Evaluation of the Broad-Range PCR-Electrospray Ionization Mass Spectrometry (PCR/ESI-MS) System and Virus Microarrays for Virus Detection

Abstract
:1. Introduction
2. Results and Discussion
2.1. Limit of Virus Detection
2.1.1. RT-PCR Assays
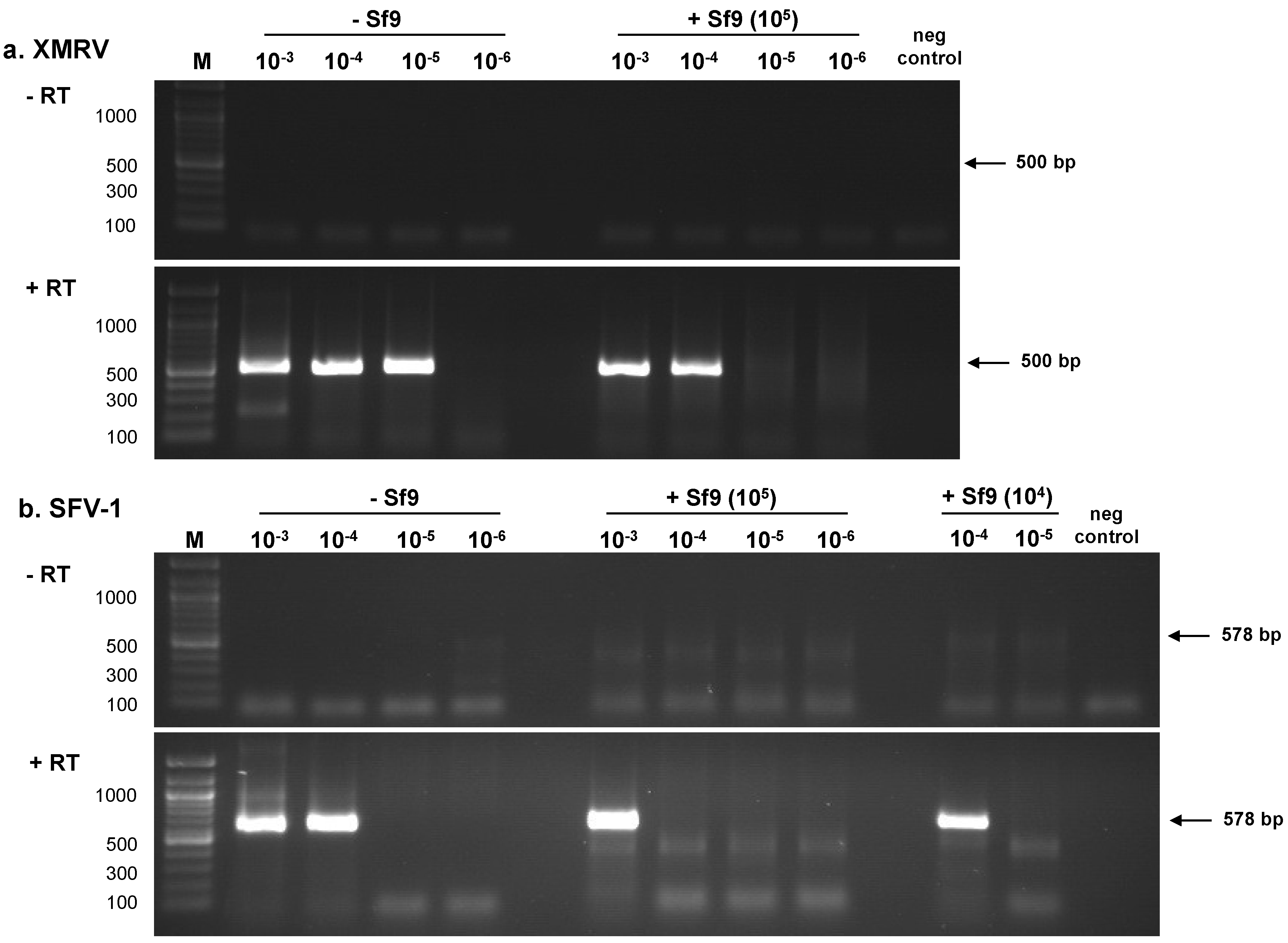

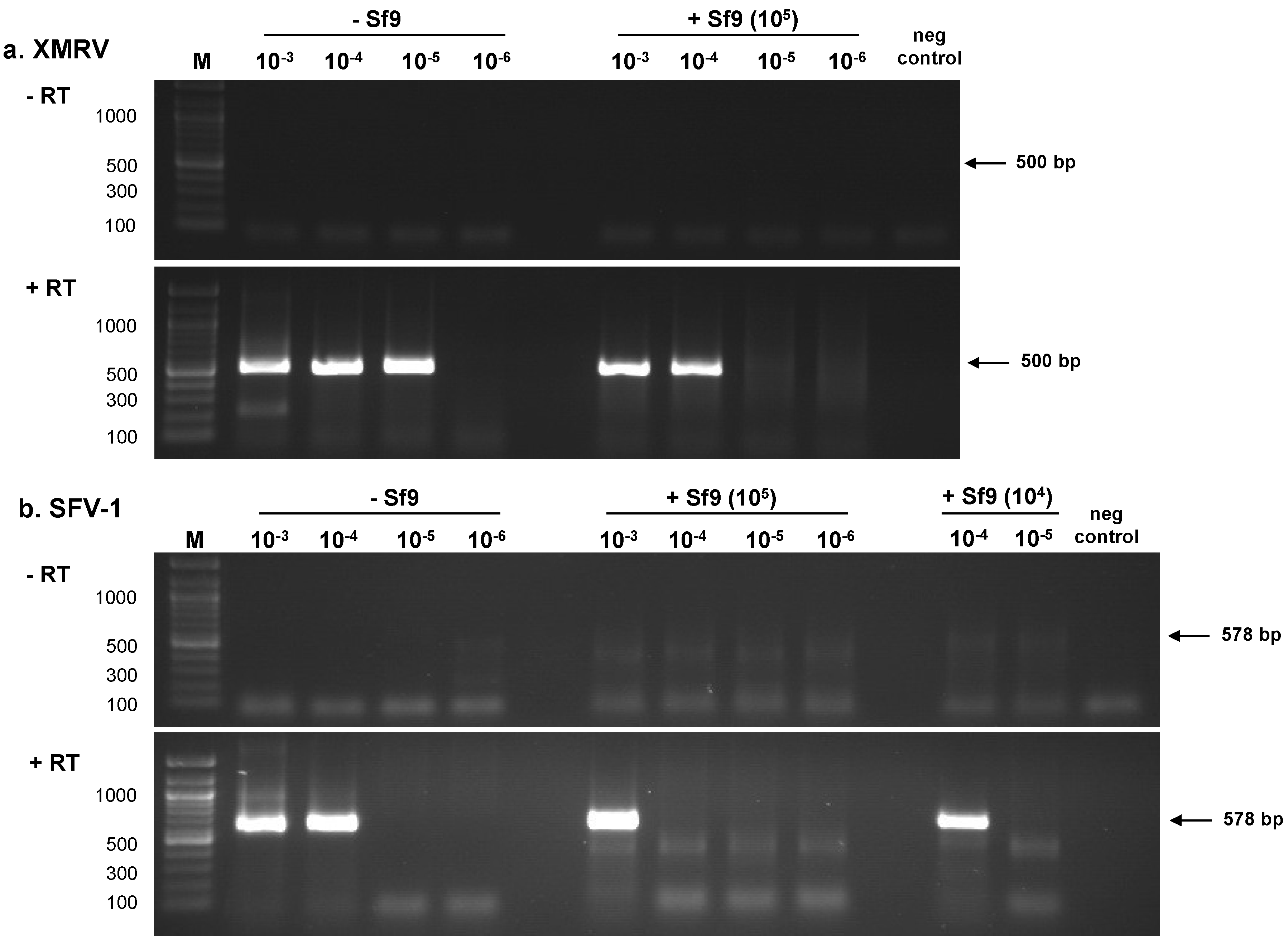{kind=link}
{kind=link}
{kind=link}
| Sample | PLEX-ID | ViroChip | RT-PCR a | ||||||
|---|---|---|---|---|---|---|---|---|---|
| RNA Dilution b | TCID50/mL | Particles/µL c | (-)Sf9 | +105 | (-)Sf9 | +105 | (-)Sf9 | (-)Sf9 | +105 |
| 10−3 | 101.5 | 312 | + | + | + | nt d | nt | + | + |
| 10−4 | 100.5 | 31.2 | + | + | + | nt | + | + | + |
| 10−5 | 10−0.5 | 3.12 | + | + | + | - | + | + | - |
| 10−6 | 10−1.5 | 0.312 | - | - | + | - | + | - | - |
| 10−7 | 10−2.5 | 0.0312 | - | - | - | nt | - | - | - |
| Sample | PLEX-ID | LLMDA | RT-PCR a | |||||
|---|---|---|---|---|---|---|---|---|
| RNA Dilution b | TCID50/mL | Particles/µL c | (-)Sf9 | (-)Sf9 | +104 | (-)Sf9 | +105 | +104 |
| 10−3 | 102.5 | 402 | nt d | nt | nt | + | + | nt |
| 10−4 | 101.5 | 40.2 | - | + | nt | + | - | + |
| 10−5 | 100.5 | 4.02 | - | + | - | - | - | - |
| 10−6 | 10−0.5 | 0.402 | - | - | - | - | - | nt |
2.1.2. PLEX-ID and Virus Arrays
