Brd4-Mediated Nuclear Retention of the Papillomavirus E2 Protein Contributes to Its Stabilization in Host Cells



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
2.1. Results
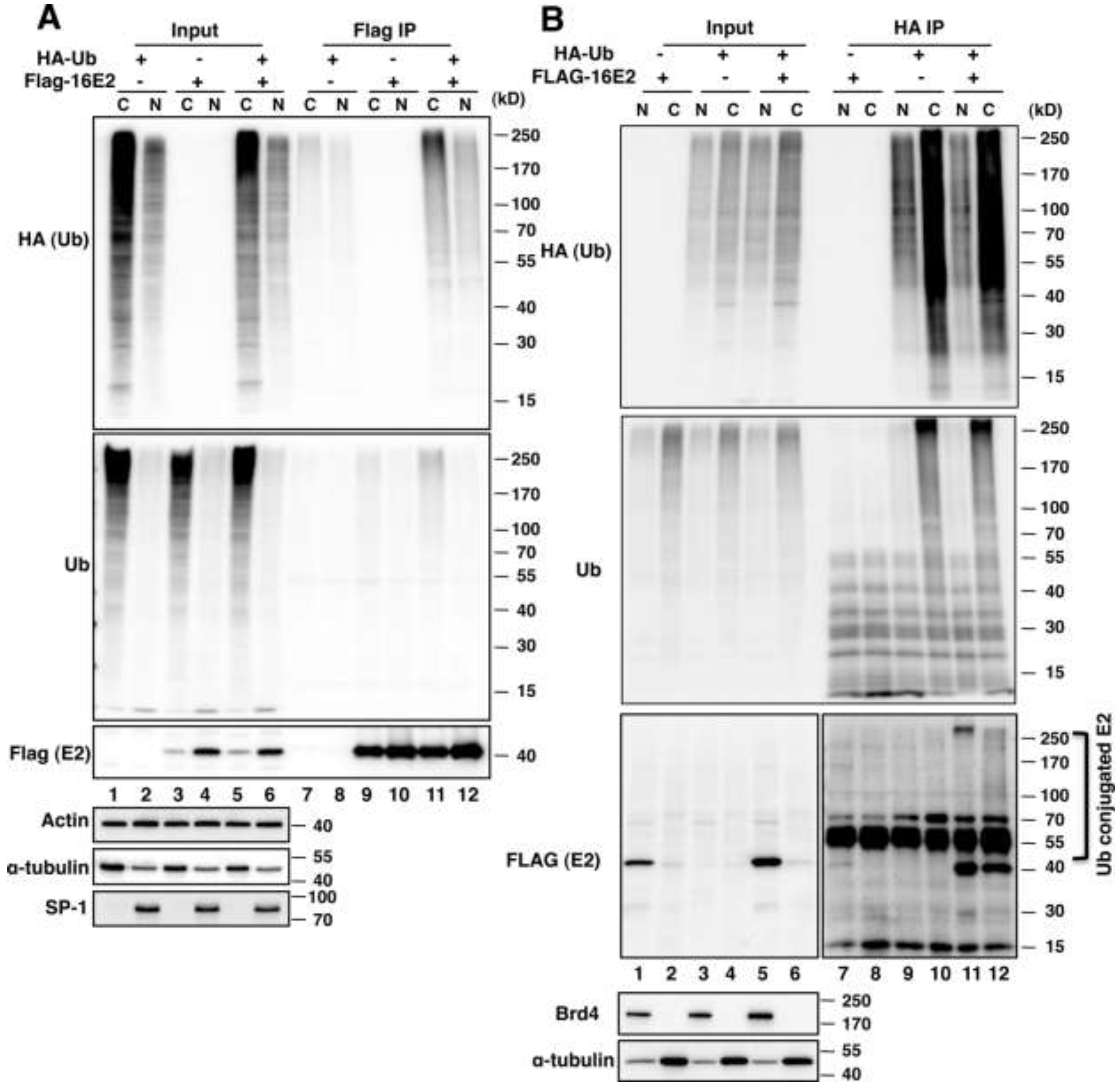
2.1.1. E2 Is Mainly Ubiquitylated in the Host Cytoplasm
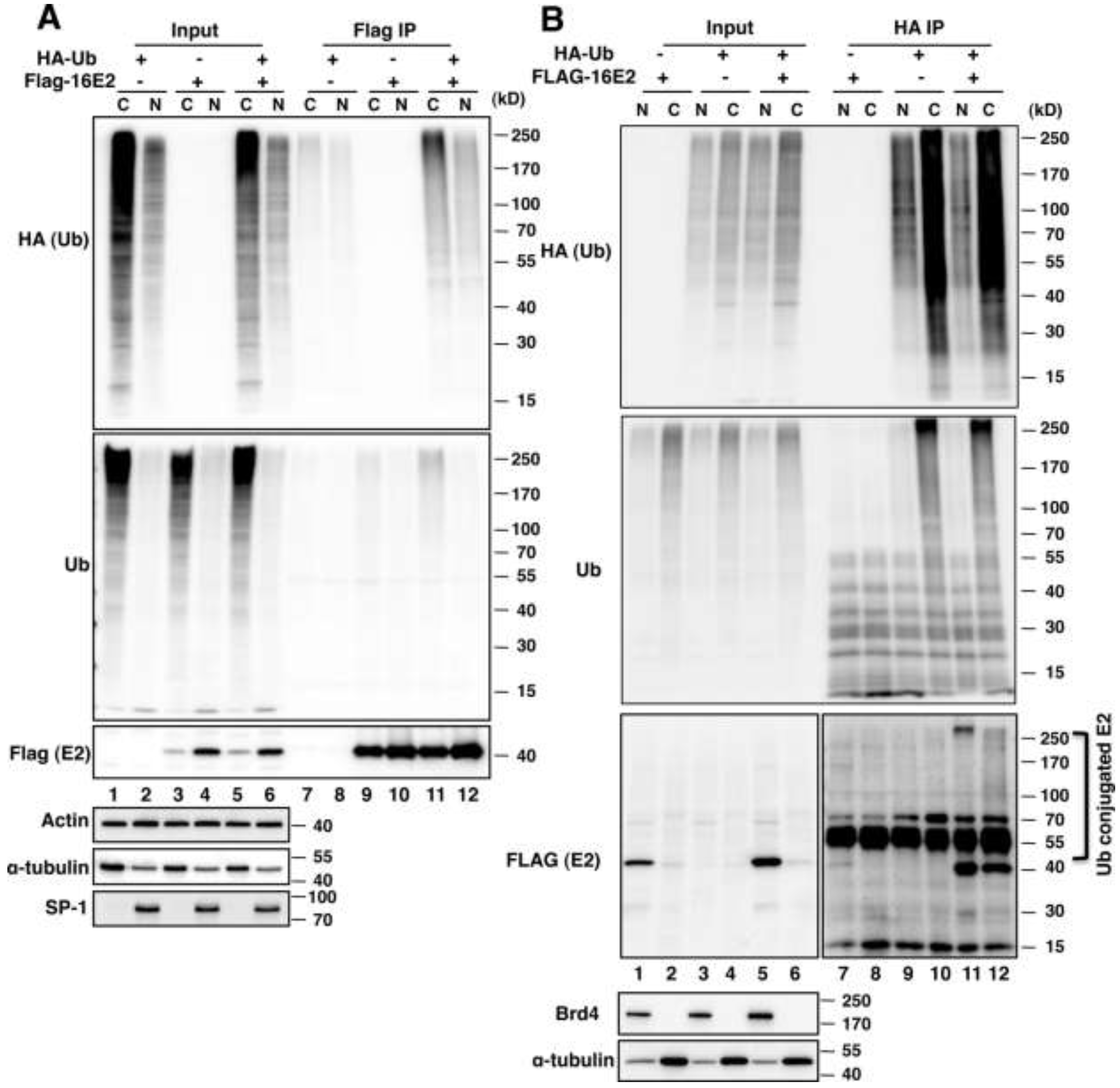
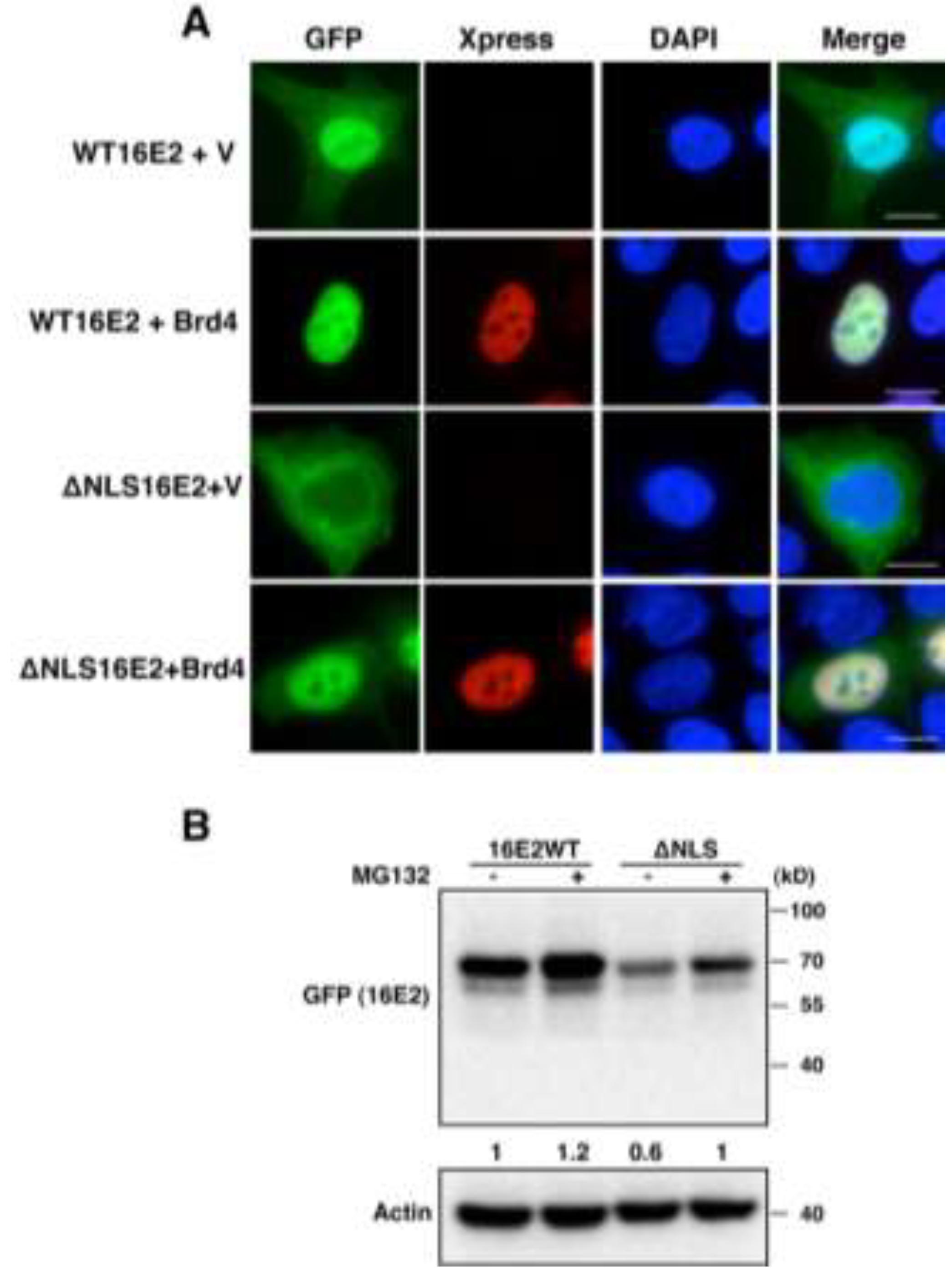
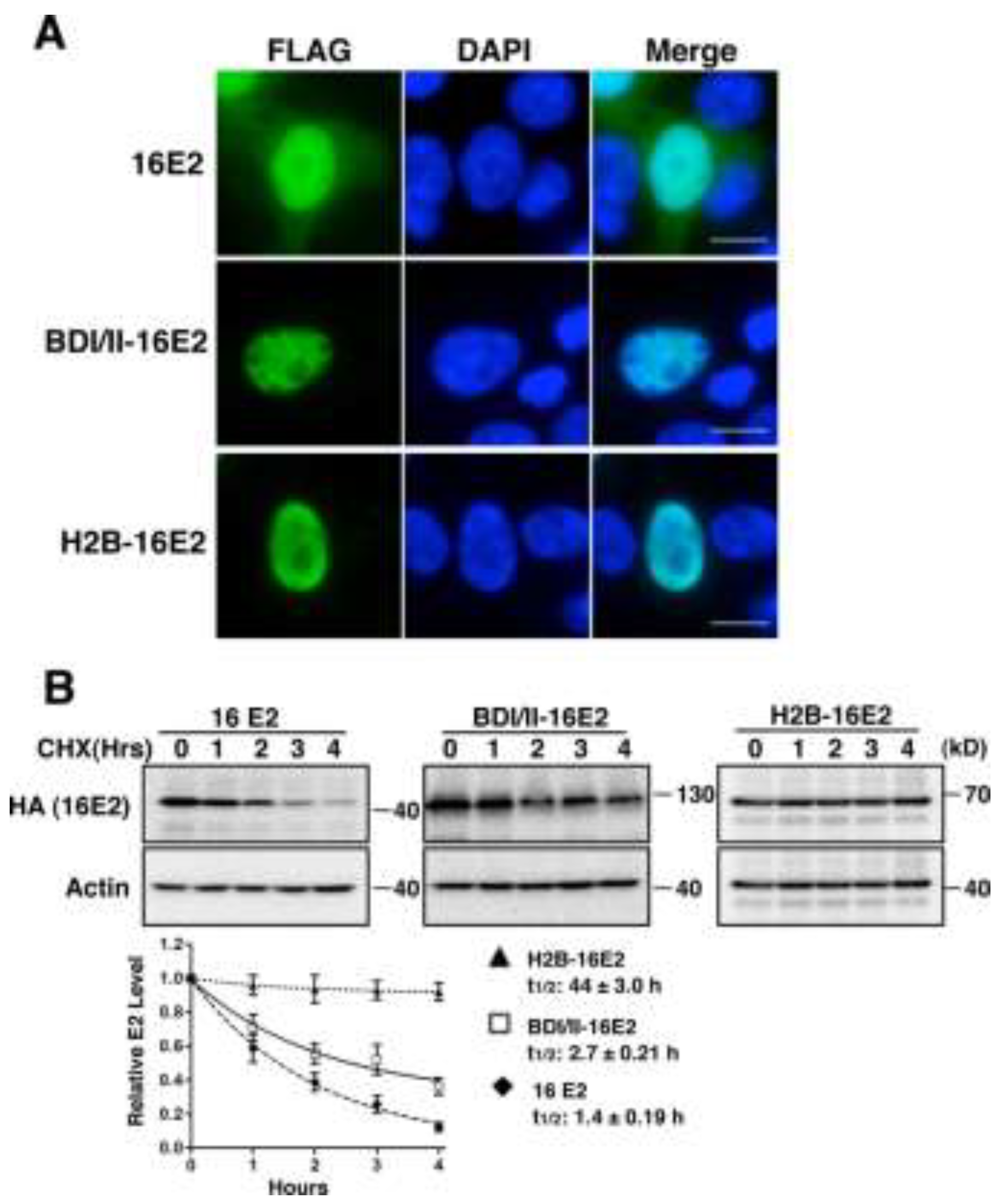
2.1.2. Brd4 Enhances E2 Nuclear Retention
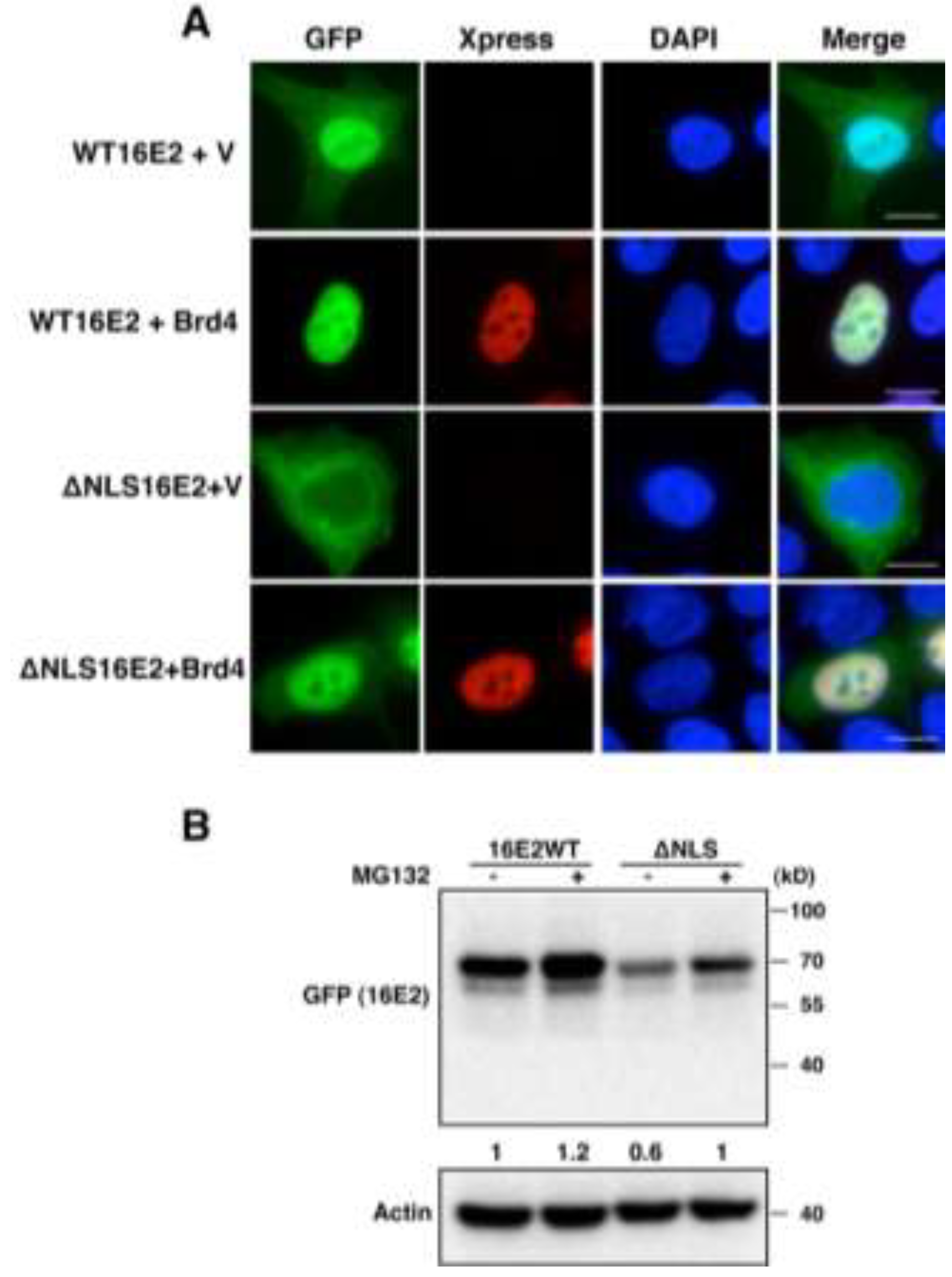
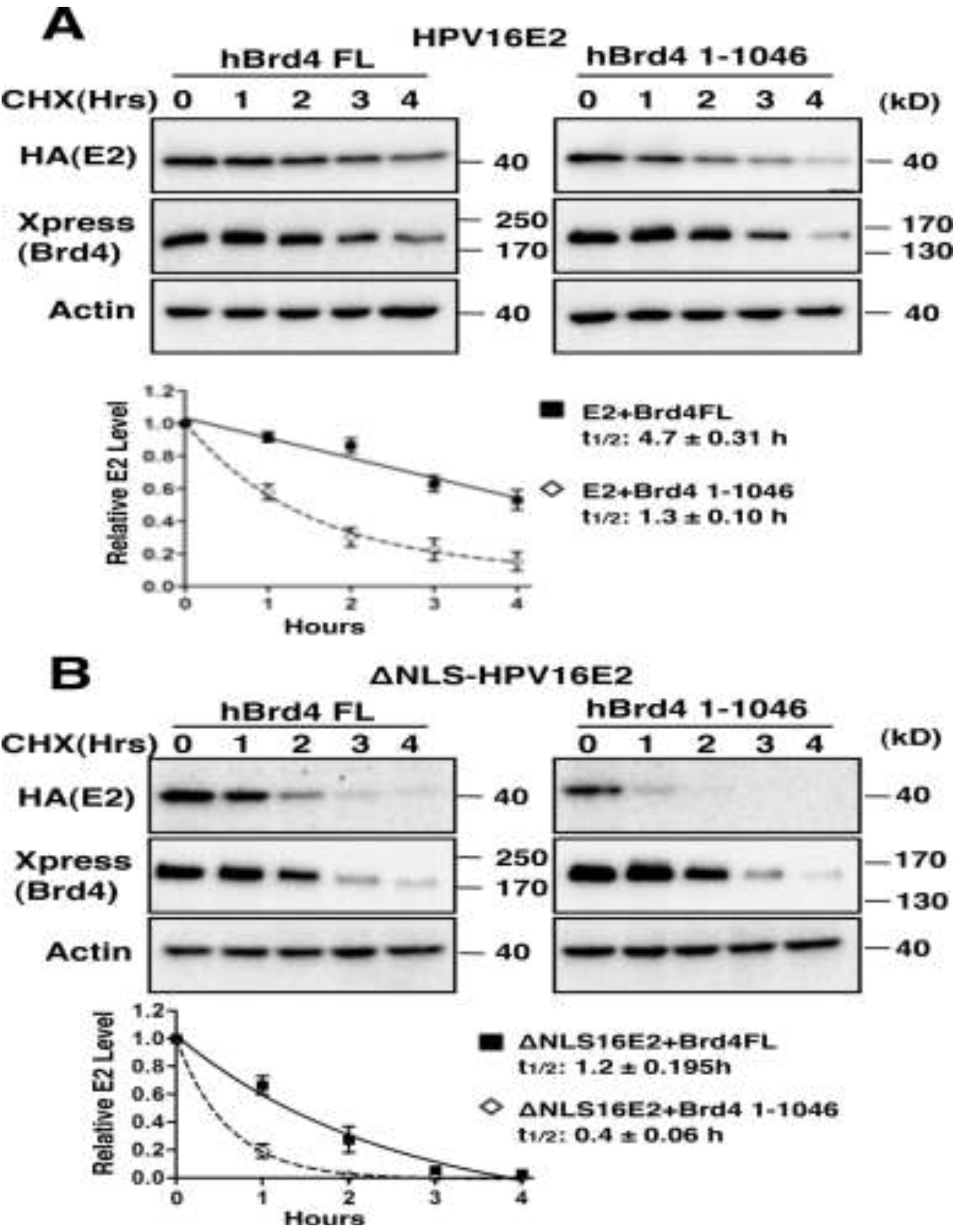
2.1.3. Brd4-Mediated E2 Nuclear Retention is Essential for E2 Stabilization
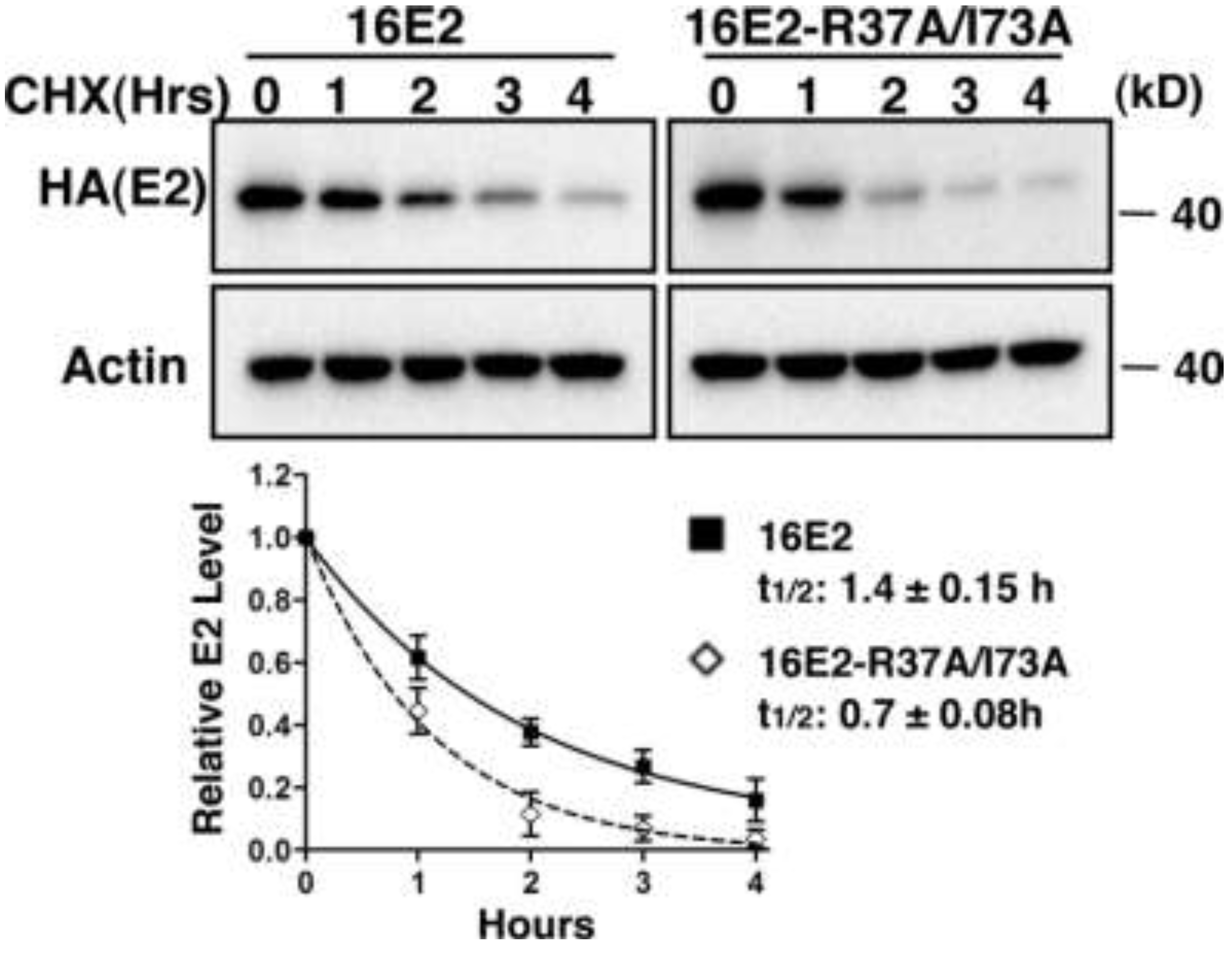
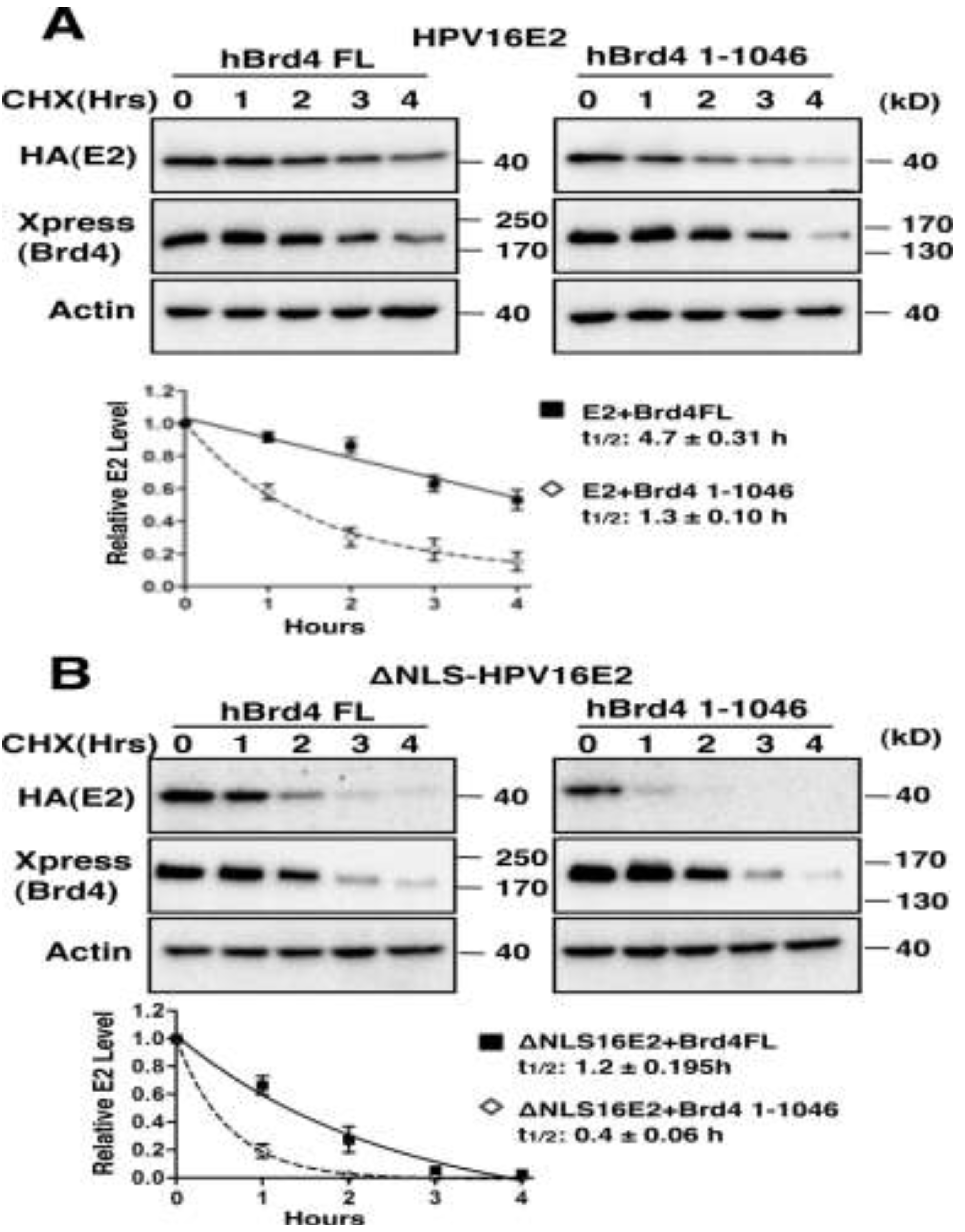
2.1.4. Interaction with Brd4 is Responsible for E2 Stabilization
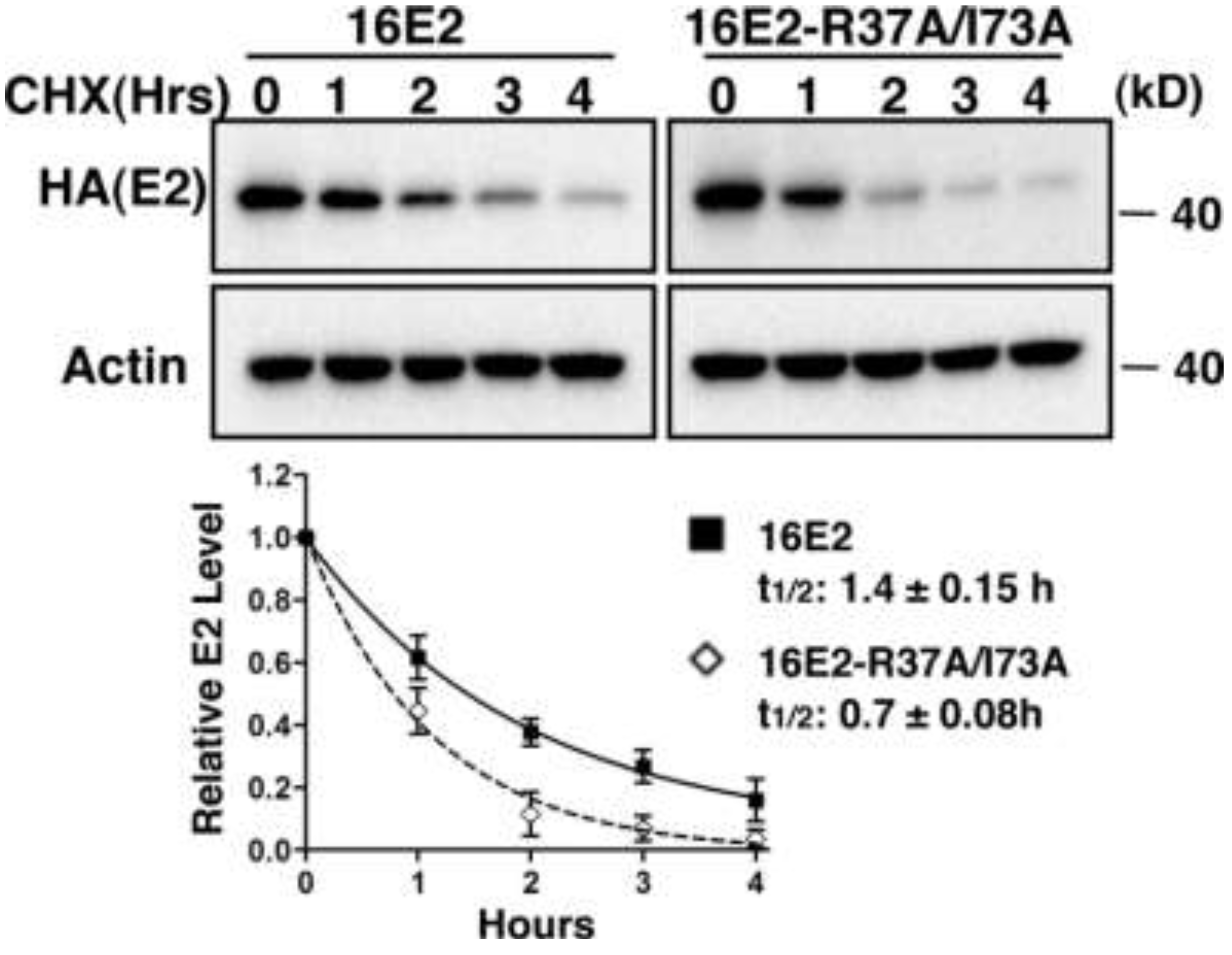
2.1.5. Tethering E2 Proteins to Host Chromatin Increases E2 Stability
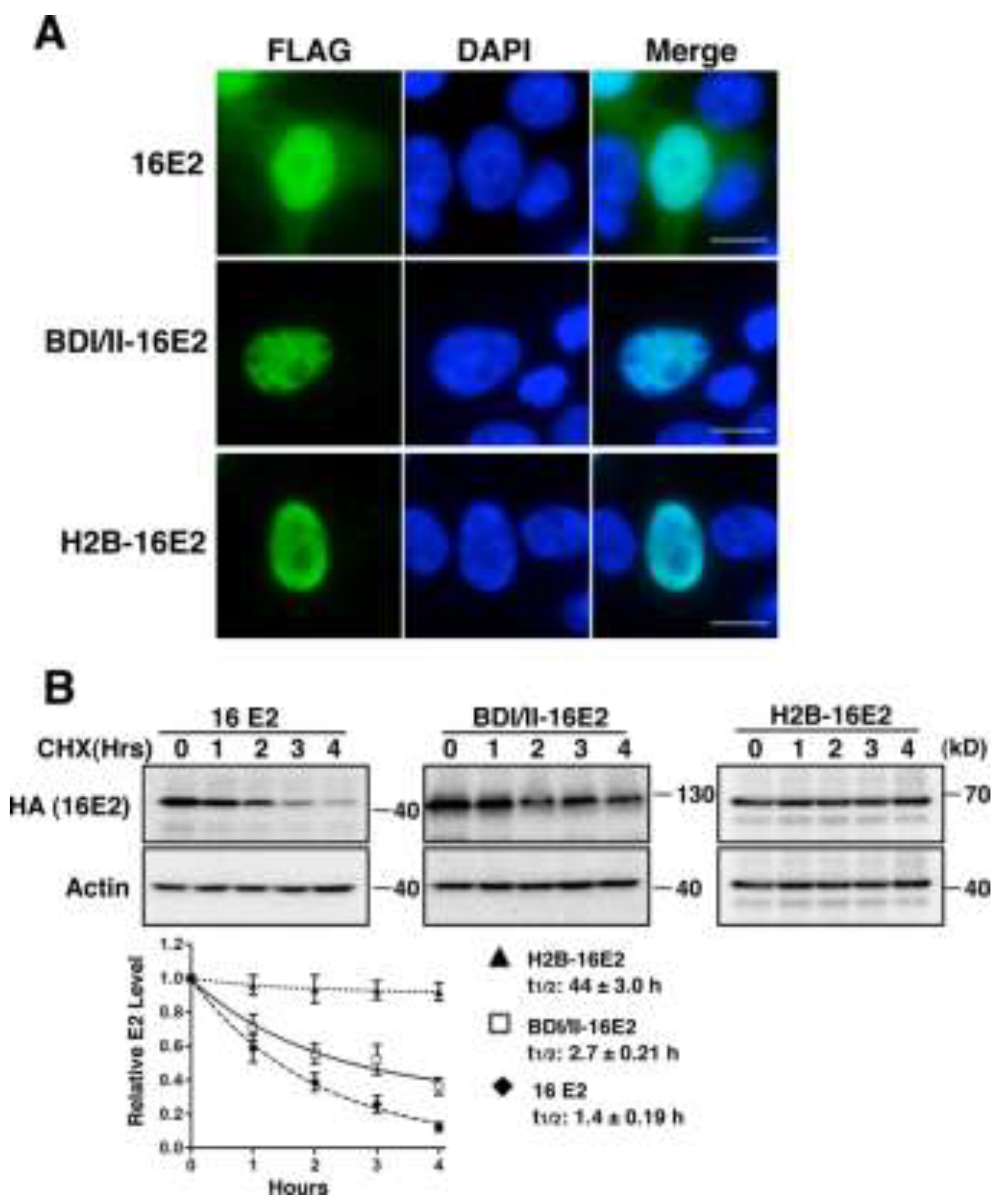
2.2. Discussion
3. Experimental Section
3.1. Cell Culture, Cell Lines, and Transfection
3.2. Recombinant Plasmid Construction
3.3. Protein Half-Life Cycloheximide Blocking/Chasing Analysis
3.4. Western Blotting
3.5. Immunoprecipitation
3.6. Immunofluorescent Staining
3.7. Statistical Analysis
