J.; Kotorashvili, A.; Nikolich, M.P. Comparative whole genome analysis of six diagnostic brucellaphages. Gene 2014, 541, 115–122. [Google Scholar] [CrossRef] [PubMed]
- Jones, L.M.; Merz, G.S.; Wilson, J.B. Phage typing reactions on Brucella species. Appl. Microbiol. 1968, 16, 1179–1190. [Google Scholar] [PubMed]
- Corbel, M.J. Brucella phages: Advances in the development of a reliable phage typing system for smooth and non-smooth Brucella isolates. Ann. Inst. Pasteur Microbiol. 1987, 138, 70–75. [Google Scholar] [CrossRef]
- Taran, I.F.; Zanina, V.M.; Liamkin, G.I.; Tsybin, B.P.; Tikhenko, N.I. Comparative evaluation of the spectrum of lytic effects of bacteriophages Tb, Wb, Fi, Bk2 and R on various Brucella species. Zhurnal Mikrobiol. Epidemiol. Immunobiol. 1983, 2, 48–52. [Google Scholar]
- Sergueev, K.V.; He, Y.; Borschel, R.H.; Nikolich, M.P.; Filippov, A.A. Rapid and sensitive detection of Yersinia pestis using amplification of plague diagnostic bacteriophages monitored by real-time PCR. PLoS ONE 2010, 5. [Google Scholar] [CrossRef] [PubMed]
- Sergueev, K.V.; Nikolich, M.P.; Filippov, A.A. Field and clinical applications of advanced bacteriophage-based detection of Yersinia pestis. Adv. Exp. Med. Biol. 2012, 954, 135–141. [Google Scholar] [CrossRef] [PubMed]
- McDuff, C.R.; Jones, L.M.; Wilson, J.B. Characteristics of brucellaphage. J. Bacteriol. 1962, 83, 324–329. [Google Scholar] [PubMed]
- Douglas, J.T.; Elberg, S.S. Isolation of Brucella melitensis phage of broad biotype and species specificity. Infect. Immun. 1976, 14, 306–308. [Google Scholar] [PubMed]
- Al Dahouk, S.; Hofer, E.; Tomaso, H.; Vergnaud, G.; Le Flèche, P.; Cloeckaert, A.; Koylass, M.S.; Whatmore, A.M.; Nöckler, K.; Scholz, H.C. Intraspecies biodiversity of the genetically homologous species Brucella microti. Appl. Environ. Microbiol. 2012, 78, 1534–1543. [Google Scholar] [CrossRef] [PubMed]
- Sanodze, L.; Bautista, C.T.; Garuchava, N.; Chubinidze, S.; Tsertsvadze, E.; Broladze, M.; Chitadze, N.; Sidamonidze, K.; Tsanava, S.; Akhvlediani, T.; et al. Expansion of brucellosis detection in the country of Georgia by screening household members of cases and neighboring community members. BMC Public Health 2015, 15. [Google Scholar] [CrossRef] [PubMed]
- OIE Manual of Diagnostic Tests and Vaccines for Terrestrial Animals 2016. Available online: http://www.oie.int/en/international-standard-setting/terrestrial-manual/access-online/ (accessed on 31 March 2017).
