Current Understanding of the Role of the Brd4 Protein in the Papillomavirus Lifecycle

Abstract

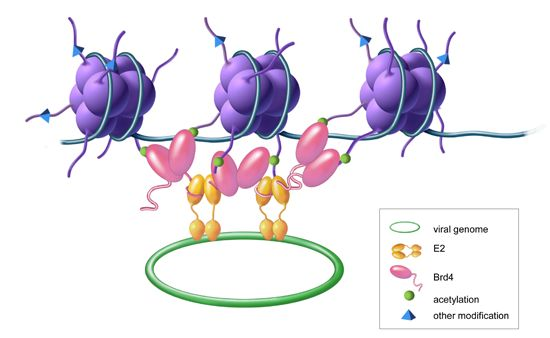{kind=link}
{kind=link}
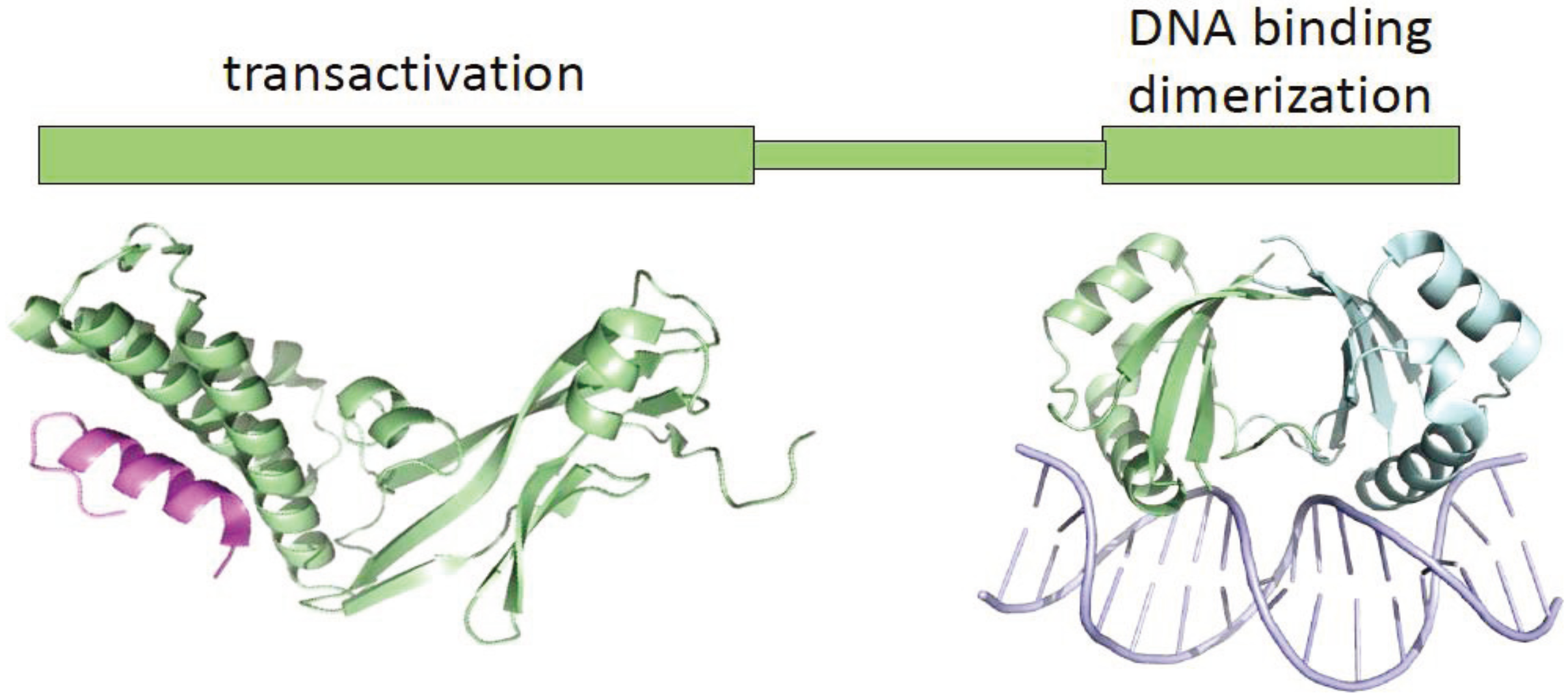{kind=link}
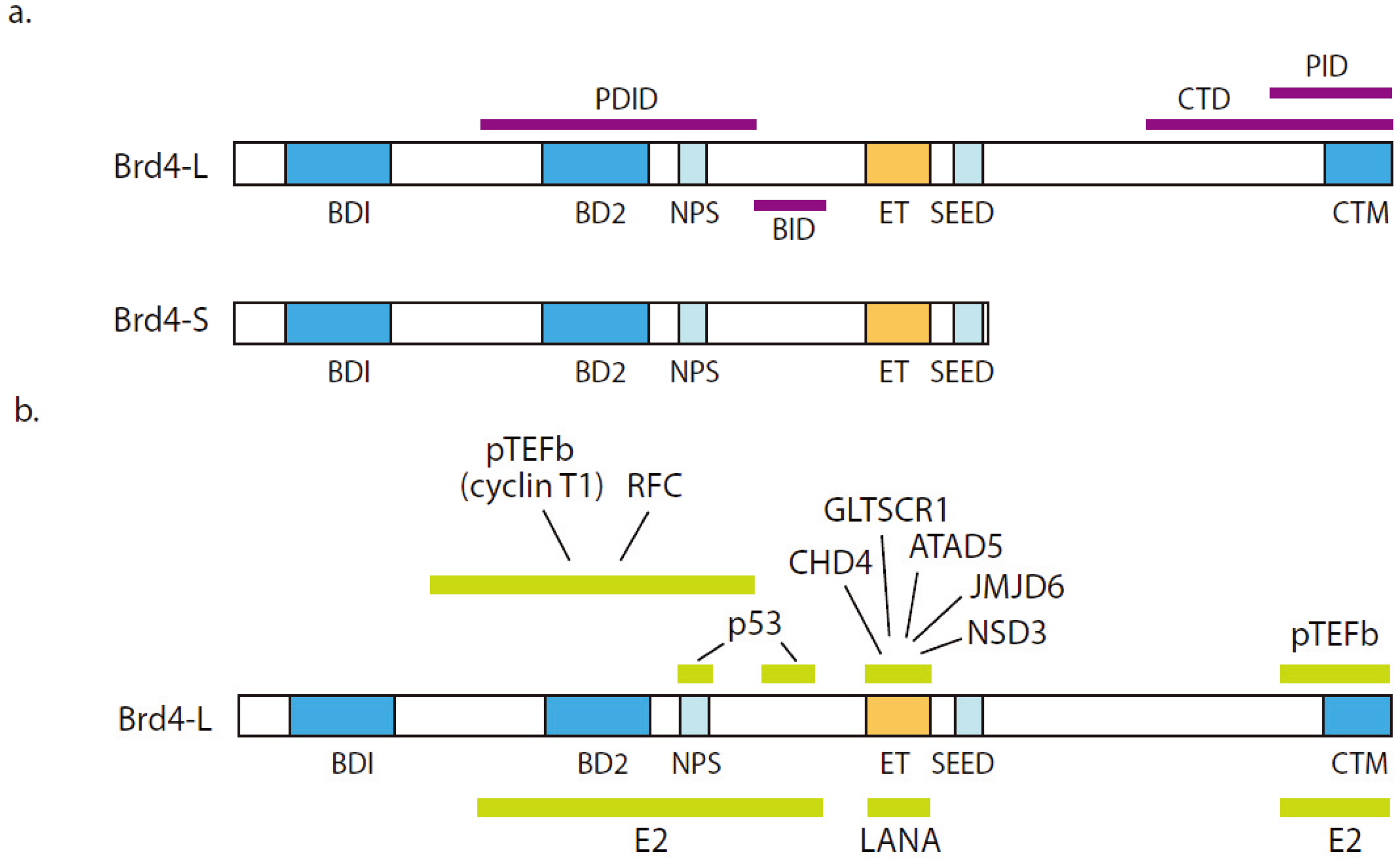{kind=link}
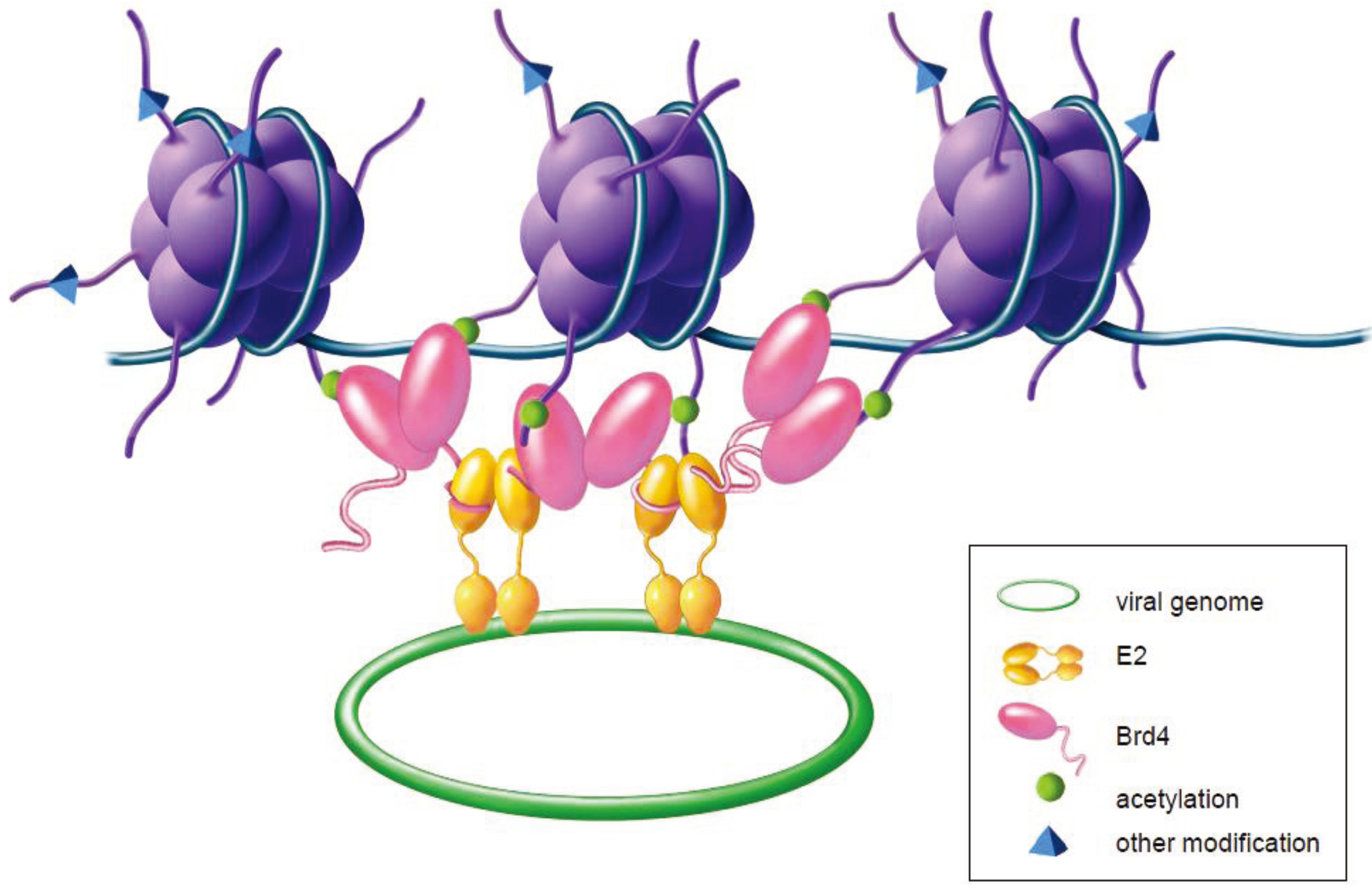{kind=link}
1. Introduction
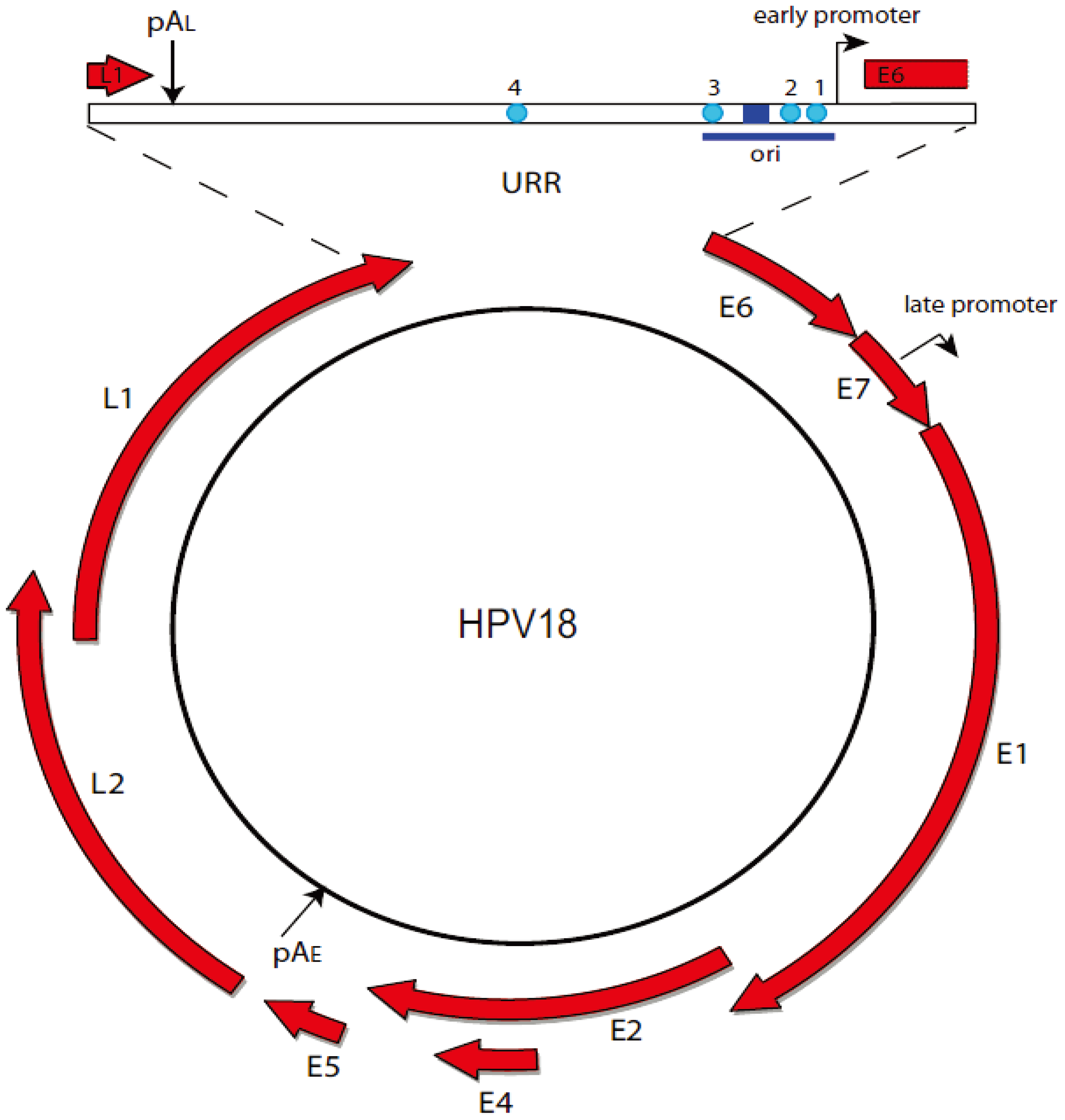
2. Papillomaviruses
2.1. The Papillomavirus Lifecycle
2.2. Viral Transcription
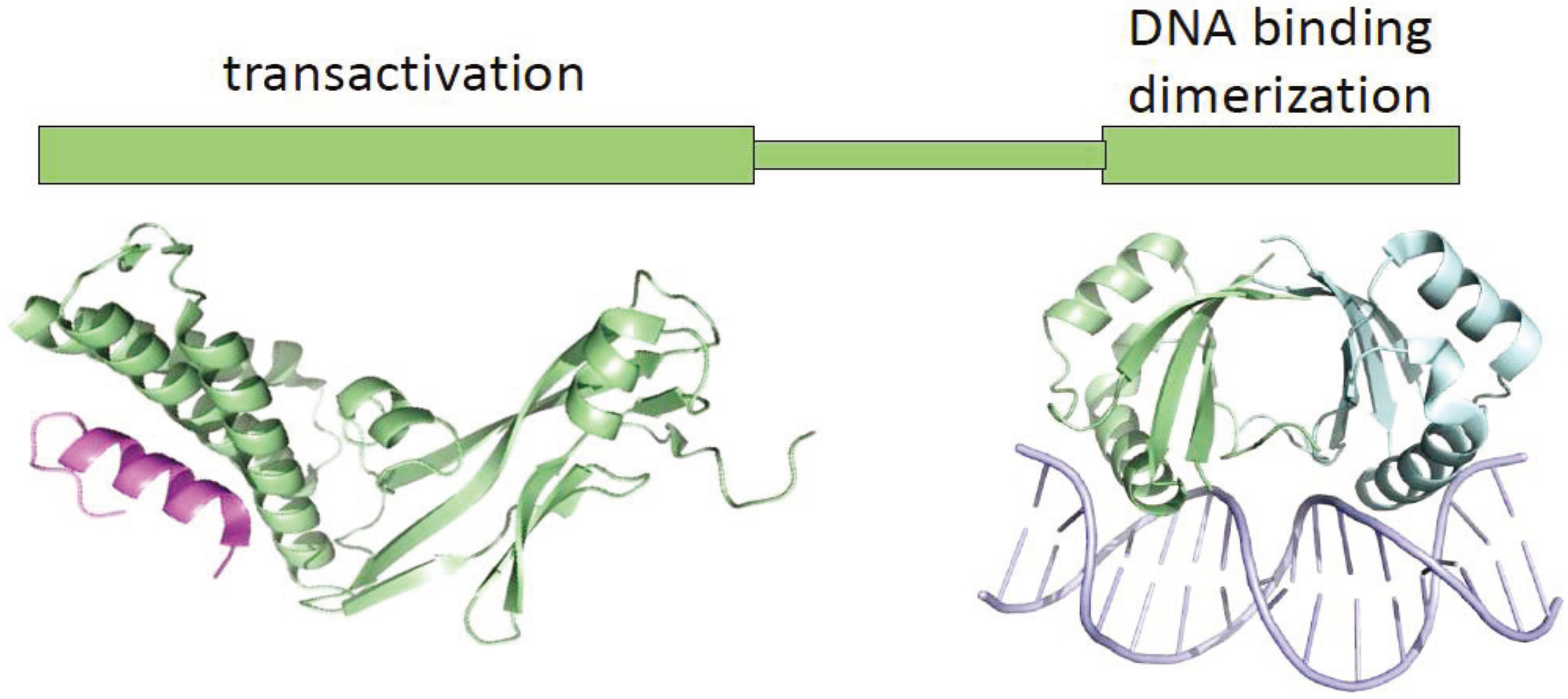
2.3. Disruption of E2 Function by HPV Integration
2.4. Viral Replication
2.5. Differences in Transcription and Replication among Papillomaviruses
3. The Brd4 Protein
