IFN-Dependent and -Independent Reduction in West Nile Virus Infectivity in Human Dermal Fibroblasts

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
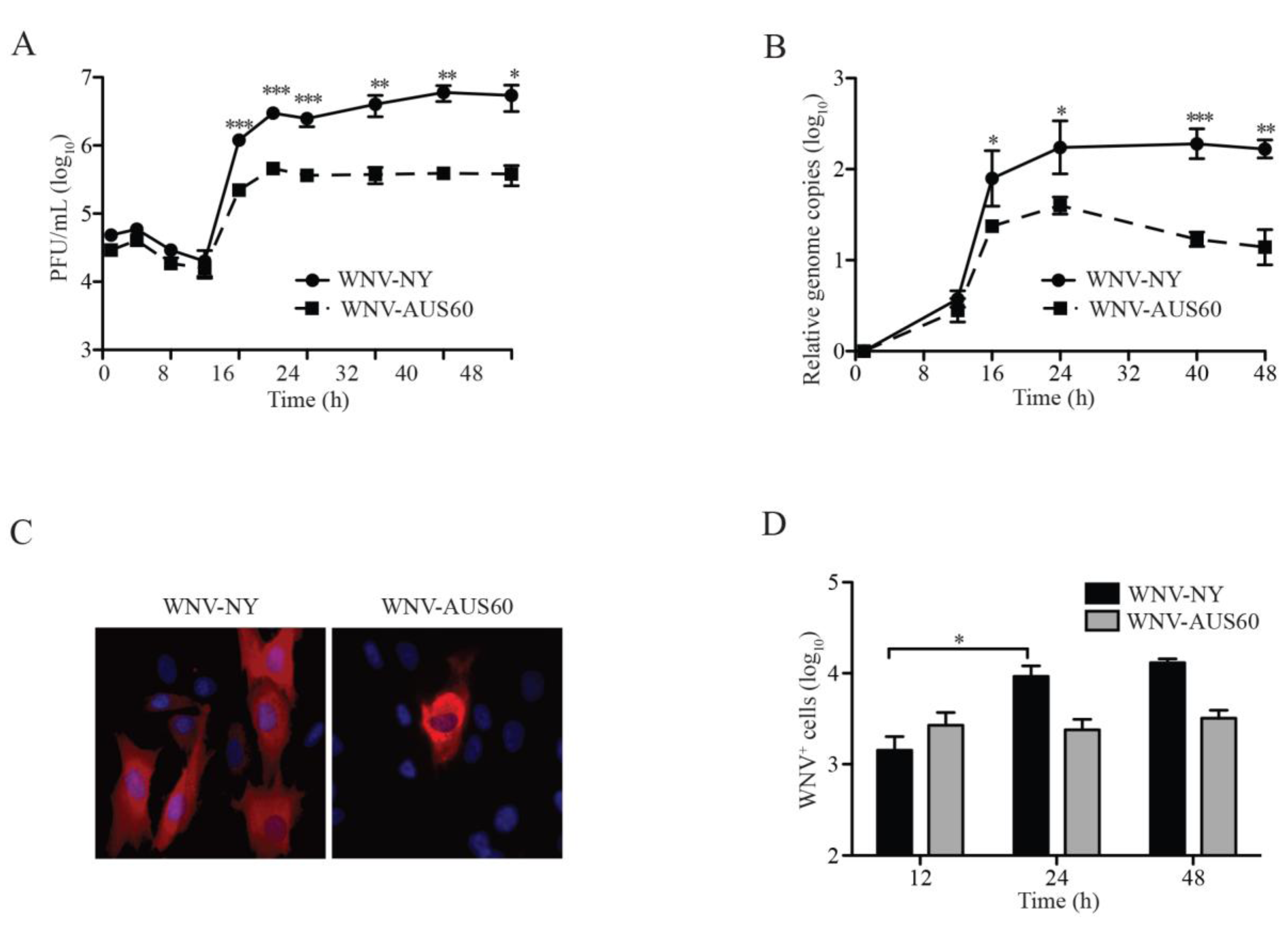
2.1. WNV Replication in Human Dermal Fibroblasts