2.2. Investigation of Cell Lines and Cell Culture Reagents
2.2.1. PLEX-ID Analysis
| Cell Lines | Alpharetrovirus-related | Betaretrovirus-related | Gammaretrovirus-related | |||||
|---|---|---|---|---|---|---|---|---|
| ALV | RAV-2 | Unidentified | SRV-1 | Unidentified | RD114 | BaEV | Undistinguishable | |
| Human | ||||||||
| A549 | + b | + | + | |||||
| Raji | + | + | + | + | ||||
| MRC-5 | + | + | + | |||||
| A204 | + | + | + | + | + | |||
| HEK293 | + | + | + | |||||
| 293T/17 a | + | + | + | |||||
| HeLa | + | + | + | |||||
| Simian | ||||||||
| VERO | + | + | ||||||
| CV-1 | + | + | ||||||
| Canine | ||||||||
| MDCK | + | + | ||||||
| Cf2Th | + | + | ||||||
2.2.2. Virus Arrays
2.3. Follow-Up Strategy for a Positive Hit by PLEX-ID
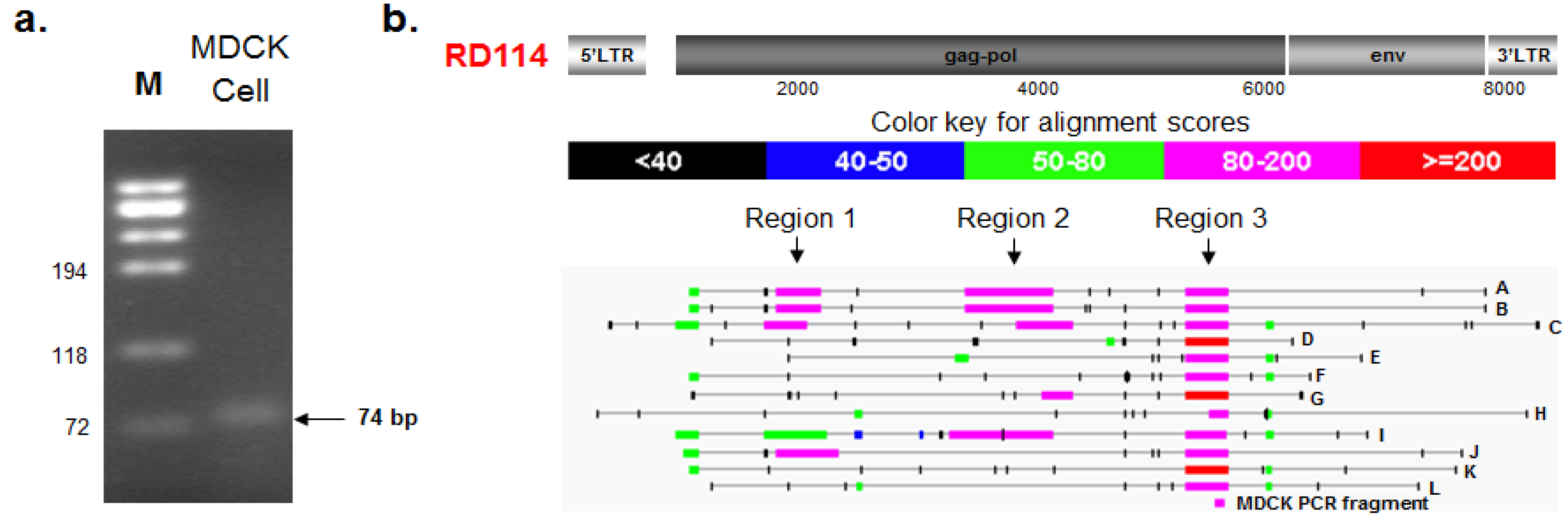
2.3.1. PCR Amplification and Nucleotide Sequencing
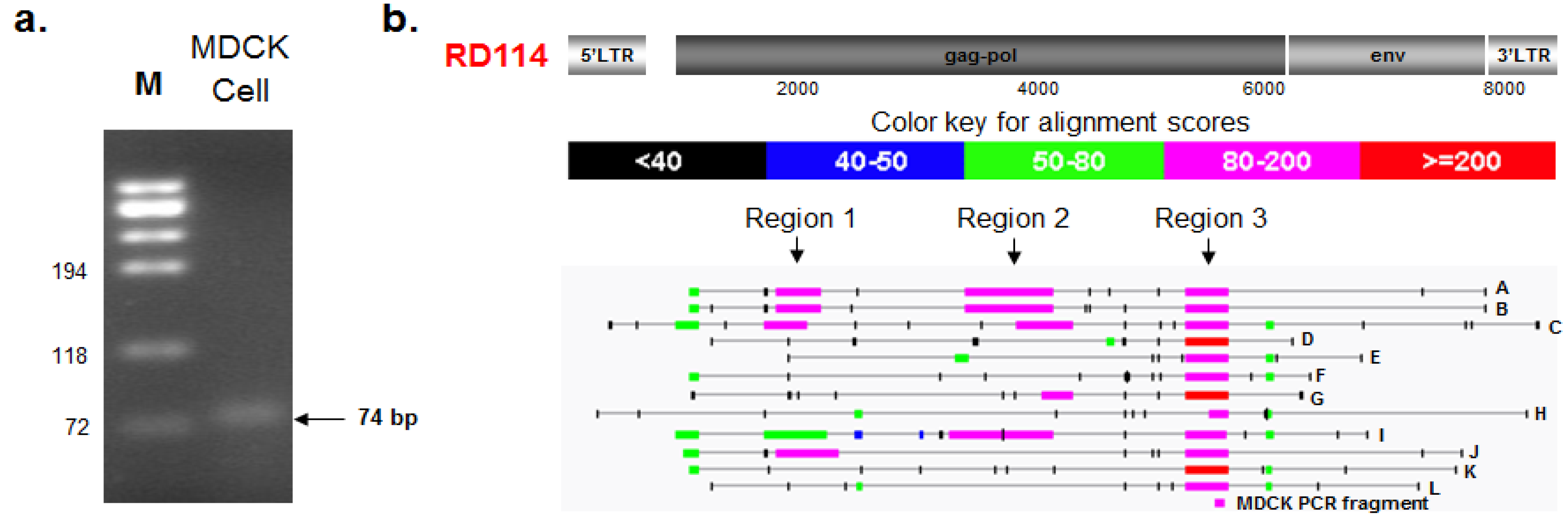

2.3.2. BLAST and RetroTector© Analysis
| Provirus ID | BAC Clone XX-(Chain) | Chr a | Accession | RetroTector Provirus Location b | Region 3 c | Provirus Motifs Detected f | |||
|---|---|---|---|---|---|---|---|---|---|
| Range d | % e | ||||||||
| CfERV-A | 73E6 (seq1) | 1 | AC184807.10 | 5394 | 14,041 | 9902 | 10,264 | 73 | 5' LTR, PBS, MA, CA, RT, IN, TM |
