Redistribution of Endosomal Membranes to the African Swine Fever Virus Replication Site


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cells, Virus and Infections
2.2. ASFV Titration
2.3. Detection and Quantitation of the ASFV Genome
2.4. Proteins Detection by Western Blot
2.5. Antibodies
2.6. Flow Cytometry
2.7. Indirect Immunofluorescence, Conventional and Confocal Microscopy
2.8. Measurement of Rab7 Endosomal Aggregates
2.9. Measurement of Viral Factories and Endosomal Aggregates
2.10. Nocodazole Treatment
2.11. Statistical Analysis
3. Results
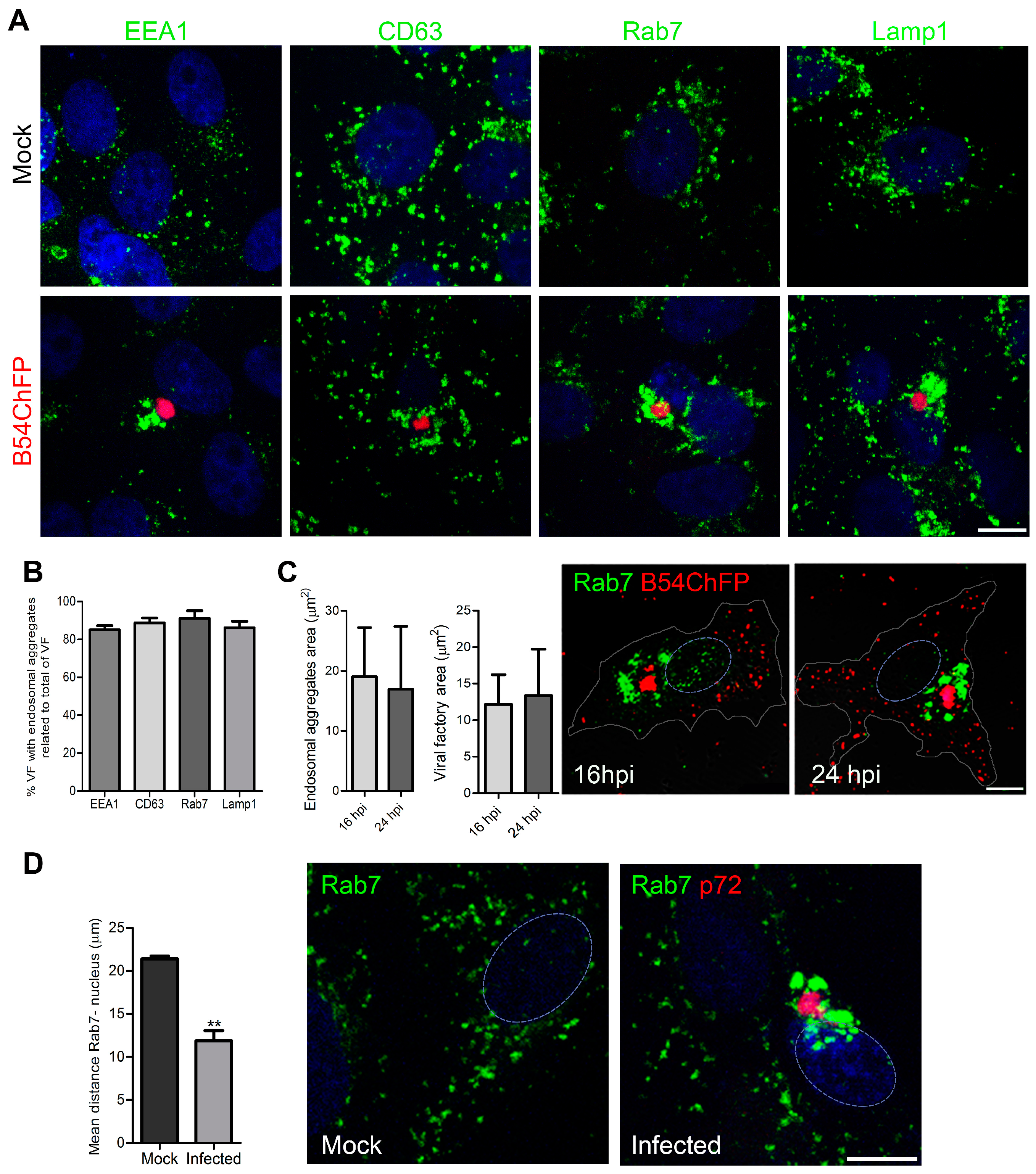
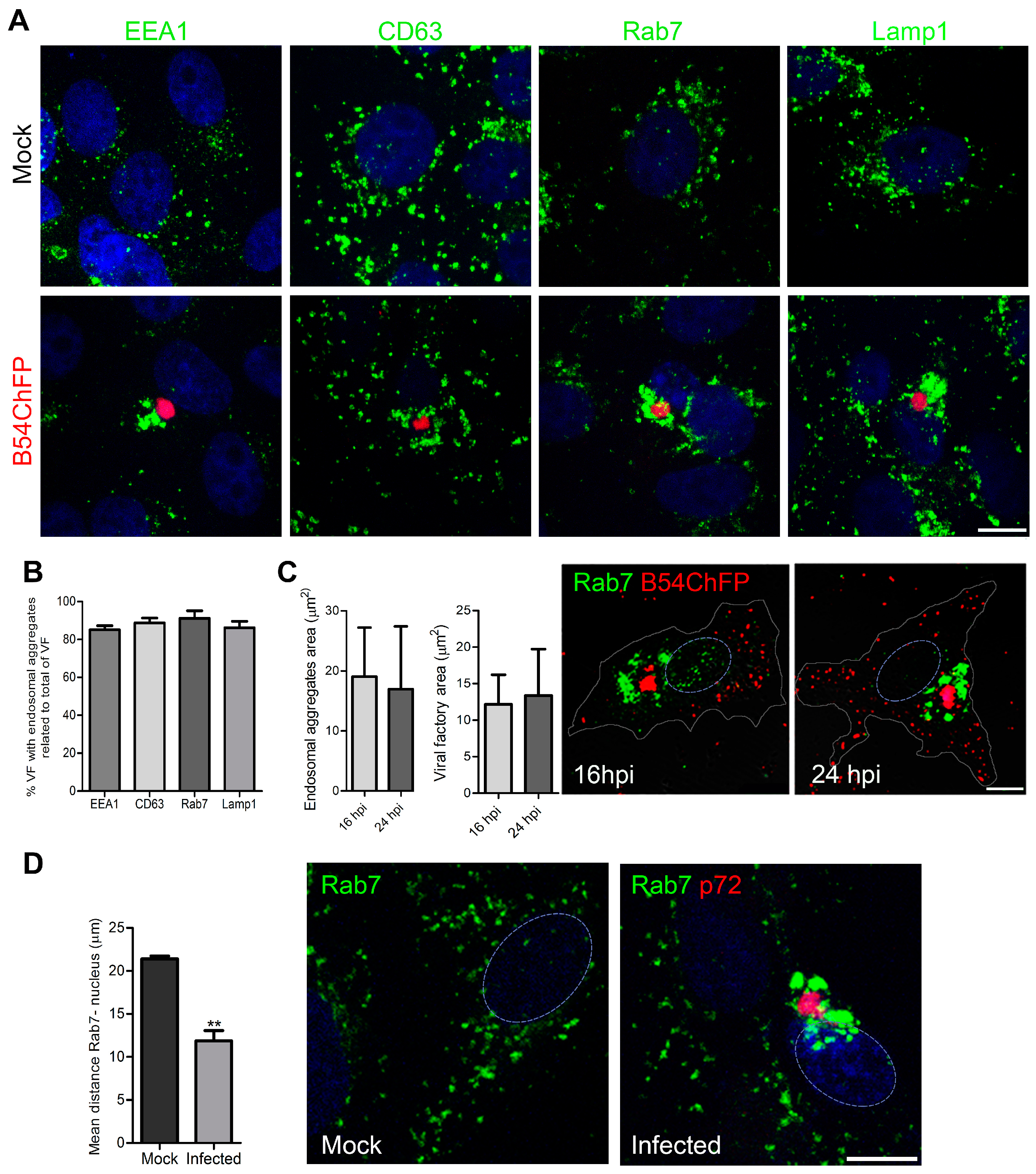
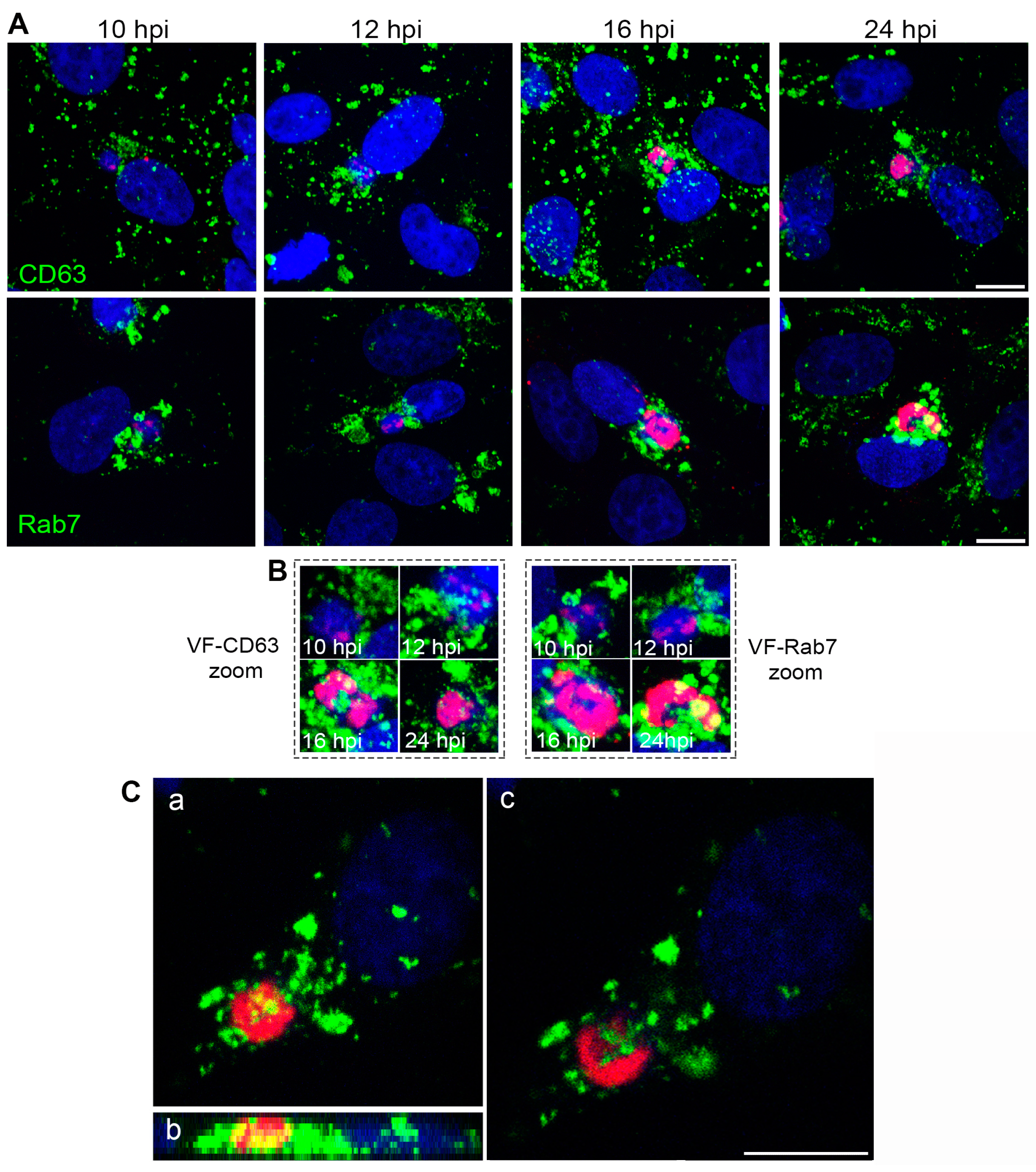
3.1. ASFV Remodels Endosomes
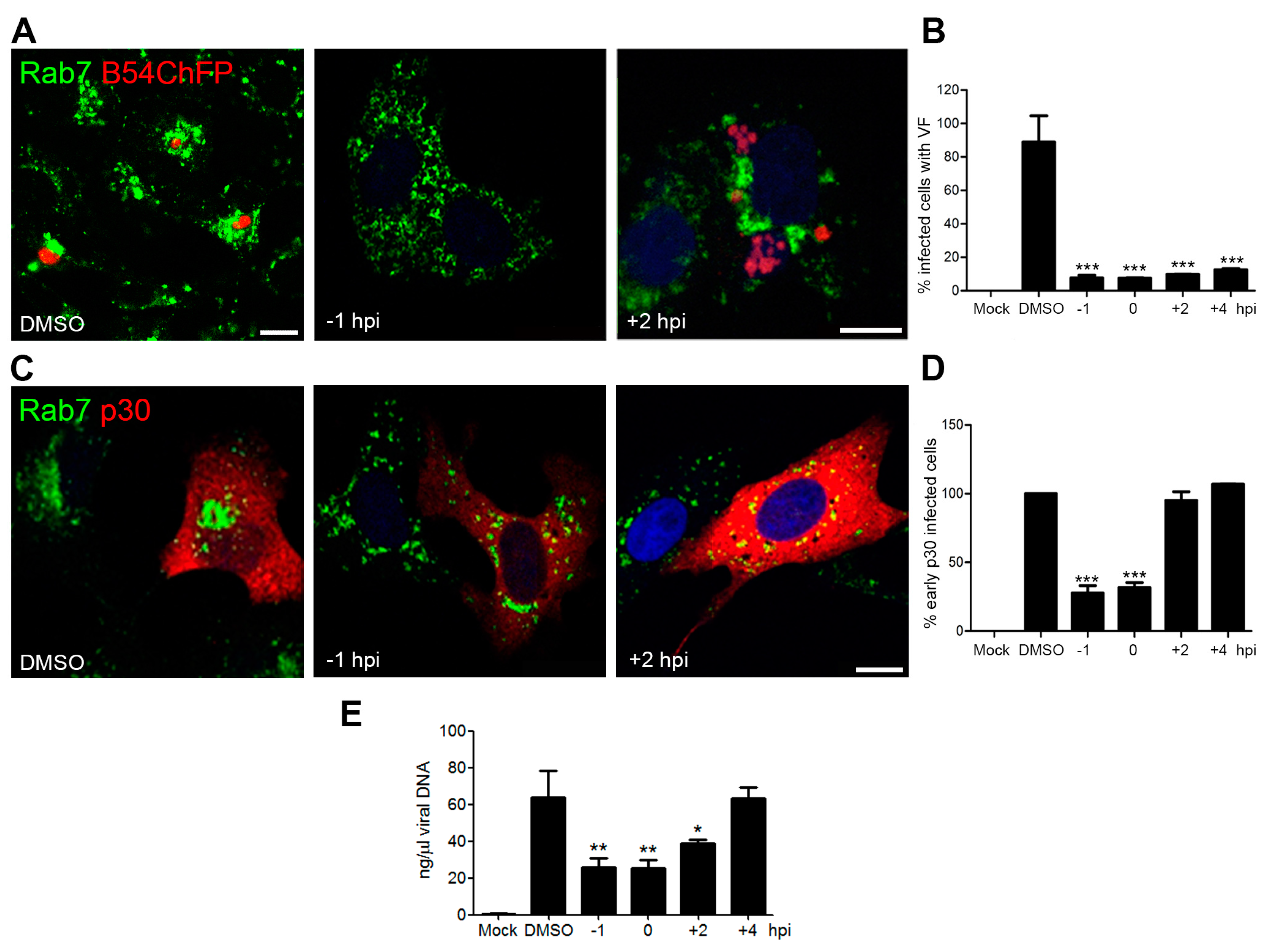
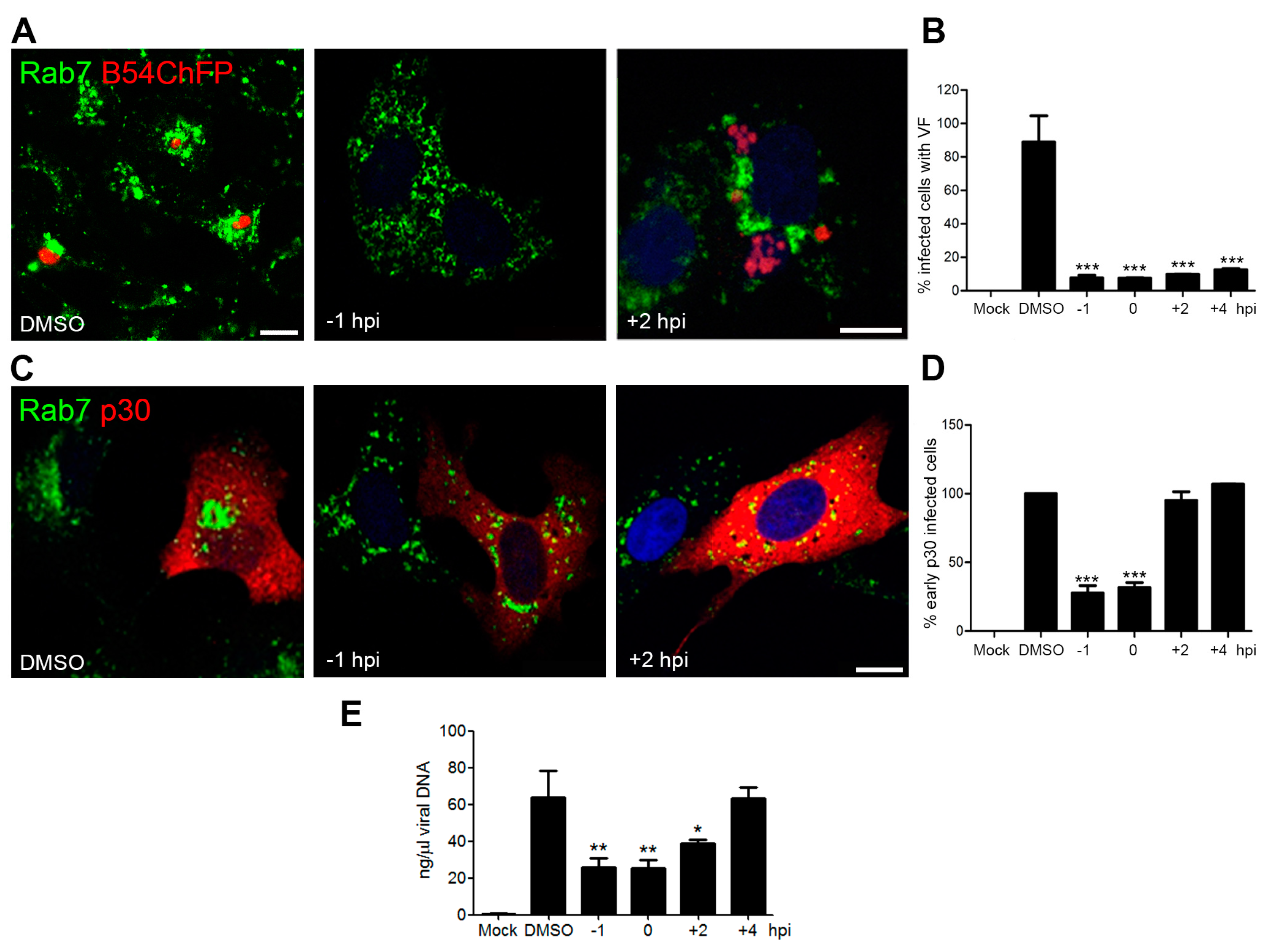
3.2. Endosomal Recruitment Relies on Viral Infection Progression
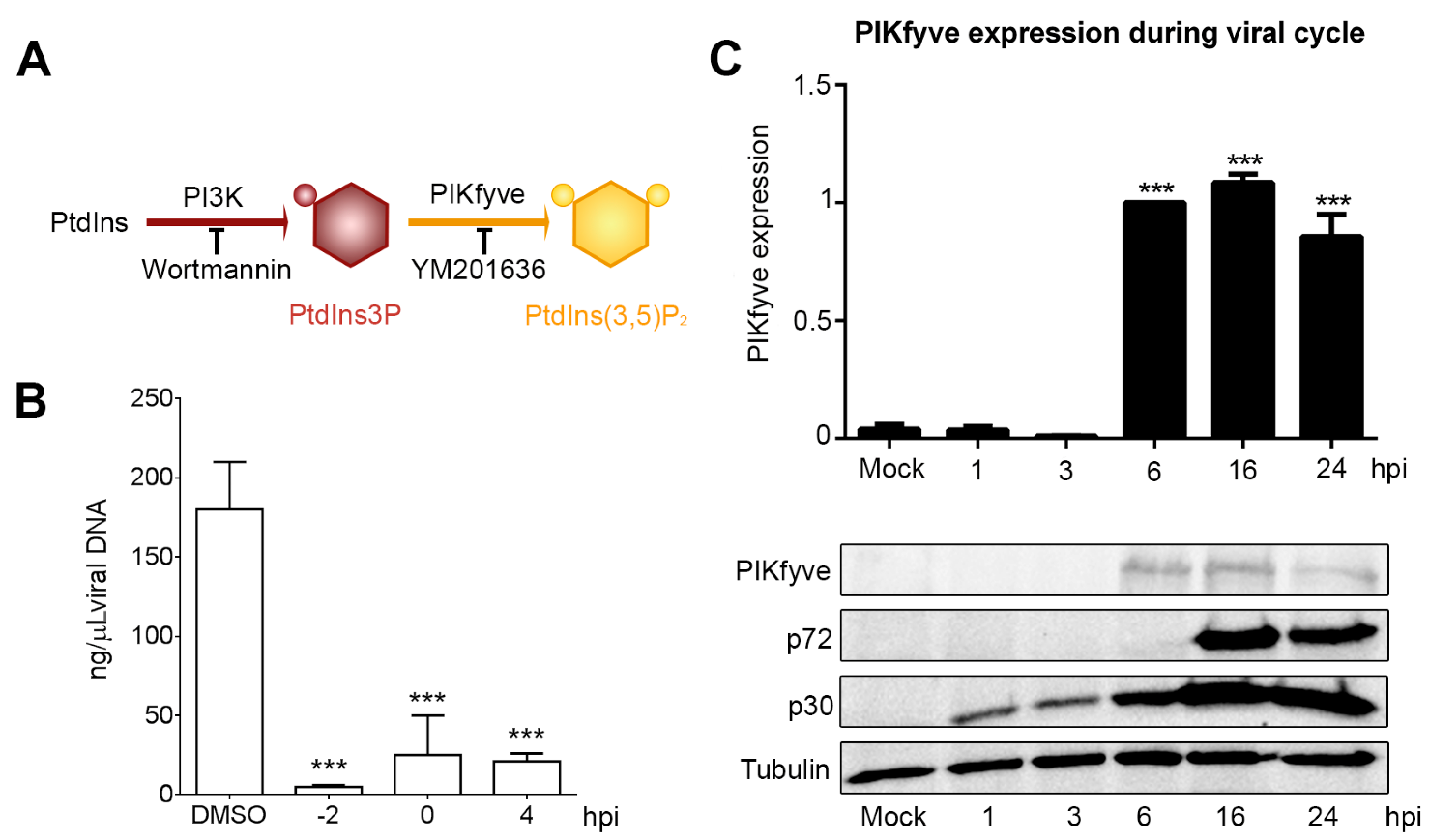
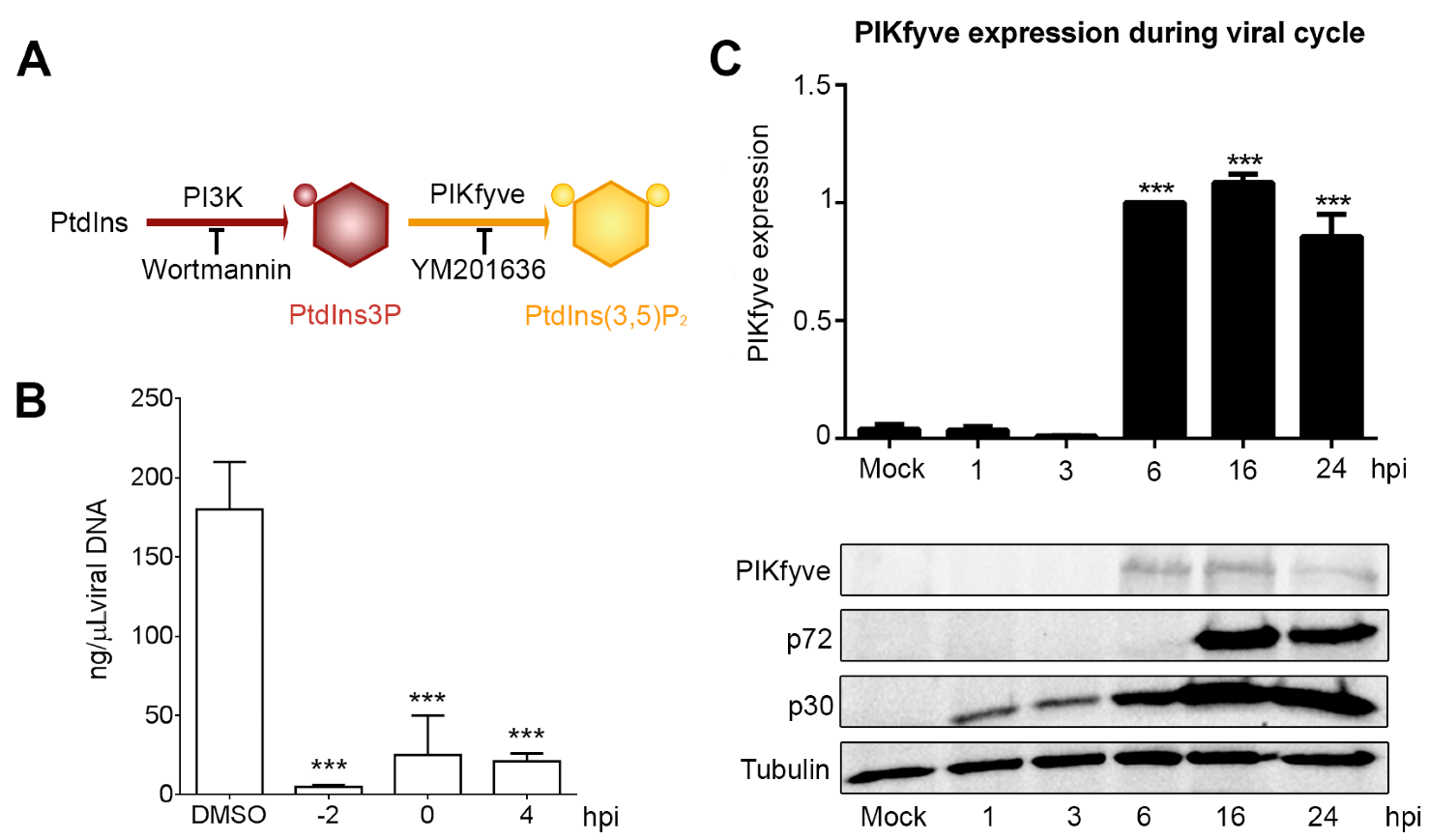
3.3. Endosome Membrane Lipids Are Essential for a Successful ASFV Infection
4. Discussion
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Huotari, J.; Helenius, A. Endosome maturation. Embo. J. 2011, 30, 3481–3500. [Google Scholar] [CrossRef] [PubMed]
- Gruenberg, J. The endocytic pathway: A mosaic of domains. Nat. Rev. Mol. Cell. Biol. 2001, 2, 721–730. [Google Scholar] [CrossRef] [PubMed]
- Bissig, C.; Lenoir, M.; Velluz, M.C.; Kufareva, I.; Abagyan, R.; Overduin, M.; Gruenberg, J. Viral infection controlled by a calcium-dependent lipid-binding module in alix. Dev. Cell 2013, 25, 364–373. [Google Scholar] [CrossRef] [PubMed]