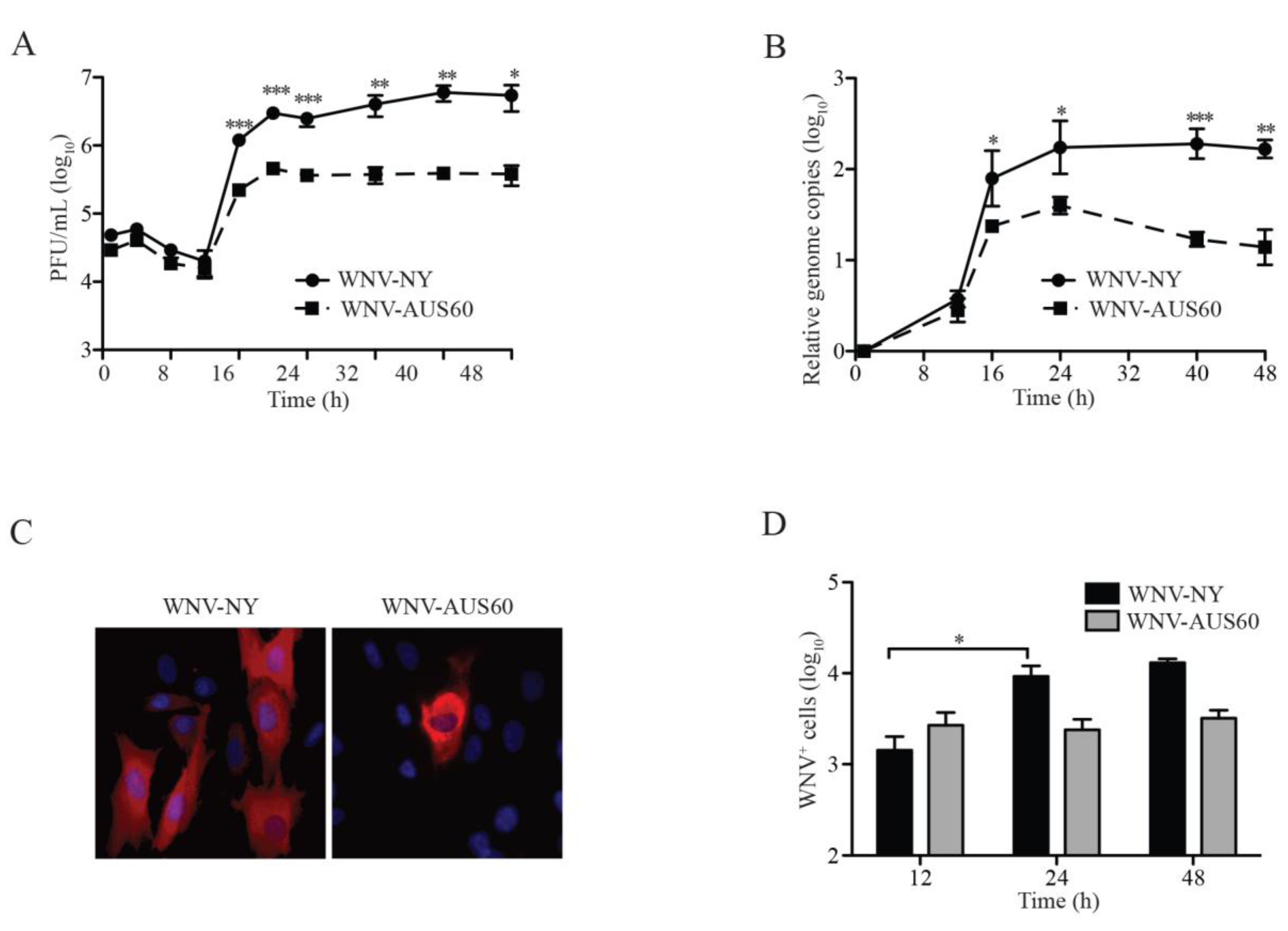
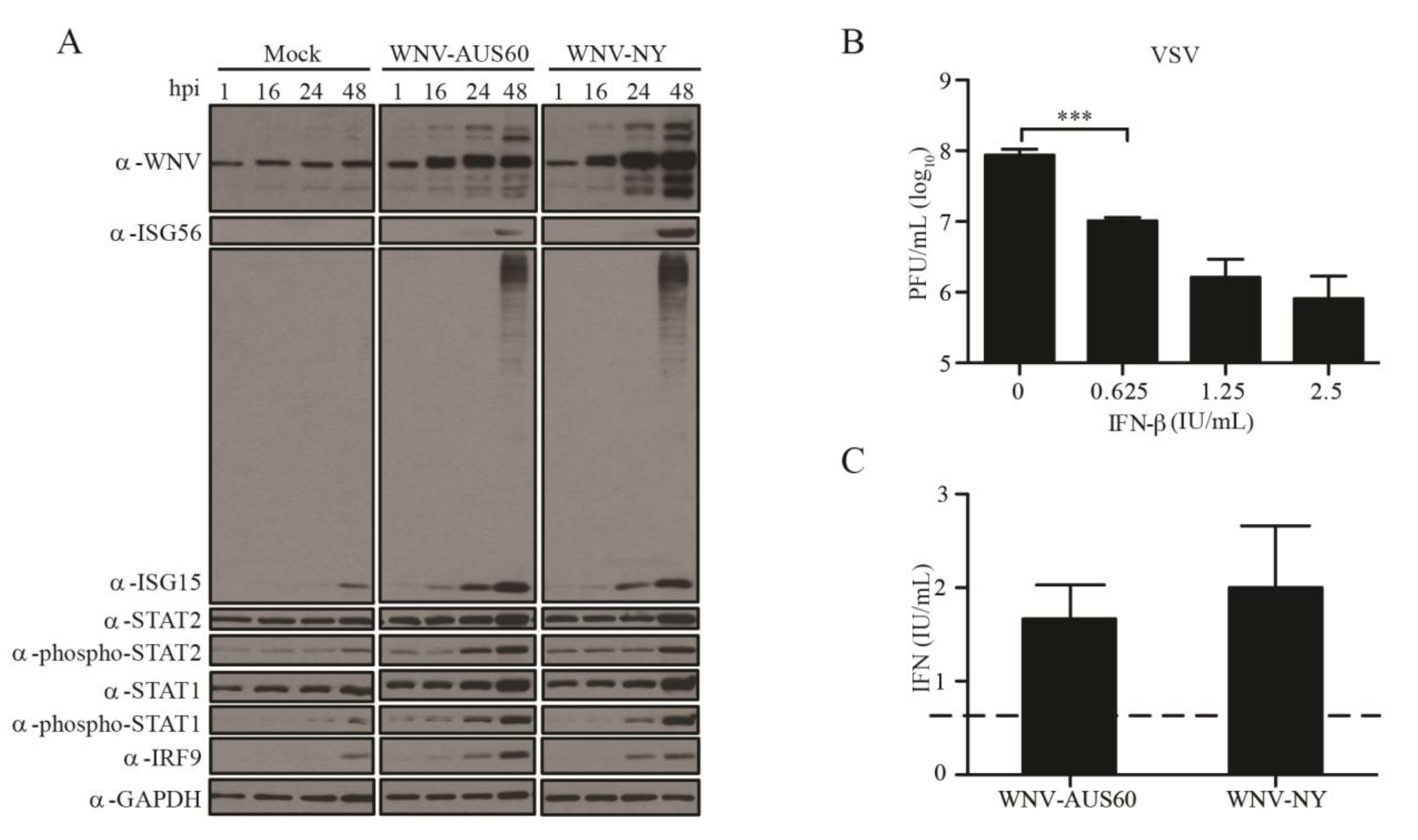
2.2. IFN Response to WNV in HDF
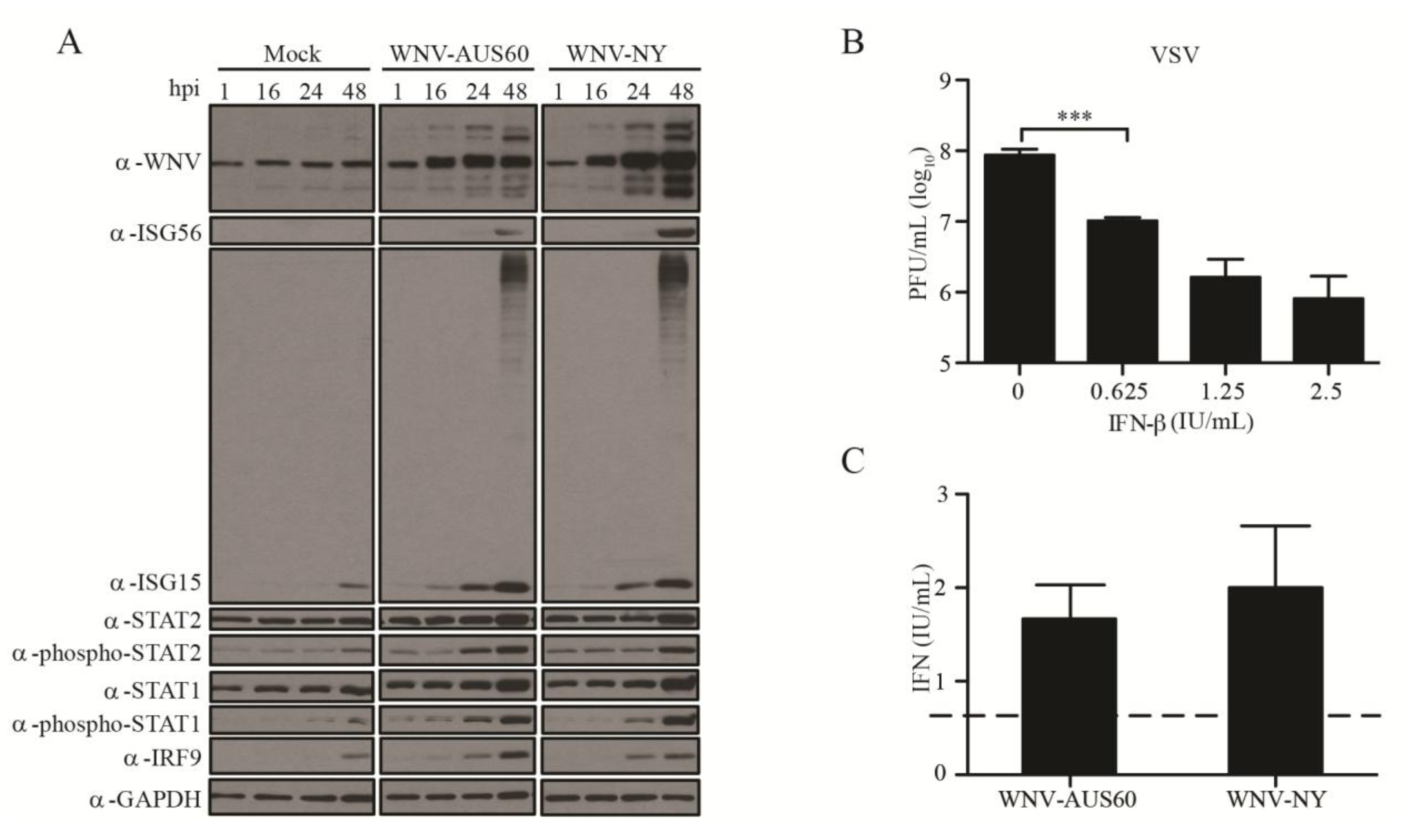

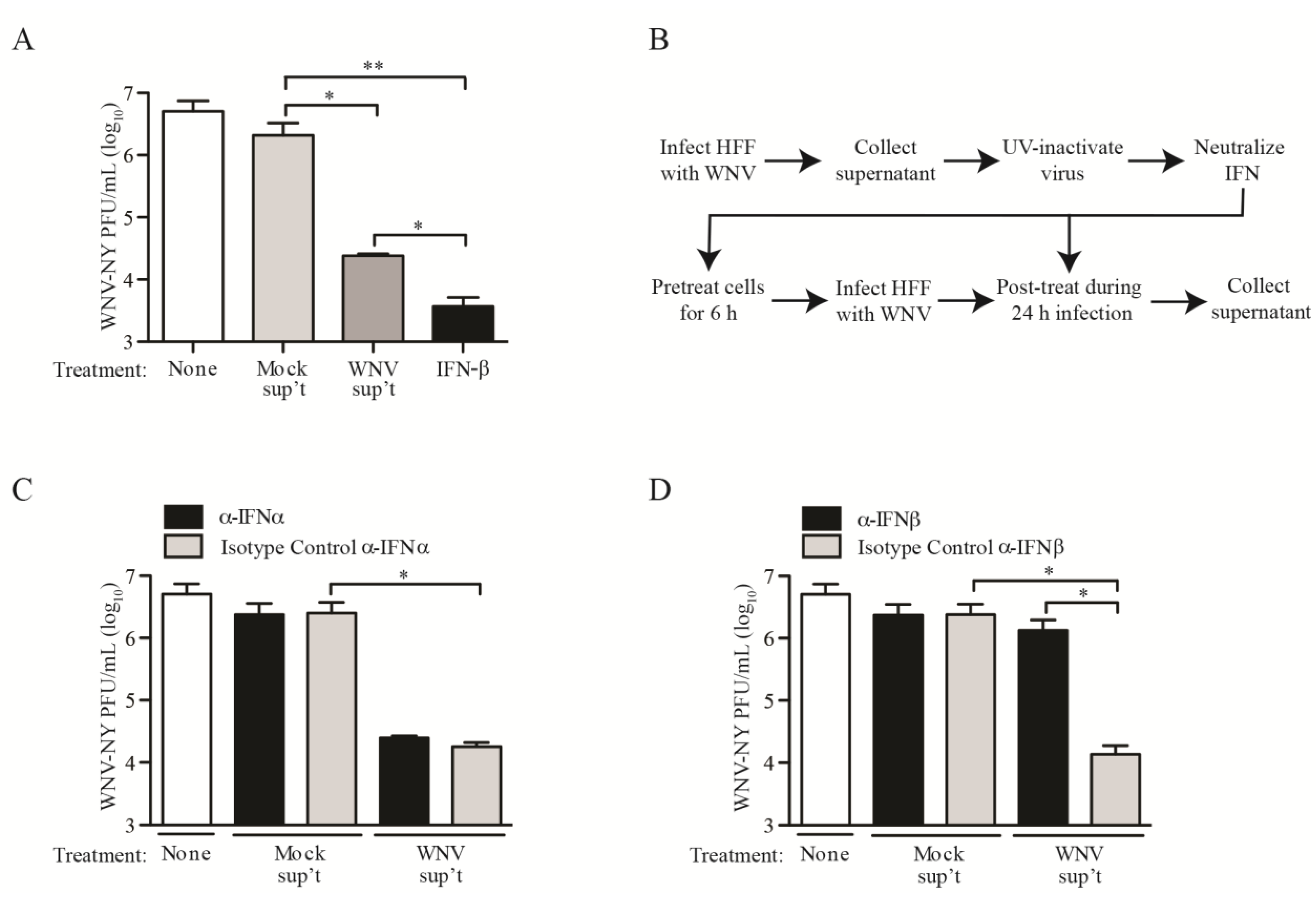
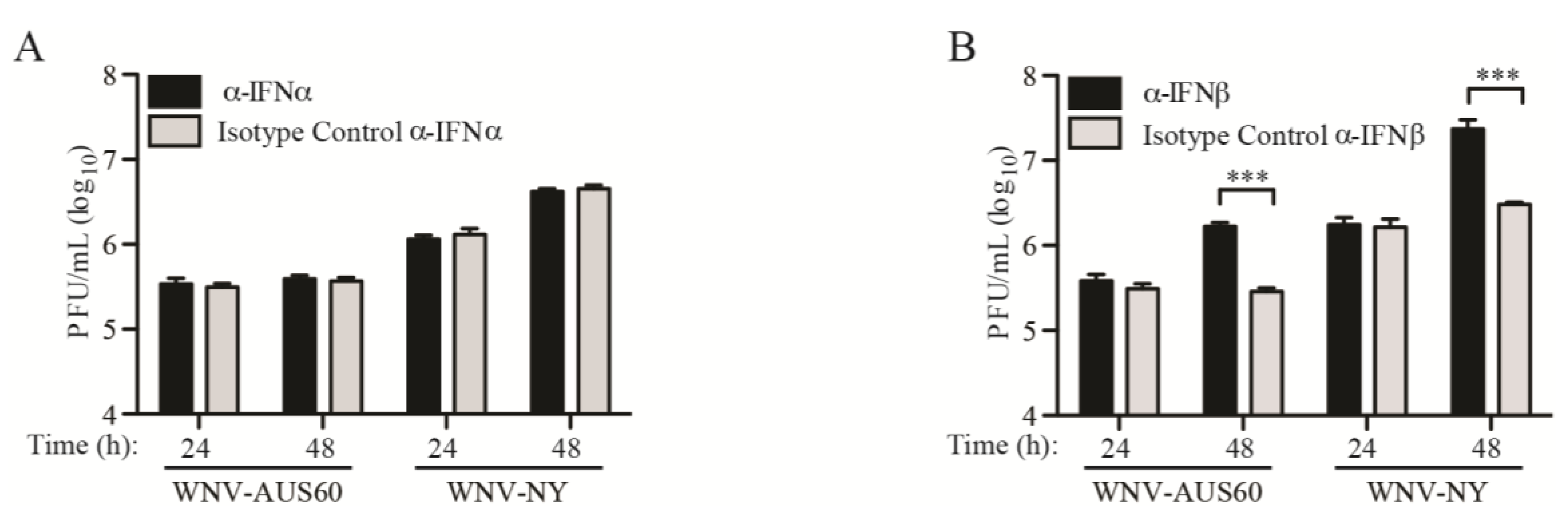