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References and Notes
- Howley, P.M.; Lowy, D.R. Papillomaviruses and their replication. In Filelds Virology, 4th ed.; Knipe, D.M., Howley, P.M., Eds.; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2001; Volume 2, p. 2197. [Google Scholar]
- Zur Hausen, H. Papillomaviruses and cancer: From basic studies to clinical application. Nat. Rev. Cancer 2002, 2, 342–350. [Google Scholar] [CrossRef]
- Hebner, C.M.; Laimins, L.A. Human papillomaviruses: Basic mechanisms of pathogenesis and oncogenicity. Rev. Med. Virol. 2006, 16, 83–97. [Google Scholar] [CrossRef]
- McBride, A.A.; Romanczuk, H.; Howley, P.M. The papillomavirus E2 regulatory proteins. J. Biol. Chem. 1991, 266, 18411–18414. [Google Scholar]
- Penrose, K.J.; McBride, A.A. Proteasome-mediated degradation of the papillomavirus E2-TA protein is regulated by phosphorylation and can modulate viral genome copy number. J. Virol. 2000, 74, 6031–6038. [Google Scholar] [CrossRef]
- Bellanger, S.; Demeret, C.; Goyat, S.; Thierry, F. Stability of the human papillomavirus type 18 E2 protein is regulated by a proteasome degradation pathway through its amino-terminal transactivation domain. J. Virol. 2001, 75, 7244–7251. [Google Scholar] [CrossRef]