- Greber, U.F. Signalling in viral entry. Cell. Mol. Life Sci. 2002, 59, 608–626. [Google Scholar] [CrossRef] [PubMed]
- Marsh, M.; Helenius, A. Virus entry: Open sesame. Cell 2006, 124, 729–740. [Google Scholar] [CrossRef] [PubMed]
- Mercer, J.; Schelhaas, M.; Helenius, A. Virus entry by endocytosis. Annu. Rev. Biochem. 2010, 79, 803–833. [Google Scholar] [CrossRef] [PubMed]
- Alonso, C.; Galindo, I.; Cuesta-Geijo, M.A.; Cabezas, M.; Hernaez, B.; Munoz-Moreno, R. African swine fever virus-cell interactions: From virus entry to cell survival. Virus Res. 2013, 173, 42–57. [Google Scholar] [CrossRef] [PubMed]
- Callaway, E. Pig fever sweeps across Russia. Nature 2012, 488, 565–566. [Google Scholar] [CrossRef] [PubMed]
- Zakaryan, H.; Revilla, Y. African swine fever virus: Current state and future perspectives in vaccine and antiviral research. Vet. Microbiol. 2016, 185, 15–19. [Google Scholar] [CrossRef] [PubMed]
- O’Donnell, V.; Holinka, L.G.; Krug, P.W.; Gladue, D.P.; Carlson, J.; Sanford, B.; Alfano, M.; Kramer, E.; Lu, Z.; Arzt, J.; et al. African swine fever virus Georgia 2007 with a deletion of virulence-associated gene 9GL (b119l), when administered at low doses, leads to virus attenuation in swine and induces an effective protection against homologous challenge. J. Virol. 2015, 89, 8556–8566. [Google Scholar] [CrossRef] [PubMed]
- Pejsak, Z.; Truszczynski, M.; Niemczuk, K.; Kozak, E.; Markowska-Daniel, I. Epidemiology of african swine fever in poland since the detection of the first case. Pol. J. Vet. Sci. 2014, 17, 665–672. [Google Scholar] [CrossRef] [PubMed]
