Hepatitis B Virus and DNA Damage Response: Interactions and Consequences for the Infection


{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Hepatitis B Virus Life Cycle
3. Brief Overview of DNA Damage Response Pathways
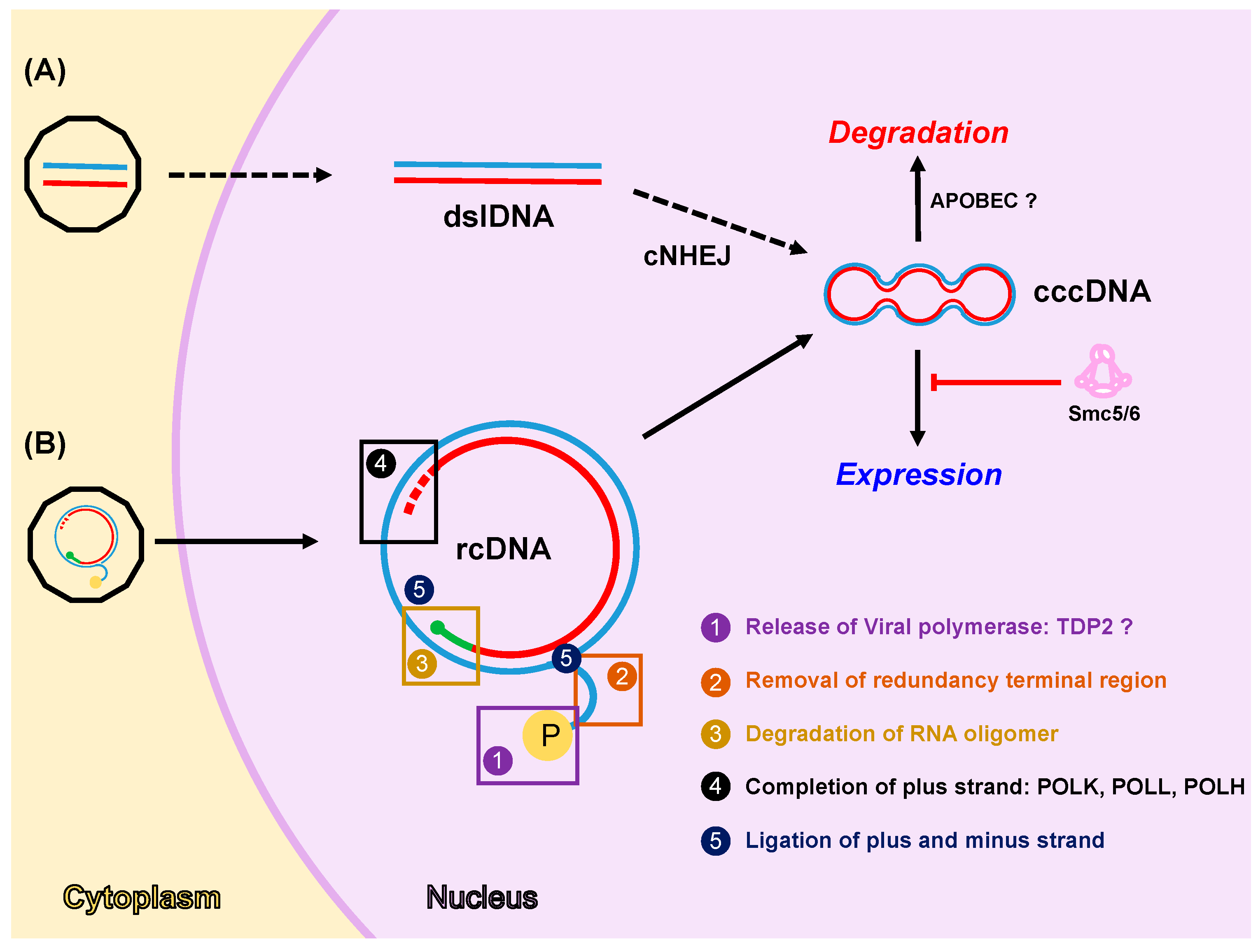
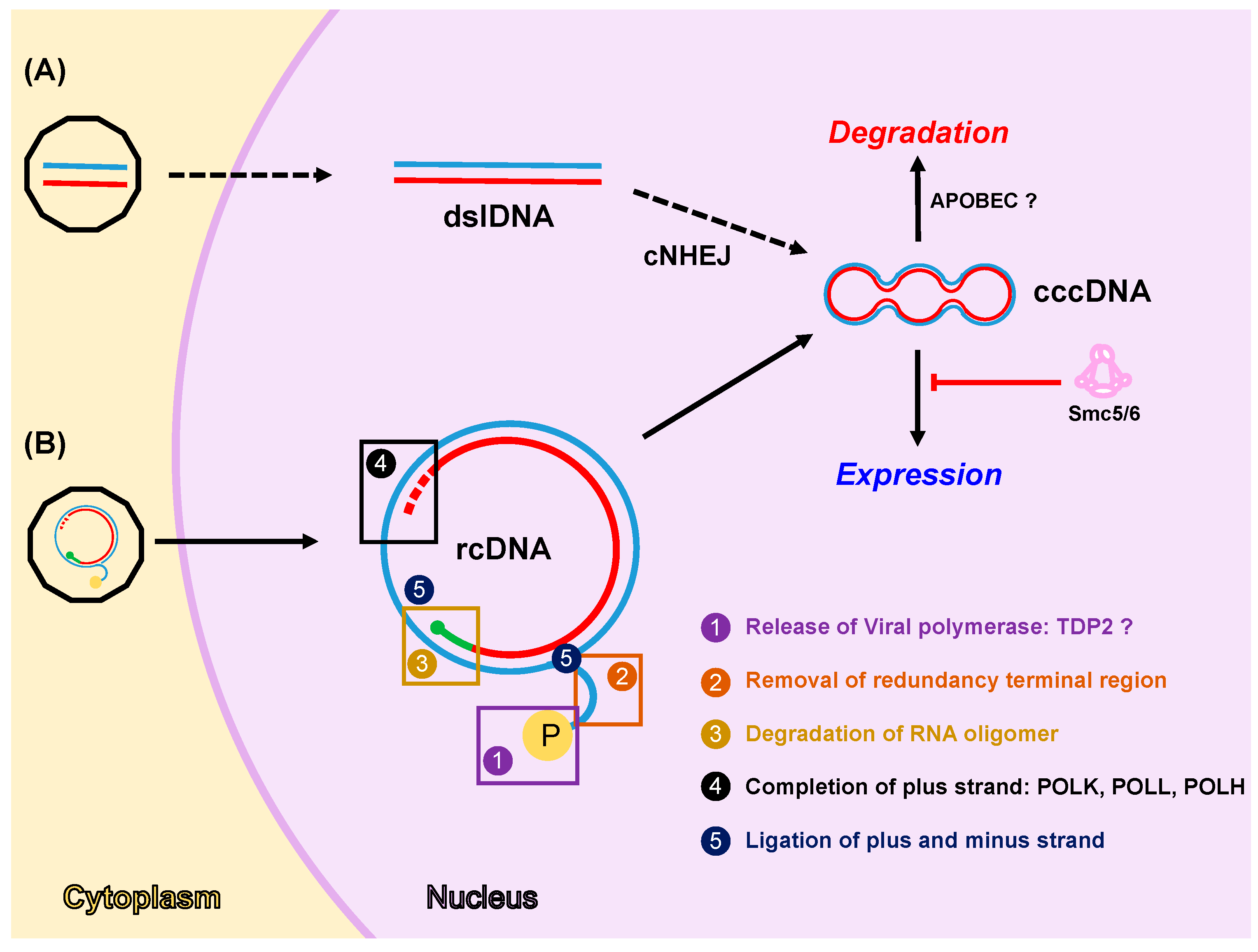
4. DDR Factors and Pathways Involved in cccDNA Formation, Expression and Degradation
4.1. Formation of cccDNA
4.2. Expression from cccDNA
4.3. Degradation of cccDNA
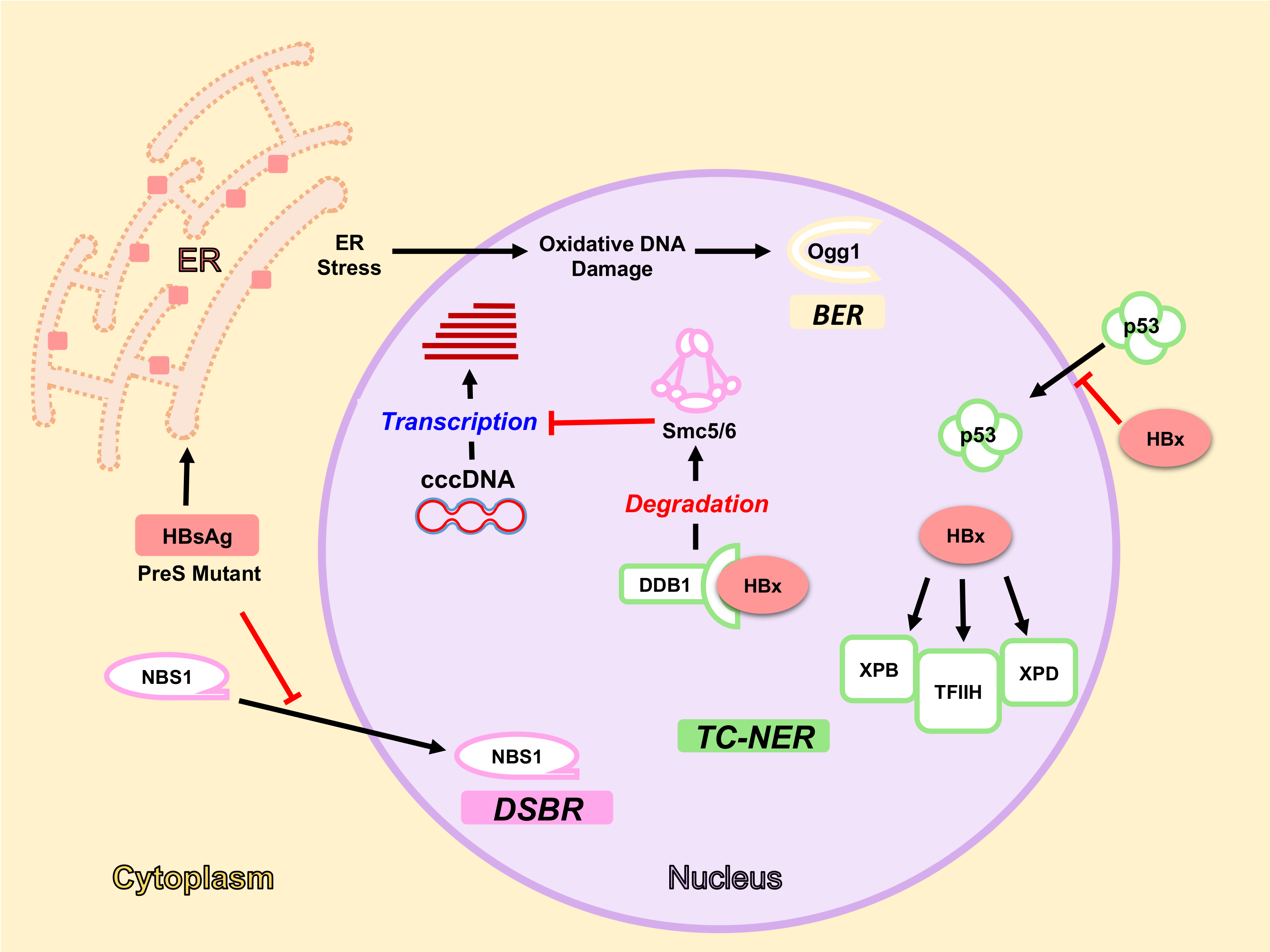
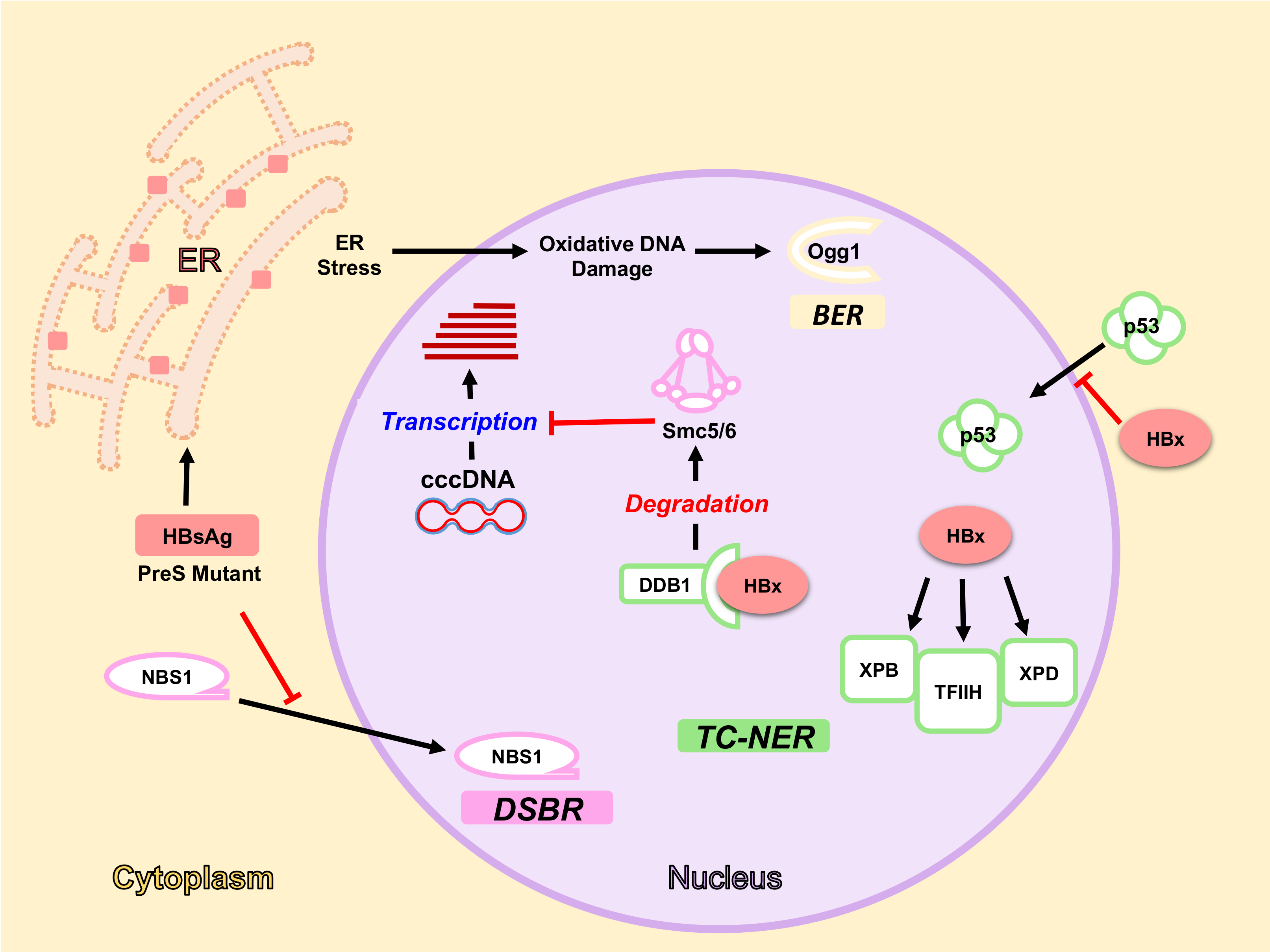
5. HBV Factors Interfere with DDR Pathways
5.1. HBx Protein and DDR
5.1.1. HBx Interaction with DDB1-Cul4 Complex
5.1.2. HBx and NER Pathway
5.2. HBsAg, ER Stress and Oxidative DNA Damage
6. Consequences of DDR Deregulation for HBV Infection
6.1. DNA Damage Response and HBV Integration
6.2. DNA Damage Response and HBV Replication
6.3. DNA Damage Response and HBV-Induced HCC
7. Final Remarks
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Chisari, F.V.; Ferrari, C. Hepatitis B virus immunopathogenesis. Annu. Rev. Immunol. 1995, 13, 29–60. [Google Scholar] [CrossRef] [PubMed]
- Perz, J.F.; Armstrong, G.L.; Farrington, L.A.; Hutin, Y.J.F.; Bell, B.P. The contributions of hepatitis B virus and hepatitis C virus infections to cirrhosis and primary liver cancer worldwide. J. Hepatol. 2006, 45, 529–538. [Google Scholar] [CrossRef] [PubMed]
- Schweitzer, A.; Horn, J.; Mikolajczyk, R.T.; Krause, G.; Ott, J.J. Estimations of worldwide prevalence of chronic hepatitis B virus infection: A systematic review of data published between 1965 and 2013. Lancet 2015, 386, 1546–1555. [Google Scholar] [CrossRef]
- Hai, H.; Tamori, A.; Kawada, N. Role of hepatitis B virus DNA integration in human hepatocarcinogenesis. World J. Gastroenterol. 2014, 20, 6236–6243. [Google Scholar] [CrossRef] [PubMed]
- Schreiner, S.; Nassal, M. A Role for the host DNA damage response in hepatitis B virus cccDNA formation-and beyond? Viruses 2017, 9, 125. [Google Scholar] [CrossRef] [PubMed]
- Seeger, C.; Mason, W.S. Molecular biology of hepatitis B virus infection. Virology 2015, 479–480, 672–686. [Google Scholar] [CrossRef] [PubMed]
- Ozer, A.; Khaoustov, V.I.; Mearns, M.; Lewis, D.E.; Genta, R.M.; Darlington, G.J.; Yoffe, B. Effect of hepatocyte proliferation and cellular DNA synthesis on hepatitis B virus replication. Gastroenterology 1996, 110, 1519–1528. [Google Scholar] [CrossRef] [PubMed]
- Summers, J.; Mason, W.S. Replication of the genome of a hepatitis B—Like virus by reverse transcription of an RNA intermediate. Cell 1982, 29, 403–415. [Google Scholar] [CrossRef]
- Schulze, A.; Gripon, P.; Urban, S. Hepatitis B virus infection initiates with a large surface protein-dependent binding to heparan sulfate proteoglycans. Hepatology 2007, 46, 1759–1768. [Google Scholar] [CrossRef] [PubMed]
- Yan, H.; Zhong, G.; Xu, G.; He, W.; Jing, Z.; Gao, Z.; Huang, Y.; Qi, Y.; Peng, B.; Wang, H.; et al. Sodium taurocholate cotransporting polypeptide is a functional receptor for human hepatitis B and D virus. Elife Sci. 2012, 1, e00049. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.-C.; Chen, C.-C.; Chang, W.-C.; Tao, M.-H.; Huang, C. Entry of hepatitis B virus into immortalized human primary hepatocytes by clathrin-dependent endocytosis. J. Virol. 2012, 86, 9443–9453. [Google Scholar] [CrossRef] [PubMed]
- Rabe, B.; Vlachou, A.; Pante, N.; Helenius, A.; Kann, M. Nuclear import of hepatitis B virus capsids and release of the viral genome. Proc. Natl. Acad. Sci. USA 2011, 100, 9849–9854. [Google Scholar] [CrossRef] [PubMed]