| CfERV-B | 309C2 (seq1) | 1 | AC194581.5 | 143,113 | 150,196 | 146,057 | 146,419 | 73 | 5' LTR, PBS, MA, CA, RT, IN, TM |
| CfERV-C | 412K16 (seq1) | 1 | AC191152.6 | 47,384 | 51,060 | 47,352 | 47,712 | 72 | PBS, MA, CA, RT, IN |
| CfERV-D | 219I19 (seq1) | 3 | AC186968.15 | 71,780 | 76,840 | 73,083 | 73,454 | 74 | 5' LTR, PBS, CA, NC, PR, IN, 3 'LTR |
| CfERV-E | 231B7 (seq3) | 8 | AC194586.17 | 211,515 | 222,372 | 220,330 | 220,712 | 70 | 5' LTR, PBS, CA, NC, PR, RT, IN, TM, 3' LTR |
| CfERV-F | 408G5 (seq1) | 13 | AC188926.6 | 167,789 | 173,976 | 170,817 | 171,150 | 68 | 5' LTR, PBS, CA, MA, NC, PR, TM |
| CfERV-G | 239F12 (seq1) | 25 | AC186972.16 | 89,015 | 94,074 | 93,745 | 94,113 | 73 | RT, IN |
| CfERV-H | 146G22 (seq2) | X | AC186068.17 | 147,356 | 151,076 | 149,410 | 149,580 | 76 | CA, NC, PR, IN |
| CfERV-I | 35E2 (seq1) | X | AC187326.11 | 106,297 | 114,944 | 109,230 | 109,569 | 72 | 5' LTR, PBS, MA, CA, NC,PR, RT, IN, TM, 3' LTR |
| CfERV-J | 306G8 (seq1) | X | AC189722.4 | 76,817 | 80,919 | 79,993 | 80,358 | 72 | PBS, MA, CA, PR, IN |
| CfERV-K | 494D7 (seq1) | 14 | AC189730.3 | 3268 | 8924 | 5573 | 5939 | 74 | 5' LTR, PBS, CA, NC, IN, TM |