- Galindo, I.; Cuesta-Geijo, M.A.; Hlavova, K.; Munoz-Moreno, R.; Barrado-Gil, L.; Dominguez, J.; Alonso, C. African swine fever virus infects macrophages, the natural host cells, via clathrin- and cholesterol-dependent endocytosis. Virus Res. 2015, 200, 45–55. [Google Scholar] [CrossRef] [PubMed]
- Hernaez, B.; Alonso, C. Dynamin- and clathrin-dependent endocytosis in african swine fever virus entry. J. Virol. 2010, 84, 2100–2109. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, E.G.; Quintas, A.; Perez-Nunez, D.; Nogal, M.; Barroso, S.; Carrascosa, A.L.; Revilla, Y. African swine fever virus uses macropinocytosis to enter host cells. PLoS Pathog. 2012, 8, e1002754. [Google Scholar] [CrossRef] [PubMed]
- Hernaez, B.; Guerra, M.; Salas, M.L.; Andres, G. African swine fever virus undergoes outer envelope disruption, capsid disassembly and inner envelope fusion before core release from multivesicular endosomes. PLoS Pathog. 2016, 12, e1005595. [Google Scholar] [CrossRef] [PubMed]
- Cuesta-Geijo, M.A.; Galindo, I.; Hernaez, B.; Quetglas, J.I.; Dalmau-Mena, I.; Alonso, C. Endosomal maturation, RAB7 GTPase and phosphoinositides in african swine fever virus entry. PLoS ONE 2012, 7, e48853. [Google Scholar] [CrossRef] [PubMed]
- Cuesta-Geijo, M.A.; Chiappi, M.; Galindo, I.; Barrado-Gil, L.; Munoz-Moreno, R.; Carrascosa, J.L.; Alonso, C. Cholesterol flux is required for endosomal progression of African swine fever virions during the initial establishment of infection. J. Virol. 2015, 90, 1534–1543. [Google Scholar] [CrossRef] [PubMed]