- Worsley, B.; Goodwin, S.; Jahans, K.; Atallah, C. First report of a strain of Brucella melitensis that was widely sensitive to brucellaphages isolated in the United Arab Emirates. Clin. Infect. Dis. 1996, 22, 190–191. [Google Scholar] [CrossRef] [PubMed]
- Köse, S.; Kiliç, S.; Ozbel, Y. Identification of Brucella species isolated from proven brucellosis patients in Izmir, Turkey. J. Basic Microbiol. 2005, 45, 323–327. [Google Scholar] [CrossRef] [PubMed]
- Foster, G.; Osterman, B.S.; Godfroid, J.; Jacques, I.; Cloeckaert, A. Brucella ceti sp. nov. and Brucella pinnipedialis sp. nov. for Brucella strains with cetaceans and seals as their preferred hosts. Int. J. Syst. Evolut. Microbiol. 2007, 57, 2688–2693. [Google Scholar] [CrossRef] [PubMed]
- Scholz, H.C.; Nöckler, K.; Göllner, C.; Bahn, P.; Vergnaud, G.; Tomaso, H.; Al Dahouk, S.; Kämpfer, P.; Cloeckaert, A.; Maquart, M.; et al. Brucella inopinata sp. nov., isolated from a breast implant infection. Int. J. Syst. Evol. Microbiol. 2010, 60, 801–808. [Google Scholar] [CrossRef] [PubMed]
- Leclerc, H. The bacteriophage titer increase reaction. Ann. Inst. Pasteur Lille 1963, 14, 145–158. [Google Scholar] [PubMed]
- Bakulov, I.A.; Kotliarov, V.M.; Kol’pikova, T.I. Sensitivity of the phage titer increase test in detecting Listeria. Zhurnal Mikrobiol. Epidemiol. Immunobiol. 1984, 9, 40–43. [Google Scholar]
- Albert, H.; Heydenrych, A.; Brookes, R.; Mole, R.J.; Harley, B.; Subotsky, E.; Henry, R.; Azevedo, V. Performance of a rapid phage-based test, FASTPlaqueTB, to diagnose pulmonary tuberculosis from sputum specimens in South Africa. Int. J. Tuberc. Lung Dis. 2002, 6, 529–537. [Google Scholar] [PubMed]
- Kiraz, N.; Et, L.; Akgun, Y.; Kasifoglu, N.; Kiremitci, A. Rapid detection of Mycobacterium tuberculosis from sputum specimens using the FASTPlaqueTB test. Int. J. Tuberc. Lung Dis. 2007, 11, 904–908. [Google Scholar] [PubMed]
- Stanley, E.C.; Mole, R.J.; Smith, R.J.; Glenn, S.M.; Barer, M.R.; McGowan, M.; Rees, C.E. Development of a new, combined rapid method using phage and PCR for detection and identification of viable Mycobacterium paratuberculosis bacteria within 48 hours. Appl. Environ. Microbiol. 2007, 73, 1851–1857. [Google Scholar] [CrossRef] [PubMed]
- Swift, B.M.; Denton, E.J.; Mahendran, S.A.; Huxley, J.N.; Rees, C.E. Development of a rapid phage-based method for the detection of viable Mycobacterium avium subsp. paratuberculosis in blood within 48 h. J. Microbiol. Methods 2013, 94, 175–179. [Google Scholar] [CrossRef] [PubMed]
- Hirsh, D.C.; Martin, L.D. Rapid detection of Salmonella spp. by using Felix-O1 bacteriophage and high-performance liquid chromatography. Appl. Environ. Microbiol. 1983, 45, 260–264. [Google Scholar] [PubMed]
- Edelman, D.C.; Barletta, J. Real-time PCR provides improved detection and titer determination of bacteriophage. Biotechniques 2003, 35, 368–375. [Google Scholar] [PubMed]
- Reiman, R.W.; Atchley, D.H.; Voorhees, K.J. Indirect detection of Bacillus anthracis using real-time PCR to detect amplified gamma phage DNA. J. Microbiol. Methods 2007, 68, 651–653. [Google Scholar] [CrossRef] [PubMed]
- Guan, J.; Chan, M.; Allain, B.; Mandeville, R.; Brooks, B.W. Detection of multiple antibiotic-resistant Salmonella enterica serovar Typhimurium DT104 by phage replication-competitive enzyme-linked immunosorbent assay. J. Food Prot. 2006, 69, 739–742. [Google Scholar] [CrossRef] [PubMed]
- Bhowmick, T.; Mirrett, S.; Reller, L.B.; Price, C.; Qi, C.; Weinstein, M.P.; Kirn, T.J. Controlled multicenter evaluation of a bacteriophage-based method for rapid detection of Staphylococcus aureus in positive blood cultures. J. Clin. Microbiol. 2013, 51, 1226–1230. [Google Scholar] [CrossRef] [PubMed]
- Kannan, P.; Yong, H.Y.; Reiman, L.; Cleaver, C.; Patel, P.; Bhagwat, A.A. Bacteriophage-based rapid and sensitive detection of Escherichia coli O157:H7 isolates from ground beef. Foodborne Pathog. Dis. 2010, 7, 1551–1558. [Google Scholar] [CrossRef] [PubMed]