| CfERV-L | 224p2 and 282j14 (seq1) | 12 | AJ630363.1 | 113,754 | 118,869 | 115,518 | 115,882 | 73 | PR, IN, TM |
| CfERV-M g | 73E6 (seq3) | 1 | 207,305 | 216,077 | n/a | n/a | 5' LTR, PBS, MA, CA, NC, IN, TM, 3' LTR | ||
| CfERV-N g | 219I19 (seq3) | 3 | 219,296 | 226,087 | n/a | n/a | 5' LTR, PBS, CA, NC, PR, RT, IN, 3' LTR | ||
| CfERV-O h | 35E2 (seq2) | X | 106,282 | 115,165 | n/a | n/a | 5' LTR, PBS, MA, CA, NC, PR, RT, IN, TM, 3' LTR | ||
3. Experimental
3.1. Cell Lines
3.2. Preparation of XMRV and SFV-1 RNA Panels with and without Background Sf9 Total Nucleic Acids and RT-PCR Assays
3.3. PCR/ESI-MS (PLEX-ID)
3.4. LLMDA and Virohip
3.5. PCR Assay for RD114-Like Virus and Nucleotide Sequencing Analysis
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Blyn, L.B.; Hall, T.A.; Libby, B.; Ranken, R.; Sampath, R.; Rudnick, K.; Moradi, E.; Desai, A.; Metzgar, D.; Russell, K.L.; et al. Rapid detection and molecular serotyping of adenovirus by use of PCR followed by electrospray ionization mass spectrometry. J. Clin. Microbiol. 2008, 46, 644–651. [Google Scholar] [CrossRef]
- Chen, E.C.; Miller, S.A.; DeRisi, J.L.; Chiu, C.Y. Using a pan-viral microarray assay (Virochip) to screen clinical samples for viral pathogens. J. Vis. Exp. 2011. [Google Scholar] [CrossRef]