3.1. Brd4 Structure and Function
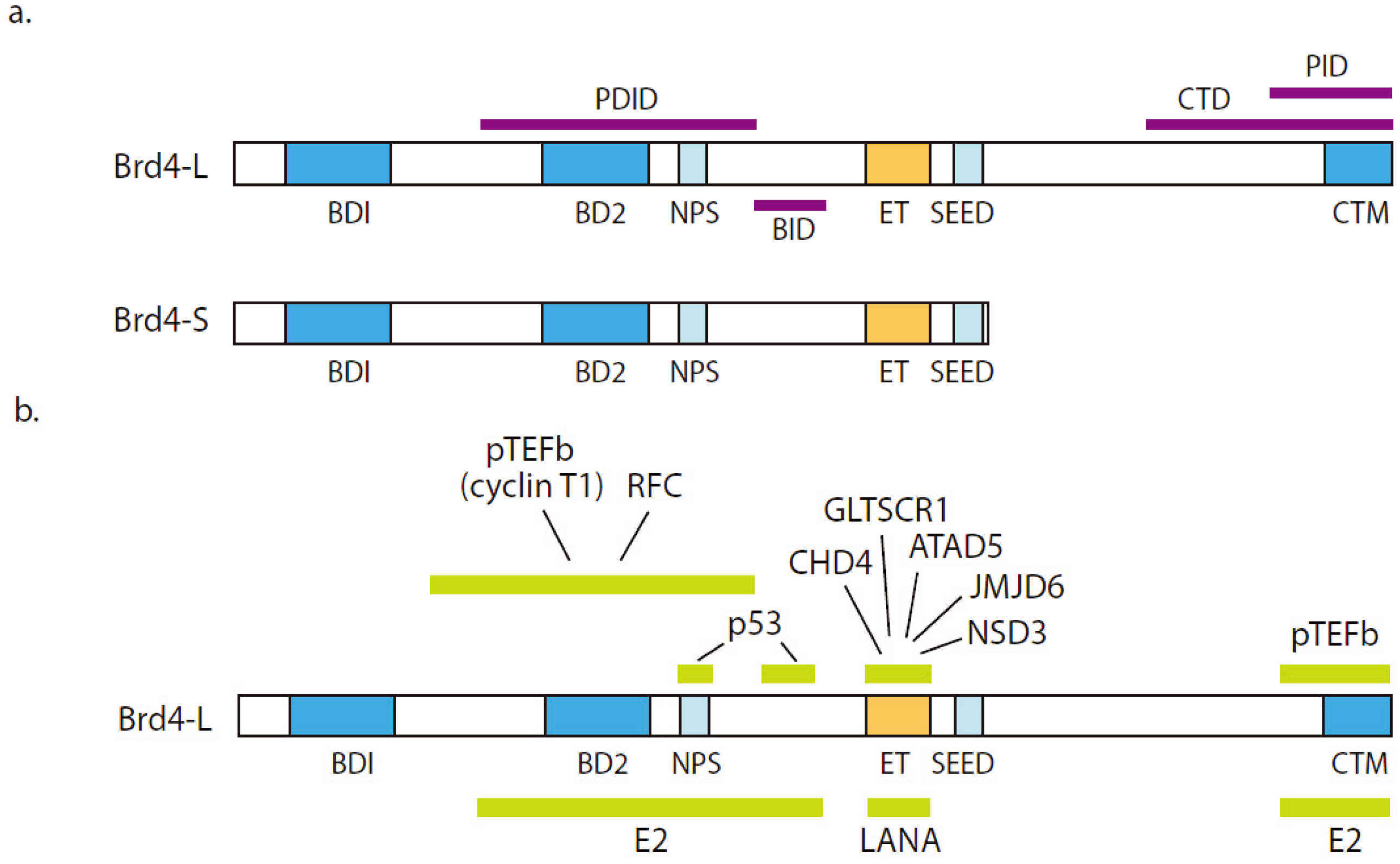
3.2. Association of Brd4 with Disease
4. Brd4 and Papillomaviruses
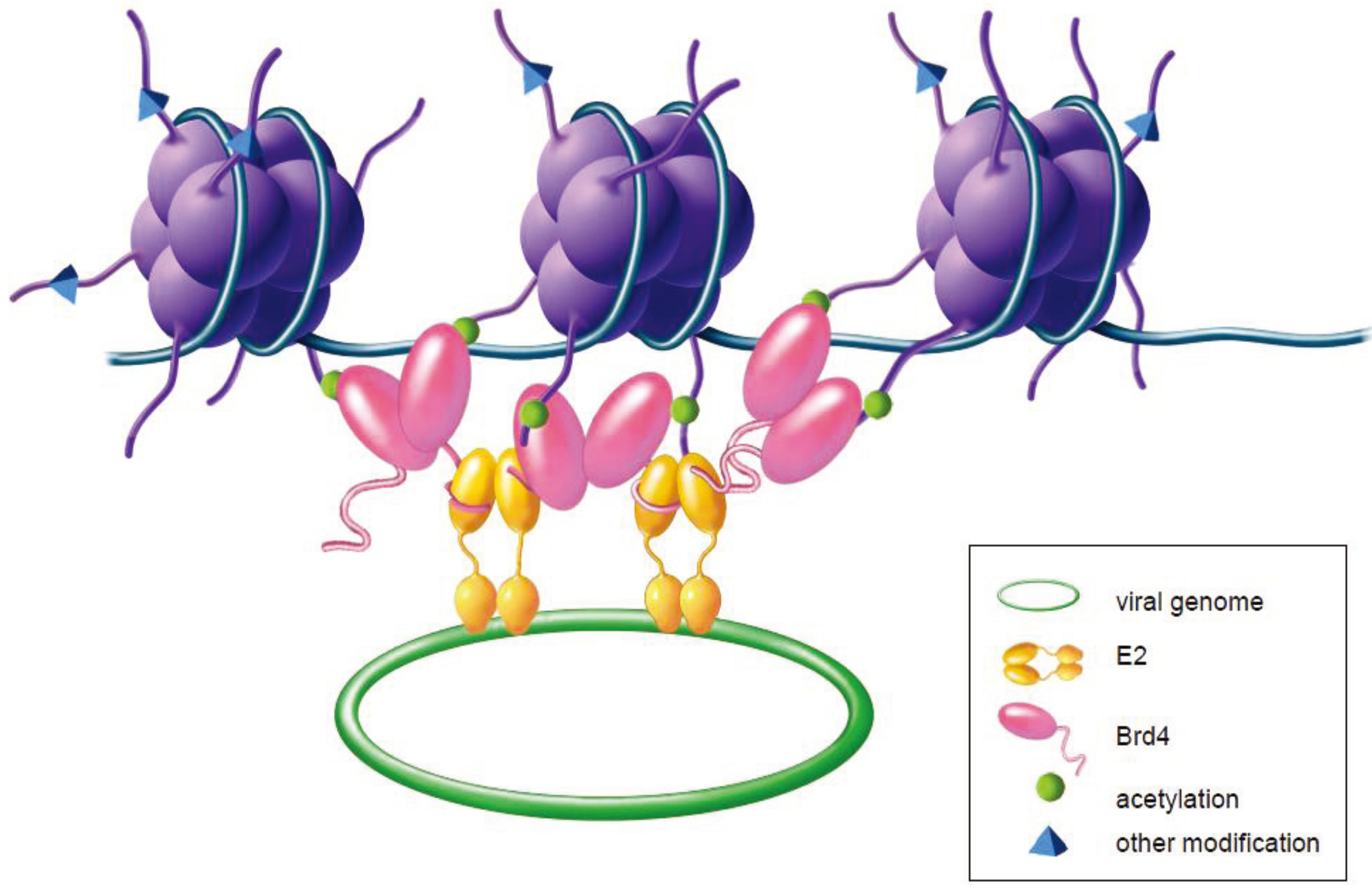
4.1. Interaction between Papillomavirus E2 and Brd4 Proteins
4.2. Brd4 Modulates the Stability of the E2 Proteins
4.3. The Role of Brd4 in Viral Transcription
4.4. The Role of Brd4 in Viral Genome Replication
4.5. The Role of Brd4 in Viral Genome Maintenance and Partitioning
4.6. Association of Brd4 with Other Viruses
5. Conclusions
Acknowledgments
Conflict of Interest
References and Notes