- Penrose, K.J.; Garcia-Alai, M.; de Prat-Gay, G.; McBride, A.A. Casein Kinase II phosphorylation-induced conformational switch triggers degradation of the papillomavirus E2 protein. J. Biol. Chem. 2004, 279, 22430–22439. [Google Scholar]
- Wu, Y.C.; Bian, X.L.; Heaton, P.R.; Deyrieux, A.F.; Wilson, V.G. Host cell sumoylation level influences papillomavirus E2 protein stability. Virology 2009, 387, 176–183. [Google Scholar] [CrossRef]
- Zheng, G.; Schweiger, M.R.; Martinez-Noel, G.; Zheng, L.; Smith, J.A.; Harper, J.W.; Howley, P.M. Brd4 regulation of papillomavirus protein E2 stability. J. Virol. 2009, 83, 8683–8692. [Google Scholar] [CrossRef]
- Bellanger, S.; Tan, C.L.; Nei, W.; He, P.P.; Thierry, F. The human papillomavirus type 18 E2 protein is a cell cycle-dependent target of the SCFSkp2 ubiquitin ligase. J. Virol. 2010, 84, 437–444. [Google Scholar] [CrossRef]
- You, J.; Croyle, J.L.; Nishimura, A.; Ozato, K.; Howley, P.M. Interaction of the bovine papillomavirus E2 protein with Brd4 tethers the viral DNA to host mitotic chromosomes. Cell 2004, 117, 349–360. [Google Scholar] [CrossRef]
