Hazard Characterization of Modified Vaccinia Virus Ankara Vector: What Are the Knowledge Gaps?

 ,
, 
Abstract
:
1. Introduction
1.1. Genetically Modified Viruses (GMVs) for Vaccination
1.2. Steps in Environmental Risk Assessment of GMVs
1.3. Poxviruses as Vaccine and Vaccine Vector
1.4. MVA Is a Safe Vector—Need for Hazard Characterization?
2. Modified Vaccinia Virus Ankara
2.1. Origin and History of Use as a Smallpox Vaccine
2.2. Major Characteristics of MVA
2.3. MVA as a Vaccine Vector
2.4. Priorities for MVA Research: Is Environmental Risk Assessment a Priority?
3. Knowledge Gaps and Omitted Research
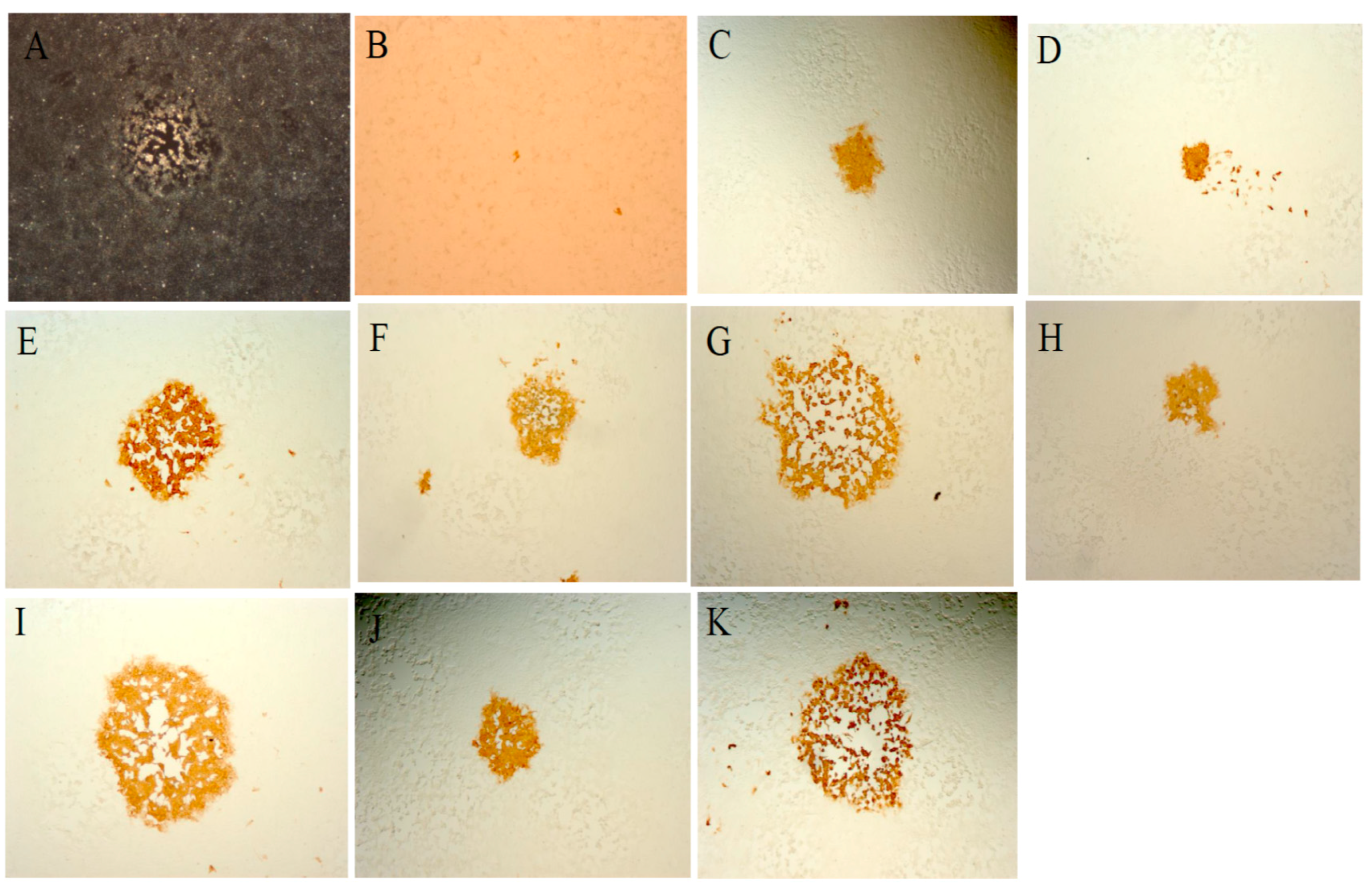
3.1. Host Cell Restriction of MVA in Human and Other Mammalian Cells
3.2. Clonal Purity and Genome Stability of MVA
3.3. Transgene and Genome Stability of MVA-Vectored Vaccine
3.4. Geographic Distribution and Occurrence of Naturally Circulating Orthopoxviruses
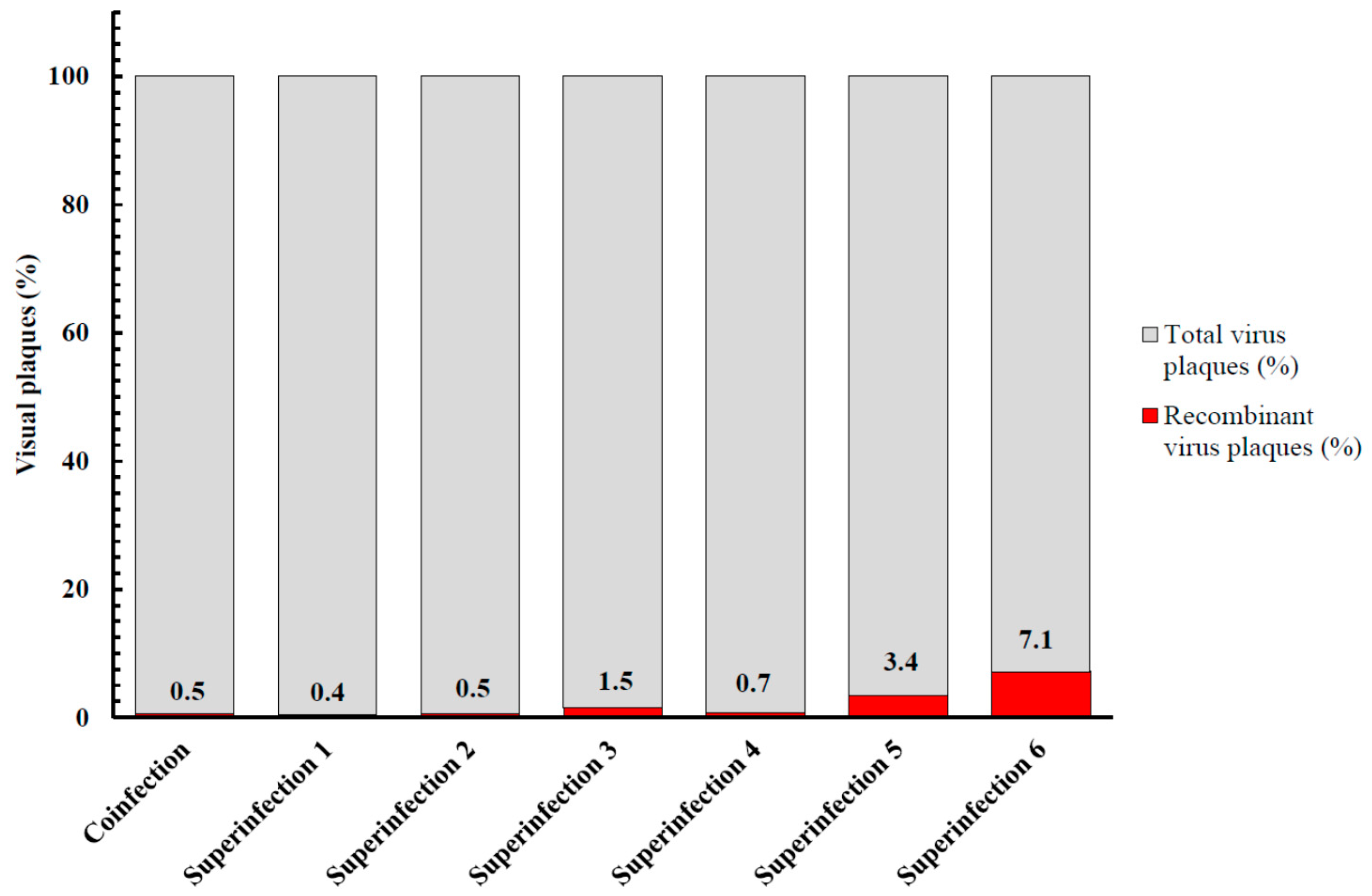
3.5. Recombination between MVA-Vectored Vaccine and Other Orthopoxviruses
3.6. Biodistribution, Shedding and Persistence of GMVs
3.6.1. Biodistribution
3.6.2. Shedding
3.6.3. Persistence of Virus Vector in the Environment
3.7. Transmission of Shed Virus Vector and Virus-Vectored Vaccine
3.8. Immune Responses: Implications for Biosafety
3.8.1. Anti-Vector Immunity
3.8.2. Effect of Transgenic Protein on Th1-Th2 Response and Cytokine Proliferation
3.8.3. Modulation of Intracellular Signaling Pathways
4. Future Perspective
4.1. Uncertainty and Uncertainty Analysis
4.2. Worst Case Scenario
4.3. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Ura, T.; Okuda, K.; Shimada, M. Developments in viral vector-based vaccines. Vaccines 2014, 2, 624–641. [Google Scholar] [CrossRef] [PubMed]
- Ramezanpour, B.; Haan, I.; Osterhaus, A.; Claassen, E. Vector-based genetically modified vaccines: Exploiting Jenner’s legacy. Vaccine 2016, 34, 6436–6448. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, S.C. Clinical development of Modified Vaccinia virus Ankara vaccines. Vaccine 2013, 31, 4241–4246. [Google Scholar] [CrossRef] [PubMed]
- Cooney, E.L.; Collier, A.C.; Greenberg, P.D.; Coombs, R.W.; Zarling, J.; Arditti, D.E.; Hoffman, M.C.; Hu, S.L.; Corey, L. Safety of and immunological response to a recombinant vaccinia virus vaccine expressing HIV envelope glycoprotein. Lancet 1991, 337, 567–572. [Google Scholar] [CrossRef]
- Arnberg, N. Adenovirus receptors: Implications for tropism, treatment and targeting. Rev. Med. Virol. 2009, 19, 165–178. [Google Scholar] [CrossRef] [PubMed]
- Roberts, D.M.; Nanda, A.; Havenga, M.J.; Abbink, P.; Lynch, D.M.; Ewald, B.A.; Liu, J.; Thorner, A.R.; Swanson, P.E.; Gorgone, D.A.; et al. Hexon-chimaeric adenovirus serotype 5 vectors circumvent pre-existing anti-vector immunity. Nature 2006, 441, 239–243. [Google Scholar] [CrossRef] [PubMed]
- Abe, S.; Okuda, K.; Ura, T.; Kondo, A.; Yoshida, A.; Yoshizaki, S.; Mizuguchi, H.; Klinman, D.; Shimada, M. Adenovirus type 5 with modified hexons induces robust transgene-specific immune responses in mice with pre-existing immunity against adenovirus type 5. J. Gene Med. 2009, 11, 570–579. [Google Scholar] [CrossRef] [PubMed]
- Ura, T.; Yoshida, A.; Xin, K.Q.; Yoshizaki, S.; Yashima, S.; Abe, S.; Mizuguchi, H.; Okuda, K. Designed recombinant adenovirus type 5 vector induced envelope-specific CD8(+) cytotoxic T lymphocytes and cross-reactive neutralizing antibodies against human immunodeficiency virus type 1. J. Gene Med. 2009, 11, 139–149. [Google Scholar] [CrossRef] [PubMed]
- Donsante, A.; Vogler, C.; Muzyczka, N.; Crawford, J.M.; Barker, J.; Flotte, T.; Campbell-Thompson, M.; Daly, T.; Sands, M.S. Observed incidence of tumorigenesis in long-term rodent studies of rAAV vectors. Gene Ther. 2001, 8, 1343–1346. [Google Scholar] [CrossRef] [PubMed]
- Lentz, T.B.; Gray, S.J.; Samulski, R.J. Viral vectors for gene delivery to the central nervous system. Neurobiol. Dis. 2012, 48, 179–188. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Dullmann, J.; Schiedlmeier, B.; Schmidt, M.; von Kalle, C.; Meyer, J.; Forster, M.; Stocking, C.; Wahlers, A.; Frank, O.; et al. Murine leukemia induced by retroviral gene marking. Science 2002, 296, 497. [Google Scholar] [CrossRef] [PubMed]
- Qasim, W.; Gaspar, H.B.; Thrasher, A.J. Gene therapy for severe combined immune deficiency. Expert Rev. Mol. Med. 2004, 6, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Moriya, C.; Horiba, S.; Kurihara, K.; Kamada, T.; Takahara, Y.; Inoue, M.; Iida, A.; Hara, H.; Shu, T.; Hasegawa, M.; et al. Intranasal Sendai viral vector vaccination is more immunogenic than intramuscular under pre-existing anti-vector antibodies. Vaccine 2011, 29, 8557–8563. [Google Scholar] [CrossRef] [PubMed]
- Directive 2001/18/EC of the European Parliament and of the Council of 12 March on the deliberate release into the environment of genetically modified organisms and repealing Council Directive 90/220/EEC. European Parliament and Council, 2001; Vol. Off J 17.04.2001.
