
Immunotherapeutic Potential of Oncolytic H-1 Parvovirus: Hints of Glioblastoma Microenvironment Conversion towards Immunogenicity

 ,
, 

Abstract
:1. Glioblastoma: Current Clinical State-Of-The-Art
2. Immune Suppression in Glioblastoma Patients
3. Immune-Targeted Therapeutic Approaches for Glioblastoma
3.1. Tumor Vaccination
3.2. Immune Checkpoint Inhibition
3.3. Adoptive T Cell Transfer
4. Need for Novel Immunotherapeutic Strategies: Oncolytic Viruses
5. Oncolytic H-1 Parvovirus
6. Immunotherapeutic Potential of Oncolytic H-1PV: Preclinical Evidence
6.1. In Tumor Models Other than Glioma
6.2. In Glioma Models
7. Immunotherapeutic Potential of H-1PV in Glioblastoma Patients: First Evidence of Immunogenic Tumor Microenvironment Conversion
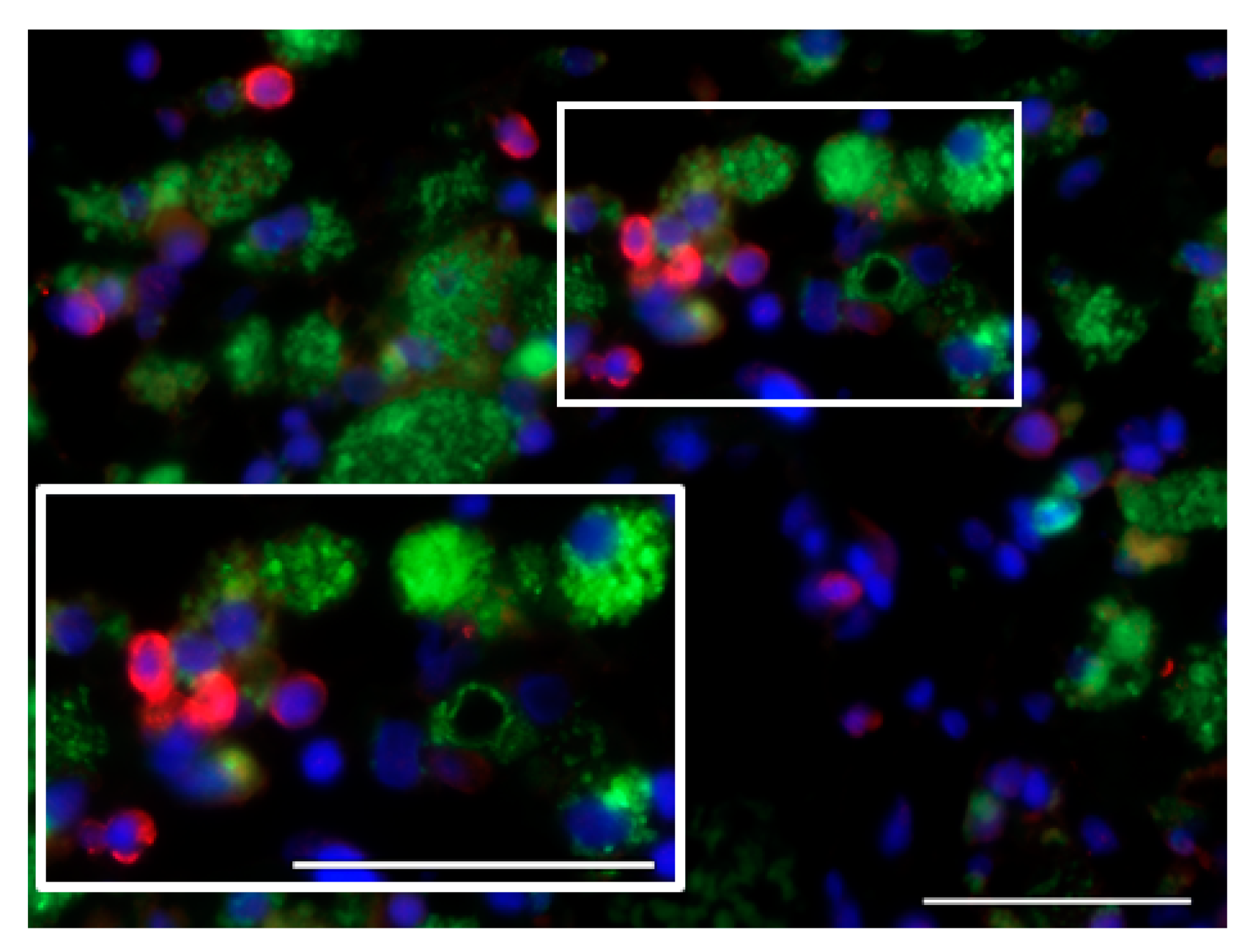
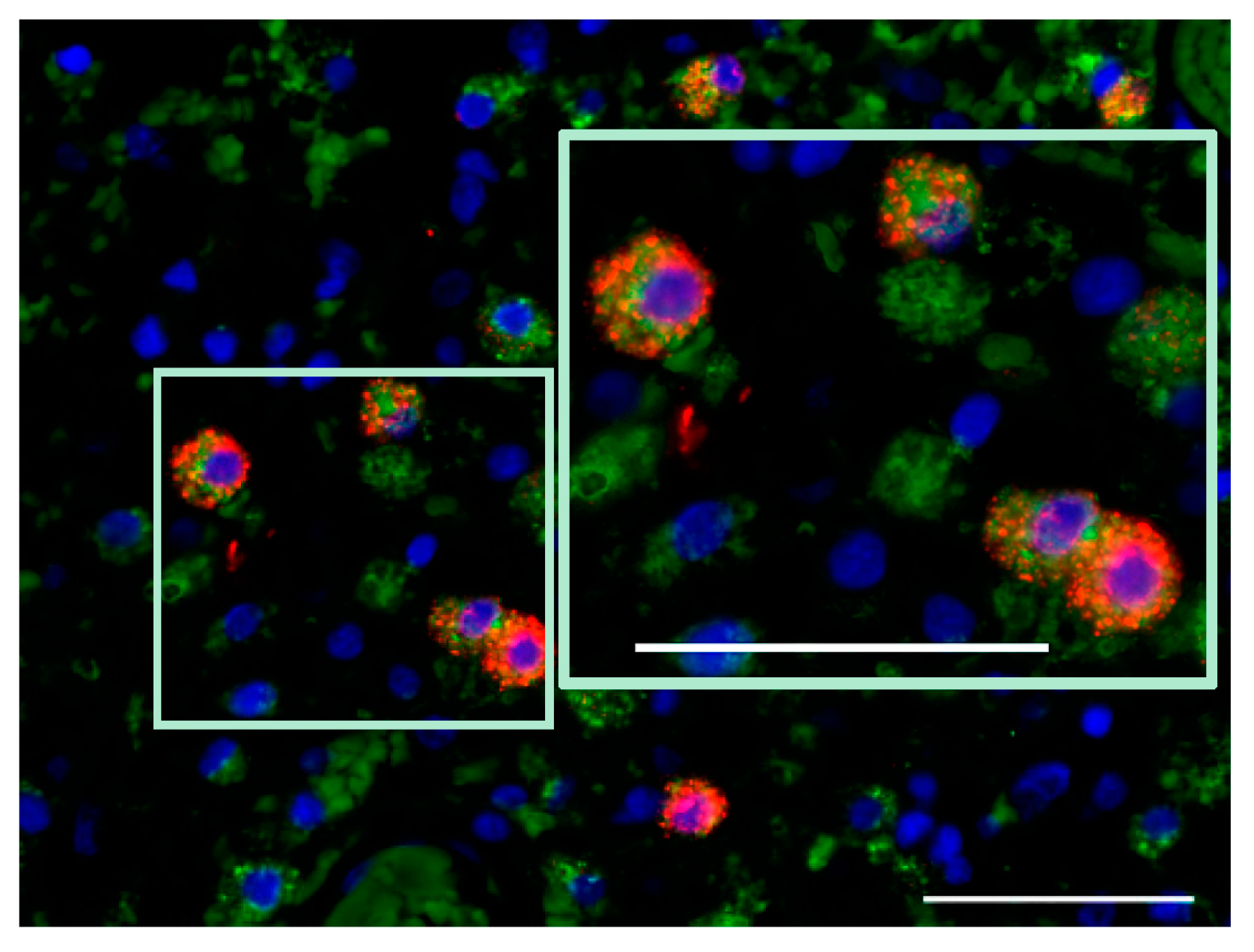
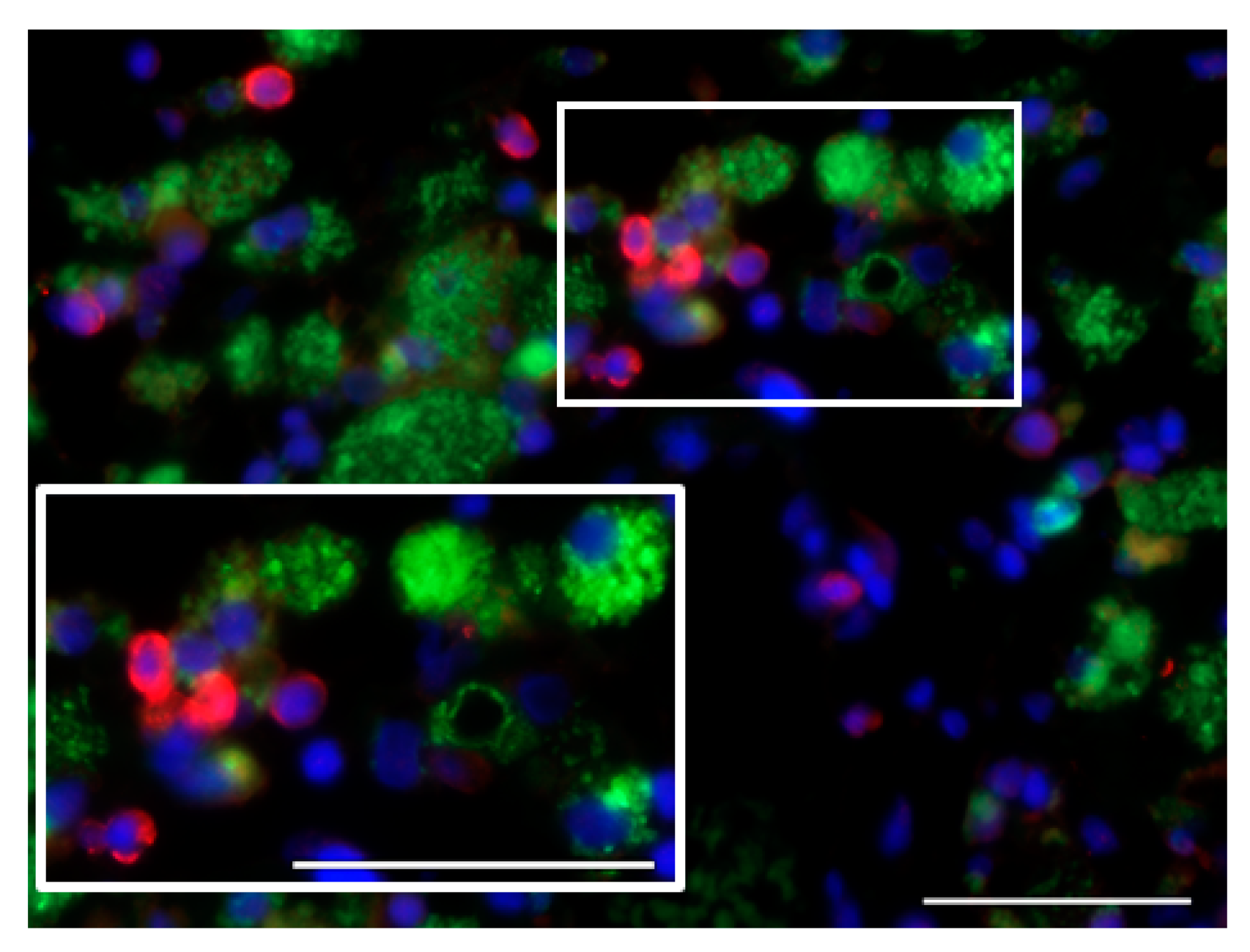
7.1. Glioblastoma Infiltration with Immune Cells
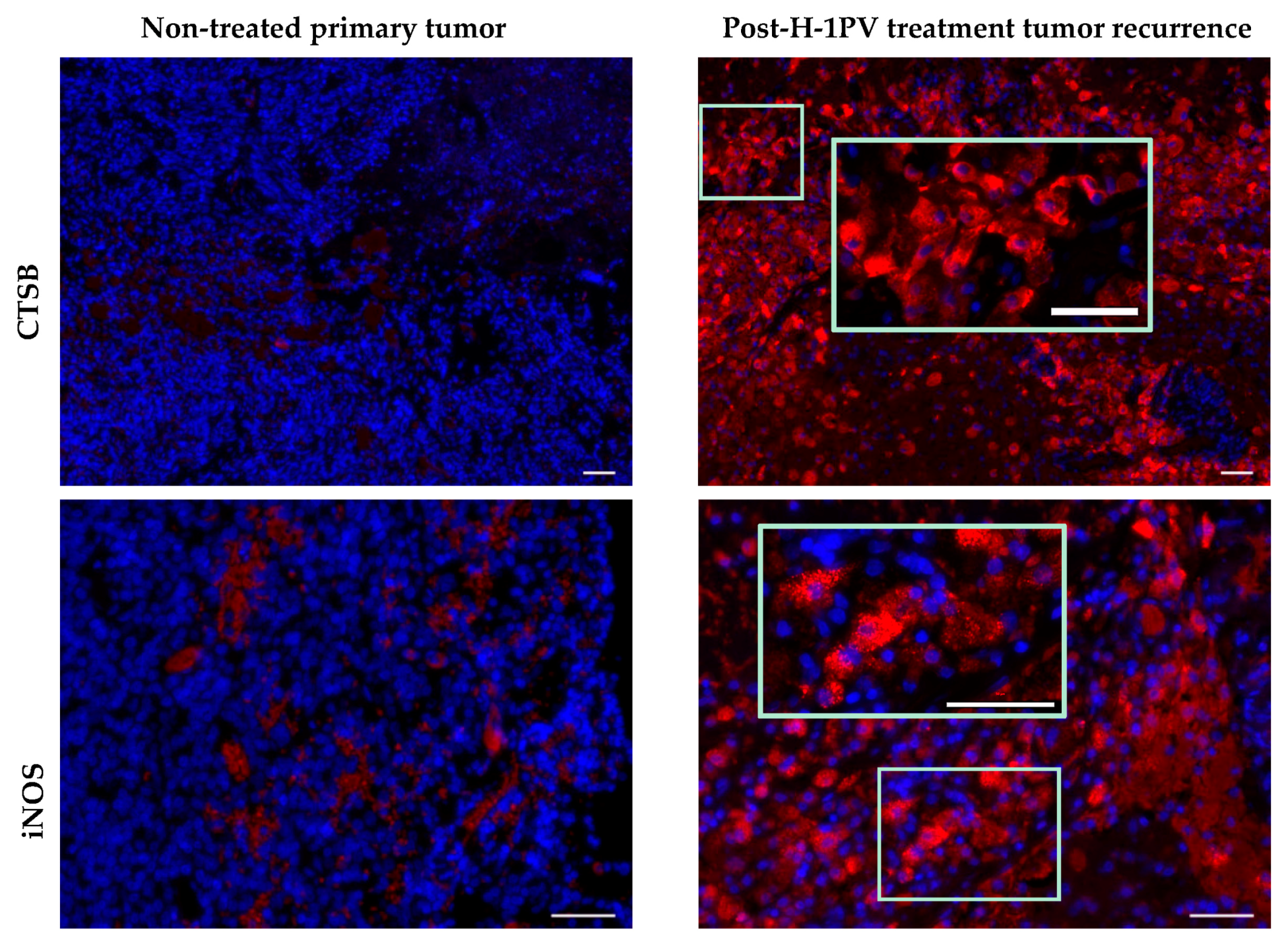
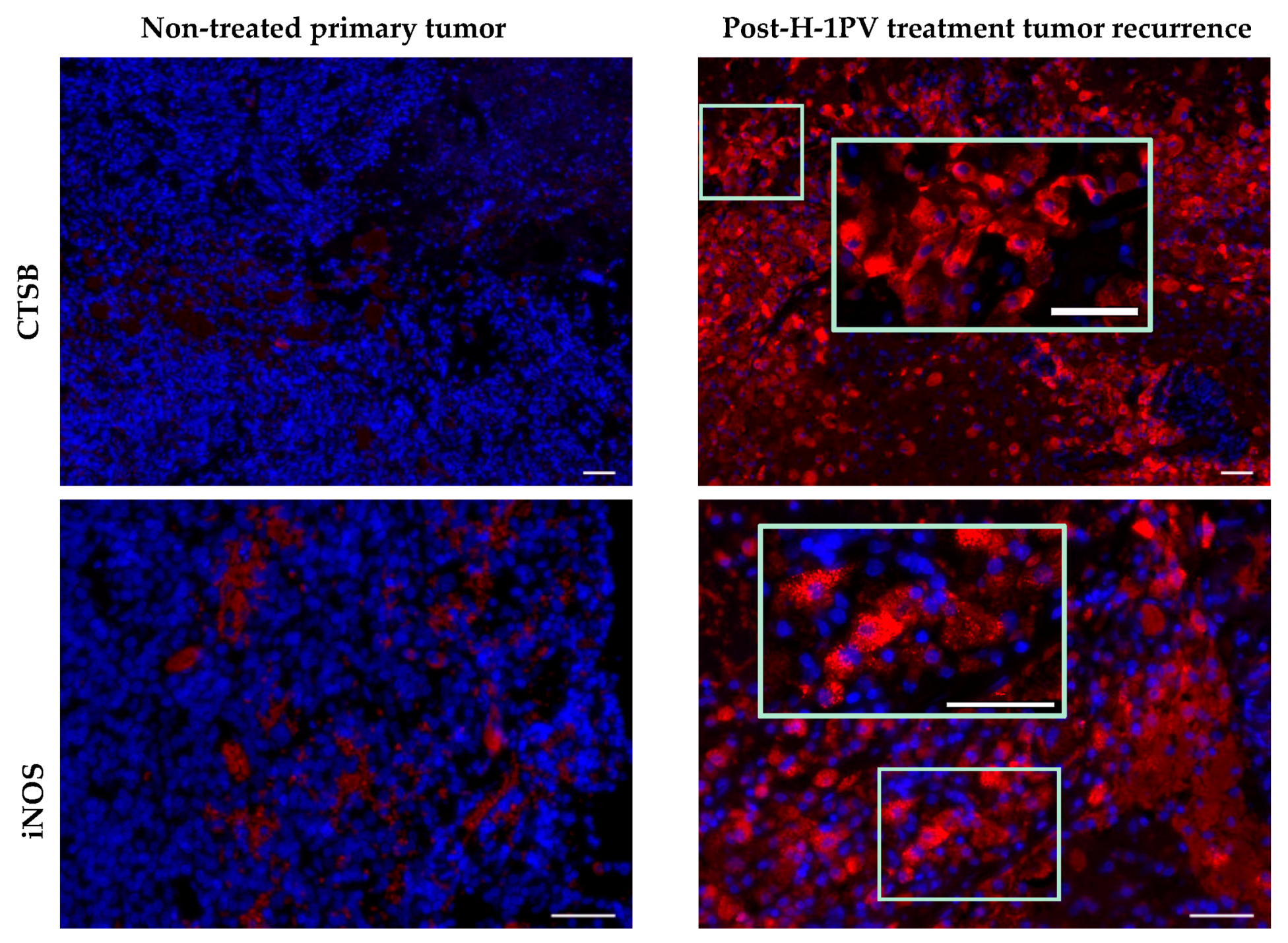
7.2. Activation Status of Glioblastoma-Infiltrating Immune Cells