- Netherton, C.L.; Wileman, T.E. African swine fever virus organelle rearrangements. Virus Res. 2013, 173, 76–86. [Google Scholar] [CrossRef] [PubMed]
- Alonso, C.; Miskin, J.; Hernaez, B.; Fernandez-Zapatero, P.; Soto, L.; Canto, C.; Rodriguez-Crespo, I.; Dixon, L.; Escribano, J.M. African swine fever virus protein p54 interacts with the microtubular motor complex through direct binding to light-chain dynein. J. Virol. 2001, 75, 9819–9827. [Google Scholar] [CrossRef] [PubMed]
- Cobbold, C.; Whittle, J.T.; Wileman, T. Involvement of the endoplasmic reticulum in the assembly and envelopment of African swine fever virus. J. Virol. 1996, 70, 8382–8390. [Google Scholar] [PubMed]
- Andres, G.; Garcia-Escudero, R.; Simon-Mateo, C.; Vinuela, E. African swine fever virus is enveloped by a two-membraned collapsed cisterna derived from the endoplasmic reticulum. J. Virol. 1998, 72, 8988–9001. [Google Scholar] [PubMed]
- Rouiller, I.; Brookes, S.M.; Hyatt, A.D.; Windsor, M.; Wileman, T. African swine fever virus is wrapped by the endoplasmic reticulum. J. Virol. 1998, 72, 2373–2387. [Google Scholar] [PubMed]
- Andres, G.; Garcia-Escudero, R.; Vinuela, E.; Salas, M.L.; Rodriguez, J.M. African swine fever virus structural protein pR120R is essential for virus transport from assembly sites to plasma membrane but not for infectivity. J. Virol. 2001, 75, 6758–6768. [Google Scholar] [CrossRef] [PubMed]