- Anson, D.S. The use of retroviral vectors for gene therapy—What are the risks? A review of retroviral pathogenesis and its relevance to retroviral vector-mediated gene delivery. Genet. Vaccines Ther. 2004, 2, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Auman, J.T. Gene therapy: Have the risks associated with viral vectors been solved? Curr. Opin. Mol. Ther. 2010, 12, 637–638. [Google Scholar] [PubMed]
- Verheust, C.; Goossens, M.; Pauwels, K.; Breyer, D. Biosafety aspects of modified vaccinia virus Ankara (MVA)-based vectors used for gene therapy or vaccination. Vaccine 2012, 30, 2623–2632. [Google Scholar] [CrossRef] [PubMed]
- Baldo, A.; van den Akker, E.; Bergmans, H.E.; Lim, F.; Pauwels, K. General considerations on the biosafety of virus-derived vectors used in gene therapy and vaccination. Curr. Gene Ther. 2013, 13, 385–394. [Google Scholar] [CrossRef] [PubMed]
- Goossens, M.; Pauwels, K.; Willemarck, N.; Breyer, D. Environmental risk assessment of clinical trials involving modified vaccinia virus Ankara (MVA)-based vectors. Curr. Gene Ther. 2013, 13, 413–420. [Google Scholar] [CrossRef] [PubMed]
- Moss, B. Poxviridae: The Viruses and Their Replication. In Fields Virology, 4th ed.; Roizman, B., Howley, P., Straus, S., Martin, M., DE, G., Lamb, R., Knipe, D., Eds.; Lippincott Williams and Wilkins: Philadelphia, PA, USA, 2001; pp. 2849–2883. [Google Scholar]
- Fenner, F.H.; Hender, D.A.; Arita, I.; Jesek, J.; Ladnyi, I.D. Smallpox and Its Eradication; World Health Organisation: Geneva, Switzerland, 1988. [Google Scholar]
- Parrino, J.; Graham, B.S. Smallpox vaccines: Past, present and future. J. Allergy Clin. Immunol. 2006, 118, 1320–1326. [Google Scholar] [CrossRef] [PubMed]
- Geddes, A.M. The history of smallpox. Clin. Dermatol. 2006, 24, 152–157. [Google Scholar] [CrossRef] [PubMed]
- Drewitt, F.D. The Life of Edward Jenner; Cambridge University Press: New York, NY, USA, 1933. [Google Scholar]
- Jenner, E. The Three Original Publications on Vaccination Agianst Smallpox; P. F. Collier and Son, 1909–1914; Bartleby.com: New York, NY, USA, 2001; p Part 4 of 8. [Google Scholar]
- Sanchez-Sampedro, L.; Perdiguero, B.; Mejias-Perez, E.; Garcia-Arriaza, J.; Di Pilato, M.; Esteban, M. The evolution of poxvirus vaccines. Viruses 2015, 7, 1726–1803. [Google Scholar] [CrossRef] [PubMed]
- Koplow, D.A. Smallpox: The Fight to Eradicate a Global Scourge; University of California Press: Berkeley, CA, USA, 2003. [Google Scholar]
- Panicali, D.; Paoletti, E. Construction of poxviruses as cloning vectors: Insertion of the thymidine kinase gene from herpes simplex virus into the DNA of infectious vaccinia virus. Proc. Natl. Acad. Sci. USA 1982, 79, 4927–4931. [Google Scholar] [CrossRef] [PubMed]
- Mackett, M.; Smith, G.L.; Moss, B. Vaccinia virus: A selectable eukaryotic cloning and expression vector. Proc. Natl. Acad. Sci. USA 1982, 79, 7415–7419. [Google Scholar] [CrossRef] [PubMed]
- Whitley, R.J. Smallpox: A potential agent of bioterrorism. Antivir. Res. 2003, 57, 7–12. [Google Scholar] [CrossRef]
- Damaso, C.R.; Esposito, J.J.; Condit, R.C.; Moussatche, N. An emergent poxvirus from humans and cattle in Rio de Janeiro State: Cantagalo virus may derive from Brazilian smallpox vaccine. Virology 2000, 277, 439–449. [Google Scholar] [CrossRef] [PubMed]
- Vorou, R.M.; Papavassiliou, V.G.; Pierroutsakos, I.N. Cowpox virus infection: An emerging health threat. Curr. Opin. Infect. Dis. 2008, 21, 153–156. [Google Scholar] [CrossRef] [PubMed]
- Reed, K.D.; Melski, J.W.; Graham, M.B.; Regnery, R.L.; Sotir, M.J.; Wegner, M.V.; Kazmierczak, J.J.; Stratman, E.J.; Li, Y.; Fairley, J.A.; et al. The detection of monkeypox in humans in the Western Hemisphere. N. Engl. J. Med. 2004, 350, 342–350. [Google Scholar] [CrossRef] [PubMed]
- Volz, A.; Sutter, G. Modified vaccinia virus ankara: History, value in basic research and current perspectives for vaccine development. Adv. Virus Res. 2017, 97, 187–243. [Google Scholar] [PubMed]
- Liu, Z.; Ravindranathan, R.; Kalinski, P.; Guo, Z.S.; Bartlett, D.L. Rational combination of oncolytic vaccinia virus and PD-L1 blockade works synergistically to enhance therapeutic efficacy. Nat. Commun. 2017, 8, 14754. [Google Scholar] [CrossRef] [PubMed]
- Izzi, V.; Buler, M.; Masuelli, L.; Giganti, M.G.; Modesti, A.; Bei, R. Poxvirus-based vaccines for cancer immunotherapy: New insights from combined cytokines/co-stimulatory molecules delivery and “uncommon” strains. Anticancer Agents Med. Chem. 2014, 14, 183–189. [Google Scholar] [CrossRef] [PubMed]
- Rupprecht, C.E.; Wiktor, T.J.; Johnston, D.H.; Hamir, A.N.; Dietzschold, B.; Wunner, W.H.; Glickman, L.T.; Koprowski, H. Oral immunization and protection of raccoons (Procyon lotor) with a vaccinia-rabies glycoprotein recombinant virus vaccine. Proc. Natl. Acad. Sci. USA 1986, 83, 7947–7950. [Google Scholar] [CrossRef] [PubMed]
- Muller, T.F.; Schroder, R.; Wysocki, P.; Mettenleiter, T.C.; Freuling, C.M. Spatio-temporal use of oral rabies vaccines in fox rabies elimination programmes in Europe. PLoS Negl. Trop. Dis. 2015, 9, e0003953. [Google Scholar] [CrossRef] [PubMed]
- Casey, C.G.; Iskander, J.K.; Roper, M.H.; Mast, E.E.; Wen, X.J.; Torok, T.J.; Chapman, L.E.; Swerdlow, D.L.; Morgan, J.; Heffelfinger, J.D.; et al. Adverse events associated with smallpox vaccination in the United States, January–October 2003. JAMA 2005, 294, 2734–2743. [Google Scholar] [CrossRef] [PubMed]
- Mempel, M.; Isa, G.; Klugbauer, N.; Meyer, H.; Wildi, G.; Ring, J.; Hofmann, F.; Hofmann, H. Laboratory acquired infection with recombinant vaccinia virus containing an immunomodulating construct. J. Investig. Dermatol. 2003, 120, 356–358. [Google Scholar] [CrossRef] [PubMed]
- Draper, S.J.; Heeney, J.L. Viruses as vaccine vectors for infectious diseases and cancer. Nat. Rev. Microbiol. 2010, 8, 62–73. [Google Scholar] [CrossRef] [PubMed]
- Cottingham, M.G.; Carroll, M.W. Recombinant MVA vaccines: Dispelling the myths. Vaccine 2013, 31, 4247–4251. [Google Scholar] [CrossRef] [PubMed]
- Altenburg, A.F.; Kreijtz, J.H.; de Vries, R.D.; Song, F.; Fux, R.; Rimmelzwaan, G.F.; Sutter, G.; Volz, A. Modified vaccinia virus ankara (MVA) as production platform for vaccines against influenza and other viral respiratory diseases. Viruses 2014, 6, 2735–2761. [Google Scholar] [CrossRef] [PubMed]
- Mayr, A.; Stickl, H.; Muller, H.K.; Danner, K.; Singer, H. The smallpox vaccination strain MVA: Marker, genetic structure, experience gained with the parenteral vaccination and behavior in organisms with a debilitated defence mechanism (author’s transl). Zent. Bakteriol. B 1978, 167, 375–390. [Google Scholar]
- Meyer, H.; Sutter, G.; Mayr, A. Mapping of deletions in the genome of the highly attenuated vaccinia virus MVA and their influence on virulence. J. Gen. Virol. 1991, 72 (Pt 5), 1031–1038. [Google Scholar] [CrossRef] [PubMed]
- Antoine, G.; Scheiflinger, F.; Dorner, F.; Falkner, F.G. The complete genomic sequence of the modified vaccinia Ankara strain: Comparison with other orthopoxviruses. Virology 1998, 244, 365–396. [Google Scholar] [CrossRef] [PubMed]
- Meisinger-Henschel, C.; Spath, M.; Lukassen, S.; Wolferstatter, M.; Kachelriess, H.; Baur, K.; Dirmeier, U.; Wagner, M.; Chaplin, P.; Suter, M.; et al. Introduction of the six major genomic deletions of modified vaccinia virus Ankara (MVA) into the parental vaccinia virus is not sufficient to reproduce an MVA-like phenotype in cell culture and in mice. J. Virol. 2010, 84, 9907–9919. [Google Scholar] [CrossRef] [PubMed]