- Ecker, D.J.; Sampath, R.; Blyn, L.B.; Eshoo, M.W.; Ivy, C.; Ecker, J.A.; Libby, B.; Samant, V.; Sannes-Lowery, K.A.; Melton, R.E.; et al. Rapid identification and strain-typing of respiratory pathogens for epidemic surveillance. Proc. Natl. Acad. Sci. USA 2005, 102, 8012–8017. [Google Scholar] [CrossRef]
- Gardner, S.N.; Jaing, C.J.; McLoughlin, K.S.; Slezak, T.R. A microbial detection array (MDA) for viral and bacterial detection. BMC Genomics 2010, 11, e668. [Google Scholar] [CrossRef]
- Victoria, J.G.; Wang, C.; Jones, M.S.; Jaing, C.; McLoughlin, K.; Gardner, S.; Delwart, E.L. Viral nucleic acids in live-attenuated vaccines: Detection of minority variants and an adventitious virus. J. Virol. 2010, 84, 6033–6040. [Google Scholar] [CrossRef]
- Onions, D.; Kolman, J. Massively parallel sequencing, a new method for detecting adventitious agents. Biologicals 2010, 38, 377–380. [Google Scholar] [CrossRef]
- European Medicines Agency. Guideline on the use of porcine trypsin used in the manufacture of human biological medicinal products. (21 Feburary 2013, draft). Available online: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2013/03/WC500139532.pdf (accessed on 25 September 2013).