- Newbold, J.E.; Xin, H.; Tencza, M.; Sherman, G.; Dean, J.; Bowden, S.; Locarnini, S. The covalently closed duplex form of the hepadnavirus genome exists in situ as a heterogeneous population of viral minichromosomes. J. Virol. 1995, 69, 3350–3357. [Google Scholar] [PubMed]
- Obert, S.; Zachmann-Brand, B.; Deindl, E.; Tucker, W.; Bartenschlager, R.; Schaller, H. A splice hepadnavirus RNA that is essential for virus replication. EMBO J. 1996, 15, 2565–2574. [Google Scholar] [PubMed]
- Soussan, P.; Tuveri, R.; Nalpas, B.; Garreau, F.; Zavala, F.; Masson, A.; Pol, S.; Brechot, C.; Kremsdorf, D. The expression of hepatitis B spliced protein (HBSP) encoded by a spliced hepatitis B virus RNA is associated with viral replication and liver fibrosis. J. Hepatol. 2003, 38, 343–348. [Google Scholar] [CrossRef]
- Bartenschlager, R.; Schaller, H. Hepadnaviral assembly is initiated by polymerase binding to the encapsidation signal in the viral RNA genome. EMBO J. 1992, 11, 3413–3420. [Google Scholar] [PubMed]
- Staprans, S.; Loeb, D.D.; Ganem, D. Mutations affecting hepadnavirus plus-strand DNA synthesis dissociate primer cleavage from translocation and reveal the origin of linear viral DNA. J. Virol. 1991, 65, 1255–1262. [Google Scholar] [PubMed]
- Tuttleman, J.S.; Pourcel, C.; Summers, J. Formation of the pool of covalently closed circular viral DNA in hepadnavirus-infected cells. Cell 1986, 47, 451–460. [Google Scholar] [CrossRef]
- Watanabe, T.; Sorensen, E.M.; Naito, A.; Schott, M.; Kim, S.; Ahlquist, P. Involvement of host cellular multivesicular body functions in hepatitis B virus budding. Proc. Natl. Acad. Sci. USA 2007, 104, 10205–10210. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.T.; Billaud, J.-N.; Sällberg, M.; Guidotti, L.G.; Chisari, F.V.; Jones, J.; Hughes, J.; Milich, D.R. A function of the hepatitis B virus precore protein is to regulate the immune response to the core antigen. Proc. Natl. Acad. Sci. USA 2004, 101, 14913–14918. [Google Scholar] [CrossRef] [PubMed]
- Vanlandschoot, P.; Leroux-Roels, G. Viral apoptotic mimicry: An immune evasion strategy developed by the hepatitis B virus? Trends Immunol. 2003, 24, 144–147. [Google Scholar] [CrossRef]
- Hollingworth, R.; Grand, R. Modulation of DNA damage and repair pathways by human tumour viruses. Viruses 2015, 7, 2542–2591. [Google Scholar] [CrossRef] [PubMed]
- Andrés-León, E.; Cases, I.; Arcas, A.; Rojas, A.M. DDRprot: A database of DNA damage response-related proteins. Database 2016, 2016, 1. [Google Scholar] [CrossRef] [PubMed]
- Postel-Vinay, S.; Vanhecke, E.; Olaussen, K.A.; Lord, C.J.; Ashworth, A.; Soria, J.-C. The potential of exploiting DNA-repair defects for optimizing lung cancer treatment. Nat. Rev. Clin. Oncol. 2012, 9, 144–155. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.-H.; Paull, T.T. ATM activation by DNA double-strand breaks through the Mre11-Rad50-Nbs1 complex. Science 2005, 308, 551–554. [Google Scholar] [CrossRef] [PubMed]
- Cimprich, K.A.; Cortez, D. ATR: An essential regulator of genome integrity. Nat. Rev. Mol. Cell Biol. 2008, 9, 616–627. [Google Scholar] [CrossRef] [PubMed]
- Davis, A.J.; Chen, B.P.C.; Chen, D.J. DNA-PK: A dynamic enzyme in a versatile DSB repair pathway. DNA Repair 2014, 17, 21–29. [Google Scholar] [CrossRef] [PubMed]
- Caldecott, K.W. Mammalian single-strand break repair: Mechanisms and links with chromatin. DNA Repair 2007, 6, 443–453. [Google Scholar] [CrossRef] [PubMed]
- Hartlerode, A.J.; Scully, R. Mechanisms of double-strand break repair in somatic mammalian cells. Biochem. J. 2009, 423, 157–168. [Google Scholar] [CrossRef] [PubMed]
- Wallace, S.S. Base excision repair: A critical player in many games. DNA Repair 2014, 19, 14–26. [Google Scholar] [CrossRef] [PubMed]