- Stickl, H.; Hochstein-Mintzel, V.; Mayr, A.; Huber, H.C.; Schafer, H.; Holzner, A. MVA vaccination against smallpox: Clinical tests with an attenuated live vaccinia virus strain (MVA) (author’s transl). Dtsch. Med. Wochenschr. 1974, 99, 2386–2392. [Google Scholar] [CrossRef] [PubMed]
- Sutter, G.; Moss, B. Nonreplicating vaccinia vector efficiently expresses recombinant genes. Proc. Natl. Acad. Sci. USA 1992, 89, 10847–10851. [Google Scholar] [CrossRef] [PubMed]
- Carroll, M.W.; Moss, B. Host range and cytopathogenicity of the highly attenuated MVA strain of vaccinia virus: Propagation and generation of recombinant viruses in a nonhuman mammalian cell line. Virology 1997, 238, 198–211. [Google Scholar] [CrossRef] [PubMed]
- Drexler, I.; Heller, K.; Wahren, B.; Erfle, V.; Sutter, G. Highly attenuated modified vaccinia virus Ankara replicates in baby hamster kidney cells, a potential host for virus propagation, but not in various human transformed and primary cells. J. Gen. Virol. 1998, 79 (Pt 2), 347–352. [Google Scholar] [CrossRef] [PubMed]
- Okeke, M.I.; Nilssen, O.; Traavik, T. Modified vaccinia virus Ankara multiplies in rat IEC-6 cells and limited production of mature virions occurs in other mammalian cell lines. J. Gen. Virol. 2006, 87, 21–27. [Google Scholar] [CrossRef] [PubMed]
- Meyer, H. Summary Report on First, Second and Third Generation Smallpox Vaccines; World Health Organization: Geneva, Switzerland, 2013. [Google Scholar]
- Slifka, M.K. The Future of smallpox vaccination: Is MVA the key? Med. Immunol. 2005, 4, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Earl, P.L.; Americo, J.L.; Wyatt, L.S.; Eller, L.A.; Whitbeck, J.C.; Cohen, G.H.; Eisenberg, R.J.; Hartmann, C.J.; Jackson, D.L.; Kulesh, D.A.; et al. Immunogenicity of a highly attenuated MVA smallpox vaccine and protection against monkeypox. Nature 2004, 428, 182–185. [Google Scholar] [CrossRef] [PubMed]
- Suter, M.; Meisinger-Henschel, C.; Tzatzaris, M.; Hulsemann, V.; Lukassen, S.; Wulff, N.H.; Hausmann, J.; Howley, P.; Chaplin, P. Modified vaccinia Ankara strains with identical coding sequences actually represent complex mixtures of viruses that determine the biological properties of each strain. Vaccine 2009, 27, 7442–7450. [Google Scholar] [CrossRef] [PubMed]
- Blanchard, T.J.; Alcami, A.; Andrea, P.; Smith, G.L. Modified vaccinia virus Ankara undergoes limited replication in human cells and lacks several immunomodulatory proteins: Implications for use as a human vaccine. J. Gen. Virol. 1998, 79 (Pt 5), 1159–1167. [Google Scholar] [CrossRef] [PubMed]
- Jordan, I.; Horn, D.; Oehmke, S.; Leendertz, F.H.; Sandig, V. Cell lines from the Egyptian fruit bat are permissive for modified vaccinia Ankara. Virus Res. 2009, 145, 54–62. [Google Scholar] [CrossRef] [PubMed]
- Dorrell, L.; Williams, P.; Suttill, A.; Brown, D.; Roberts, J.; Conlon, C.; Hanke, T.; McMichael, A. Safety and tolerability of recombinant modified vaccinia virus Ankara expressing an HIV-1 gag/multiepitope immunogen (MVA.HIVA) in HIV-1-infected persons receiving combination antiretroviral therapy. Vaccine 2007, 25, 3277–3283. [Google Scholar] [CrossRef] [PubMed]
- Hanke, T.; McMichael, A.J.; Dennis, M.J.; Sharpe, S.A.; Powell, L.A.; McLoughlin, L.; Crome, S.J. Biodistribution and persistence of an MVA-vectored candidate HIV vaccine in SIV-infected rhesus macaques and SCID mice. Vaccine 2005, 23, 1507–1514. [Google Scholar] [CrossRef] [PubMed]
- Darsow, U.; Sbornik, M.; Rombold, S.; Katzer, K.; von Sonnenburg, F.; Behrendt, H.; Ring, J. Long-term safety of replication-defective smallpox vaccine (MVA-BN) in atopic eczema and allergic rhinitis. J. Eur. Acad. Dermatol. Venereol. 2016, 30, 1971–1977. [Google Scholar] [CrossRef] [PubMed]
- Overton, E.T.; Stapleton, J.; Frank, I.; Hassler, S.; Goepfert, P.A.; Barker, D.; Wagner, E.; von Krempelhuber, A.; Virgin, G.; Meyer, T.P.; et al. Safety and Immunogenicity of Modified Vaccinia Ankara-Bavarian Nordic Smallpox Vaccine in Vaccinia-Naive and Experienced Human Immunodeficiency Virus-Infected Individuals: An Open-Label, Controlled Clinical Phase II Trial. Open Forum Infect. Dis. 2015, 2, ofv040. [Google Scholar] [CrossRef] [PubMed]
- Earl, P.L.; Americo, J.L.; Wyatt, L.S.; Espenshade, O.; Bassler, J.; Gong, K.; Lin, S.; Peters, E.; Rhodes, L., Jr.; Spano, Y.E.; et al. Rapid protection in a monkeypox model by a single injection of a replication-deficient vaccinia virus. Proc. Natl. Acad. Sci USA 2008, 105, 10889–10894. [Google Scholar] [CrossRef] [PubMed]
- Guerra, S.; Gonzalez, J.M.; Climent, N.; Reyburn, H.; Lopez-Fernandez, L.A.; Najera, J.L.; Gomez, C.E.; Garcia, F.; Gatell, J.M.; Gallart, T.; et al. Selective induction of host genes by MVA-B, a candidate vaccine against HIV/AIDS. J. Virol. 2010, 84, 8141–8152. [Google Scholar] [CrossRef] [PubMed]
- Joachim, A.; Nilsson, C.; Aboud, S.; Bakari, M.; Lyamuya, E.F.; Robb, M.L.; Marovich, M.A.; Earl, P.; Moss, B.; Ochsenbauer, C.; et al. Potent functional antibody responses elicited by HIV-I DNA priming and boosting with heterologous HIV-1 recombinant MVA in healthy Tanzanian adults. PLoS ONE 2015, 10, e0118486. [Google Scholar] [CrossRef] [PubMed]
- Munseri, P.J.; Kroidl, A.; Nilsson, C.; Joachim, A.; Geldmacher, C.; Mann, P.; Moshiro, C.; Aboud, S.; Lyamuya, E.; Maboko, L.; et al. Priming with a simplified intradermal HIV-1 DNA vaccine regimen followed by boosting with recombinant HIV-1 MVA vaccine is safe and immunogenic: A phase IIa randomized clinical trial. PLoS ONE 2015, 10, e0119629. [Google Scholar] [CrossRef] [PubMed]
- Nilsson, C.; Godoy-Ramirez, K.; Hejdeman, B.; Brave, A.; Gudmundsdotter, L.; Hallengard, D.; Currier, J.R.; Wieczorek, L.; Hasselrot, K.; Earl, P.L.; et al. Broad and potent cellular and humoral immune responses after a second late HIV-modified vaccinia virus ankara vaccination in HIV-DNA-primed and HIV-modified vaccinia virus Ankara-boosted Swedish vaccinees. AIDS Res. Hum. Retrovir. 2014, 30, 299–311. [Google Scholar] [CrossRef] [PubMed]
- Tameris, M.D.; Hatherill, M.; Landry, B.S.; Scriba, T.J.; Snowden, M.A.; Lockhart, S.; Shea, J.E.; McClain, J.B.; Hussey, G.D.; Hanekom, W.A.; et al. Safety and efficacy of MVA85A, a new tuberculosis vaccine, in infants previously vaccinated with BCG: A randomised, placebo-controlled phase 2b trial. Lancet 2013, 381, 1021–1028. [Google Scholar] [CrossRef]
- Hodgson, S.H.; Ewer, K.J.; Bliss, C.M.; Edwards, N.J.; Rampling, T.; Anagnostou, N.A.; de Barra, E.; Havelock, T.; Bowyer, G.; Poulton, I.D.; et al. Evaluation of the efficacy of ChAd63-MVA vectored vaccines expressing circumsporozoite protein and ME-TRAP against controlled human malaria infection in malaria-naive individuals. J. Infect. Dis. 2015, 211, 1076–1086. [Google Scholar] [CrossRef] [PubMed]
- Biswas, S.; Choudhary, P.; Elias, S.C.; Miura, K.; Milne, K.H.; de Cassan, S.C.; Collins, K.A.; Halstead, F.D.; Bliss, C.M.; Ewer, K.J.; et al. Assessment of humoral immune responses to blood-stage malaria antigens following ChAd63-MVA immunization, controlled human malaria infection and natural exposure. PLoS ONE 2014, 9, e107903. [Google Scholar] [CrossRef] [PubMed]
- Sebastian, S.; Gilbert, S.C. Recombinant modified vaccinia virus Ankara-based malaria vaccines. Expert Rev. Vaccin. 2016, 15, 91–103. [Google Scholar] [CrossRef] [PubMed]
- Milligan, I.D.; Gibani, M.M.; Sewell, R.; Clutterbuck, E.A.; Campbell, D.; Plested, E.; Nuthall, E.; Voysey, M.; Silva-Reyes, L.; McElrath, M.J.; et al. Safety and Immunogenicity of Novel Adenovirus Type 26- and Modified Vaccinia Ankara-Vectored Ebola Vaccines: A Randomized Clinical Trial. JAMA 2016, 315, 1610–1623. [Google Scholar] [CrossRef] [PubMed]