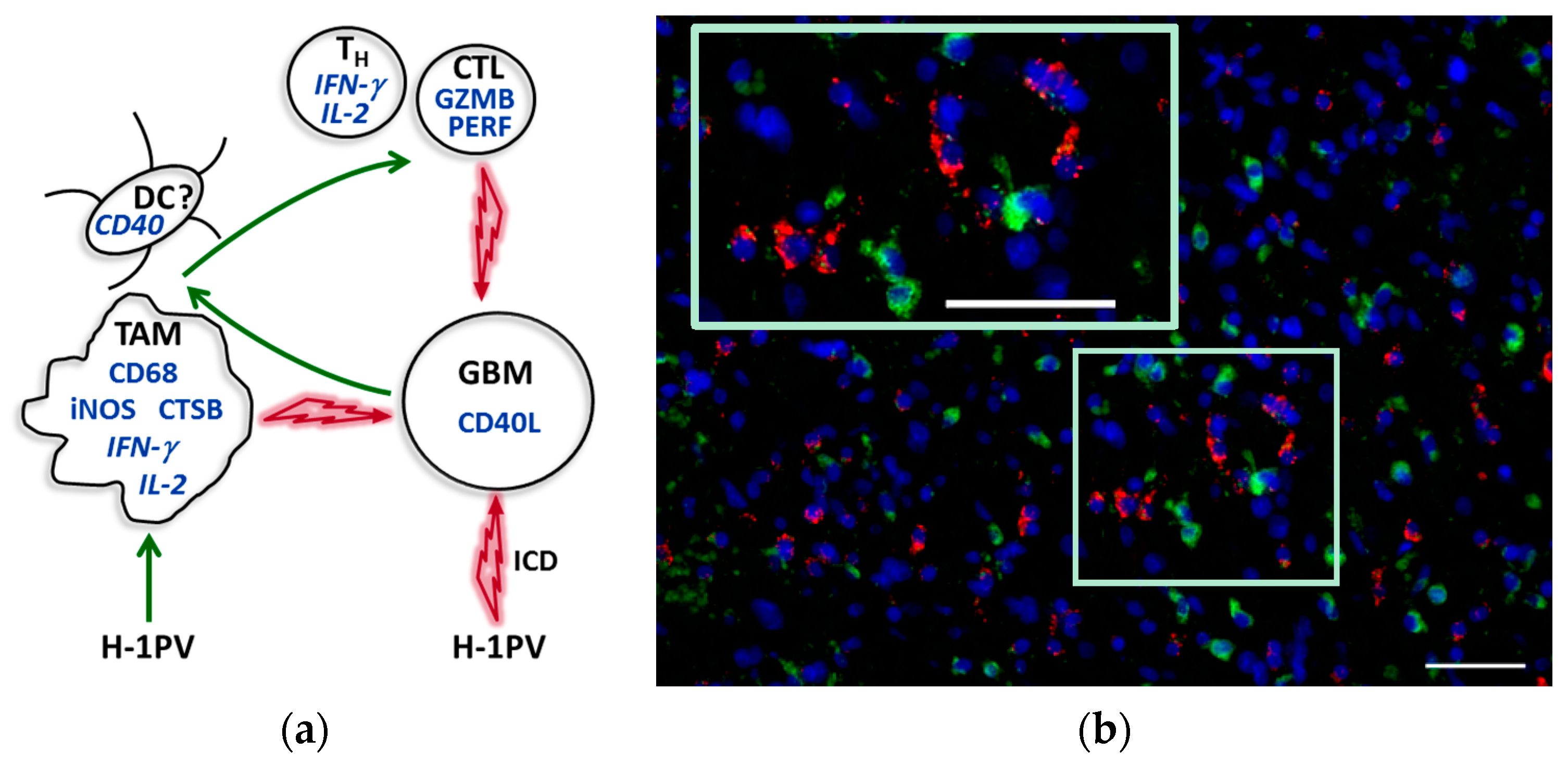
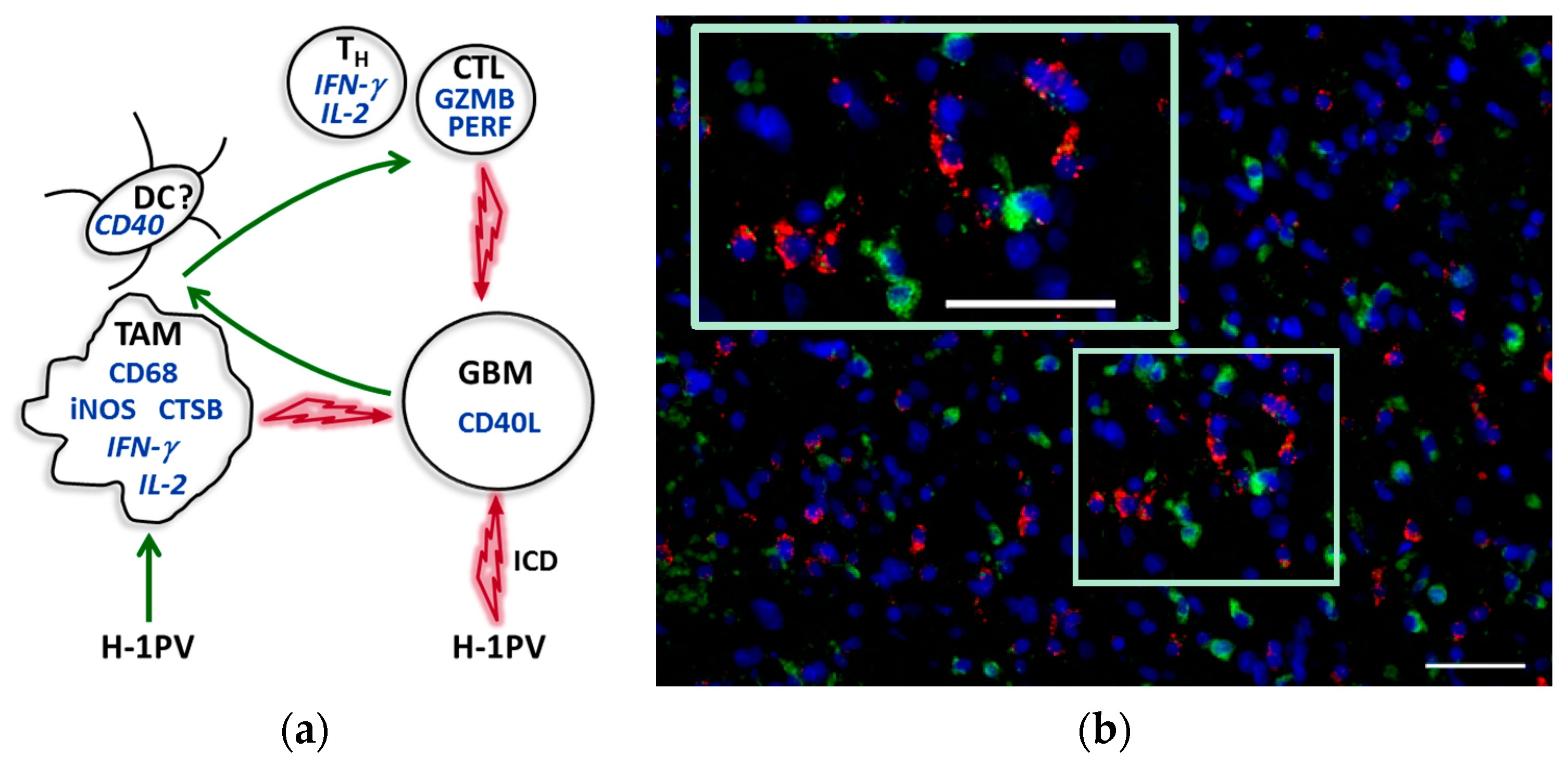
7.3. CD40L Expression by Glioblastoma Cells
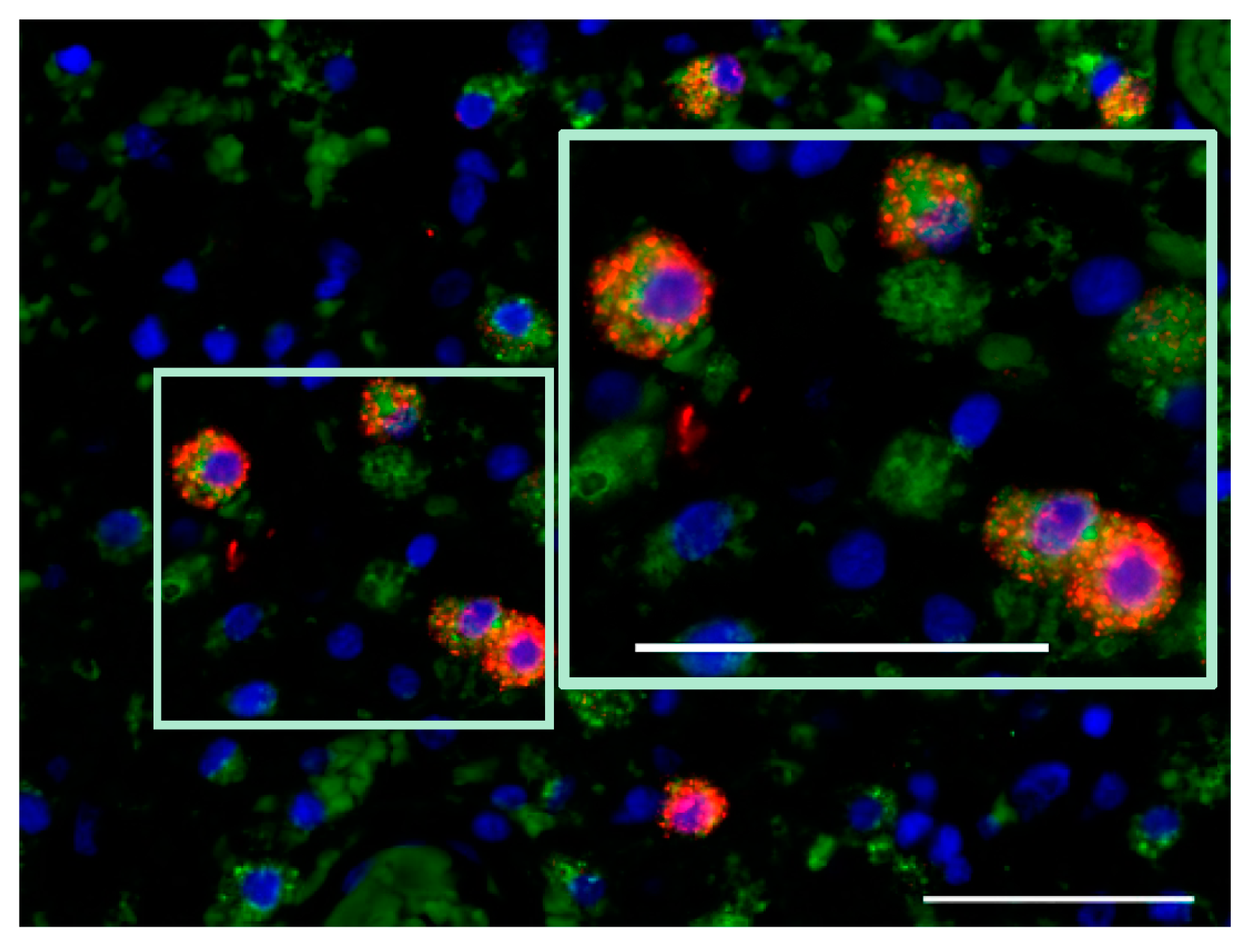
7.4. Proinflammatory Cytokine Production in Glioblastoma Microenvironment
7.5. Induction of Cathepsin B Expression
8. Other Oncolytic Viruses and Tumor Microenvironment Modulation
8.1. Preclinical Studies
8.2. Clinical Studies
9. Tumor Microenvironment Modulation by H-1PV: Current Hypothesis and Perspective
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Wilson, T.A.; Karajanis, M.A.; Harter, D.H. Glioblastoma multiforme: State of the art and future therapeutics. Surg. Neurol. Int. 2014, 5, 64. [Google Scholar] [CrossRef] [PubMed]
- Stupp, R.; Mason, W.P.; van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.; Mirimanoff, R.O. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef] [PubMed]
- National Comprehensive Cancer Network Clinical Practice Guidelines in Oncology: Central Nervous System Cancers, Version 1.2015. 2015. Available online: https://www.nccn.org/professionals/physician_gls/pdf/cns.pdf (accessed on 17 October 2017).