- Ma, H.; Khan, A.S. Detection of latent retroviruses in vaccine-related cell substrates: Investigation of RT activity produced by chemical induction of vero cells. PDA J. Pharm. Sci. Technol. 2011, 65, 685–689. [Google Scholar] [CrossRef]
- Onions, D.; Cote, C.; Love, B.; Toms, B.; Koduri, S.; Armstrong, A.; Chang, A.; Kolman, J. Ensuring the safety of vaccine cell substrates by massively parallel sequencing of the transcriptome. Vaccine 2011, 29, 7117–7121. [Google Scholar] [CrossRef]
- Sampath, R.; Blyn, L.B.; Ecker, D.J. Rapid molecular assays for microbial contaminant monitoring in the bioprocess industry. PDA J. Pharm. Sci. Technol. 2010, 64, 458–464. [Google Scholar]
- FDA. Characterization and qualification of cell substrates and other biological materials used in the production of viral vaccines for infectious disease indications. February 2010. Available online: http://www.fda.gov/downloads/biologicsbloodvaccines/guidancecomplianceregulatoryinformation/guidances/vaccines/ucm202439.pdf (accessed on 25 September 2013). [Google Scholar]
- Khan, A.S. Testing considerations for novel cell substrates: A regulatory perspective. PDA J. Pharm. Sci. Technol. 2010, 64, 426–431. [Google Scholar]
- Khan, A.S.; Ma, W.; Ma, Y.; Kumar, A.; Williams, D.K.; Muller, J.; Ma, H.; Galvin, T.A. Proposed algorithm to investigate latent and occult viruses in vaccine cell substrates by chemical induction. Biologicals 2009, 37, 196–201. [Google Scholar] [CrossRef]
- Eshoo, M.W.; Whitehouse, C.A.; Zoll, S.T.; Massire, C.; Pennella, T.T.; Blyn, L.B.; Sampath, R.; Hall, T.A.; Ecker, J.A.; Desai, A.; et al. Direct broad-range detection of alphaviruses in mosquito extracts. Virology 2007, 368, 286–295. [Google Scholar] [CrossRef]
- Hofstadler, S.A.; Sampath, R.; Blyn, L.B.; Eshoo, M.W.; Hall, T.A.; Jiang, Y.; Drader, J.J.; Hannis, J.C.; Sannes-Lowery, K.A.; Cummins, L.L.; et al. TIGER: The universal biosensor. Int. J. Mass Spectrom. 2005, 242, 23–41. [Google Scholar] [CrossRef]
- Ecker, D.J.; Sampath, R.; Willett, P.; Samant, V.; Massire, C.; Hall, T.A.; Hari, K.; McNeil, J.A.; Buchen-Osmond, C.; Budowle, B. The Microbial Rosetta Stone database: A common structure for microbial biosecurity threat agents. J. Forensic. Sci. 2005, 50, 1380–1385. [Google Scholar]
- Ecker, D.J.; Sampath, R.; Massire, C.; Blyn, L.B.; Hall, T.A.; Eshoo, M.W.; Hofstadler, S.A. Ibis T5000: A universal biosensor approach for microbiology. Nat. Rev. Microbiol. 2008, 6, 553–558. [Google Scholar] [CrossRef]
- Sampath, R.; Mulholland, N.; Blyn, L.B.; Massire, C.; Whitehouse, C.A.; Waybright, N.; Harter, C.; Bogan, J.; Miranda, M.S.; Smith, D.; et al. Comprehensive biothreat cluster identification by PCR/electrospray-ionization mass spectrometry. PLoS One 2012, 7, e36528. [Google Scholar]
- Sampath, R.; Hofstadler, S.A.; Blyn, L.B.; Eshoo, M.W.; Hall, T.A.; Massire, C.; Levene, H.M.; Hannis, J.C.; Harrell, P.M.; Neuman, B.; et al. Rapid identification of emerging pathogens: Coronavirus. Emerg. Infect. Dis. 2005, 11, 373–379. [Google Scholar] [CrossRef]
- Athogen, Ibis Biosciences, Carlsbad, CA, USA. Available online: http://www.athogen.com/consulting-services/biopharmaceutical-tests.html (accessed on 20 September 2013).