- Jouvenet, N.; Monaghan, P.; Way, M.; Wileman, T. Transport of African swine fever virus from assembly sites to the plasma membrane is dependent on microtubules and conventional kinesin. J. Virol. 2004, 78, 7990–8001. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, Z.G.; de Matos, A.P.; Rodrigues-Pousada, C. Association of African swine fever virus with the cytoskeleton. Virus Res. 1988, 11, 175–192. [Google Scholar] [CrossRef]
- De Matos, A.P.; Carvalho, Z.G. African swine fever virus interaction with microtubules. Biol. Cell 1993, 78, 229–234. [Google Scholar] [CrossRef]
- Hernaez, B.; Escribano, J.M.; Alonso, C. Visualization of the African swine fever virus infection in living cells by incorporation into the virus particle of green fluorescent protein-p54 membrane protein chimera. Virology 2006, 350, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Enjuanes, L.; Carrascosa, A.L.; Moreno, M.A.; Vinuela, E. Titration of African swine fever (ASF) virus. J. Gen. Virol. 1976, 32, 471–477. [Google Scholar] [CrossRef] [PubMed]
- Yanez, R.J.; Rodriguez, J.M.; Nogal, M.L.; Yuste, L.; Enriquez, C.; Rodriguez, J.F.; Vinuela, E. Analysis of the complete nucleotide sequence of African swine fever virus. Virology 1995, 208, 249–278. [Google Scholar] [CrossRef] [PubMed]
- King, D.P.; Reid, S.M.; Hutchings, G.H.; Grierson, S.S.; Wilkinson, P.J.; Dixon, L.K.; Bastos, A.D.; Drew, T.W. Development of a TaqMan PCR assay with internal amplification control for the detection of African swine fever virus. J. Virol Methods 2003, 107, 53–61. [Google Scholar] [CrossRef]