- Weller, M.; Cloughesy, T.; Perry, J.R.; Wick, W. Standards of care for treatment of recurrent glioblastoma—Are we there yet? Neuro Oncol. 2013, 15, 4–27. [Google Scholar] [CrossRef] [PubMed]
- Suchorska, B.; Weller, M.; Tabatabai, G.; Senft, C.; Hau, P.; Sabel, M.C.; Herrlinger, U.; Ketter, R.; Schlegel, U.; Marosi, C.; et al. Complete resection of contrast-enhancing tumor volume is associated with improved survival in recurrent glioblastoma—Results from the DIRECTOR trial. Neuro Oncol. 2016, 18, 549–556. [Google Scholar] [CrossRef] [PubMed]
- Franceschi, E.; Tosoni, A.; Bartolini, S.; Mazzocchi, V.; Fioravanti, A.; Brandes, A.A. Treatment options for recurrent glioblastoma: Pittfalls and future trends. Expert Rev. Anticancer Ther. 2009, 9, 613–619. [Google Scholar] [CrossRef] [PubMed]
- Cohen, M.H.; Shen, Y.L.; Keegan, P.; Pazdur, R. FDA drug approval summary: Bevacizumab (Avastin) as treatment of recurrent glioblastoma multiforme. Oncologist 2009, 14, 1131–1138. [Google Scholar] [CrossRef] [PubMed]