2.3. IFN Suppresses WNV Infectious Particle Production at Late Times Post-Infection
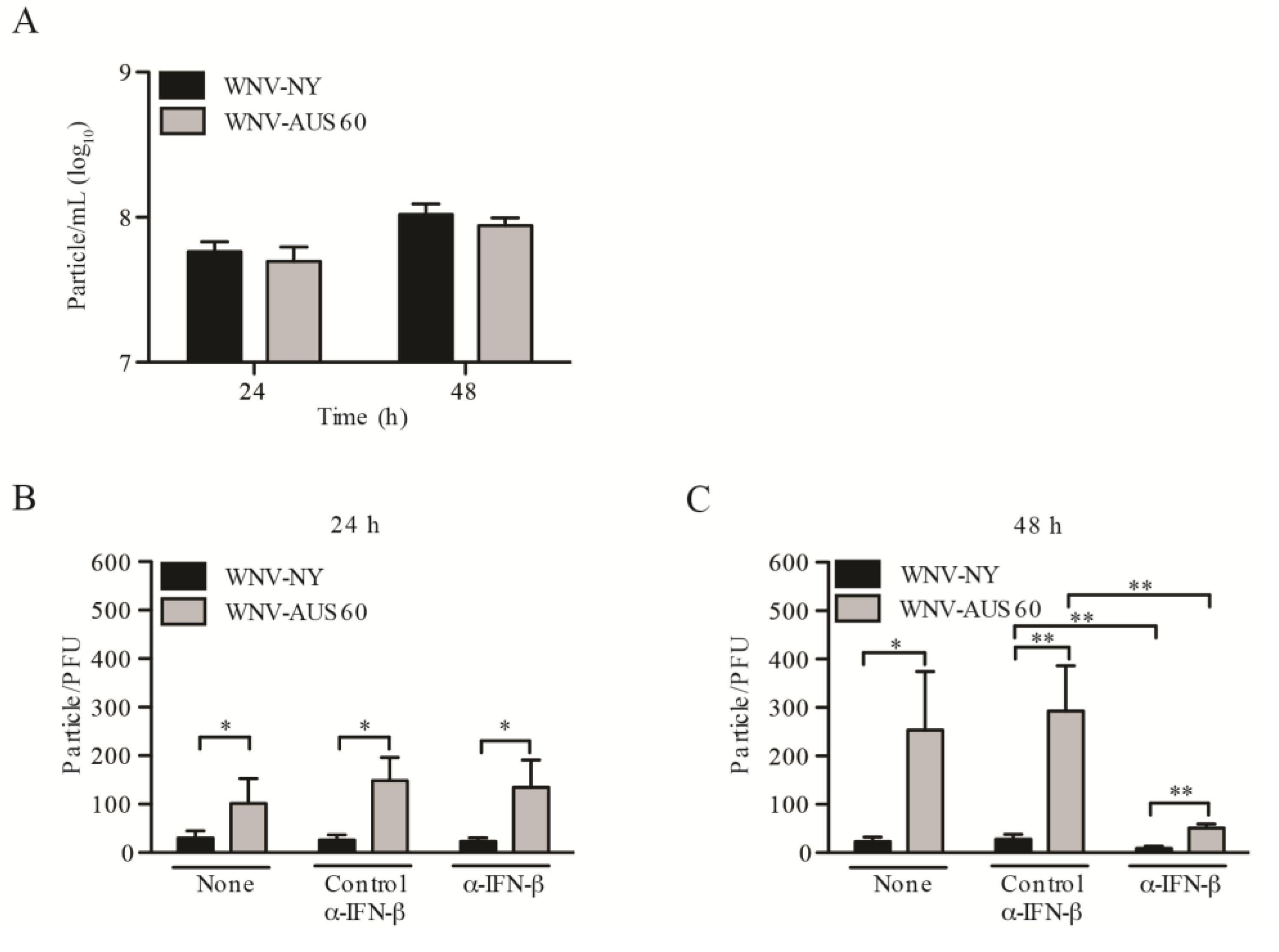
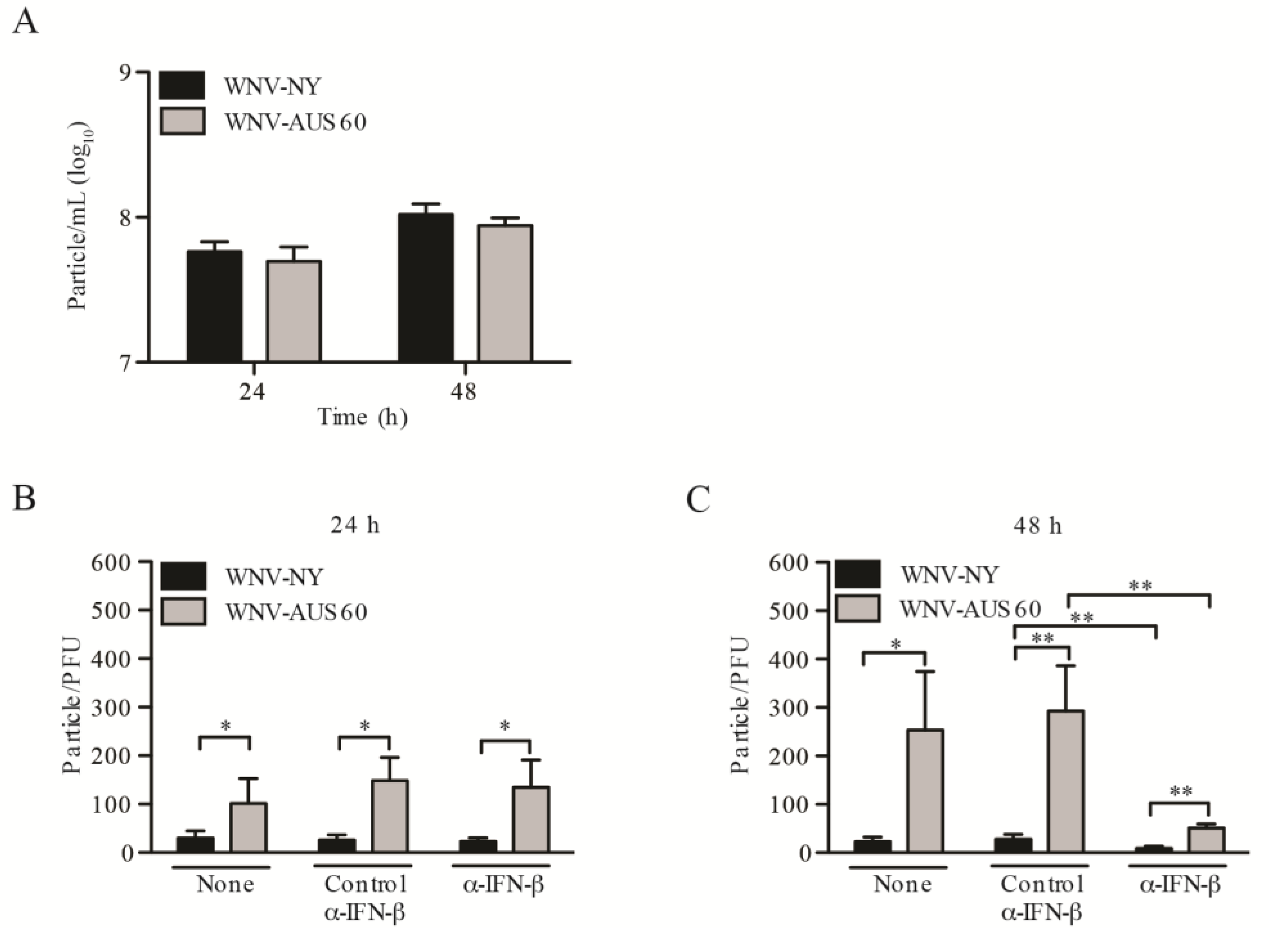
2.4. WNV Strain Variation in Particle Infectivity during HDF Infection
3. Experimental Section
3.1. Cells and Viruses
3.2. Focus-Forming Assays
3.3. Virus Growth Curves
3.4. Plaque Assays
3.5. Quantitative Real-Time PCR
3.6. Immunofluorescence Assay (IFA)
3.7. Flow Cytometry
3.8. Immunoblot Analysis
3.9. UV-Inactivation
3.10. Interferon Bioassay
3.11. Neutralization of Type-I IFN
3.12. Detection and Enumeration of Total Virus Particles
3.13. Statistical Analysis
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References and Notes
- CDC. Available online: http://www.cdc.gov/westnile/index.html/ (accessed on 1 November 2013).