- Tapia, M.D.; Sow, S.O.; Lyke, K.E.; Haidara, F.C.; Diallo, F.; Doumbia, M.; Traore, A.; Coulibaly, F.; Kodio, M.; Onwuchekwa, U.; et al. Use of ChAd3-EBO-Z Ebola virus vaccine in Malian and US adults and boosting of Malian adults with MVA-BN-Filo: A phase 1, single-blind, randomised trial, a phase 1b, open-label and double-blind, dose-escalation trial and a nested, randomised, double-blind, placebo-controlled trial. Lancet Infect. Dis. 2016, 16, 31–42. [Google Scholar] [PubMed]
- Kreijtz, J.H.; Goeijenbier, M.; Moesker, F.M.; van den Dries, L.; Goeijenbier, S.; De Gruyter, H.L.; Lehmann, M.H.; Mutsert, G.; van de Vijver, D.A.; Volz, A.; et al. Safety and immunogenicity of a modified-vaccinia-virus-Ankara-based influenza A H5N1 vaccine: A randomised, double-blind phase 1/2a clinical trial. Lancet Infect. Dis. 2014, 14, 1196–1207. [Google Scholar] [CrossRef]
- Lillie, P.J.; Berthoud, T.K.; Powell, T.J.; Lambe, T.; Mullarkey, C.; Spencer, A.J.; Hamill, M.; Peng, Y.; Blais, M.E.; Duncan, C.J.; et al. Preliminary assessment of the efficacy of a T-cell-based influenza vaccine, MVA-NP+M1, in humans. Clin. Infect. Dis. 2012, 55, 19–25. [Google Scholar] [CrossRef] [PubMed]
- Cavenaugh, J.S.; Awi, D.; Mendy, M.; Hill, A.V.; Whittle, H.; McConkey, S.J. Partially randomized, non-blinded trial of DNA and MVA therapeutic vaccines based on hepatitis B virus surface protein for chronic HBV infection. PLoS ONE 2011, 6, e14626. [Google Scholar] [CrossRef] [PubMed]
- Volz, A.; Sutter, G. Protective efficacy of Modified Vaccinia virus Ankara in preclinical studies. Vaccine 2013, 31, 4235–4240. [Google Scholar] [CrossRef] [PubMed]
- Cappuccini, F.; Pollock, E.; Stribbling, S.; Hill, A.V.S.; Redchenko, I. 5T4 oncofoetal glycoprotein: An old target for a novel prostate cancer immunotherapy. Oncotarget 2017. [Google Scholar] [CrossRef] [PubMed]
- Husseini, F.; Delord, J.P.; Fournel-Federico, C.; Guitton, J.; Erbs, P.; Homerin, M.; Halluard, C.; Jemming, C.; Orange, C.; Limacher, J.M.; et al. Vectorized gene therapy of liver tumors: Proof-of-concept of TG4023 (MVA-FCU1) in combination with flucytosine. Ann. Oncol. 2017, 28, 169–174. [Google Scholar] [CrossRef] [PubMed]
- Schaedler, E.; Remy-Ziller, C.; Hortelano, J.; Kehrer, N.; Claudepierre, M.C.; Gatard, T.; Jakobs, C.; Preville, X.; Carpentier, A.F.; Rittner, K. Sequential administration of a MVA-based MUC1 cancer vaccine and the TLR9 ligand Litenimod (Li28) improves local immune defense against tumors. Vaccine 2017, 35, 577–585. [Google Scholar] [CrossRef] [PubMed]
- Rowe, J.; Cen, P. TroVax in colorectal cancer. Hum. Vaccin. Immunother. 2014, 10, 3196–3200. [Google Scholar] [CrossRef] [PubMed]
- Alberca, B.; Bachanek-Bankowska, K.; Cabana, M.; Calvo-Pinilla, E.; Viaplana, E.; Frost, L.; Gubbins, S.; Urniza, A.; Mertens, P.; Castillo-Olivares, J. Vaccination of horses with a recombinant modified vaccinia Ankara virus (MVA) expressing African horse sickness (AHS) virus major capsid protein VP2 provides complete clinical protection against challenge. Vaccine 2014, 32, 3670–3674. [Google Scholar] [CrossRef] [PubMed]
- Haagmans, B.L.; van den Brand, J.M.; Raj, V.S.; Volz, A.; Wohlsein, P.; Smits, S.L.; Schipper, D.; Bestebroer, T.M.; Okba, N.; Fux, R.; et al. An orthopoxvirus-based vaccine reduces virus excretion after MERS-CoV infection in dromedary camels. Science 2016, 351, 77–81. [Google Scholar] [CrossRef] [PubMed]
- Lopera-Madrid, J.; Osorio, J.E.; He, Y.; Xiang, Z.; Adams, L.G.; Laughlin, R.C.; Mwangi, W.; Subramanya, S.; Neilan, J.; Brake, D.; et al. Safety and immunogenicity of mammalian cell derived and Modified Vaccinia Ankara vectored African swine fever subunit antigens in swine. Vet. Immunol. Immunopathol. 2017, 185, 20–33. [Google Scholar] [CrossRef] [PubMed]
- Del Medico Zajac, M.P.; Zanetti, F.A.; Esusy, M.S.; Federico, C.R.; Zabal, O.; Valera, A.R.; Calamante, G. Induction of Both Local Immune Response in Mice and Protection in a Rabbit Model by Intranasal Immunization with Modified Vaccinia Ankara Virus Expressing a Secreted Form of Bovine Herpesvirus 1 Glycoprotein D. Viral. Immunol. 2017, 30, 70–76. [Google Scholar] [CrossRef] [PubMed]
- Volz, A.; Fux, R.; Langenmayer, M.C.; Sutter, G. Modified vaccinia virus ankara (MVA)—Development as recombinant vaccine and prospects for use in veterinary medicine. Berl. Munch. Tierarztl. Wochenschr. 2015, 128, 464–472. [Google Scholar] [PubMed]
- Falivene, J.; del Medico Zajac, M.P.; Pascutti, M.F.; Rodriguez, A.M.; Maeto, C.; Perdiguero, B.; Gomez, C.E.; Esteban, M.; Calamante, G.; Gherardi, M.M. Improving the MVA vaccine potential by deleting the viral gene coding for the IL-18 binding protein. PLoS ONE 2012, 7, e32220. [Google Scholar] [CrossRef] [PubMed]
- Rehm, K.E.; Roper, R.L. Deletion of the A35 gene from Modified Vaccinia Virus Ankara increases immunogenicity and isotype switching. Vaccine 2011, 29, 3276–3283. [Google Scholar] [CrossRef] [PubMed]
- Legrand, F.A.; Verardi, P.H.; Jones, L.A.; Chan, K.S.; Peng, Y.; Yilma, T.D. Induction of potent humoral and cell-mediated immune responses by attenuated vaccinia virus vectors with deleted serpin genes. J. Virol. 2004, 78, 2770–2779. [Google Scholar] [CrossRef] [PubMed]
- Mandl, S.J.; Rountree, R.B.; Dalpozzo, K.; Do, L.; Lombardo, J.R.; Schoonmaker, P.L.; Dirmeier, U.; Steigerwald, R.; Giffon, T.; Laus, R.; et al. Immunotherapy with MVA-BN(R)-HER2 induces HER-2-specific Th1 immunity and alters the intratumoral balance of effector and regulatory T cells. Cancer Immunol. Immunother. 2012, 61, 19–29. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Arriaza, J.; Esteban, M. Enhancing poxvirus vectors vaccine immunogenicity. Hum. Vaccin. Immunother. 2014, 10, 2235–2244. [Google Scholar] [CrossRef] [PubMed]
- Baur, K.; Brinkmann, K.; Schweneker, M.; Patzold, J.; Meisinger-Henschel, C.; Hermann, J.; Steigerwald, R.; Chaplin, P.; Suter, M.; Hausmann, J. Immediate-early expression of a recombinant antigen by modified vaccinia virus ankara breaks the immunodominance of strong vector-specific B8R antigen in acute and memory CD8 T-cell responses. J. Virol. 2010, 84, 8743–8752. [Google Scholar] [CrossRef] [PubMed]
- Wennier, S.T.; Brinkmann, K.; Steinhausser, C.; Maylander, N.; Mnich, C.; Wielert, U.; Dirmeier, U.; Hausmann, J.; Chaplin, P.; Steigerwald, R. A novel naturally occurring tandem promoter in modified vaccinia virus ankara drives very early gene expression and potent immune responses. PLoS ONE 2013, 8, e73511. [Google Scholar] [CrossRef] [PubMed]
- Di Pilato, M.; Mejias-Perez, E.; Gomez, C.E.; Perdiguero, B.; Sorzano, C.O.; Esteban, M. New vaccinia virus promoter as a potential candidate for future vaccines. J. Gen. Virol. 2013, 94, 2771–2776. [Google Scholar] [CrossRef] [PubMed]
- Hansen, H.; Okeke, M.I.; Nilssen, O.; Traavik, T. Recombinant viruses obtained from co-infection in vitro with a live vaccinia-vectored influenza vaccine and a naturally occurring cowpox virus display different plaque phenotypes and loss of the transgene. Vaccine 2004, 23, 499–506. [Google Scholar] [CrossRef] [PubMed]
- Okeke, M.I.; Nilssen, O.; Moens, U.; Tryland, M.; Traavik, T. In vitro host range, multiplication and virion forms of recombinant viruses obtained from co-infection in vitro with a vaccinia-vectored influenza vaccine and a naturally occurring cowpox virus isolate. Virol. J. 2009, 6, 55. [Google Scholar] [CrossRef] [PubMed]
- European Medicines Agency. Available online: http://www.ema.europa.eu/ema/ (accessed 10 July 2017).