- Marteijn, J.A.; Lans, H.; Vermeulen, W.; Hoeijmakers, J.H. Understanding nucleotide excision repair and its roles in cancer and ageing. Nat. Rev. Mol. Cell Biol. 2014, 15, 465–481. [Google Scholar] [CrossRef] [PubMed]
- Li, G.-M. New insights and challenges in mismatch repair: Getting over the chromatin hurdle. DNA Repair 2014, 19, 48–54. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; D’Andrea, A.D. Regulation of DNA cross-link repair by the Fanconi anemia/BRCA pathway. Genes Dev. 2012, 26, 1393–1408. [Google Scholar] [CrossRef] [PubMed]
- Fridman, J.S.; Lowe, S.W. Control of apoptosis by p53. Oncogene 2003, 22, 9030–9040. [Google Scholar] [CrossRef] [PubMed]
- Sancar, A.; Lindsey-Boltz, L.A.; Unsal-Kaçmaz, K.; Linn, S. Molecular mechanisms of mammalian DNA repair and the DNA damage checkpoints. Annu. Rev. Biochem. 2004, 73, 39–85. [Google Scholar] [CrossRef] [PubMed]
- Qi, Y.; Gao, Z.; Xu, G.; Peng, B.; Liu, C.; Yan, H.; Yao, Q.; Sun, G.; Liu, Y.; Tang, D.; et al. DNA polymerase κ is a key cellular factor for the formation of covalently closed circular DNA of hepatitis B virus. PLoS Pathog. 2016, 12, e1005893. [Google Scholar] [CrossRef] [PubMed]
- Königer, C.; Wingert, I.; Marsmann, M.; Rösler, C.; Beck, J.; Nassal, M. Involvement of the host DNA-repair enzyme TDP2 in formation of the covalently closed circular DNA persistence reservoir of hepatitis B viruses. Proc. Natl. Acad. Sci. USA 2014, 111, E4244–E4253. [Google Scholar] [CrossRef] [PubMed]
- Kinoshita, W.; Ogura, N.; Watashi, K.; Wakita, T. Host factor PRPF31 is involved in cccDNA production in HBV-replicating cells. Biochem. Biophys. Res. Commun. 2017, 482, 638–644. [Google Scholar] [CrossRef] [PubMed]
- Cui, X.; McAllister, R.; Boregowda, R.; Sohn, J.A.; Ledesma, F.C.; Caldecott, K.W.; Seeger, C.; Hu, J. Does tyrosyl DNA phosphodiesterase-2 play a role in hepatitis B virus genome repair? PLoS ONE 2015, 10, e0128401. [Google Scholar] [CrossRef] [PubMed]
- Köck, J.; Rösler, C.; Zhang, J.-J.; Blum, H.E.; Nassal, M.; Thoma, C. Generation of covalently closed circular DNA of hepatitis B viruses via intracellular recycling is regulated in a virus specific manner. PLoS Pathog. 2010, 6, e1001082. [Google Scholar] [CrossRef] [PubMed]
- Moser, J.; Kool, H.; Giakzidis, I.; Caldecott, K.; Mullenders, L.H.; Fousteri, M.I. Sealing of chromosomal DNA nicks during nucleotide excision repair requires XRCC1 and DNA ligase III α in a cell-cycle-specific manner. Mol. Cell 2007, 27, 311–323. [Google Scholar] [CrossRef] [PubMed]
- Ogi, T.; Limsirichaikul, S.; Overmeer, R.M.; Volker, M.; Takenaka, K.; Cloney, R.; Nakazawa, Y.; Niimi, A.; Miki, Y.; Jaspers, N.G.; et al. Three DNA polymerases, recruited by different mechanisms, carry out NER repair synthesis in human cells. Mol. Cell 2010, 37, 714–727. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Summers, J. Illegitimate replication of linear hepadnavirus DNA through nonhomologous recombination. J. Virol. 1995, 69, 4029–4036. [Google Scholar] [PubMed]
- Yang, W.; Summers, J. Infection of ducklings with virus particles containing linear double-stranded duck hepatitis B virus DNA: Illegitimate replication and reversion. J. Virol. 1998, 72, 8710–8717. [Google Scholar] [PubMed]
- Yang, W.; Summers, J. Integration of hepadnavirus DNA in infected liver: Evidence for a linear precursor. J. Virol. 1999, 73, 9710–9717. [Google Scholar] [PubMed]
- Guo, H.; Xu, C.; Zhou, T.; Block, T.M.; Guo, J.-T. Characterization of the host factors required for hepadnavirus covalently closed circular (ccc) DNA formation. PLoS ONE 2012, 7, e43270. [Google Scholar] [CrossRef] [PubMed]