- Bondre, V.P.; Jadi, R.S.; Mishra, A.C.; Yergolkar, P.N.; Arankalle, V.A. West Nile virus isolates from India: Evidence for a distinct genetic lineage. J. Gen. Virol. 2007, 88, 875–884. [Google Scholar] [CrossRef]
- Lanciotti, R.S.; Ebel, G.D.; Deubel, V.; Kerst, A.J.; Murri, S.; Meyer, R.; Bowen, M.; McKinney, N.; Morrill, W.E.; Crabtree, M.B.; et al. Complete genome sequences and phylogenetic analysis of West Nile virus strains isolated from the United States, Europe, and the Middle East. Virology 2002, 298, 96–105. [Google Scholar] [CrossRef]
- Bakonyi, T.; Ferenczi, E.; Erdélyi, K.; Kutasi, O.; Csörgő, T.; Seidel, B.; Weissenböck, H.; Brugger, K.; Bán, E.; Nowotny, N. Explosive spread of a neuroinvasive lineage 2 West Nile virus in Central Europe, 2008/2009. Vet. Microbiol. 2013, 165, 61–70. [Google Scholar] [CrossRef]
- Sirbu, A.; Ceianu, C.S.; Panculescu-Gatej, R.I.; Vazquez, A.; Tenorio, A.; Rebreanu, R.; Niedrig, M.; Nicolescu, G.; Pistol, A. Outbreak of West Nile virus infection in humans, Romania, July to October 2010. Eurosurveillance 2011, 16. pii:19762. [Google Scholar]
- Danis, K.; Papa, A.; Theocharopoulos, G.; Dougas, G.; Athanasiou, M.; Detsis, M.; Baka, A.; Lytras, T.; Mellou, K. Outbreak of West Nile virus infection in Greece, 2010. Emerg. Infect. Dis. 2011, 17, 1868–1872. [Google Scholar] [CrossRef]
- Platonov, A.E.; Karan, L.S.; Shopenskaia, T.A.; Fedorova, M.V.; Koliasnikova, N.M.; Rusakova, N.M.; Shishkina, L.V.; Arshba, T.E.; Zhuravlev, V.I.; Govorukhina, M.V.; et al. Genotyping of West Nile fever virus strains circulating in Southern Russia as an epidemiological investigation method: Principles and results. Zh Mikrobiol. Epidemiol. Immunobiol. 2011, Mar-Apr, 29–37. [Google Scholar]
- Frost, M.J.; Zhang, J.; Edmonds, J.H.; Prow, N.A.; Gu, X.; Davis, R.; Hornitzky, C.; Arzey, K.E.; Finlaison, D.; Hick, P.; et al. Characterization of virulent West Nile virus kunjin strain, Australia, 2011. Emerg. Infect. Dis. 2012, 18, 792–800. [Google Scholar]
- Venter, M.; Human, S.; Zaayman, D.; Gerdes, G.H.; Williams, J.; Steyl, J.; Leman, P.A.; Paweska, J.T.; Setzkorn, H.; Rous, G.; et al. Lineage 2 West Nile virus as cause of fatal neurologic disease in horses, South Africa. Emerg. Infect. Dis. 2009, 15, 877–884. [Google Scholar] [CrossRef]
- Venter, M.; Swanepoel, R. West Nile virus lineage 2 as a cause of zoonotic neurological disease in humans and horses in Southern Africa. Vector Borne Zoonotic Dis. 2010, 10, 659–664. [Google Scholar] [CrossRef]
- Kutasi, O.; Bakonyi, T.; Lecollinet, S.; Biksi, I.; Ferenczi, E.; Bahuon, C.; Sardi, S.; Zientara, S.; Szenci, O. Equine encephalomyelitis outbreak caused by a genetic lineage 2 West Nile virus in Hungary. J. Vet. Intern. Med. 2011, 25, 586–591. [Google Scholar] [CrossRef] [Green Version]
- Magurano, F.; Remoli, M.E.; Baggieri, M.; Fortuna, C.; Marchi, A.; Fiorentini, C.; Bucci, P.; Benedetti, E.; Ciufolini, M.G.; Rizzo, C.; et al. Circulation of West Nile virus lineage 1 and 2 during an outbreak in Italy. Clin. Microbiol. Infect. 2012, 18, E545–E547. [Google Scholar]
- Brault, A.C.; Langevin, S.A.; Bowen, R.A.; Panella, N.A.; Biggerstaff, B.J.; Miller, B.R.; Komar, N. Differential virulence of West Nile strains for American crows. Emerg. Infect. Dis. 2004, 10, 2161–2168. [Google Scholar] [CrossRef]
- Langevin, S.A.; Brault, A.C.; Panella, N.A.; Bowen, R.A.; Komar, N. Variation in virulence of West Nile virus strains for house sparrows (Passer domesticus). Am. J. Trop. Med. Hyg. 2005, 72, 99–102. [Google Scholar]
- Botha, E.M.; Markotter, W.; Wolfaardt, M.; Paweska, J.T.; Swanepoel, R.; Palacios, G.; Nel, L.H.; Venter, M. Genetic determinants of virulence in pathogenic lineage 2 West Nile virus strains. Emerg. Infect. Dis. 2008, 14, 222–230. [Google Scholar] [CrossRef]
- Beasley, D.W.C.; Li, L.; Suderman, M.T.; Barrett, A.D.T. Mouse neuroinvasive phenotype of West Nile virus strains varies depending upon virus genotype. Virology 2002, 296, 17–23. [Google Scholar] [CrossRef]
- Beasley, D.W.C.; Davis, C.T.; Whiteman, M.; Granwehr, B.; Kinney, R.M.; Barrett, A.D.T. Molecular determinants of virulence of West Nile virus in North America. Arch. Virol. Suppl. 2004, 18, 35–41. [Google Scholar]
- Colpitts, T.M.; Conway, M.J.; Montgomery, R.R.; Fikrig, E. West Nile virus: Biology, transmission, and human infection. Clin. Microbiol. Rev. 2012, 25, 635–648. [Google Scholar] [CrossRef]
- Machain-Williams, C.; Padilla-Paz, S.E.; Weber, M.; Cetina-Trejo, R.; Juarez-Ordaz, J.A.; Loroño-Pino, M.A.; Ulloa, A.; Wang, C.; Garcia-Rejon, J.; Blitvich, B.J. Antibodies to West Nile virus in wild and farmed crocodiles in Southeastern Mexico. J. Wildl. Dis. 2013, 49, 690–693. [Google Scholar] [CrossRef]
- Reagan, K.L.; Machain-Williams, C.; Wang, T.; Blair, C.D. Immunization of mice with recombinant mosquito salivary protein D7 enhances mortality from subsequent West Nile virus infection via mosquito bite. PLoS Negl. Trop. Dis. 2012, 6, e1935. [Google Scholar] [CrossRef]
- Styer, L.M.; Kent, K.A.; Albright, R.G.; Bennett, C.J.; Kramer, L.D.; Bernard, K.A. Mosquitoes inoculate high doses of West Nile virus as they probe and feed on live hosts. PLoS Pathog. 2007, 3, 1262–1270. [Google Scholar]