- Greninger, A.L.; Chen, E.C.; Sittler, T.; Scheinerman, A.; Roubinian, N.; Yu, G.; Kim, E.; Pillai, D.R.; Guyard, C.; Mazzulli, T.; et al. A metagenomic analysis of pandemic influenza A (2009 H1N1) infection in patients from North America. PLoS One 2010, 5, e13381. [Google Scholar] [CrossRef]
- Lee, D.; das Gupta, J.; Gaughan, C.; Steffen, I.; Tang, N.; Luk, K.C.; Qiu, X.; Urisman, A.; Fischer, N.; Molinaro, R.; et al. In-depth investigation of archival and prospectively collected samples reveals no evidence for XMRV infection in prostate cancer. PLoS One 2012, 7, e44954. [Google Scholar] [CrossRef]
- Galvin, T.A.; Ahmed, I.A.; Shahabuddin, M.; Bryan, T.; Khan, A.S. Identification of recombination in the envelope gene of simian foamy virus sertype 2 isolated from Macaca cyclopis. J. Virol. 2013, 87, 8792–8797. [Google Scholar] [CrossRef]
- Rose, N.; Opriessnig, T.; Grasland, B.; Jestin, A. Epidemiology and transmission of porcine circovirus type 2 (PCV2). Virus Res. 2012, 164, 78–79. [Google Scholar] [CrossRef]
- Van Regenmortel, M.H.V.; Fauquet, C.M.; Bishop, D.H.L.; Carstens, E.B.; Estes, M.K.; Lemon, S.M.; Maniloff, J.; Mayo, M.A.; McGeoch, D.J.; Pringle, C.R.; et al. Virus Taxonomy, Classification and Nomenclature of Viruses: Seventh Report of the International Committee on Taxonomy of Viruses; Academic Press: San Diego, CA, USA, 2000. [Google Scholar]
- Gifford, R.; Tristem, M. The evolution, distribution and diversity of endogenous retroviruses. Virus Genes 2003, 26, 291–315. [Google Scholar] [CrossRef]
- Ma, H.; Ma, Y.; Ma, W.; Williams, D.K.; Galvin, T.A.; Khan, A.S. Chemical induction of endogenous retrovirus particles from the vero cell line of African green monkeys. J. Virol. 2011, 85, 6579–6588. [Google Scholar] [CrossRef]
- DuBridge, R.B.; Tang, P.; Hsia, H.C.; Leong, P.M.; Miller, J.H.; Calos, M.P. Analysis of mutation in human cells by using an Epstein-Barr virus shuttle system. Mol. Cell. Biol. 1987, 7, 379–387. [Google Scholar]
- Graham, F.L.; Smiley, J.; Russell, W.C.; Nairn, R. Characteristics of a human cell line transformed by DNA from human adenovirus type 5. J. Gen. Virol. 1977, 36, 59–74. [Google Scholar] [CrossRef]
- Lindahl, T.; Adams, A.; Bjursell, G.; Bornkamm, G.W.; Kaschka-Dierich, C.; Jehn, U. Covalently closed circular duplex DNA of Epstein-Barr virus in a human lymphoid cell line. J. Mol. Biol. 1976, 102, 511–530. [Google Scholar] [CrossRef]
- Meissner, J.D. Nucleotide sequences and further characterization of human papillomavirus DNA present in the CaSki, SiHa and HeLa cervical carcinoma cell lines. J. Gen. Virol. 1999, 80, 1725–1733. [Google Scholar]
- Menzel, T.; Rohrmann, G.F. Diversity of errantivirus (retrovirus) sequences in two cell lines used for baculovirus expression, Spodoptera frugiperda and Trichoplusia ni. Virus Genes 2008, 36, 583–586. [Google Scholar] [CrossRef]
- Ma, H.; Galvin, T.A.; Glasner, D.R.; Shaheduzzaman, S.; Khan, A.S. Identification of a novel rhabdovirus in Spodoptera frugiperda cell lines. J. Virol. 2014. [Google Scholar] [CrossRef]
- Kimura, M. A simple method for estimating evolutionary rates of base substitutions through comparative studies of nucleotide sequences. J. Mol. Evol. 1980, 16, 111–120. [Google Scholar] [CrossRef]