- Niu, C.; Livingston, C.M.; Li, L.; Beran, R.K.; Daffis, S.; Ramakrishnan, D.; Burdette, D.; Peiser, L.; Salas, E.; Ramos, H.; et al. The Smc5/6 complex restricts HBV when localized to ND10 without inducing an innate immune response and is counteracted by the HBV X protein shortly after infection. PLoS ONE 2017, 12, e0169648. [Google Scholar] [CrossRef] [PubMed]
- Leupin, O.; Bontron, S.; Schaeffer, C.; Strubin, M. Hepatitis B virus X protein stimulates viral genome replication via a DDB1-dependent pathway distinct from that leading to cell death. J. Virol. 2005, 79, 4238–4245. [Google Scholar] [CrossRef] [PubMed]
- Lucifora, J.; Arzberger, S.; Durantel, D.; Belloni, L.; Strubin, M.; Levrero, M.; Zoulim, F.; Hantz, O.; Protzer, U. Hepatitis B virus X protein is essential to initiate and maintain virus replication after infection. J. Hepatol. 2011, 55, 996–1003. [Google Scholar] [CrossRef] [PubMed]
- Angers, S.; Li, T.; Yi, X.; MacCoss, M.J.; Moon, R.T.; Zheng, N. Molecular architecture and assembly of the DDB1-CUL4A ubiquitin ligase machinery. Nature 2006, 443, 590–593. [Google Scholar] [CrossRef] [PubMed]
- Decorsière, A.; Mueller, H.; van Breugel, P.C.; Abdul, F.; Gerossier, L.; Beran, R.K.; Livingston, C.M.; Niu, C.; Fletcher, S.P.; Hantz, O.; et al. Hepatitis B virus X protein identifies the Smc5/6 complex as a host restriction factor. Nature 2016, 531, 386–389. [Google Scholar] [CrossRef] [PubMed]
- Murphy, C.M.; Xu, Y.; Li, F.; Nio, K.; Reszka-Blanco, N.; Li, X.; Wu, Y.; Yu, Y.; Xiong, Y.; Su, L. Hepatitis B virus X protein promotes degradation of SMC5/6 to enhance HBV replication. Cell Rep. 2016, 16, 2846–2854. [Google Scholar] [CrossRef] [PubMed]
- Van Breugel, P.C.; Robert, E.I.; Mueller, H.; Decorsière, A.; Zoulim, F.; Hantz, O.; Strubin, M. Hepatitis B virus X protein stimulates gene expression selectively from extrachromosomal DNA templates. Hepatology 2012, 56, 2116–2124. [Google Scholar] [CrossRef] [PubMed]
- Guidotti, L.G.; Rochford, R.; Chung, J.; Shapiro, M.; Purcell, R.; Chisari, F.V. Viral clearance without destruction of infected cells during acute HBV infection. Science 1999, 284, 825–829. [Google Scholar] [CrossRef] [PubMed]
- Lucifora, J.; Xia, Y.; Reisinger, F.; Zhang, K.; Stadler, D.; Cheng, X.; Sprinzl, M.F.; Koppensteiner, H.; Makowska, Z.; Volz, T.; et al. Specific and nonhepatotoxic degradation of nuclear hepatitis B virus cccDNA. Science 2014, 343, 1221–1228. [Google Scholar] [CrossRef] [PubMed]
- Meier, M.A.; Suslov, A.; Ketterer, S.; Heim, M.H.; Wieland, S.F. Hepatitis B virus covalently closed circular DNA homeostasis is independent of the lymphotoxin pathway during chronic HBV infection. J. Viral. Hepat. 2017, 24, 662–671. [Google Scholar] [CrossRef] [PubMed]
- Seeger, C.; Sohn, J.A. Complete spectrum of CRISPR/Cas9-induced mutations on HBV cccDNA. Mol. Ther. 2016, 24, 1258–1266. [Google Scholar] [CrossRef] [PubMed]
- Rösler, C.; Köck, J.; Kann, M.; Malim, M.H.; Blum, H.E.; Baumert, T.F.; von Weizsäcker, F. APOBEC-mediated interference with hepadnavirus production. Hepatology 2005, 42, 301–309. [Google Scholar] [CrossRef] [PubMed]
- Liang, G.; Kitamura, K.; Wang, Z.; Liu, G.; Chowdhury, S.; Fu, W.; Koura, M.; Wakae, K.; Honjo, T.; Muramatsu, M. RNA editing of hepatitis B virus transcripts by activation-induced cytidine deaminase. Proc. Natl. Acad. Sci. USA 2013, 110, 2246–2251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin-Vilchez, S.; Lara-Pezzi, E.; Trapero-Marugán, M.; Moreno-Otero, R.; Sanz-Cameno, P. The molecular and pathophysiological implications of hepatitis B X antigen in chronic hepatitis B virus infection. Rev. Med. Virol. 2011, 21, 315–329. [Google Scholar] [CrossRef] [PubMed]