- Sutter, G.; Ramsey-Ewing, A.; Rosales, R.; Moss, B. Stable expression of the vaccinia virus K1L gene in rabbit cells complements the host range defect of a vaccinia virus mutant. J. Virol. 1994, 68, 4109–4116. [Google Scholar] [PubMed]
- Wyatt, L.S.; Carroll, M.W.; Czerny, C.P.; Merchlinsky, M.; Sisler, J.R.; Moss, B. Marker rescue of the host range restriction defects of modified vaccinia virus Ankara. Virology 1998, 251, 334–342. [Google Scholar] [CrossRef] [PubMed]
- Mayr, A.; Munz, E. Changes in the vaccinia virus through continuing passages in chick embryo fibroblast cultures. Zent. Bakteriol. Orig. 1964, 195, 24–35. [Google Scholar]
- Qin, L.; Upton, C.; Hazes, B.; Evans, D.H. Genomic analysis of the vaccinia virus strain variants found in Dryvax vaccine. J. Virol. 2011, 85, 13049–13060. [Google Scholar] [CrossRef] [PubMed]
- Burgers, W.A.; Shephard, E.; Monroe, J.E.; Greenhalgh, T.; Binder, A.; Hurter, E.; van Harmelen, J.H.; Williamson, C.; Williamson, A.L. Construction, characterization and immunogenicity of a multigene modified vaccinia Ankara (MVA) vaccine based on HIV type 1 subtype C. AIDS Res. Hum. Retrovir. 2008, 24, 195–206. [Google Scholar] [CrossRef] [PubMed]
- Wyatt, L.S.; Belyakov, I.M.; Earl, P.L.; Berzofsky, J.A.; Moss, B. Enhanced cell surface expression, immunogenicity and genetic stability resulting from a spontaneous truncation of HIV Env expressed by a recombinant MVA. Virology 2008, 372, 260–272. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Martinez, J.; Zhou, W.; La Rosa, C.; Srivastava, T.; Dasgupta, A.; Rawal, R.; Li, Z.; Britt, W.J.; Diamond, D. Modified H5 promoter improves stability of insert genes while maintaining immunogenicity during extended passage of genetically engineered MVA vaccines. Vaccine 2010, 28, 1547–1557. [Google Scholar] [CrossRef] [PubMed]
- Timm, A.; Enzinger, C.; Felder, E.; Chaplin, P. Genetic stability of recombinant MVA-BN. Vaccine 2006, 24, 4618–4621. [Google Scholar] [CrossRef] [PubMed]
- Manuel, E.R.; Wang, Z.; Li, Z.; La Rosa, C.; Zhou, W.; Diamond, D.J. Intergenic region 3 of modified vaccinia ankara is a functional site for insert gene expression and allows for potent antigen-specific immune responses. Virology 2010, 403, 155–162. [Google Scholar] [CrossRef] [PubMed]
- Alharbi, N.K.; Spencer, A.J.; Salman, A.M.; Tully, C.M.; Chinnakannan, S.K.; Lambe, T.; Yamaguchi, Y.; Morris, S.J.; Orubu, T.; Draper, S.J.; et al. Enhancing cellular immunogenicity of MVA-vectored vaccines by utilizing the F11L endogenous promoter. Vaccine 2016, 34, 49–55. [Google Scholar] [CrossRef] [PubMed]
- Orubu, T.; Alharbi, N.K.; Lambe, T.; Gilbert, S.C.; Cottingham, M.G. Expression and cellular immunogenicity of a transgenic antigen driven by endogenous poxviral early promoters at their authentic loci in MVA. PLoS ONE 2012, 7, e40167. [Google Scholar] [CrossRef] [PubMed]
- Di Pilato, M.; Sanchez-Sampedro, L.; Mejias-Perez, E.; Sorzano, C.O.; Esteban, M. Modification of promoter spacer length in vaccinia virus as a strategy to control the antigen expression. J. Gen. Virol. 2015, 96, 2360–2371. [Google Scholar] [CrossRef] [PubMed]
- Chakrabarti, S.; Sisler, J.R.; Moss, B. Compact, synthetic, vaccinia virus early/late promoter for protein expression. Biotechniques 1997, 23, 1094–1097. [Google Scholar] [PubMed]
- Wyatt, L.S.; Earl, P.L.; Xiao, W.; Americo, J.L.; Cotter, C.A.; Vogt, J.; Moss, B. Elucidating and minimizing the loss by recombinant vaccinia virus of human immunodeficiency virus gene expression resulting from spontaneous mutations and positive selection. J. Virol. 2009, 83, 7176–7184. [Google Scholar] [CrossRef] [PubMed]
- Franke, A.; Pfaff, F.; Jenckel, M.; Hoffmann, B.; Hoper, D.; Antwerpen, M.; Meyer, H.; Beer, M.; Hoffmann, D. Classification of Cowpox Viruses into Several Distinct Clades and Identification of a Novel Lineage. Viruses 2017, 9. [Google Scholar] [CrossRef] [PubMed]
- Meyer, H.; Schay, C.; Mahnel, H.; Pfeffer, M. Characterization of orthopoxviruses isolated from man and animals in Germany. Arch. Virol. 1999, 144, 491–501. [Google Scholar] [CrossRef] [PubMed]
- Dabrowski, P.W.; Radonic, A.; Kurth, A.; Nitsche, A. Genome-wide comparison of cowpox viruses reveals a new clade related to Variola virus. PLoS ONE 2013, 8, e79953. [Google Scholar] [CrossRef] [PubMed]
- Mauldin, M.R.; Antwerpen, M.; Emerson, G.L.; Li, Y.; Zoeller, G.; Carroll, D.S.; Meyer, H. Cowpox virus: What’s in a Name? Viruses 2017, 9. [Google Scholar] [CrossRef] [PubMed]
- Kaysser, P.; von Bomhard, W.; Dobrzykowski, L.; Meyer, H. Genetic diversity of feline cowpox virus, Germany 2000–2008. Vet. Microbiol. 2010, 141, 282–288. [Google Scholar] [CrossRef] [PubMed]
- Chantrey, J.; Meyer, H.; Baxby, D.; Begon, M.; Bown, K.J.; Hazel, S.M.; Jones, T.; Montgomery, W.I.; Bennett, M. Cowpox: Reservoir hosts and geographic range. Epidemiol. Infect. 1999, 122, 455–460. [Google Scholar] [CrossRef] [PubMed]
- Crouch, A.C.; Baxby, D.; McCracken, C.M.; Gaskell, R.M.; Bennett, M. Serological evidence for the reservoir hosts of cowpox virus in British wildlife. Epidemiol. Infect. 1995, 115, 185–191. [Google Scholar] [CrossRef] [PubMed]
- Hansen, H.; Okeke, M.I.; Nilssen, O.; Traavik, T. Comparison and phylogenetic analysis of cowpox viruses isolated from cats and humans in Fennoscandia. Arch. Virol. 2009, 154, 1293–1302. [Google Scholar] [CrossRef] [PubMed]
- Tryland, M.; Sandvik, T.; Arnemo, J.M.; Stuve, G.; Olsvik, O.; Traavik, T. Antibodies against orthopoxviruses in wild carnivores from Fennoscandia. J. Wildl. Dis. 1998, 34, 443–450. [Google Scholar] [CrossRef] [PubMed]
- Tryland, M.; Sandvik, T.; Mehl, R.; Bennett, M.; Traavik, T.; Olsvik, O. Serosurvey for orthopoxviruses in rodents and shrews from Norway. J. Wildl. Dis. 1998, 34, 240–250. [Google Scholar] [CrossRef] [PubMed]
- Tryland, M.; Okeke, M.I.; Af Segerstad, C.H.; Morner, T.; Traavik, T.; Ryser-Degiorgis, M.P. Orthopoxvirus DNA in Eurasian lynx, Sweden. Emerg. Infect. Dis. 2011, 17, 626–632. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okeke, M.I.; Hansen, H.; Traavik, T. A naturally occurring cowpox virus with an ectromelia virus A-type inclusion protein gene displays atypical A-type inclusions. Infect. Genet. Evol. 2012, 12, 160–168. [Google Scholar] [CrossRef] [PubMed]
- Kinnunen, P.M.; Henttonen, H.; Hoffmann, B.; Kallio, E.R.; Korthase, C.; Laakkonen, J.; Niemimaa, J.; Palva, A.; Schlegel, M.; Ali, H.S.; et al. Orthopox virus infections in Eurasian wild rodents. Vector Borne Zoonotic Dis. 2011, 11, 1133–1140. [Google Scholar] [CrossRef] [PubMed]
- Emerson, G.L.; Li, Y.; Frace, M.A.; Olsen-Rasmussen, M.A.; Khristova, M.L.; Govil, D.; Sammons, S.A.; Regnery, R.L.; Karem, K.L.; Damon, I.K.; et al. The phylogenetics and ecology of the orthopoxviruses endemic to North America. PLoS ONE 2009, 4, e7666. [Google Scholar] [CrossRef] [PubMed]
- Springer, Y.P.; Hsu, C.H.; Werle, Z.R.; Olson, L.E.; Cooper, M.P.; Castrodale, L.J.; Fowler, N.; McCollum, A.M.; Goldsmith, C.S.; Emerson, G.L.; et al. Novel Orthopoxvirus Infection in an Alaska Resident. Clin. Infect. Dis. 2017, 64, 1737–1741. [Google Scholar] [CrossRef] [PubMed]
- Fleischauer, C.; Upton, C.; Victoria, J.; Jones, G.J.; Roper, R.L. Genome sequence and comparative virulence of raccoonpox virus: The first North American poxvirus sequence. J. Gen. Virol. 2015, 96, 2806–2821. [Google Scholar] [CrossRef] [PubMed]