- Razavi, S.-M.; Lee, K.E.; Jin, B.E.; Aujla, P.S.; Gholamin, S.; Li, G. Immune evasion strategies of glioblastoma. Front. Surg. 2016, 3, 11. [Google Scholar] [CrossRef] [PubMed]
- Gustafson, M.P.; Lin, Y.; New, K.C.; Bulur, P.A.; O’Neill, B.P.; Gastineau, D.A.; Dietz, A.B. Systemic immune suppression in glioblastoma: The interplay between CD14+HLA-DRlo/neg monocytes, tumor factors, and dexamethasone. Neuro Oncol. 2010, 12, 631–644. [Google Scholar] [CrossRef] [PubMed]
- Fecci, P.E.; Mitchell, D.A.; Whitesides, J.F.; Xie, W.; Friedman, A.H.; Archer, G.E.; Herndon, J.E.; Bigner, D.D.; Dranoff, G.; Sampson, J.H. Increased regulatory T cell fraction amidst a diminished CD4 compartment explains cellular immune defects in patients with malignant glioma. Cancer Res. 2006, 66, 3294–3302. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, J.C.; Gonzalez, G.C.; Zhang, L.; Ibrahim, G.; Kelly, J.J.; Gustafson, M.P.; Lin, Y.; Dietz, A.B.; Forsyth, P.A.; Wee Yong, V.; et al. Normal human monocytes exposed to glioma cells acquire myeloid-derived suppressor cell-like properties. Neuro Oncol. 2010, 12, 351–365. [Google Scholar] [CrossRef] [PubMed]