- Su, Q.; Schröder, C.H.; Hofmann, W.J.; Otto, G.; Pichlmayr, R.; Bannasch, P. Expression of hepatitis B virus X protein in HBV-infected human livers and hepatocellular carcinomas. Hepatology 1998, 27, 1109–1120. [Google Scholar] [CrossRef] [PubMed]
- Levrero, M.; Stemler, M.; Pasquinelli, C.; Alberti, A.; Jean-Jean, O.; Franco, A.; Balsano, C.; Diop, D.; Brechot, C.; Melegari, M. Significance of anti-HBx antibodies in hepatitis B virus infection. Hepatology 1991, 13, 143–149. [Google Scholar] [CrossRef] [PubMed]
- Peng, Z.; Zhang, Y.; Gu, W.; Wang, Z.; Li, D.; Zhang, F.; Qiu, G.; Xie, K. Integration of the hepatitis B virus X fragment in hepatocellular carcinoma and its effects on the expression of multiple molecules: A key to the cell cycle and apoptosis. Int. J. Oncol. 2005, 26, 467–473. [Google Scholar] [CrossRef] [PubMed]
- Kremsdorf, D.; Soussan, P.; Paterlini-Brechot, P.; Brechot, C. Hepatitis B virus-related hepatocellular carcinoma: Paradigms for viral-related human carcinogenesis. Oncogene 2006, 25, 3823–3833. [Google Scholar] [CrossRef] [PubMed]
- Lee, T.H.; Elledge, S.J.; Butel, J.S. Hepatitis B virus X protein interacts with a probable cellular DNA repair protein. J. Virol. 1995, 69, 1107–1114. [Google Scholar] [PubMed]
- Capovilla, A.; Carmona, S.; Arbuthnot, P. Hepatitis B virus X-protein binds damaged DNA and sensitizes liver cells to ultraviolet irradiation. Biochem. Biophys. Res. Commun. 1997, 232, 255–260. [Google Scholar] [CrossRef] [PubMed]
- Martin Lluesma, S.; Schaeffer, C.; Robert, E.I.; van Breugel, P.C.; Leupin, O.; Hantz, O.; Strubin, M. Hepatitis B virus X protein affects S phase progression leading to chromosome segregation defects by binding to damaged DNA binding protein 1. Hepatology 2008, 48, 1467–1476. [Google Scholar] [CrossRef] [PubMed]
- Uhlmann, F. SMC complexes: From DNA to chromosomes. Nat. Rev. Mol. Cell Biol. 2016, 17, 399–412. [Google Scholar] [CrossRef] [PubMed]
- Chang, H.R.; Munkhjargal, A.; Kim, M.-J.; Park, S.Y.; Jung, E.; Ryu, J.-H.; Yang, Y.; Lim, J.-S.; Kim, Y. The functional roles of PML nuclear bodies in genome maintenance. Mutat. Res. 2017. [Google Scholar] [CrossRef] [PubMed]
- Guo, L.; Wang, X.; Ren, L.; Zeng, M.; Wang, S.; Weng, Y.; Tang, Z.; Wang, X.; Tang, Y.; Hu, H.; et al. HBx affects CUL4-DDB1 function in both positive and negative manners. Biochem. Biophys. Res. Commun. 2014, 450, 1492–1497. [Google Scholar] [CrossRef] [PubMed]
- Benhenda, S.; Ducroux, A.; Rivière, L.; Sobhian, B.; Ward, M.D.; Dion, S.; Hantz, O.; Protzer, U.; Michel, M.-L.; Benkirane, M.; et al. Methyltransferase PRMT1 is a binding partner of HBx and a negative regulator of hepatitis B virus transcription. J. Virol. 2013, 87, 4360–4371. [Google Scholar] [CrossRef] [PubMed]
- Minor, M.; Slagle, B. Hepatitis B Virus HBx protein interactions with the ubiquitin proteasome system. Viruses 2014, 6, 4683–4702. [Google Scholar] [CrossRef] [PubMed]
- Keeney, S.; Eker, A.P.; Brody, T.; Vermeulen, W.; Bootsma, D.; Hoeijmakers, J.H.; Linn, S. Correction of the DNA repair defect in xeroderma pigmentosum group E by injection of a DNA damage-binding protein. Proc. Natl. Acad. Sci. USA 1994, 91, 4053–4056. [Google Scholar] [CrossRef] [PubMed]
- Keeney, S.; Chang, G.J.; Linn, S. Characterization of a human DNA damage binding protein implicated in xeroderma pigmentosum E. J. Biol. Chem. 1993, 268, 21293–21300. [Google Scholar] [PubMed]
- Becker, S.A.; Lee, T.H.; Butel, J.S.; Slagle, B.L. Hepatitis B virus X protein interferes with cellular DNA repair. J. Virol. 1998, 72, 266–272. [Google Scholar] [PubMed]
- Jia, L.; Wang, X.W.; Harris, C.C. Hepatitis B virus X protein inhibits nucleotide excision repair. Int. J. Cancer 1999, 80, 875–879. [Google Scholar] [CrossRef]