- Neufeld, T.; Schwartz-Mittelmann, A.; Biran, D.; Ron, E.Z.; Rishpon, J. Combined phage typing and amperometric detection of released enzymatic activity for the specific identification and quantification of bacteria. Anal. Chem. 2003, 75, 580–585. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, W.R., Jr.; Barletta, R.G.; Udani, R.; Chan, J.; Kalkut, G.; Sosne, G.; Kieser, T.; Sarkis, G.J.; Hatfull, G.F.; Bloom, B.R. Rapid assessment of drug susceptibilities of Mycobacterium tuberculosis by means of luciferase reporter phages. Science 1993, 260, 819–822. [Google Scholar] [CrossRef] [PubMed]
- Dusthackeer, A.; Kumar, V.; Subbian, S.; Sivaramakrishnan, G.; Zhu, G.; Subramanyam, B.; Hassan, S.; Nagamaiah, S.; Chan, J.; Paranji Rama, N. Construction and evaluation of luciferase reporter phages for the detection of active and non-replicating tubercle bacilli. J. Microbiol. Methods 2008, 73, 18–25. [Google Scholar] [CrossRef] [PubMed]
- Schofield, D.A.; Molineux, I.J.; Westwater, C. Diagnostic bioluminescent phage for detection of Yersinia pestis. J. Clin. Microbiol. 2009, 47, 3887–3894. [Google Scholar] [CrossRef] [PubMed]
- Schofield, D.A.; Westwater, C. Phage-mediated bioluminescent detection of Bacillus anthracis. J. Appl. Microbiol. 2009, 107, 1468–1478. [Google Scholar] [CrossRef] [PubMed]
- Awais, R.; Fukudomi, H.; Miyanaga, K.; Unno, H.; Tanji, Y. A recombinant bacteriophage-based assay for the discriminative detection of culturable and viable but nonculturable Escherichia coli O157:H7. Biotechnol. Prog. 2006, 22, 853–859. [Google Scholar] [CrossRef] [PubMed]
- Wallach, J.C.; Ferrero, M.C.; Victoria Delpino, M.; Fossati, C.A.; Baldi, P.C. Occupational infection due to Brucella abortus S19 among workers involved in vaccine production in Argentina. Clin. Microbiol. Infect. 2008, 14, 805–807. [Google Scholar] [CrossRef] [PubMed]
- Elsaghir, A.A.; James, E.A. Misidentification of Brucella melitensis as Ochrobactrum anthropi by API 20NE. J. Med. Microbiol. 2003, 52, 441–442. [Google Scholar] [CrossRef] [PubMed]
- Horvat, R.T.; El Atrouni, W.; Hammoud, K.; Hawkinson, D.; Cowden, S. Ribosomal RNA sequence analysis of Brucella infection misidentified as Ochrobactrum anthropi infection. J. Clin. Microbiol. 2011, 49, 1165–1168. [Google Scholar] [CrossRef] [PubMed]
- Carrington, M.; Choe, U.; Ubillos, S.; Stanek, D.; Campbell, M.; Wansbrough, L.; Lee, P.; Churchwell, G.; Rosas, K.; Zaki, S.R.; et al. Fatal case of brucellosis misdiagnosed in early stages of Brucella suis infection in a 46-year-old patient with Marfan syndrome. J. Clin. Microbiol. 2012, 50, 2173–2175. [Google Scholar] [CrossRef] [PubMed]
- Ahvonen, P.; Jansson, E.; Aho, K. Marked crossagglutination between brucellae and a subtype of Yersinia enterocolitica. Acta Pathol. Microbiol. Scand. 1969, 75, 291–295. [Google Scholar] [PubMed]
- Bundle, D.R.; Gidney, M.A.J.; Perry, M.B.; Duncan, J.R.; Cherwonogrodzky, J.W. Serological confirmation of Brucella abortus and Yersinia enterocolitica O:9 O-antigens by monoclonal antibodies. Infect. Immun. 1984, 46, 389–393. [Google Scholar] [PubMed]
- Chenais, E.; Bagge, E.; Lambertz, S.T.; Artursson, K. Yersinia enterocolitica serotype O:9 cultured from Swedish sheep showing serologically false-positive reactions for Brucella melitensis. Infect. Ecol. Epidemiol. 2012, 2, 19027. [Google Scholar] [CrossRef] [PubMed]
- Drancourt, M.; Brouqui, P.; Raoult, D. Afipia clevelandensis antibodies and cross-reactivity with Brucella spp. and Yersinia enterocolitica O:9. Clin. Diagn. Lab. Immunol. 1997, 4, 748–752. [Google Scholar] [PubMed]
- Probert, W.S.; Schrader, K.N.; Khuong, N.Y.; Bystrom, S.L.; Graves, M.H. Real-time multiplex PCR assay for detection of Brucella spp., B. abortus, and B. melitensis. J. Clin. Microbiol. 2004, 42, 1290–1293. [Google Scholar] [CrossRef] [PubMed]
- Bogdanovich, T.; Skurnik, M.; Lübeck, P.S.; Ahrens, P.; Hoorfar, J. Validated 5′ nuclease PCR assay for rapid identification of the genus Brucella. J. Clin. Microbiol. 2004, 42, 2261–2263. [Google Scholar] [CrossRef] [PubMed]