- Styer, L.M.; Lim, P.-Y.; Louie, K.L.; Albright, R.G.; Kramer, L.D.; Bernard, K.A. Mosquito saliva causes enhancement of West Nile virus infection in mice. J. Virol. 2011, 85, 1517–1527. [Google Scholar] [CrossRef]
- Schneider, B.S.; McGee, C.E.; Jordan, J.M.; Stevenson, H.L.; Soong, L.; Higgs, S. Prior exposure to uninfected mosquitoes enhances mortality in naturally-transmitted West Nile virus infection. PLoS One 2007, 2, e1171. [Google Scholar] [CrossRef]
- Johnston, L.J.; Halliday, G.M.; King, N.J. Phenotypic changes in Langerhans’ cells after infection with arboviruses: A role in the immune response to epidermally acquired viral infection? J. Virol. 1996, 70, 4761–4766. [Google Scholar]
- Limon-Flores, A.Y.; Perez-Tapia, M.; Estrada-Garcia, I.; Vaughan, G.; Escobar-Gutierrez, A.; Calderon-Amador, J.; Herrera-Rodriguez, S.E.; Brizuela-Garcia, A.; Heras-Chavarria, M.; Flores-Langarica, A.; et al. Dengue virus inoculation to human skin explants: An effective approach to assess in situ the early infection and the effects on cutaneous dendritic cells. Int. J. Clin. Exp. Pathol. 2005, 86, 323–334. [Google Scholar] [CrossRef]
- Palmer, D.R.; Sun, P.; Celluzzi, C.; Pang, S.; Sun, W.; Marovich, M.A.; Burgess, T.; Bisbing, J. Differential effects of dengue virus on infected and bystander dendritic cells. J. Virol. 2005, 79, 2432–2439. [Google Scholar] [CrossRef]
- Wu, S.J.; Grouard-Vogel, G.; Sun, W.; Mascola, J.R.; Brachtel, E.; Putvatana, R.; Louder, M.K.; Filgueira, L.; Marovich, M.A.; Wong, H.K.; et al. Human skin Langerhans cells are targets of dengue virus infection. Nat. Med. 2000, 6, 816–820. [Google Scholar] [CrossRef]
- Byrne, S.N.; Halliday, G.M.; Johnston, L.J.; King, N.J. Interleukin-1beta but not tumor necrosis factor is involved in West Nile virus-induced Langerhans cell migration from the skin in C57BL/6 mice. J. Investig. Dermatol. 2001, 117, 702–709. [Google Scholar] [CrossRef]
- Johnston, B.L.; Conly, J.M. West Nile virus—Where did it come from and where might it go? Can. J. Infect. Dis. Med. Microbiol. 2000, 11, 175–178. [Google Scholar]
- Lim, P.-Y.; Behr, M.J.; Chadwick, C.M.; Shi, P.-Y.; Bernard, K.A. Keratinocytes are cell targets of West Nile virus in vivo. J. Virol. 2011, 85, 5197–5201. [Google Scholar] [CrossRef]
- Kurane, I.; Janus, J.; Ennis, F.A. Dengue virus infection of human skin fibroblasts in vitro production of IFN-beta, IL-6 and GM-CSF. Arch. Virol. 1992, 124, 21–30. [Google Scholar] [CrossRef]
- Shi, P.Y.; Tilgner, M.; Lo, M.K.; Kent, K.A.; Bernard, K.A. Infectious cDNA clone of the epidemic West Nile virus from New York City. J. Virol. 2002, 76, 5847–5856. [Google Scholar] [CrossRef]
- Lazear, H.M.; Pinto, A.K.; Vogt, M.R.; Gale, M., Jr.; Diamond, M.S. Beta interferon controls West Nile virus infection and pathogenesis in mice. J. Virol. 2011, 85, 7186–7194. [Google Scholar] [CrossRef]
- Suthar, M.S.; Brassil, M.M.; Blahnik, G.; McMillan, A.; Ramos, H.J.; Proll, S.C.; Belisle, S.E.; Katze, M.G.; Gale, M., Jr. A systems biology approach reveals that tissue tropism to West Nile virus is regulated by antiviral genes and innate immune cellular processes. PLoS Pathog. 2013, 9, e1003168. [Google Scholar] [CrossRef]
- Samuel, M.A.; Diamond, M.S. Alpha/beta interferon protects against lethal West Nile virus infection by restricting cellular tropism and enhancing neuronal survival. J. Virol. 2005, 79, 13350–13361. [Google Scholar] [CrossRef]
- Maheshwari, R.K.; Friedman, R.M. Production of vesicular stomatitis virus with low infectivity by interferon-treated cells. J. Gen. Virol. 1979, 44, 261–264. [Google Scholar] [CrossRef]
- Wong, P.K.; Yuen, P.H.; MacLeod, R.; Chang, E.H.; Myers, M.W.; Friedman, R.M. The effect of interferon on de novo infection of Moloney murine leukemia virus. Cell 1977, 10, 245–252. [Google Scholar] [CrossRef]
- Chang, E.H.; Mims, S.J.; Triche, T.J.; Friedman, R.M. Interferon inhibits mouse leukaemia virus release: An electron microscope study. J. Gen. Virol. 1977, 34, 363–367. [Google Scholar] [CrossRef]
- Munoz, A.; Carrasco, L. Formation of non-infective herpesvirus particles in cultured cells treated with human interferon. J. Gen. Virol. 1984, 65, 1069–1078. [Google Scholar] [CrossRef]
- Babe, L.M.; Unal, A.; Craik, C.S. Obstruction of HIV-1 particle release by interferon-alpha occurs before viral protease processing and is independent of envelope glycoprotein. J. Interf. Cytokine Res. 1997, 17, 287–293. [Google Scholar]
- Hansen, B.D.; Nara, P.L.; Maheshwari, R.K.; Sidhu, G.S.; Bernbaum, J.G.; Hoekzema, D.; Meltzer, M.S.; Gendelman, H.E. Loss of infectivity by progeny virus from alpha interferon-treated human immunodeficiency virus type 1-infected T cells is associated with defective assembly of envelope gp120. J. Virol. 1992, 66, 7543–7548. [Google Scholar]
- Rozen-Gagnon, K.; Stapleford, K.A.; Mongelli, V.; Blanc, H.; Failloux, A.-B.; Saleh, M.-C.; Vignuzzi, M. Alphavirus mutator variants present host-specific defects and attenuation in mammalian and insect models. PLoS Pathog. 2014, 10, e1003877. [Google Scholar] [CrossRef]
- Rodenhuis-Zybert, I.A.; van der Schaar, H.M.; da Silva Voorham, J.M.; van der Ende-Metselaar, H.; Lei, H.-Y.; Wilschut, J.; Smit, J.M. Immature dengue virus: A veiled pathogen? PLoS Pathog. 2010, 6, e1000718. [Google Scholar] [CrossRef]