- Hernaez, B.; Escribano, J.M.; Alonso, C. African swine fever virus protein p30 interaction with heterogeneous nuclear ribonucleoprotein k (HNRNP-K) during infection. FEBS Lett. 2008, 582, 3275–3280. [Google Scholar] [CrossRef] [PubMed]
- Afonso, C.L.; Alcaraz, C.; Brun, A.; Sussman, M.D.; Onisk, D.V.; Escribano, J.M.; Rock, D.L. Characterization of p30, a highly antigenic membrane and secreted protein of African swine fever virus. Virology 1992, 189, 368–373. [Google Scholar] [CrossRef]
- Rojo, G.; Chamorro, M.; Salas, M.L.; Vinuela, E.; Cuezva, J.M.; Salas, J. Migration of mitochondria to viral assembly sites in African swine fever virus-infected cells. J. Virol. 1998, 72, 7583–7588. [Google Scholar] [PubMed]
- Netherton, C.L.; McCrossan, M.C.; Denyer, M.; Ponnambalam, S.; Armstrong, J.; Takamatsu, H.H.; Wileman, T.E. African swine fever virus causes microtubule-dependent dispersal of the trans-golgi network and slows delivery of membrane protein to the plasma membrane. J. Virol. 2006, 80, 11385–11392. [Google Scholar] [CrossRef] [PubMed]
- Stefanovic, S.; Windsor, M.; Nagata, K.I.; Inagaki, M.; Wileman, T. Vimentin rearrangement during African swine fever virus infection involves retrograde transport along microtubules and phosphorylation of vimentin by calcium calmodulin kinase II. J. Virol. 2005, 79, 11766–11775. [Google Scholar] [CrossRef] [PubMed]
- Novoa, R.R.; Calderita, G.; Arranz, R.; Fontana, J.; Granzow, H.; Risco, C. Virus factories: Associations of cell organelles for viral replication and morphogenesis. Biol. Cell 2005, 97, 147–172. [Google Scholar] [CrossRef] [PubMed]