- Gopaul, K.K.; Koylass, M.S.; Smith, C.J.; Whatmore, A.M. Rapid identification of Brucella isolates to the species level by real time PCR based single nucleotide polymorphism (SNP) analysis. BMC Microbiol. 2008, 8. [Google Scholar] [CrossRef] [PubMed]
- Bounaadja, L.; Albert, D.; Chénais, B.; Hénault, S.; Zygmunt, M.S.; Poliak, S.; Garin-Bastuji, B. Real-time PCR for identification of Brucella spp.: A comparative study of IS711, bcsp31 and per target genes. Vet. Microbiol. 2009, 137, 156–164. [Google Scholar] [CrossRef] [PubMed]
- Winchell, J.M.; Wolff, B.J.; Tiller, R.; Bowen, M.D.; Hoffmaster, A.R. Rapid identification and discrimination of Brucella isolates by use of real-time PCR and high-resolution melt analysis. J. Clin. Microbiol. 2010, 48, 697–702. [Google Scholar] [CrossRef] [PubMed]
- Hänsel, C.; Mertens, K.; Elschner, M.C.; Melzer, F. Novel real-time PCR detection assay for Brucella suis. Vet. Rec. Open 2015, 2. [Google Scholar] [CrossRef] [PubMed]
- Kattar, M.M.; Zalloua, P.A.; Araj, G.F.; Samaha-Kfoury, J.; Shbaklo, H.; Kanj, S.S.; Khalife, S.; Deeb, M. Development and evaluation of real-time polymerase chain reaction assays on whole blood and paraffin-embedded tissues for rapid diagnosis of human brucellosis. Diagn. Microbiol. Infect. Dis. 2007, 59, 23–32. [Google Scholar] [CrossRef] [PubMed]
- Gwida, M.M.; El-Gohary, A.H.; Melzer, F.; Tomaso, H.; Rösler, U.; Wernery, U.; Wernery, R.; Elschner, M.C.; Khan, I.; Eickhoff, M.; et al. Comparison of diagnostic tests for the detection of Brucella spp. in camel sera. BMC Res. Notes 2011, 4. [Google Scholar] [CrossRef] [PubMed]
- Queipo-Ortuño, M.I.; Colmenero, J.D.; Reguera, J.M.; García-Ordoñez, M.A.; Pachón, M.E.; Gonzalez, M.; Morata, P. Rapid diagnosis of human brucellosis by SYBR Green I-based real-time PCR assay and melting curve analysis in serum samples. Clin. Microbiol. Infect. 2005, 11, 713–718. [Google Scholar] [CrossRef] [PubMed]
- Popkhadze, M.Z.; Abashidze, T.G. Characterization of a Brucella Phage isolated at Tbilisi Research Institute for Vaccines and Sera, Abstracts of Inter-Institute Science Conference on Bacteriophagy; Tbilisi University Press: Tbilisi, USSR, 1957; p. 40. [Google Scholar]
- Corbel, M.J.; Thomas, E.L. Description of a new phage lytic for several Brucella species. J. Biol. Stand. 1976, 4, 195–201. [Google Scholar] [CrossRef]
- Graves, R.R. The story of John M. Buck’s and Matilda’s contribution to the cattle industry. J. Am. Vet. Med. Assoc. 1943, 102, 193–195. [Google Scholar]
- Holmes, B.; Popoff, M.; Kiredjian, M.; Kersters, K. Ochrobactrum anthropi gen. nov., sp. nov. from human clinical specimens and previously known as group Vd. Int. J. Syst. Bacteriol. 1988, 38, 406–416. [Google Scholar] [CrossRef]
- Brenner, D.J.; Hollis, D.G.; Moss, C.W.; English, C.K.; Hall, G.S.; Vincent, J.; Radosevic, J.; Birkness, K.A.; Bibb, W.F.; Quinn, F.D.; et al. Proposal of Afipia gen. nov., with Afipia felis sp. nov. (formerly the cat scratch disease bacillus), Afipia clevelandensis sp. nov. (formerly the Cleveland Clinic Foundation strain), Afipia broomeae sp. nov., and three unnamed genospecies. J. Clin. Microbiol. 1991, 29, 2450–2460. [Google Scholar] [PubMed]
- Sterne, M. The use of anthrax vaccines prepared from avirulent (uncapsulated) variants of Bacillus anthracis. Onderstepoort J. Vet. Sci. Anim. Ind. 1939, 13, 307–312. [Google Scholar]
- Appleyard, R.K. Segregation of new lysogenic types during growth of a doubly lysogenic strain derived from Escherichia coli K-12. Genetics 1954, 39, 440–452. [Google Scholar] [PubMed]
- Sambrook, J.; Fritsch, E.F.; Maniatis, T. Molecular Cloning: A Laboratory Manual, 2nd ed.; Cold Spring Harbor Laboratory Press: New York, NY, USA, 1989. [Google Scholar]
- Etemadi, H.; Raissadat, A.; Pickett, M.J.; Zafari, Y.; Vahedifar, P. Isolation of Brucella spp. from clinical specimens. J. Clin. Microbiol. 1984, 20, 586. [Google Scholar] [PubMed]
- NetPrimer, Free Primer Analysis Software, Premier Biosoft Int. Available online: http://www.premierbiosoft.com/netprimer/index.html (accessed on 25 January 2017).