- Dey, A.; Ellenberg, J.; Farina, A.; Coleman, A.E.; Maruyama, T.; Sciortino, S.; Lippincott-Schwartz, J.; Ozato, K. A bromodomain protein, MCAP, associates with mitotic chromosomes and affects G(2)-to-M transition. Mol. Cell. Biol. 2000, 20, 6537–6549. [Google Scholar] [CrossRef]
- Dey, A.; Chitsaz, F.; Abbasi, A.; Misteli, T.; Ozato, K. The double bromodomain protein Brd4 binds to acetylated chromatin during interphase and mitosis. Proc. Natl. Acad. Sci. USA 2003, 100, 8758–8763. [Google Scholar] [CrossRef]
- Abbate, E.A.; Voitenleitner, C.; Botchan, M.R. Structure of the papillomavirus DNA-tethering complex E2:Brd4 and a peptide that ablates HPV chromosomal association. Mol. Cell 2006, 24, 877–889. [Google Scholar] [CrossRef]
- Baxter, M.K.; McPhillips, M.G.; Ozato, K.; McBride, A.A. The mitotic chromosome binding activity of the papillomavirus E2 protein correlates with interaction with the cellular chromosomal protein, Brd4. J. Virol. 2005, 79, 4806–4818. [Google Scholar] [CrossRef]
- Brannon, A.R.; Maresca, J.A.; Boeke, J.D.; Basrai, M.A.; McBride, A.A. Reconstitution of papillomavirus E2-mediated plasmid maintenance in Saccharomyces cerevisiae by the Brd4 bromodomain protein. Proc. Natl. Acad. Sci. USA 2005, 102, 2998–3003. [Google Scholar] [CrossRef]