- Elshuber, S.; Allison, S.L.; Heinz, F.X.; Mandl, C.W. Cleavage of protein prM is necessary for infection of BHK-21 cells by tick-borne encephalitis virus. J. Gen. Virol. 2003, 84, 183–191. [Google Scholar] [CrossRef]
- Heinz, F.X.; Stiasny, K.; Puschner-Auer, G.; Holzmann, H.; Allison, S.L.; Mandl, C.W.; Kunz, C. Structural changes and functional control of the tick-borne encephalitis virus glycoprotein E by the heterodimeric association with protein prM. Virology 1994, 198, 109–117. [Google Scholar] [CrossRef]
- Liu, W.J.; Chen, H.B.; Wang, X.J.; Huang, H.; Khromykh, A.A. Analysis of Adaptive Mutations in Kunjin Virus Replicon RNA Reveals a Novel Role for the Flavivirus Nonstructural Protein NS2A in Inhibition of Beta Interferon Promoter-Driven Transcription. J. Virol. 2004, 78, 12225–12235. [Google Scholar] [CrossRef] [Green Version]
- Liu, W.J.; Wang, X.J.; Mokhonov, V.V.; Shi, P.; Randall, R.; Khromykh, A.A. Inhibition of Interferon Signaling by the New York 99 Strain and Kunjin Subtype of West Nile Virus Involves Blockage of STAT1 and STAT2 Activation by Nonstructural Proteins. J. Virol. 2005, 79, 1934–1942. [Google Scholar] [CrossRef]
- Liu, W.J.; Wang, X.J.; Clark, D.C.; Lobigs, M.; Hall, R.A.; Khromykh, A.A. A Single Amino Acid Substitution in the West Nile Virus Nonstructural Protein NS2A Disables Its Ability To Inhibit Alpha/Beta Interferon Induction and Attenuates Virus Virulence in Mice. J. Virol. 2006, 80, 2396–2404. [Google Scholar] [CrossRef]
- Wilson, J.R.; de Sessions, P.F.; Leon, M.; Scholle, F. West Nile virus nonstructural protein 1 inhibits TLR3 signal transduction. J. Virol. 2008, 82, 8262–8271. [Google Scholar] [CrossRef]
- Arjona, A.; Ledizet, M.; Anthony, K.; Bonafé, N.; Modis, Y.; Town, T.; Fikrig, E. West Nile virus envelope protein inhibits dsRNA-induced innate immune responses. J. Immunol. 2007, 179, 8403–8409. [Google Scholar]
- Baronti, C.; Sire, J.; de Lamballerie, X.; Quérat, G. Nonstructural NS1 proteins of several mosquito-borne Flavivirus do not inhibit TLR3 signaling. Virology 2010, 404, 319–330. [Google Scholar] [CrossRef]
- Best, S.M.; Morris, K.L.; Shannon, J.G.; Robertson, J.; Mitzel, D.N.; Park, G.S.; Boer, E.; Wolfinbarger, J.B.; Bloom, M.E.; Robertson, S.J. Inhibition of Interferon-Stimulated JAK-STAT Signaling by a Tick-Borne Flavivirus and Identification of NS5 as an Interferon Antagonist. J. Virol. 2005, 79, 12828–12839. [Google Scholar] [CrossRef]
- Guo, J.; Hayashi, J.; Seeger, C. West Nile Virus Inhibits the Signal Transduction Pathway of Alpha Interferon. J. Virol. 2005, 79, 1343–1350. [Google Scholar] [CrossRef]
- Muñoz-Jordan, J.L.; Sánchez-Burgos, G.G.; Laurent-Rolle, M.; García-Sastre, A. Inhibition of interferon signaling by dengue virus. Proc. Natl. Acad. Sci. USA 2003, 100, 14333–14338. [Google Scholar] [CrossRef]
- Diamond, M.S. Mechanisms of evasion of the type I interferon antiviral response by flaviviruses. J. Interf. Cytokine Res. 2009, 29, 521–530. [Google Scholar] [CrossRef]
- Fredericksen, B.L.; Smith, M.; Katze, M.G.; Shi, P.; Gale, M. The Host Response to West Nile Virus Infection Limits Viral Spread through the Activation of the Interferon Regulatory Factor 3 Pathway. J. Virol. 2004, 78, 7737–7747. [Google Scholar] [CrossRef]
- Cho, H.; Proll, S.C.; Szretter, K.J.; Katze, M.G.; Gale, M., Jr.; Diamond, M.S. Differential innate immune response programs in neuronal subtypes determine susceptibility to infection in the brain by positive-stranded RNA viruses. Nat. Med. 2013, 19, 458–464. [Google Scholar] [CrossRef]
- Venter, M.; Myers, T.G.; Wilson, M.A.; Kindt, T.J.; Pawaska, J.T.; Burt, F.J.; Leman, P.A.; Swanepoel, R. Gene expression in mice infected with West Nile virus strains of different neurovirulence. Virology 2005, 342, 119–140. [Google Scholar]
- Scherbik, S.V.; Stockman, B.M.; Brinton, M.A. Differential Expression of Interferon (IFN) Regulatory Factors and IFN-Stimulated Genes at Early Times after West Nile Virus Infection of Mouse Embryo Fibroblasts. J. Virol. 2007, 81, 12005–12018. [Google Scholar] [CrossRef]
- Hoenen, A.; Gillespie, L.; Morgan, G.; Heide, P.; Khromykh, A.; Mackensie, J. The West Nile virus assembly process evades the conserved antiviral mechanism of the interferon-induced MxA protein. Virology 2014, 448, 104–116. [Google Scholar] [CrossRef]
- Szretter, K.J.; Daniels, B.P.; Cho, H.; Gainey, M.D.; Yokoyama, W.M.; Gale, M.; Virgin, H.W.; Klein, R.S.; Sen, G.C.; Diamond, M.S. 2′-O Methylation of the Viral mRNA Cap by West Nile Virus Evades Ifit1-Dependent and -Independent Mechanisms of Host Restriction In Vivo. PLoS Pathog. 2012, 8, e1002698. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Hoover, L.I.; Fredericksen, B.L. IFN-Dependent and -Independent Reduction in West Nile Virus Infectivity in Human Dermal Fibroblasts. Viruses 2014, 6, 1424-1441. https://doi.org/10.3390/v6031424
Hoover LI, Fredericksen BL. IFN-Dependent and -Independent Reduction in West Nile Virus Infectivity in Human Dermal Fibroblasts. Viruses. 2014; 6(3):1424-1441. https://doi.org/10.3390/v6031424
Chicago/Turabian StyleHoover, Lisa I., and Brenda L. Fredericksen. 2014. "IFN-Dependent and -Independent Reduction in West Nile Virus Infectivity in Human Dermal Fibroblasts" Viruses 6, no. 3: 1424-1441. https://doi.org/10.3390/v6031424