- Ogden, A.T.; Horgan, D.; Waziri, A.; Anderson, D.; Louca, J.; McKhann, G.M.; Sisti, M.B.; Parsa, A.T.; Bruce, J.N. Defective receptor expression and dendritic cell differentiation of monocytes in glioblastomas. Neurosurgery 2006, 59, 902–910. [Google Scholar] [CrossRef] [PubMed]
- Nduom, E.K.; Weller, M.; Heimberger, A.B. Immunosuppressive mechanisms in glioblastoma. Neuro Oncol. 2015, 17, vii9–vii14. [Google Scholar] [CrossRef] [PubMed]
- Huang, B.; Zhang, H.; Gu, L.; Ye, B.; Jian, Z.; Stary, C.; Xiong, X. Advances in immunotherapy for glioblastoma multiforme. J. Immunol. Res. 2017, 2017, 3597613. [Google Scholar] [CrossRef] [PubMed]
- Weiss, T.; Weller, M.; Roth, P. Immunotherapy for glioblastoma: Concepts and challenges. Curr. Opin. Neurol. 2015, 28, 639–646. [Google Scholar] [CrossRef] [PubMed]
- Marucci, A. Rindopeptimut Misses OS Endpoint in Phase III Glioblastoma Trial; OncLive: Cranbury, NJ, USA, 2016; Available online: http://www.onclive.com/web-exclusives/rindopepimut-misses-os-endpoint-in-phase-iii-glioblastoma-trial (accessed on 20 October 2017).