- Jaitovich-Groisman, I.; Benlimame, N.; Slagle, B.L.; Perez, M.H.; Alpert, L.; Song, D.J.; Fotouhi-Ardakani, N.; Galipeau, J.; Alaoui-Jamali, M.A. Transcriptional regulation of the TFIIH transcription repair components XPB and XPD by the hepatitis B virus x protein in liver cells and transgenic liver tissue. J. Biol. Chem. 2001, 276, 14124–14132. [Google Scholar] [CrossRef] [PubMed]
- Takada, S.; Kaneniwa, N.; Tsuchida, N.; Koike, K. Cytoplasmic retention of the p53 tumor suppressor gene product is observed in the hepatitis B virus X gene-transfected cells. Oncogene 1997, 15, 1895–1901. [Google Scholar] [CrossRef] [PubMed]
- Capovilla, A.; Arbuthnot, P. Hepatitis B virus X protein does not influence essential steps of nucleotide excision repair effected by human liver extracts. Biochem. Biophys. Res. Commun. 2003, 312, 806–810. [Google Scholar] [CrossRef] [PubMed]
- Guo, Z.; Heydari, A.R.; Wu, W.; Yang, H.; Sabia, M.R.; Richardson, A. Characterization of gene-specific DNA repair by primary cultures of rat hepatocytes. J. Cell Physiol. 1998, 176, 314–322. [Google Scholar] [CrossRef]
- Hsu, H.C.; Wu, T.T.; Sheu, J.C.; Wu, C.Y.; Chiou, T.J.; Lee, C.S.; Chen, D.S. Biologic significance of the detection of HBsAg and HBcAg in liver and tumor from 204 HBsAg-positive patients with primary hepatocellular carcinoma. Hepatology 1989, 9, 747–750. [Google Scholar] [CrossRef] [PubMed]
- Fan, Y.F.; Lu, C.C.; Chen, W.C.; Yao, W.J.; Wang, H.C.; Chang, T.T.; Lei, H.Y.; Shiau, A.L.; Su, I.J. Prevalence and significance of hepatitis B virus (HBV) pre-S mutants in serum and liver at different replicative stages of chronic HBV infection. Hepatology 2001, 33, 277–286. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, Y.H.; Su, I.J.; Wang, H.-C.; Chang, W.-W.; Lei, H.-Y.; Lai, M.-D.; Chang, W.-T.; Huang, W. Pre-S mutant surface antigens in chronic hepatitis B virus infection induce oxidative stress and DNA damage. Carcinogenesis 2004, 25, 2023–2032. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, Y.H.; Chang, Y.Y.; Su, I.J.; Yen, C.J.; Liu, Y.R.; Liu, R.J.; Hsieh, W.C.; Tsai, H.W.; Wang, L.H.; Huang, W. Hepatitis B virus pre-S2 mutant large surface protein inhibits DNA double-strand break repair and leads to genome instability in hepatocarcinogenesis. J. Pathol. 2015, 236, 337–347. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, Y.H.; Su, I.J.; Wang, H.-C.; Tsai, J.-H.; Huang, Y.-J.; Chang, W.-W.; Lai, M.-D.; Lei, H.-Y.; Huang, W. Hepatitis B virus pre-S2 mutant surface antigen induces degradation of cyclin-dependent kinase inhibitor p27Kip1 through c-Jun activation domain-binding protein 1. Mol. Cancer Res. 2007, 5, 1063–1072. [Google Scholar] [CrossRef] [PubMed]
- Luan, F.; Liu, H.; Gao, L.; Liu, J.; Sun, Z.; Ju, Y.; Hou, N.; Guo, C.; Liang, X.; Zhang, L.; et al. Hepatitis B virus protein preS2 potentially promotes HCC development via its transcriptional activation of hTERT. Gut 2009, 58, 1528–1537. [Google Scholar] [CrossRef] [PubMed]
- Gwak, G.-Y.; Lee, D.H.; Moon, T.G.; Choi, M.S.; Lee, J.H.; Koh, K.C.; Paik, S.W.; Park, C.K.; Joh, J.-W.; Yoo, B.C. The correlation of hepatitis B virus pre-S mutation with cellular oxidative DNA damage in hepatocellular carcinoma. Hepatogastroenterology 2008, 55, 2028–2032. [Google Scholar] [PubMed]
- Tu, T.; Budzinska, M.A.; Shackel, N.A.; Urban, S. HBV DNA integration: Molecular mechanisms and clinical implications. Viruses 2017, 9, 75. [Google Scholar] [CrossRef] [PubMed]
- Dandri, M.; Burda, M.R.; Burkle, A.; Zuckerman, D.M.; Will, H.; Rogler, C.E.; Greten, H.; Petersen, J. Increase in de novo HBV DNA integrations in response to oxidative DNA damage or inhibition of poly(ADP-ribosyl)ation. Hepatology 2002, 35, 217–223. [Google Scholar] [CrossRef] [PubMed]