- Miller, S.; Krijnse-Locker, J. Modification of intracellular membrane structures for virus replication. Nat. Rev. Microbiol. 2008, 6, 363–374. [Google Scholar] [CrossRef] [PubMed]
- Das, S.; Vasanji, A.; Pellett, P.E. Three-dimensional structure of the human cytomegalovirus cytoplasmic virion assembly complex includes a reoriented secretory apparatus. J. Virol. 2007, 81, 11861–11869. [Google Scholar] [CrossRef] [PubMed]
- Alwine, J.C. The human cytomegalovirus assembly compartment: A masterpiece of viral manipulation of cellular processes that facilitates assembly and egress. PLoS Pathog. 2012, 8, e1002878. [Google Scholar] [CrossRef] [PubMed]
- Kerr, M.C.; Wang, J.T.; Castro, N.A.; Hamilton, N.A.; Town, L.; Brown, D.L.; Meunier, F.A.; Brown, N.F.; Stow, J.L.; Teasdale, R.D. Inhibition of the PtdIns(5) kinase PIKfyve disrupts intracellular replication of Salmonella. EMBO J. 2010, 29, 1331–1347. [Google Scholar] [CrossRef] [PubMed]
- Weber, S.S.; Ragaz, C.; Reus, K.; Nyfeler, Y.; Hilbi, H. Legionella pneumophila exploits PI(4)P to anchor secreted effector proteins to the replicative vacuole. PLoS Pathog. 2006, 2, e46. [Google Scholar] [CrossRef] [PubMed]
- Hsu, N.Y.; Ilnytska, O.; Belov, G.; Santiana, M.; Chen, Y.H.; Takvorian, P.M.; Pau, C.; van der Schaar, H.; Kaushik-Basu, N.; Balla, T.; et al. Viral reorganization of the secretory pathway generates distinct organelles for RNA replication. Cell 2010, 141, 799–811. [Google Scholar] [CrossRef] [PubMed]