- Bouvard, V.; Baan, R.; Straif, K.; Grosse, Y.; Secretan, B.; El, G.F.; Benbrahim-Tallaa, L.; Guha, N.; Freeman, C.; Galichet, L.; et al. A review of human carcinogens—Part B: Biological agents. Lancet Oncol. 2009, 10, 321–322. [Google Scholar] [CrossRef]
- Walboomers, J.M.; Jacobs, M.V.; Manos, M.M.; Bosch, F.X.; Kummer, J.A.; Shah, K.V.; Snijders, P.J.; Peto, J.; Meijer, C.J.; Muñoz, N. Human papillomavirus is a necessary cause of invasive cervical cancer worldwide. J. Pathol. 1999, 189, 12–19. [Google Scholar] [CrossRef]
- Gillison, M.L.; Lowy, D.R. A causal role for human papillomavirus in head and neck cancer. Lancet 2004, 363, 1488–1489. [Google Scholar] [CrossRef]
- Johansson, C.; Schwartz, S. Regulation of human papillomavirus gene expression by splicing and polyadenylation. Nat. Rev. Microbiol. 2013, 11, 239–251. [Google Scholar] [CrossRef]
- Spalholz, B.A.; Yang, Y.C.; Howley, P.M. Transactivation of a bovine papilloma virus transcriptional regulatory element by the E2 gene product. Cell 1985, 42, 183–191. [Google Scholar] [CrossRef]
- McBride, A.A.; Byrne, J.C.; Howley, P.M. E2 polypeptides encoded by bovine papillomavirus type 1 form dimers through the common carboxyl-terminal domain: Transactivation is mediated by the conserved amino-terminal domain. Proc. Natl. Acad. Sci. USA 1989, 86, 510–514. [Google Scholar] [CrossRef]