- Felsenstein, J. Confidence limits on phylogenies: An approach using the bootstrap. Evolution 1985, 39, 783–791. [Google Scholar] [CrossRef]
- Tamura, K.; Peterson, D.; Peterson, N.; Stecher, G.; Nei, M.; Kumar, S. MEGA5: Molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol. Biol. Evol. 2011, 28, 2731–2739. [Google Scholar] [CrossRef]
- Barrio, A.M.; Ekerljung, M.; Jern, P.; Benachenhou, F.; Sperber, G.O.; Bongcam-Rudloff, E.; Blomberg, J.; Andersson, G. The first sequenced carnivore genome shows complex host-endogenous retrovirus relationships. PLoS One 2011, 6, e19832. [Google Scholar] [CrossRef]
- Sperber, G.; Lovgren, A.; Eriksson, N.E.; Benachenhou, F.; Blomberg, J. RetroTector online, a rational tool for analysis of retroviral elements in small and medium size vertebrate genomic sequences. BMC Bioinf. 2009, 10, S4. [Google Scholar]
- Williams, D.K.; Galvin, T.A.; Gao, Y.; O’Neill, C.; Glasner, D.; Khan, A.S. No evidence of xenotropic murine leukemia virus-related virus transmission by blood transfusion from infected rhesus macaques. J. Virol. 2013, 87, 2278–2286. [Google Scholar] [CrossRef]
- Ma, Y.K.; Khan, A.S. Evaluation of different RT enzyme standards for quantitation of retroviruses using the single-tube fluorescent product-enhanced reverse transcriptase assay. J. Virol. Methods 2009, 157, 133–140. [Google Scholar] [CrossRef]
- Williams, D.K.; Galvin, T.A.; Ma, H.; Khan, A.S. Investigation of xenotropic murine leukemia virus-related virus (XMRV) in human and other cell lines. Biologicals 2011, 39, 378–383. [Google Scholar] [CrossRef]
- Sampath, R.; Russell, K.L.; Massire, C.; Eshoo, M.W.; Harpin, V.; Blyn, L.B.; Melton, R.; Ivy, C.; Pennella, T.; Li, F.; et al. Global surveillance of emerging Influenza virus genotypes by mass spectrometry. PLoS One 2007, 2, e489. [Google Scholar] [CrossRef]
- Coffin, J.M. Endogenous retroviruses. In RNA Tumor Viruses, 2nd ed.; Weiss, R., Teich, N., Varmus, H., Coffin, J., Eds.; Cold Spring Harbor Laboratory: Cold Spring Harbor, NY, USA, 1984; pp. 1109–1203. [Google Scholar]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Taliaferro, L.P.; Galvin, T.A.; Ma, H.; Shaheduzzaman, S.; Williams, D.K.; Glasner, D.R.; Khan, A.S. Evaluation of the Broad-Range PCR-Electrospray Ionization Mass Spectrometry (PCR/ESI-MS) System and Virus Microarrays for Virus Detection. Viruses 2014, 6, 1876-1896. https://doi.org/10.3390/v6051876
Taliaferro LP, Galvin TA, Ma H, Shaheduzzaman S, Williams DK, Glasner DR, Khan AS. Evaluation of the Broad-Range PCR-Electrospray Ionization Mass Spectrometry (PCR/ESI-MS) System and Virus Microarrays for Virus Detection. Viruses. 2014; 6(5):1876-1896. https://doi.org/10.3390/v6051876
Chicago/Turabian StyleTaliaferro, Lanyn P., Teresa A. Galvin, Hailun Ma, Syed Shaheduzzaman, Dhanya K. Williams, Dustin R. Glasner, and Arifa S. Khan. 2014. "Evaluation of the Broad-Range PCR-Electrospray Ionization Mass Spectrometry (PCR/ESI-MS) System and Virus Microarrays for Virus Detection" Viruses 6, no. 5: 1876-1896. https://doi.org/10.3390/v6051876
APA StyleTaliaferro, L. P., Galvin, T. A., Ma, H., Shaheduzzaman, S., Williams, D. K., Glasner, D. R., & Khan, A. S. (2014). Evaluation of the Broad-Range PCR-Electrospray Ionization Mass Spectrometry (PCR/ESI-MS) System and Virus Microarrays for Virus Detection. Viruses, 6(5), 1876-1896. https://doi.org/10.3390/v6051876