- Ilves, I.; Maemets, K.; Silla, T.; Janikson, K.; Ustav, M. Brd4 is involved in multiple processes of the bovine papillomavirus type 1 life cycle. J. Virol. 2006, 80, 3660–3665. [Google Scholar] [CrossRef]
- McPhillips, M.G.; Ozato, K.; McBride, A.A. Interaction of bovine papillomavirus E2 protein with Brd4 stabilizes its association with chromatin. J. Virol. 2005, 79, 8920–8932. [Google Scholar] [CrossRef]
- McPhillips, M.G.; Oliveira, J.G.; Spindler, J.E.; Mitra, R.; McBride, A.A. Brd4 is required for e2-mediated transcriptional activation but not genome partitioning of all papillomaviruses. J. Virol. 2006, 80, 9530–9543. [Google Scholar] [CrossRef]
- Schweiger, M.R.; You, J.; Howley, P.M. Bromodomain protein 4 mediates the papillomavirus E2 transcriptional activation function. J. Virol. 2006, 80, 4276–4285. [Google Scholar] [CrossRef]
- Wu, S.Y.; Chiang, C.M. The double bromodomain-containing chromatin adaptor Brd4 and transcriptional regulation. J. Biol. Chem. 2007, 282, 13141–13145. [Google Scholar] [CrossRef]
- Wu, S.Y.; Lee, A.Y.; Hou, S.Y.; Kemper, J.K.; Erdjument-Bromage, H.; Tempst, P.; Chiang, C.M. Brd4 links chromatin targeting to HPV transcriptional silencing. Genes Dev. 2006, 20, 2383–2396. [Google Scholar] [CrossRef]