- Pelloski, C.E.; Ballman, K.V.; Furth, A.F.; Zhang, L.; Lin, E.; Sulman, E.P.; Bhat, K.; McDonald, J.M.; Yung, W.K.; Colman, H.; et al. Epidermal growth factor receptor variant III status defines clinically distinct subtypes of glioblastoma. J. Clin. Oncol. 2007, 25, 2288–2294. [Google Scholar] [CrossRef] [PubMed]
- Dutoit, V.; Herold-Mende, C.; Hilf, N.; Schoor, O.; Beckhove, P.; Bucher, J.; Dorsch, K.; Flohr, S.; Fritsche, J.; Lewandrowski, P.; et al. Exploiting the glioblastoma peptidome to discover novel tumour-associated antigens for immunotherapy. Brain 2012, 135, 1042–1054. [Google Scholar] [CrossRef] [PubMed]
- Hodi, F.S.; O’Day, S.J.; McDermot, D.F.; Weber, R.W.; Sosman, J.A.; Haanen, J.B.; Gonzalez, R.; Robert, C.; Schadendorf, D.; Hassel, J.C.; et al. Improved survival with ipilimimab in patients with metastatic melanoma. N. Engl. J. Med. 2010, 363, 711–723. [Google Scholar] [CrossRef] [PubMed]
- Ribas, A.; Kefford, R.; Marshall, M.A. Phase III randomized clinical trial comparing tremelimumab with standard-of-care chemotherapy in patients with advanced melanoma. J. Clin. Oncol. 2013, 31, 616–622. [Google Scholar] [CrossRef] [PubMed]
- Schuessler, A.; Smith, C.; Beagley, L.; Boyle, G.M.; Rehan, S.; Matthews, K.; Jones, L.; Crough, T.; Dasari, V.; Klein, K.; et al. Autologous T-cell therapy for cytomegalovirus as a consolidative treatment for recurrent glioblastoma. Cancer Res. 2014, 74, 3466–3476. [Google Scholar] [CrossRef] [PubMed]
- Gelao, L.; Criscitiello, C.; Esposito, A.; Goldhirsch, A.; Curigliano, G. Immune checkpoint blockade in cancer treatment: A double-edged sword cross-targeting the host as an “innocent bystander”. Toxins 2014, 6, 914–933. [Google Scholar] [CrossRef] [PubMed]
- Hartmann, J.; Schüßler-Lenz, M.; Bondanza, A.; Buchholz, C.J. Clinical development of CAR T cells—Challenges and opportunities in translating innovative treatment concepts. EMBO Mol. Med. 2017, 9, 1183–1197. [Google Scholar] [CrossRef] [PubMed]
- Chiocca, E.A.; Rabkin, S.D. Oncolytic viruses and their application to cancer immunotherapy. Cancer Immunol. Res. 2014, 2, 295–300. [Google Scholar] [CrossRef] [PubMed]
- Lawler, S.E.; Chiocca, E.A. Oncolytic virus-mediated immunotherapy: A combinatorial approach for cancer treatment. J. Clin. Oncol. 2015, 33, 2812–2814. [Google Scholar] [CrossRef] [PubMed]
- Kaufman, H.L.; Kohlhapp, F.J.; Zloza, A. Oncolytic viruses: A new class of immunotherapy drugs. Cancer Immunother. 2015, 14, 642–662. [Google Scholar] [CrossRef] [PubMed]
- Prestwich, R.J.; Harrington, K.J.; Pandha, H.S.; Vile, R.G.; Melcher, A.A.; Errington, F. Oncolytic viruses: A novel form of immunotherapy. Expert Rev. Anticancer Ther. 2008, 8, 1581–1588. [Google Scholar] [CrossRef] [PubMed]
- Newman, S.J.; McCallin, P.F.; Sever, J.L. Attempts to isolate H-1 virus from spontaneous human abortions: A negative report. Teratology 1970, 3, 279–281. [Google Scholar] [CrossRef] [PubMed]
- Rommelaere, L.; Geletneky, K.; Angelova, A.L.; Daeffler, L.; Dinsart, C.; Kiprianova, I.; Schlehofer, J.R.; Raykov, Z. Oncolytic parvoviruses as cancer therapeutics. Cytokine Growth Factor Rev. 2010, 21, 185–195. [Google Scholar] [CrossRef] [PubMed]
- Marchini, A.; Bonifati, S.; Scott, E.M.; Angelova, A.L.; Rommelaere, J. Oncolytic parvoviruses: From basic virology to clinical applications. Virol. J. 2015, 12, 6. [Google Scholar] [CrossRef] [PubMed]
- Angelova, A.L.; Geletneky, K.; Nüesch, J.P.F.; Rommelaere, J. Tumor selectivity of oncolytic parvoviruses: From in vitro and animal models to cancer patients. Front. Bioeng. Biotechnol. 2015, 3, 55. [Google Scholar] [CrossRef] [PubMed]
- Geletneky, K.; Nüesch, J.P.F.; Angelova, A.; Kiprianova, I.; Rommelaere, J. Double-faceted mechanism of parvoviral oncosuppression. Curr. Opin. Virol. 2015, 13, 17–24. [Google Scholar] [CrossRef] [PubMed]
- Moehler, M.; Goepfert, K.; Heinrich, B.; Breitbach, C.J.; Delic, M.; Galle, P.R.; Rommelaere, J. Oncolytic virotherapy as emerging immunotherapeutic modality: Potential of parvovirus H-1. Front. Oncol. 2014, 4, 92. [Google Scholar] [CrossRef] [PubMed]
- Raykov, Z.; Grekova, S.; Galabov, A.S.; Balboni, G.; Koch, U.; Aprahamian, M.; Rommelaere, J. Combined oncolytic and vaccination activities of parvovirus H-1 in a metastatic tumor model. Oncol. Rep. 2007, 17, 1493–1499. [Google Scholar] [CrossRef] [PubMed]
- Moehler, M.H.; Zeidler, M.; Wilsberg, V.; Cornelis, J.J.; Woelfel, T.; Rommelaere, J.; Galle, P.R.; Heike, M. Parvovirus H-1-induced tumor cell death enhances human immune response in vitro via increased phagocytosis, maturation, and cross-presentation by dendritic cells. Hum. Gene Ther. 2005, 16, 996–1005. [Google Scholar] [CrossRef] [PubMed]
- Grekova, S.; Aprahamian, M.; Giese, N.; Schmitt, S.; Giese, T.; Falk, C.S.; Daeffler, L.; Cziepluch, C.; Rommelaere, J.; Raykov, Z. Immune cells participate in the oncosuppressive activity of parvovirus H-1PV and are activated as a result of their abortive infection. Cancer Biol. Ther. 2010, 10, 1280–1289. [Google Scholar] [CrossRef] [PubMed]