- Smithson, C.; Tang, N.; Sammons, S.; Frace, M.; Batra, D.; Li, Y.; Emerson, G.L.; Carroll, D.S.; Upton, C. The genomes of three North American orthopoxviruses. Virus Genes 2017, 53, 21–34. [Google Scholar] [CrossRef] [PubMed]
- Miranda, J.B.; Borges, I.A.; Campos, S.P.S.; Vieira, F.N.; de Azara, T.M.F.; Marques, F.A.; Costa, G.B.; Luis, A.; de Oliveira, J.S.; Ferreira, P.C.P.; et al. Serologic and Molecular Evidence of Vaccinia Virus Circulation among Small Mammals from Different Biomes, Brazil. Emerg. Infect. Dis. 2017, 23, 931–938. [Google Scholar] [CrossRef] [PubMed]
- Costa, G.B.; Augusto, L.T.; Leite, J.A.; Ferreira, P.C.; Bonjardim, C.A.; Abrahao, J.S.; Kroon, E.G.; Moreno, E.C.; Trindade Gde, S. Seroprevalence of Orthopoxvirus in rural Brazil: Insights into anti-OPV immunity status and its implications for emergent zoonotic OPV. Virol. J. 2016, 13, 121. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, G.; Assis, F.; Almeida, G.; Albarnaz, J.; Lima, M.; Andrade, A.C.; Calixto, R.; Oliveira, C.; Diomedes Neto, J.; Trindade, G.; et al. From lesions to viral clones: Biological and molecular diversity amongst autochthonous Brazilian vaccinia virus. Viruses 2015, 7, 1218–1237. [Google Scholar] [CrossRef] [PubMed]
- Moussatche, N.; Damaso, C.R.; McFadden, G. When good vaccines go wild: Feral Orthopoxvirus in developing countries and beyond. J. Infect. Dev. Ctries 2008, 2, 156–173. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.K.; Hosamani, M.; Balamurugan, V.; Bhanuprakash, V.; Rasool, T.J.; Yadav, M.P. Buffalopox: An emerging and re-emerging zoonosis. Anim. Health Res. Rev. 2007, 8, 105–114. [Google Scholar] [CrossRef] [PubMed]
- Bhanuprakash, V.; Venkatesan, G.; Balamurugan, V.; Hosamani, M.; Yogisharadhya, R.; Gandhale, P.; Reddy, K.V.; Damle, A.S.; Kher, H.N.; Chandel, B.S.; et al. Zoonotic infections of buffalopox in India. Zoonoses Public Health 2010, 57, e149–e155. [Google Scholar] [CrossRef] [PubMed]
- Bera, B.C.; Shanmugasundaram, K.; Barua, S.; Venkatesan, G.; Virmani, N.; Riyesh, T.; Gulati, B.R.; Bhanuprakash, V.; Vaid, R.K.; Kakker, N.K.; et al. Zoonotic cases of camelpox infection in India. Vet. Microbiol. 2011, 152, 29–38. [Google Scholar] [CrossRef] [PubMed]
- Afolabi, M.O.; Tiono, A.B.; Adetifa, U.J.; Yaro, J.B.; Drammeh, A.; Nebie, I.; Bliss, C.; Hodgson, S.H.; Anagnostou, N.A.; Sanou, G.S.; et al. Safety and Immunogenicity of ChAd63 and MVA ME-TRAP in West African Children and Infants. Mol. Ther. 2016, 24, 1470–1477. [Google Scholar] [CrossRef] [PubMed]
- Mensah, V.A.; Gueye, A.; Ndiaye, M.; Edwards, N.J.; Wright, D.; Anagnostou, N.A.; Syll, M.; Ndaw, A.; Abiola, A.; Bliss, C.; et al. Safety, Immunogenicity and Efficacy of Prime-Boost Vaccination with ChAd63 and MVA Encoding ME-TRAP against Plasmodium falciparum Infection in Adults in Senegal. PLoS ONE 2016, 11, e0167951. [Google Scholar] [CrossRef] [PubMed]
- Excler, J.L.; Michael, N.L. Lessons from HIV-1 vaccine efficacy trials. Curr. Opin. HIV AIDS 2016, 11, 607–613. [Google Scholar] [CrossRef] [PubMed]
- Brochier, B.M.; Languet, B.; Blancou, J.; Kieny, M.P.; Lecocq, J.P.; Costy, F.; Desmettre, P.; Pastoret, P.P. Use of recombinant vaccinia-rabies virus for oral vaccination of fox cubs (Vulpes vulpes, L) against rabies. Vet. Microbiol. 1988, 18, 103–108. [Google Scholar] [CrossRef]
- Freuling, C.M.; Hampson, K.; Selhorst, T.; Schroder, R.; Meslin, F.X.; Mettenleiter, T.C.; Muller, T. The elimination of fox rabies from Europe: Determinants of success and lessons for the future. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2013, 368, 20120142. [Google Scholar] [CrossRef] [PubMed]
- Ball, L.A. High-frequency homologous recombination in vaccinia virus DNA. J. Virol. 1987, 61, 1788–1795. [Google Scholar] [PubMed]
- Fathi, Z.; Dyster, L.M.; Seto, J.; Condit, R.C.; Niles, E.G. Intragenic and intergenic recombination between temperature-sensitive mutants of vaccinia virus. J. Gen. Virol. 1991, 72 (Pt 11), 2733–2737. [Google Scholar] [CrossRef] [PubMed]
- Block, W.; Upton, C.; McFadden, G. Tumorigenic poxviruses: Genomic organization of malignant rabbit virus, a recombinant between Shope fibroma virus and myxoma virus. Virology 1985, 140, 113–124. [Google Scholar] [CrossRef]
- Qin, L.; Evans, D.H. Genome scale patterns of recombination between coinfecting vaccinia viruses. J. Virol. 2014, 88, 5277–5286. [Google Scholar] [CrossRef] [PubMed]
- Staib, C.; Drexler, I.; Sutter, G. Construction and isolation of recombinant MVA. Methods Mol. Biol. 2004, 269, 77–100. [Google Scholar] [PubMed]
- Lin, Y.C.; Evans, D.H. Vaccinia virus particles mix inefficiently and in a way that would restrict viral recombination, in co-infected cells. J. Virol. 2010, 84, 2432–2443. [Google Scholar] [CrossRef] [PubMed]
- Doceul, V.; Hollinshead, M.; van der Linden, L.; Smith, G.L. Repulsion of superinfecting virions: A mechanism for rapid virus spread. Science 2010, 327, 873–876. [Google Scholar] [CrossRef] [PubMed]
- Laliberte, J.P.; Moss, B. A novel mode of poxvirus superinfection exclusion that prevents fusion of the lipid bilayers of viral and cellular membranes. J. Virol. 2014, 88, 9751–9768. [Google Scholar] [CrossRef] [PubMed]
- Paszkowski, P.; Noyce, R.S.; Evans, D.H. Live-Cell Imaging of Vaccinia Virus Recombination. PLoS Pathog. 2016, 12, e1005824. [Google Scholar] [CrossRef] [PubMed]
- Nitsche, A.; Kurth, A.; Pauli, G. Viremia in human Cowpox virus infection. J. Clin. Virol. 2007, 40, 160–162. [Google Scholar] [CrossRef] [PubMed]
- Sutter, G.; Wyatt, L.S.; Foley, P.L.; Bennink, J.R.; Moss, B. A recombinant vector derived from the host range-restricted and highly attenuated MVA strain of vaccinia virus stimulates protective immunity in mice to influenza virus. Vaccine 1994, 12, 1032–1040. [Google Scholar] [CrossRef]
- Lee, S.W.; Markham, P.F.; Coppo, M.J.; Legione, A.R.; Markham, J.F.; Noormohammadi, A.H.; Browning, G.F.; Ficorilli, N.; Hartley, C.A.; Devlin, J.M. Attenuated vaccines can recombine to form virulent field viruses. Science 2012, 337, 188. [Google Scholar] [CrossRef] [PubMed]
- Ramirez, J.C.; Gherardi, M.M.; Esteban, M. Biology of attenuated modified vaccinia virus Ankara recombinant vector in mice: Virus fate and activation of B- and T-cell immune responses in comparison with the Western Reserve strain and advantages as a vaccine. J. Virol. 2000, 74, 923–933. [Google Scholar] [CrossRef] [PubMed]
- Ramirez, J.C.; Gherardi, M.M.; Rodriguez, D.; Esteban, M. Attenuated modified vaccinia virus Ankara can be used as an immunizing agent under conditions of preexisting immunity to the vector. J. Virol. 2000, 74, 7651–7655. [Google Scholar] [CrossRef] [PubMed]
- Stittelaar, K.J.; Kuiken, T.; de Swart, R.L.; van Amerongen, G.; Vos, H.W.; Niesters, H.G.; van Schalkwijk, P.; van der Kwast, T.; Wyatt, L.S.; Moss, B.; et al. Safety of modified vaccinia virus Ankara (MVA) in immune-suppressed macaques. Vaccine 2001, 19, 3700–3709. [Google Scholar] [CrossRef]
- Stittelaar, K.J.; Osterhaus, A.D. MVA: A cuckoo in the vaccine nest? Vaccine 2001, 19, V–VI. [Google Scholar] [CrossRef]
- Van den Akker, E.; van der Vlugt, C.J.; Bleijs, D.A.; Bergmans, H.E. Environmental risk assessment of replication competent viral vectors applied in clinical trials: Potential effects of inserted sequences. Curr. Gene Ther. 2013, 13, 395–412. [Google Scholar] [CrossRef] [PubMed]