- Yan, J.; Li, Q.; Lievens, S.; Tavernier, J.; You, J. Abrogation of the Brd4-positive transcription elongation factor B complex by papillomavirus E2 protein contributes to viral oncogene repression. J. Virol. 2010, 84, 76–87. [Google Scholar] [CrossRef]
- You, J.; Schweiger, M.R.; Howley, P.M. Inhibition of E2 binding to Brd4 enhances viral genome loss and phenotypic reversion of bovine papillomavirus-transformed cells. J. Virol. 2005, 79, 14956–14961. [Google Scholar] [CrossRef]
- Lee, A.Y.; Chiang, C.M. Chromatin adaptor Brd4 modulates E2 transcription activity and protein stability. J. Biol. Chem. 2009, 284, 2778–2786. [Google Scholar]
- Gagnon, D.; Joubert, S.; Senechal, H.; Fradet-Turcotte, A.; Torre, S.; Archambault, J. Proteasomal degradation of the papillomavirus E2 protein is inhibited by overexpression of bromodomain-containing protein 4. J. Virol. 2009, 83, 4127–4139. [Google Scholar] [CrossRef]
- Blachon, S.; Bellanger, S.; Demeret, C.; Thierry, F. Nucleo-cytoplasmic shuttling of high risk human Papillomavirus E2 proteins induces apoptosis. J. Biol. Chem. 2005, 280, 36088–36098. [Google Scholar] [CrossRef]