- Heinrich, B.; Goepfert, K.; Delic, M.; Galle, P.R.; Moehler, M. Influence of the oncolytic parvovirus H-1, CTLA-4 antibody tremelimumab and cytostatic drugs on the human immune system in a human in vitro model of colorectal cancer cells. Onco Targets Ther. 2013, 6, 1119–1127. [Google Scholar] [CrossRef] [PubMed]
- Bhat, R.; Dempe, S.; Dinsart, C.; Rommelaere, J. Enhancement of NK cell antitumor responses using an oncolytic parvovirus. Int. J. Cancer 2011, 128, 908–919. [Google Scholar] [CrossRef] [PubMed]
- Moralés, O.; Richard, A.; Martin, N.; Mrizak, D.; Sénéchal, M.; Miroux, C.; Pancré, V.; Rommelaere, J.; Caillet-Fauquet, P.; de Lanoit, Y.; et al. Activation of a helper but not regulatory human CD4+ T cell response by oncolytic H-1 parvovirus. PLoS ONE 2012, 7, e32197. [Google Scholar] [CrossRef] [PubMed]
- Grekova, S.P.; Raykov, Z.; Zawatzky, R.; Rommelaere, J.; Koch, U. Activation of a glioma-specific immune response by oncolytic parvovirus Minute Virus of Mice infection. Cancer Gene Ther. 2012, 19, 468–475. [Google Scholar] [CrossRef] [PubMed]
- Geletneky, K.; Kiprianova, I.; Ayache, A.; Koch, R.; Herrero Y Calle, M.; Deleu, L.; Sommer, C.; Thomas, N.; Rommelaere, J.; Schlehofer, J. Regression of advanced rat and human gliomas by local or systemic treatment with oncolytic parvovirus H-1 in rat models. Neuro Oncol. 2010, 12, 804–814. [Google Scholar] [CrossRef] [PubMed]
- Chiocca, E.A.; Lamfers, M.L. Clinical trials of oncolytic viruses for gliomas. In Current Clinical Oncology: High-Grade Gliomas: Diagnosis and Treatment; Barnett, G.H., Ed.; Humana Press Inc.: Totowa, NJ, USA, 2007; pp. 391–403. ISBN 978-1-59745-185-7. [Google Scholar]
- Duffy, M.R.; Fisher, K.; Seymour, L.W. Making oncolytic virotherapy a clinical reality: The European contribution. Hum. Gene Ther. 2017, 28, 1033–1046. [Google Scholar] [CrossRef] [PubMed]
- Geletneky, K.; Huesing, J.; Rommelaere, J.; Schlehofer, J.R.; Leuchs, B.; Dahm, M.; Krebs, O.; von Knebel Doeberitz, M.; Huber, B.; Hajda, J. Phase I/IIa study of intratumoral/intracerebral or intravenous/intracerebral administration of parvovirus H-1 (ParvOryx) in patients with progressive primary or recurrent glioblastoma multiforme: ParvOryx01 protocol. BMC Cancer 2012, 12, 99. [Google Scholar] [CrossRef] [PubMed]
- Geletneky, K.; Hajda, J.; Angelova, A.L.; Leuchs, B.; Capper, D.; Bartsch, A.; Neumann, J.-O.; Schöning, T.; Hüsing, J.; Beelte, B.; et al. Oncolytic H-1 parvovirus shows safety and signs of immunogenic activity in a first phase I/IIa glioblastoma trial. Mol. Ther. 2017, 25, 2620–2634. [Google Scholar] [CrossRef] [PubMed]
- Chonan, M.; Saito, R.; Shoji, T.; Shibahara, I.; Kanamori, M.; Sonoda, Y.; Watanabe, M.; Kikushi, T.; Ishii, N.; Tominaga, T. CD40/CD40L expression correlates with the survival of patients with glioblastomas and an augmentation in CD40 signaling enhances the efficacy of vaccinations against glioma models. Neuro Oncol. 2015, 17, 1453–1462. [Google Scholar] [CrossRef] [PubMed]
- Di Piazza, M.; Mader, C.; Geletneky, K.; Herrero Y Calle, M.; Weber, E.; Schlehofer, J.; Deleu, L.; Rommelaere, J. Cytosolic activation of cathepsins mediates parvovirus H-1-induced killing of cisplatin and TRAIL-resistant glioma cells. J. Virol. 2007, 81, 4186–4198. [Google Scholar] [CrossRef] [PubMed]
- Kingham, P.J.; Pocock, J.M. Microglial secreted cathepsin B induces neuronal apoptosis. J. Neurochem. 2001, 76, 1475–1484. [Google Scholar] [CrossRef] [PubMed]
- Hwang, S.Y.; Yoo, B.C.; Jung, J.W.; Oh, E.S.; Hwang, J.S.; Shin, J.A.; Kim, S.Y.; Cha, S.H.; Han, I.O. Induction of glioma apoptosis by microglia-secreted molecules: The role of nitric oxide and cathepsin B. Biochim. Biophys. Acta 2009, 1793, 1656–1668. [Google Scholar] [CrossRef] [PubMed]
- Rayet, B.; Lopez-Guerrero, J.-A.; Rommelaere, J.; Dinsart, C. Induction of programmed cell death by parvovirus H-1 in U937 cells: Connection with the tumor necrosis factor α signalling pathway. J. Virol. 1998, 72, 8893–8903. [Google Scholar] [PubMed]
- Lisi, L.; Ciotti, G.M.; Braun, D.; Kalinin, S.; Currò, D.; Dello Russo, C.; Coli, A.; Mangiola, A.; Anile, C.; Feinstein, D.L.; et al. Expression of iNOS, CD163 and ARG-1 taken as M1 and M2 markers of microglial polarization in human glioblastoma and the surrounding normal parenchyma. Neurosci. Lett. 2017, 645, 106–112. [Google Scholar] [CrossRef] [PubMed]
- De Vries, C.R.; Kaufman, H.L.; Lattime, E.C. Oncolytic viruses: Focusing on the tumor microenvironment. Cancer Gene Ther. 2015, 22, 169–171. [Google Scholar] [CrossRef] [PubMed]
- Zamarin, D.; Holmgaard, R.B.; Subudhi, S.K.; Park, J.S.; Mansour, M.; Palese, P.; Merghoub, T.; Wolchok, J.D.; Allison, J.P. Localized oncolytic virotherapy overcomes systemic tumor resistance to immune checkpoint blockade immunotherapy. Sci. Transl. Med. 2014, 6, 22ra32. [Google Scholar] [CrossRef] [PubMed]
- Qiao, J.; Dey, M.; Chang, A.L.; Kim, J.W.; Miska, J.; LIng, A.; Nettlebeck, D.M.; Han, Y.; Zhang, L.; Lesniak, M.S. Intratumoral oncolytic adenoviral treatment modulates the glioma microenvironment and facilitates systemic tumor-antigen-specific T cell therapy. Oncoimmunology 2015, 4, e1022302. [Google Scholar] [CrossRef] [PubMed]