- Basic Local Alignment Search Tool (BLAST). Available online: https://blast.ncbi.nlm.nih.gov/Blast.cgi (accessed on 25 January 2017).
- Hammerl, J.A.; Al Dahouk, S.; Nöckler, K.; Göllner, C.; Appel, B.; Hertwig, S. F1 and Tbilisi are closely related brucellaphages exhibiting some distinct nucleotide variations which determine the host specificity. Genome Announc. 2014, 2. [Google Scholar] [CrossRef] [PubMed]
- VassarStats: Website for Statistical Computation/ANOVA/One-Way Analysis of Variance for Independent or Correlated Samples. Available online: http://faculty.vassar.edu/lowry/anova1u.html (accessed on 24 January 2017).


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Bacteriophage | Titer After Propagation, PFU/mL * | Number of Genome Equivalents per mL ** |
|---|---|---|
| Tb | 2 × 1010 | 2.8 × 1012 |
| S708 | 2 × 109 | 6 × 1011 |
| Fz | 1 × 1011 | 2.5 × 1012 |
| Wb | 1 × 1010 | 1 × 1012 |
| Bk | 1 × 108 | 2 × 1012 |
| Bacteriophage or Bacterial Strain | Source | Reference |
|---|---|---|
| Brucellaphages: | ||
| Tb (Tbilisi) | d’Hérelle Phage Collection * | [29,30,80] |
| S708 | d’Hérelle Phage Collection * | [30,81] |
| Fz (Firenze) | d’Hérelle Phage Collection * | [30,81] |
| Wb (Weybridge) | d’Hérelle Phage Collection * | [30,81] |
| Bk (Berkeley) | d’Hérelle Phage Collection * | [30,37] |
| Bacterial strains: | ||
| Brucella abortus S19 | Laboratory collection | [82] |
| Ochrobactrum anthropi ATCC 49187 | ATCC** | [83] |
| Afipia felis ATCC 53690 | ATCC | [84] |
| Bacillus anthracis Sterne | Laboratory collection | [85] |
| Escherichia coli C600 | Laboratory collection | [86] |
| Yersinia enterocolitica 2516-87 (O:9) | Laboratory collection | [34] |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sergueev, K.V.; Filippov, A.A.; Nikolich, M.P. Highly Sensitive Bacteriophage-Based Detection of Brucella abortus in Mixed Culture and Spiked Blood. Viruses 2017, 9, 144. https://doi.org/10.3390/v9060144
Sergueev KV, Filippov AA, Nikolich MP. Highly Sensitive Bacteriophage-Based Detection of Brucella abortus in Mixed Culture and Spiked Blood. Viruses. 2017; 9(6):144. https://doi.org/10.3390/v9060144
Chicago/Turabian StyleSergueev, Kirill V., Andrey A. Filippov, and Mikeljon P. Nikolich. 2017. "Highly Sensitive Bacteriophage-Based Detection of Brucella abortus in Mixed Culture and Spiked Blood" Viruses 9, no. 6: 144. https://doi.org/10.3390/v9060144
APA StyleSergueev, K. V., Filippov, A. A., & Nikolich, M. P. (2017). Highly Sensitive Bacteriophage-Based Detection of Brucella abortus in Mixed Culture and Spiked Blood. Viruses, 9(6), 144. https://doi.org/10.3390/v9060144