- Sparkes, J.D.; Fenje, P. The effect of residual moisture in lyophilized smallpox vaccine on its stability at different temperatures. Bull. World Health Organ. 1972, 46, 729–734. [Google Scholar] [PubMed]
- Rheinbaben, F.G.J.; Exner, M.; Schmidt, M.; Mercer, A.A.; Schmidt, A.; Weber, O. Environmental Resistance Disinfection and Sterilization of Poxviruses; Birkh user Verlag: Basel, Switzerland, 2007. [Google Scholar]
- Gallina, L.; Scagliarini, A. Virucidal efficacy of common disinfectants against orf virus. Vet. Rec. 2010, 166, 725–726. [Google Scholar] [CrossRef] [PubMed]
- De Oliveira, T.M.; Rehfeld, I.S.; Coelho Guedes, M.I.; Ferreira, J.M.; Kroon, E.G.; Lobato, Z.I. Susceptibility of Vaccinia virus to chemical disinfectants. Am. J. Trop. Med. Hyg. 2011, 85, 152–157. [Google Scholar] [CrossRef] [PubMed]
- Butcher, W.; Ulaeto, D. Contact inactivation of orthopoxviruses by household disinfectants. J. Appl. Microbiol. 2005, 99, 279–284. [Google Scholar] [CrossRef] [PubMed]
- England, L.S.; Holmes, S.B.; Trevors, J.T. Persistence of viruses and DNA in soil. World J. Microb. Biot. 1998, 14, 163–169. [Google Scholar] [CrossRef]
- Woo, H.J.; Reifman, J. A quantitative quasispecies theory-based model of virus escape mutation under immune selection. Proc. Natl. Acad. Sci. USA 2012, 109, 12980–12985. [Google Scholar] [CrossRef] [PubMed]
- Thomas, S.; Redfern, J.B.; Lidbury, B.A.; Mahalingam, S. Antibody-dependent enhancement and vaccine development. Expert Rev. Vaccin. 2006, 5, 409–412. [Google Scholar] [CrossRef] [PubMed]
- Johnson, T.R.; Graham, B.S. Secreted respiratory syncytial virus G glycoprotein induces interleukin-5 (IL-5), IL-13 and eosinophilia by an IL-4-independent mechanism. J. Virol. 1999, 73, 8485–8495. [Google Scholar] [PubMed]
- Wyatt, L.S.; Earl, P.L.; Eller, L.A.; Moss, B. Highly attenuated smallpox vaccine protects mice with and without immune deficiencies against pathogenic vaccinia virus challenge. Proc. Natl. Acad. Sci. USA 2004, 101, 4590–4595. [Google Scholar] [CrossRef] [PubMed]
- Price, P.J.; Torres-Dominguez, L.E.; Brandmuller, C.; Sutter, G.; Lehmann, M.H. Modified Vaccinia virus Ankara: Innate immune activation and induction of cellular signalling. Vaccine 2013, 31, 4231–4234. [Google Scholar] [CrossRef] [PubMed]
- Smith, G.L.; Benfield, C.T.; Maluquer de Motes, C.; Mazzon, M.; Ember, S.W.; Ferguson, B.J.; Sumner, R.P. Vaccinia virus immune evasion: Mechanisms, virulence and immunogenicity. J. Gen. Virol. 2013, 94, 2367–2392. [Google Scholar] [CrossRef] [PubMed]
- Clark, R.H.; Kenyon, J.C.; Bartlett, N.W.; Tscharke, D.C.; Smith, G.L. Deletion of gene A41L enhances vaccinia virus immunogenicity and vaccine efficacy. J. Gen. Virol. 2006, 87, 29–38. [Google Scholar] [CrossRef] [PubMed]
- Staib, C.; Kisling, S.; Erfle, V.; Sutter, G. Inactivation of the viral interleukin 1beta receptor improves CD8+ T-cell memory responses elicited upon immunization with modified vaccinia virus Ankara. J. Gen. Virol. 2005, 86, 1997–2006. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Arriaza, J.; Arnaez, P.; Gomez, C.E.; Sorzano, C.O.; Esteban, M. Improving Adaptive and Memory Immune Responses of an HIV/AIDS Vaccine Candidate MVA-B by Deletion of Vaccinia Virus Genes (C6L and K7R) Blocking Interferon Signaling Pathways. PLoS ONE 2013, 8, e66894. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Arriaza, J.; Gomez, C.E.; Sorzano, C.O.; Esteban, M. Deletion of the vaccinia virus N2L gene encoding an inhibitor of IRF3 improves the immunogenicity of modified vaccinia virus Ankara expressing HIV-1 antigens. J. Virol. 2014, 88, 3392–3410. [Google Scholar] [CrossRef] [PubMed]
- Chavan, R.; Marfatia, K.A.; An, I.C.; Garber, D.A.; Feinberg, M.B. Expression of CCL20 and granulocyte-macrophage colony-stimulating factor, but not Flt3-L, from modified vaccinia virus ankara enhances antiviral cellular and humoral immune responses. J. Virol. 2006, 80, 7676–7687. [Google Scholar] [CrossRef] [PubMed]
- Kannanganat, S.; Wyatt, L.S.; Gangadhara, S.; Chamcha, V.; Chea, L.S.; Kozlowski, P.A.; LaBranche, C.C.; Chennareddi, L.; Lawson, B.; Reddy, P.B.; et al. High Doses of GM-CSF Inhibit Antibody Responses in Rectal Secretions and Diminish Modified Vaccinia Ankara/Simian Immunodeficiency Virus Vaccine Protection in TRIM5α-Restrictive Macaques. J. Immunol. 2016, 197, 3586–3596. [Google Scholar] [CrossRef] [PubMed]
- Backes, S.; Sperling, K.M.; Zwilling, J.; Gasteiger, G.; Ludwig, H.; Kremmer, E.; Schwantes, A.; Staib, C.; Sutter, G. Viral host-range factor C7 or K1 is essential for modified vaccinia virus Ankara late gene expression in human and murine cells, irrespective of their capacity to inhibit protein kinase R-mediated phosphorylation of eukaryotic translation initiation factor 2α. J. Gen. Virol. 2010, 91, 470–482. [Google Scholar] [PubMed]
- Rojas, J.J.; Sampath, P.; Bonilla, B.; Ashley, A.; Hou, W.; Byrd, D.; Thorne, S.H. Manipulating TLR Signaling Increases the Anti-tumor T Cell Response Induced by Viral Cancer Therapies. Cell Rep. 2016, 15, 264–273. [Google Scholar] [CrossRef] [PubMed]
- Berger, A. Th1 and Th2 responses: What are they? BMJ 2000, 321, 424. [Google Scholar] [CrossRef] [PubMed]
- Raphael, I.; Nalawade, S.; Eagar, T.N.; Forsthuber, T.G. T cell subsets and their signature cytokines in autoimmune and inflammatory diseases. Cytokine 2015, 74, 5–17. [Google Scholar] [CrossRef] [PubMed]
- Holgado, M.P.; Falivene, J.; Maeto, C.; Amigo, M.; Pascutti, M.F.; Vecchione, M.B.; Bruttomesso, A.; Calamante, G.; del Medico-Zajac, M.P.; Gherardi, M.M. Deletion of A44L, A46R and C12L Vaccinia Virus Genes from the MVA Genome Improved the Vector Immunogenicity by Modifying the Innate Immune Response Generating Enhanced and Optimized Specific T-Cell Responses. Viruses 2016, 8. [Google Scholar] [CrossRef] [PubMed]
- Cottingham, M.G.; Andersen, R.F.; Spencer, A.J.; Saurya, S.; Furze, J.; Hill, A.V.; Gilbert, S.C. Recombination-mediated genetic engineering of a bacterial artificial chromosome clone of modified vaccinia virus Ankara (MVA). PLoS ONE 2008, 3, e1638. [Google Scholar] [CrossRef] [PubMed]
- Perdiguero, B.; Gomez, C.E.; Najera, J.L.; Sorzano, C.O.; Delaloye, J.; Gonzalez-Sanz, R.; Jimenez, V.; Roger, T.; Calandra, T.; Pantaleo, G.; et al. Deletion of the viral anti-apoptotic gene F1L in the HIV/AIDS vaccine candidate MVA-C enhances immune responses against HIV-1 antigens. PLoS ONE 2012, 7, e48524. [Google Scholar] [CrossRef] [PubMed]
- Jackson, R.J.; Ramsay, A.J.; Christensen, C.D.; Beaton, S.; Hall, D.F.; Ramshaw, I.A. Expression of mouse interleukin-4 by a recombinant ectromelia virus suppresses cytolytic lymphocyte responses and overcomes genetic resistance to mousepox. J. Virol. 2001, 75, 1205–1210. [Google Scholar] [CrossRef] [PubMed]
- Stanford, M.M.; McFadden, G. The supervirus? Lessons from IL-4-expressing poxviruses. Trends Immunol. 2005, 26, 339–345. [Google Scholar] [CrossRef] [PubMed]
- Bonjardim, C.A. Viral exploitation of the MEK/ERK pathway—A tale of vaccinia virus and other viruses. Virology 2017, 507, 267–275. [Google Scholar] [CrossRef] [PubMed]
- Soares, J.A.; Leite, F.G.; Andrade, L.G.; Torres, A.A.; de Sousa, L.P.; Barcelos, L.S.; Teixeira, M.M.; Ferreira, P.C.; Kroon, E.G.; Souto-Padron, T.; et al. Activation of the PI3K/Akt pathway early during vaccinia and cowpox virus infections is required for both host survival and viral replication. J. Virol. 2009, 83, 6883–6899. [Google Scholar] [CrossRef] [PubMed]