- Androphy, E.J.; Lowy, D.R.; Schiller, J.T. Bovine papillomavirus E2 trans-activating gene product binds to specific sites in papillomavirus DNA. Nature 1987, 325, 70–73. [Google Scholar] [CrossRef]
- Cripe, T.P.; Haugen, T.H.; Turk, J.P.; Tabatabai, F.; Schmid, P.G., 3rd.; Dürst, M.; Gissmann, L.; Roman, A.; Turek, L.P. Transcriptional regulation of the human papillomavirus- 16 E6-E7 promoter by a keratinocyte-dependent enhancer, and by viral E2 trans-activator and repressor gene products: Implications for cervical carcinogenesis. EMBO J. 1987, 6, 3745–3753. [Google Scholar]
- Chin, M.T.; Hirochika, R.; Hirochika, H.; Broker, T.R.; Chow, L.T. Regulation of human papillomavirus type 11 enhancer and E6 promoter by activating and repressing proteins from the E2 open reading frame: Functional and biochemical studies. J. Virol. 1988, 62, 2994–3002. [Google Scholar]
- Steger, G.; Corbach, S. Dose-dependent regulation of the early promoter of human papillomavirus type 18 by the viral E2 protein. J. Virol. 1997, 71, 50–58. [Google Scholar]
- Thierry, F.; Yaniv, M. The BPV1-E2 trans-acting protein can be either an activator or a repressor of the HPV18 regulatory region. EMBO J. 1987, 6, 3391–3397. [Google Scholar]
- Bernard, B.A.; Bailly, C.; Lenoir, M.C.; Darmon, M.; Thierry, F.; Yaniv, M. The human papillomavirus type 18 (HPV18) E2 gene product is a repressor of the HPV18 regulatory region in human keratinocytes. J. Virol. 1989, 63, 4317–4324. [Google Scholar]