- Ray Chaudhuri, A.; Nussenzweig, A. The multifaceted roles of PARP1 in DNA repair and chromatin remodelling. Nat. Rev. Mol. Cell Biol. 2017, 18, 610–621. [Google Scholar] [CrossRef] [PubMed]
- Bill, C.A.; Summers, J. Genomic DNA double-strand breaks are targets for hepadnaviral DNA integration. Proc. Natl. Acad. Sci. USA 2004, 101, 11135–11140. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.-H.; Liu, X.; Yan, H.-X.; Li, W.-Y.; Zeng, X.; Yang, Y.; Zhao, J.; Liu, S.-P.; Zhuang, X.-H.; Lin, C.; et al. Genomic and oncogenic preference of HBV integration in hepatocellular carcinoma. Nat. Commun. 2016, 7, 12992. [Google Scholar] [CrossRef] [PubMed]
- Hu, Z.; Zhu, D.; Wang, W.; Li, W.; Jia, W.; Zeng, X.; Ding, W.; Yu, L.; Wang, X.; Wang, L.; et al. Genome-wide profiling of HPV integration in cervical cancer identifies clustered genomic hot spots and a potential microhomology-mediated integration mechanism. Nat. Genet. 2015, 47, 158–163. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.-Q.; Wang, L.-W.; Yan, S.-N.; Gong, Z.-J. Effects of cell cycle on telomerase activity and on hepatitis B virus replication in HepG2 2.2.15 cells. Hepatobiliary Pancreat. Dis. Int. 2004, 3, 543–547. [Google Scholar] [PubMed]
- Glebe, D.; Berting, A.; Broehl, S.; Naumann, H.; Schuster, R.; Fiedler, N.; Tolle, T.K.; Nitsche, S.; Seifer, M.; Gerlich, W.H.; et al. Optimised conditions for the production of hepatitis B virus from cell culture. Intervirology 2001, 44, 370–378. [Google Scholar] [CrossRef] [PubMed]
- Cohen, D.; Adamovich, Y.; Reuven, N.; Shaul, Y. Hepatitis B virus activates deoxynucleotide synthesis in nondividing hepatocytes by targeting the R2 gene. Hepatology 2010, 51, 1538–1546. [Google Scholar] [CrossRef] [PubMed]
- Ricardo-Lax, I.; Ramanan, V.; Michailidis, E.; Shamia, T.; Reuven, N.; Rice, C.M.; Shlomai, A.; Shaul, Y. Hepatitis B virus induces RNR-R2 expression via DNA damage response activation. J. Hepatol. 2015, 63, 789–796. [Google Scholar] [CrossRef] [PubMed]
- Lubelsky, Y.; Reuven, N.; Shaul, Y. Autorepression of RFX1 gene expression: Functional conservation from yeast to humans in response to DNA replication arrest. Mol. Cell Biol. 2005, 25, 10665–10673. [Google Scholar] [CrossRef] [PubMed]
- Urist, M.; Tanaka, T.; Poyurovsky, M.V.; Prives, C. p73 induction after DNA damage is regulated by checkpoint kinases Chk1 and Chk2. Genes Dev. 2004, 18, 3041–3054. [Google Scholar] [CrossRef] [PubMed]
- Tudzarova-Trajkovska, S.; Wesierska-Gadek, J. Strong induction of p73 protein in vivo coincides with the onset of apoptosis in rat liver after treatment with the hepatocarcinogen N-nitrosomorpholine (NNM). J. Cell Biochem. 2003, 90, 837–855. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.-H.; Hullinger, R.L.; Andrisani, O.M. Hepatitis B virus X protein via the p38MAPK pathway induces E2F1 release and ATR kinase activation mediating p53 apoptosis. J. Biol. Chem. 2008, 283, 25455–25467. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.-H.; Studach, L.L.; Andrisani, O.M. Proteins ZNF198 and SUZ12 are down-regulated in hepatitis B virus (HBV) X protein-mediated hepatocyte transformation and in HBV replication. Hepatology 2011, 53, 1137–1147. [Google Scholar] [CrossRef] [PubMed]
- Koida, N.; Ozaki, T.; Yamamoto, H.; Ono, S.; Koda, T.; Ando, K.; Okoshi, R.; Kamijo, T.; Omura, K.; Nakagawara, A. Inhibitory role of Plk1 in the regulation of p73-dependent apoptosis through physical interaction and phosphorylation. J. Biol. Chem. 2008, 283, 8555–8563. [Google Scholar] [CrossRef] [PubMed]