- Seo, G.J.; Yang, A.; Tan, B.; Kim, S.; Liang, Q.; Choi, Y.; Yuan, W.; Feng, P.; Park, H.S.; Jung, J.U. Akt Kinase-Mediated Checkpoint of cGAS DNA Sensing Pathway. Cell Rep. 2015, 13, 440–449. [Google Scholar] [CrossRef] [PubMed]
- Schweneker, M.; Lukassen, S.; Spath, M.; Wolferstatter, M.; Babel, E.; Brinkmann, K.; Wielert, U.; Chaplin, P.; Suter, M.; Hausmann, J. The vaccinia virus O1 protein is required for sustained activation of extracellular signal-regulated kinase 1/2 and promotes viral virulence. J. Virol. 2012, 86, 2323–2336. [Google Scholar] [CrossRef] [PubMed]
- Pereira, A.C.; Leite, F.G.; Brasil, B.S.; Soares-Martins, J.A.; Torres, A.A.; Pimenta, P.F.; Souto-Padron, T.; Traktman, P.; Ferreira, P.C.; Kroon, E.G.; et al. A vaccinia virus-driven interplay between the MKK4/7-JNK1/2 pathway and cytoskeleton reorganization. J. Virol. 2012, 86, 172–184. [Google Scholar] [CrossRef] [PubMed]
- Hu, W.; Hofstetter, W.; Guo, W.; Li, H.; Pataer, A.; Peng, H.H.; Guo, Z.S.; Bartlett, D.L.; Lin, A.; Swisher, S.G.; et al. JNK-deficiency enhanced oncolytic vaccinia virus replication and blocked activation of double-stranded RNA-dependent protein kinase. Cancer Gene Ther. 2008, 15, 616–624. [Google Scholar] [CrossRef] [PubMed]
- Gedey, R.; Jin, X.L.; Hinthong, O.; Shisler, J.L. Poxviral regulation of the host NF-κB response: The vaccinia virus M2L protein inhibits induction of NF-κB activation via an ERK2 pathway in virus-infected human embryonic kidney cells. J. Virol. 2006, 80, 8676–8685. [Google Scholar] [CrossRef] [PubMed]
- Caceres, A.; Perdiguero, B.; Gomez, C.E.; Cepeda, M.V.; Caelles, C.; Sorzano, C.O.; Esteban, M. Involvement of the cellular phosphatase DUSP1 in vaccinia virus infection. PLoS Pathog. 2013, 9, e1003719. [Google Scholar] [CrossRef] [PubMed]
- Knol, A.B.; Petersen, A.C.; van der Sluijs, J.P.; Lebret, E. Dealing with uncertainties in environmental burden of disease assessment. Environ. Health 2009, 8, 21. [Google Scholar] [CrossRef] [PubMed]
- Skinner, D.J.C.; Rocks, S.A.; Pollard, S.J.T. Where do uncertainties reside within environmental risk assessments? Testing UnISERA, a guide for uncertainty assessment. Environ. Pollut. 2017, 225, 390–402. [Google Scholar] [CrossRef] [PubMed]
- Wynne, B. Uncertainty and Environmental Learning—Reconceiving Science and Policy in the Preventive Paradigm. Glob. Environ. Chang. 1992, 2, 111–127. [Google Scholar] [CrossRef]
- Liu, B.F.; Bartz, L.; Duke, N. Communicating crisis uncertainty: A review of the knowledge gaps. Public Relat. Rev. 2016, 42, 479–487. [Google Scholar] [CrossRef]
- Skinner, D.J.C.; Rocks, S.A.; Pollard, S.J.T.; Drew, G.H. Identifying Uncertainty in Environmental Risk Assessments: The Development of a Novel Typology and Its Implications for Risk Characterization. Hum. Ecol. Risk Assess. 2014, 20, 607–640. [Google Scholar] [CrossRef] [Green Version]
- Skinner, D.J.C.; Rocks, S.A.; Pollard, S.J.T. A review of uncertainty in environmental risk: Characterising potential natures, locations and levels. J. Risk Res. 2014, 17, 195–219. [Google Scholar] [CrossRef]
- Mhyr, A.I.; Traavik, T. Genetically enginneered virus-vectored vaccines—Environmental risk assessment and management challenges. In Genetic Engineering—Basics, New Applications and Responsibilities; Barrera-Saldana, H.A., Ed.; InTech: Rijeka, Croatia, 2012; pp. 1–20. [Google Scholar]
- Wolt, J.D.; Keese, P.; Raybould, A.; Fitzpatrick, J.W.; Burachik, M.; Gray, A.; Olin, S.S.; Schiemann, J.; Sears, M.; Wu, F. Problem formulation in the environmental risk assessment for genetically modified plants. Transgenic Res. 2010, 19, 425–436. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
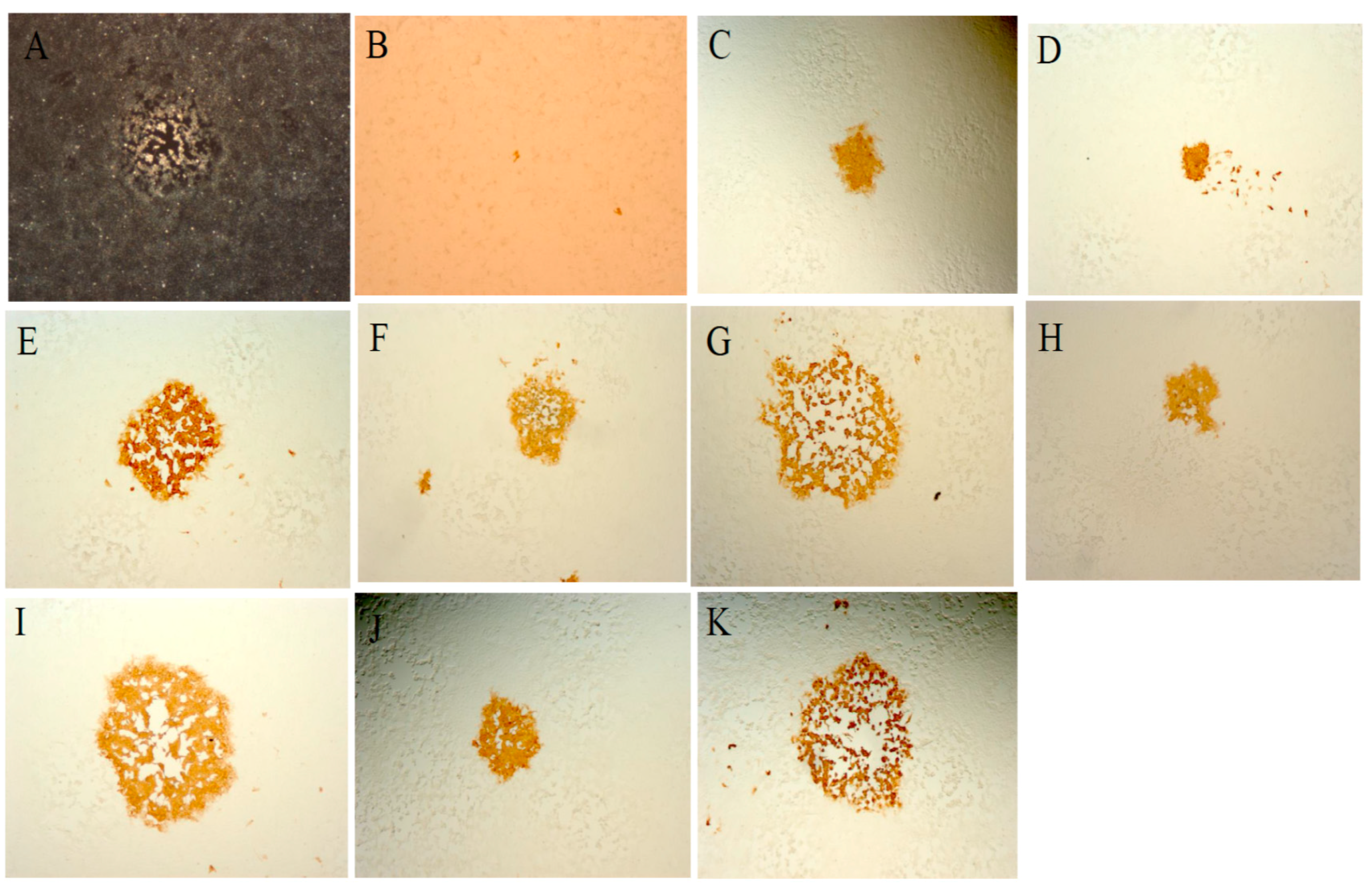{kind=link}
| Cell Lines | Species | Multiplication of MVA Strains and Variants a | References | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| MVA b | MVA-II/85 | MVA-VR1508 | MVA-BN | MVA-B | MVA-572 | MVA-1721 | MVA-574 | MVA-LZ | |||
| MDCK | Canine; kidney | SP | NP | [45,50] | |||||||
| Ederm | Equine; skin | NP | [45] | ||||||||
| RO5R | Fruit bat Egyptian | P | [58] | ||||||||
| RO5T | Fruit bat Egyptian | P | [58] | ||||||||
| RO6E | Fruit bat Egyptian | P | [58] | ||||||||
| CHL | Hamster Chinese; lung | NP | [50] | ||||||||
| CHO | Hamster Chinese; ovaries | NP | NP | [50,52] | |||||||
| BHK-21 | Hamster Syrian; kidney | P | P | P | P | P | [47,49,50,51,52] | ||||
| HEK293 | Human, kidney | NP | NP | NP | NP | NP | P | SP | [47,50,51,52,56] | ||
| Hela | Human; cervix | NP | SP | NP | NP | NP | P | NP | SP | [45,47,50,51,56,57] | |
| SW839 | Human; kidney | NP | [50] | ||||||||
| TK− 143B | Human; bone | P | SP | NP | NP | P | [56,57] | ||||
| MRC-5 | Human; lung | NP | NP | NP | NP | [45,47,57] | |||||
| FS-2 | Human; skin | NP | [57] | ||||||||
| Caco-2 | Human; colorectal | NP | [52] | ||||||||
| FHs74int | Human; esophagus | NP | [52] | ||||||||
| Hutu-80 | Human; small intestine | NP | [52] | ||||||||
| A549 | Human; lung | SP | [52] | ||||||||
| HaCaT | Human; skin | NP | SP | P | [56] | ||||||
| HRT18 | Human; colon | NP | [45] | ||||||||
| Hep-2 | Human; larynx | NP | [45] | ||||||||
| SK 29 MEL 1 | Human; skin | NP | [51] | ||||||||
| LC5 | Human; lung | NP | [51] | ||||||||
| 85 HG 66 | Human; brain | NP | [51] | ||||||||
| U138 | Human; brain | NP | [51] | ||||||||
| C8166 | Human; blood (T-cell) | NP | [51] | ||||||||
| HUT 78 | Human; blood (T-cell) | NP | [51] | ||||||||
| SY9287 | Human; blood (B-cell) | NP | [51] | ||||||||
| MA104 | Monkey; kidney | P | [45] | ||||||||
| MIB | Monkey; blood (B-cell) | NP | [51] | ||||||||
| BSC-1 | Monkey African Green; kidney | SP | [50] | ||||||||
| CV1 | Monkey African Green, kidney | SP, P | P | [50,51] | |||||||
| Vero | Monkey African Green; kidney | SP | SP | SP | SP | SP | [45,47,50,52] | ||||
| FRhK-4 | Monkey Rhesus; kidney | NP | [51] | ||||||||
| Balb3t3 | Mouse; embryonal fibroblast | NP | NP | [56,57] | |||||||
| NMULI | Mouse; glandular epithelial | SP | [52] | ||||||||
| AG101 | Mouse; skin | NP | NP | NP | [56] | ||||||
| DBT | Mouse; brain | NP | [45] | ||||||||
| PK(15) | Pig; kidney | NP | NP | [50,52] | |||||||
| BEL | Pig; lung | SP | [45] | ||||||||
| MDBK | Pig; kidney | NP | [45] | ||||||||
| RK13 | Rabbit; kidney | NP | NP | NP | [45,50,52] | ||||||
| RAB-9 | Rabbit; skin | NP | [50] | ||||||||
| SIRC | Rabbit; cornea | NP | [50] | ||||||||
| IEC-6 | Rat; small intestine | P | SP | [47,52] | |||||||
| H4IIE | Rat; liver | NP | [52] | ||||||||
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Okeke, M.I.; Okoli, A.S.; Diaz, D.; Offor, C.; Oludotun, T.G.; Tryland, M.; Bøhn, T.; Moens, U. Hazard Characterization of Modified Vaccinia Virus Ankara Vector: What Are the Knowledge Gaps? Viruses 2017, 9, 318. https://doi.org/10.3390/v9110318
Okeke MI, Okoli AS, Diaz D, Offor C, Oludotun TG, Tryland M, Bøhn T, Moens U. Hazard Characterization of Modified Vaccinia Virus Ankara Vector: What Are the Knowledge Gaps? Viruses. 2017; 9(11):318. https://doi.org/10.3390/v9110318
Chicago/Turabian StyleOkeke, Malachy I., Arinze S. Okoli, Diana Diaz, Collins Offor, Taiwo G. Oludotun, Morten Tryland, Thomas Bøhn, and Ugo Moens. 2017. "Hazard Characterization of Modified Vaccinia Virus Ankara Vector: What Are the Knowledge Gaps?" Viruses 9, no. 11: 318. https://doi.org/10.3390/v9110318