- Brown, M.C.; Holl, E.K.; Boczkowski, D.; Dobrikova, E.; Mosaheb, M.; Chandramohan, V.; Bigner, D.D.; Gromeier, M.; Nair, S.K. Cancer immunotherapy with recombinant poliovirus induces IFN-dominant activation of dendritic cells and tumor antigen-specific CTLs. Sci. Transl. Med. 2017, 9, eaan4220. [Google Scholar] [CrossRef] [PubMed]
- Mastrangelo, M.J.; Maguire, H.C., Jr.; Eisenlohr, L.C.; Laughlin, C.E.; Monken, C.E.; McCue, P.A.; Kovatich, A.J.; Lattime, E.C. Intratumoral recombinant GM-CSF-encoding virus as gene therapy in patients with cutaneous melanoma. Cancer Gene Ther. 1999, 6, 409–422. [Google Scholar] [CrossRef] [PubMed]
- Kaufman, H.L.; DeRaffele, G.; Divito, J.; Hörig, H.; Lee, D.; Panicali, D.; Voulo, M. A phase I trial of intralesional rV-Tricom vaccine in the tratment of malignant melanoma. Hum. Gene Ther. 2001, 12, 1459–1480. [Google Scholar] [PubMed]
- Kaufman, H.L.; Kim, D.W.; DeRaffele, G.; Mitcham, J.; Coffin, R.S.; Kim-Schulze, S. Local and distant immunity induced by intralesional vaccination with an oncolytic herpes virus encoding GM-CSF in patients with stage IIIc and IV melanoma. Ann. Surg. Oncol. 2010, 17, 718–730. [Google Scholar] [CrossRef] [PubMed]
- Heinzerling, L.; Künzi, V.; Oberholzer, P.A.; Kündig, T.; Naim, H.; Dummer, R. Oncolytic measles virus in cutaneous T-cell lymphomas mounts antitumor immune responses in vivo and targets interferon-resistant tumor cells. Blood 2005, 106, 2287–2294. [Google Scholar] [CrossRef] [PubMed]
- Senzer, N.N.; Kaufman, H.L.; Amatruda, T.; Nemunaitis, M.; Reid, T.; Daniels, G.; Gonzalez, R.; Glaspy, J.; Whitman, E.; Harrington, K.; et al. Phase II clinical trial of a granulocyte-macrophage colony-stimulating factor-encoding, second-generation oncolytic herpesvirus in patients with unresectable metastatic melanoma. J. Clin. Oncol. 2009, 27, 5763–5771. [Google Scholar] [CrossRef] [PubMed]
- Aitken, A.S.; Roy, D.G.; Bourgeois-Daigneault, M.-C. Taking a stab at cancer; oncolytic virus-mediated anti-cancer vaccination strategies. Biomedicines 2017, 5, 3. [Google Scholar] [CrossRef] [PubMed]
- Kees, T.; Lohr, J.; Noack, J.; Mora, R.; Gdinya, G.; Tödt, G.; Ernst, A.; Radlwimmer, B.; Falk, C.S.; Herold-Mende, C.; et al. Microglia isolated from patients with glioma gain antitumor activities on poly (I:C) stimulation. Neuro Oncol. 2012, 14, 64–78. [Google Scholar] [CrossRef] [PubMed]
- Rutledge, W.C.; Kong, J.; Gao, J.; Gutman, D.A.; Cooper, L.A.D.; Appin, C.; Park, Y.; Scarpace, L.; Mikkelsen, T.; Cohen, M.L.; et al. Tumor-infiltrating lymphocytes in glioblastoma are associated with specific genomic alterations and related to transcriptional class. Clin. Cancer Res. 2013, 19, 4951–4960. [Google Scholar] [CrossRef] [PubMed]
- El Andaloussi, A.; Lesniak, M.S. An increase in CD4+CD25+FOXP3+ regulatory T cells in tumor-infiltrating lymphocytes of human glioblastoma multiforme. Neuro Oncol. 2006, 8, 234–243. [Google Scholar] [CrossRef] [PubMed]
- Saskar, S.; Döring, A.; Zemp, F.J.; Silva, C.; Lun, X.; Wang, Y.; Kelly, J.; Hader, W.; Hamilton, M.; Mercier, P.; et al. Therapeutic activation of macrophages and microglia to suppress brain tumor-initiating cells. Nat. Neurosci. 2014, 17, 46–55. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Glioblastoma Microenvironment (Literature Data) | Glioblastoma Microenvironment (First Parvovirus Clinical Trial) |
|---|---|
| sparse inflammatory infiltrates (except the mesenchymal transcriptional class) [63] | tumor infiltration with CD8+/granzyme B+ T cells [45] |
| tumor-infiltrating lymphocyte involvement in inhibitory (e.g., via PD-1) interactions [8] | PD-1-negative tumor-infiltrating T cells |
| increased numbers of tumor-infiltrating Treg cells [64] | Treg cells only scarcely detected [45] |
| M2 tumor-supportive tumor-associated microglia/macrophage (TAM) phenotype [51] | detection of markers of M1 TAM polarization (CD68, CTSB, iNOS) [45] |
| release of immunosuppressive factors and anti-inflammatory cytokines [8] | detection of proinflammatory cytokines (IFN-γ, IL-2) [45] |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Angelova, A.L.; Barf, M.; Geletneky, K.; Unterberg, A.; Rommelaere, J. Immunotherapeutic Potential of Oncolytic H-1 Parvovirus: Hints of Glioblastoma Microenvironment Conversion towards Immunogenicity. Viruses 2017, 9, 382. https://doi.org/10.3390/v9120382
Angelova AL, Barf M, Geletneky K, Unterberg A, Rommelaere J. Immunotherapeutic Potential of Oncolytic H-1 Parvovirus: Hints of Glioblastoma Microenvironment Conversion towards Immunogenicity. Viruses. 2017; 9(12):382. https://doi.org/10.3390/v9120382
Chicago/Turabian StyleAngelova, Assia L., Milena Barf, Karsten Geletneky, Andreas Unterberg, and Jean Rommelaere. 2017. "Immunotherapeutic Potential of Oncolytic H-1 Parvovirus: Hints of Glioblastoma Microenvironment Conversion towards Immunogenicity" Viruses 9, no. 12: 382. https://doi.org/10.3390/v9120382
APA StyleAngelova, A. L., Barf, M., Geletneky, K., Unterberg, A., & Rommelaere, J. (2017). Immunotherapeutic Potential of Oncolytic H-1 Parvovirus: Hints of Glioblastoma Microenvironment Conversion towards Immunogenicity. Viruses, 9(12), 382. https://doi.org/10.3390/v9120382