- Diab, A.M.; Foca, A.; Fusil, F.; Lahlali, T.; Jalaguier, P.; Amirache, F.; N’Guyen, L.; Isorce, N.; Cosset, F.-L.; Zoulim, F.; et al. Polo-like-kinase 1 is a proviral host-factor for hepatitis B virus replication. Hepatology 2017. [Google Scholar] [CrossRef] [PubMed]
- Parkin, D.M. The global health burden of infection-associated cancers in the year 2002. Int. J. Cancer 2006, 118, 3030–3044. [Google Scholar] [CrossRef] [PubMed]
- Farber, E.; Sarma, D.S. Hepatocarcinogenesis: A dynamic cellular perspective. Lab. Investig. 1987, 56, 4–22. [Google Scholar] [PubMed]
- Nakamoto, Y.; Guidotti, L.G.; Kuhlen, C.V.; Fowler, P.; Chisari, F.V. Immune pathogenesis of hepatocellular carcinoma. J. Exp. Med. 1998, 188, 341–350. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.-C.; Huang, W.; Lai, M.-D.; Su, I.J. Hepatitis B virus pre-S mutants, endoplasmic reticulum stress and hepatocarcinogenesis. Cancer Sci. 2006, 97, 683–688. [Google Scholar] [CrossRef] [PubMed]
- Summers, J.; Jilbert, A.R.; Yang, W.; Aldrich, C.E.; Saputelli, J.; Litwin, S.; Toll, E.; Mason, W.S. Hepatocyte turnover during resolution of a transient hepadnaviral infection. Proc. Natl. Acad. Sci. USA 2003, 100, 11652–11659. [Google Scholar] [CrossRef] [PubMed]
- Sung, W.-K.; Zheng, H.; Li, S.; Chen, R.; Liu, X.; Li, Y.; Lee, N.P.; Lee, W.H.; Ariyaratne, P.N.; Tennakoon, C.; et al. Genome-wide survey of recurrent HBV integration in hepatocellular carcinoma. Nat. Genet. 2012, 44, 765–769. [Google Scholar] [CrossRef] [PubMed]
- Mason, W.S.; Low, H.C.; Xu, C.; Aldrich, C.E.; Scougall, C.A.; Grosse, A.; Clouston, A.; Chavez, D.; Litwin, S.; Peri, S.; et al. Detection of clonally expanded hepatocytes in chimpanzees with chronic hepatitis B virus infection. J. Virol. 2009, 83, 8396–8408. [Google Scholar] [CrossRef] [PubMed]
- Paterlini-Bréchot, P.; Saigo, K.; Murakami, Y.; Chami, M.; Gozuacik, D.; Mugnier, C.; Lagorce, D.; Brechot, C. Hepatitis B virus-related insertional mutagenesis occurs frequently in human liver cancers and recurrently targets human telomerase gene. Oncogene 2003, 22, 3911–3916. [Google Scholar] [CrossRef] [PubMed]
- Wong, D.K.-H.; Seto, W.-K.; Fung, J.; Ip, P.; Huang, F.-Y.; Lai, C.-L.; Yuen, M.-F. Reduction of hepatitis B surface antigen and covalently closed circular DNA by nucleos(t)ide analogues of different potency. Clin. Gastroenterol. Hepatol. 2013, 11, 1004–1010. [Google Scholar] [CrossRef] [PubMed]
- Werle-Lapostolle, B.; Bowden, S.; Locarnini, S.; Wursthorn, K.; Petersen, J.; Lau, G.; Trepo, C.; Marcellin, P.; Goodman, Z.; Delaney, W.E.; et al. Persistence of cccDNA during the natural history of chronic hepatitis B and decline during adefovir dipivoxil therapy. Gastroenterology 2004, 126, 1750–1758. [Google Scholar] [CrossRef] [PubMed]
- Kumar, R.; Pérez-del-Pulgar, S.; Testoni, B.; Lebossé, F.; Zoulim, F. Clinical relevance of the study of hepatitis B virus covalently closed circular DNA. Liver Int. 2016, 36, 72–77. [Google Scholar] [CrossRef] [PubMed]
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gómez-Moreno, A.; Garaigorta, U. Hepatitis B Virus and DNA Damage Response: Interactions and Consequences for the Infection. Viruses 2017, 9, 304. https://doi.org/10.3390/v9100304
Gómez-Moreno A, Garaigorta U. Hepatitis B Virus and DNA Damage Response: Interactions and Consequences for the Infection. Viruses. 2017; 9(10):304. https://doi.org/10.3390/v9100304
Chicago/Turabian StyleGómez-Moreno, Andoni, and Urtzi Garaigorta. 2017. "Hepatitis B Virus and DNA Damage Response: Interactions and Consequences for the Infection" Viruses 9, no. 10: 304. https://doi.org/10.3390/v9100304
APA StyleGómez-Moreno, A., & Garaigorta, U. (2017). Hepatitis B Virus and DNA Damage Response: Interactions and Consequences for the Infection. Viruses, 9(10), 304. https://doi.org/10.3390/v9100304