- Klucevsek, K.; Wertz, M.; Lucchi, J.; Leszczynski, A.; Moroianu, J. Characterization of the nuclear localization signal of high risk HPV16 E2 protein. Virology 2007, 360, 191–198. [Google Scholar] [CrossRef]
- Mochizuki, K.; Nishiyama, A.; Jang, M.K.; Dey, A.; Ghosh, A.; Tamura, T.; Natsume, H.; Yao, H.; Ozato, K. The bromodomain protein Brd4 stimulates G1 gene transcription and promotes progression to S phase. J. Biol. Chem. 2008, 283, 9040–9048. [Google Scholar] [CrossRef]
- Yang, Z.; He, N.; Zhou, Q. Brd4 recruits P-TEFb to chromosomes at late mitosis to promote G1 gene expression and cell cycle progression. Mol. Cell. Biol. 2008, 28, 967–976. [Google Scholar] [CrossRef]
- Johansson, C.; Graham, S.V.; Dornan, E.S.; Morgan, I.M. The human papillomavirus 16 E2 protein is stabilised in S phase. Virology 2009, 394, 194–199. [Google Scholar] [CrossRef]
- Wang, X.; Naidu, S.R.; Sverdrup, F.; Androphy, E.J. Tax1BP1 interacts with papillomavirus E2 and regulates E2-dependent transcription and stability. J. Virol. 2009, 83, 2274–2284. [Google Scholar] [CrossRef]
- Chin, K.T.; Chun, A.C.; Ching, Y.P.; Jeang, K.T.; Jin, D.Y. Human T-cell leukemia virus oncoprotein tax represses nuclear receptor-dependent transcription by targeting coactivator TAX1BP1. Cancer Res. 2007, 67, 1072–1081. [Google Scholar] [CrossRef]