- Tait, S.W.; Reid, E.B.; Greaves, D.R.; Wileman, T.E.; Powell, P.P. Mechanism of inactivation of NF-κb by a viral homologue of i kappa b alpha. Signal-induced release of i kappa b alpha results in binding of the viral homologue to NF-κb. J. Biol. Chem. 2000, 275, 34656–34664. [Google Scholar] [CrossRef] [PubMed]
- Rizopoulos, Z.; Balistreri, G.; Kilcher, S.; Martin, C.K.; Syedbasha, M.; Helenius, A.; Mercer, J. Vaccinia virus infection requires maturation of macropinosomes. Traffic 2015, 16, 814–831. [Google Scholar] [CrossRef] [PubMed]
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cuesta-Geijo, M.Á.; Barrado-Gil, L.; Galindo, I.; Muñoz-Moreno, R.; Alonso, C. Redistribution of Endosomal Membranes to the African Swine Fever Virus Replication Site. Viruses 2017, 9, 133. https://doi.org/10.3390/v9060133
Cuesta-Geijo MÁ, Barrado-Gil L, Galindo I, Muñoz-Moreno R, Alonso C. Redistribution of Endosomal Membranes to the African Swine Fever Virus Replication Site. Viruses. 2017; 9(6):133. https://doi.org/10.3390/v9060133
Chicago/Turabian StyleCuesta-Geijo, Miguel Ángel, Lucía Barrado-Gil, Inmaculada Galindo, Raquel Muñoz-Moreno, and Covadonga Alonso. 2017. "Redistribution of Endosomal Membranes to the African Swine Fever Virus Replication Site" Viruses 9, no. 6: 133. https://doi.org/10.3390/v9060133
APA StyleCuesta-Geijo, M. Á., Barrado-Gil, L., Galindo, I., Muñoz-Moreno, R., & Alonso, C. (2017). Redistribution of Endosomal Membranes to the African Swine Fever Virus Replication Site. Viruses, 9(6), 133. https://doi.org/10.3390/v9060133