- Longworth, M.S.; Laimins, L.A. Pathogenesis of human papillomaviruses in differentiating epithelia. MicroBiol. Mol. Biol. Rev. 2004, 68, 362–372. [Google Scholar] [CrossRef]
- Schreiber, E.; Matthias, P.; Muller, M.M.; Schaffner, W. Rapid detection of octamer binding proteins with ‘mini-extracts’, prepared from a small number of cells. Nucleic Acids Res. 1989, 17, 6419. [Google Scholar] [CrossRef]
Supplementary Files
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Li, J.; Li, Q.; Diaz, J.; You, J. Brd4-Mediated Nuclear Retention of the Papillomavirus E2 Protein Contributes to Its Stabilization in Host Cells. Viruses 2014, 6, 319-335. https://doi.org/10.3390/v6010319
Li J, Li Q, Diaz J, You J. Brd4-Mediated Nuclear Retention of the Papillomavirus E2 Protein Contributes to Its Stabilization in Host Cells. Viruses. 2014; 6(1):319-335. https://doi.org/10.3390/v6010319
Chicago/Turabian StyleLi, Jing, Qing Li, Jason Diaz, and Jianxin You. 2014. "Brd4-Mediated Nuclear Retention of the Papillomavirus E2 Protein Contributes to Its Stabilization in Host Cells" Viruses 6, no. 1: 319-335. https://doi.org/10.3390/v6010319