- Romanczuk, H.; Thierry, F.; Howley, P.M. Mutational analysis of cis elements involved in E2 modulation of human papillomavirus type 16 P 97 and type 18 P 105 promoters. J. Virol. 1990, 64, 2849–2859. [Google Scholar]
- Tan, S.H.; Gloss, B.; Bernard, H.U. During negative regulation of the human papillomavirus-16 E6 promoter, the viral E2 protein can displace Sp1 from a proximal promoter element. Nucleic Acids Res. 1992, 20, 251–256. [Google Scholar] [CrossRef]
- Dong, G.; Broker, T.R.; Chow, L.T. Human papillomavirus type 11 E2 proteins repress the homologous E6 promoter by interfering with the binding of host transcription factors to adjacent elements. J. Virol. 1994, 68, 1115–1127. [Google Scholar]
- Tan, S.H.; Leong, L.E.; Walker, P.A.; Bernard, H.U. The human papillomavirus type 16 E2 transcription factor binds with low cooperativity to two flanking sites and represses the E6 promoter through displacement of Sp1 and TFIID. J. Virol. 1994, 68, 6411–6420. [Google Scholar]
- Nishimura, A.; Ono, T.; Ishimoto, A.; Dowhanick, J.J.; Frizzell, M.A.; Howley, P.M.; Sakai, H. Mechanisms of human papillomavirus E2-mediated repression of viral oncogene expression and cervical cancer cell growth inhibition. J. Virol. 2000, 74, 3752–3760. [Google Scholar] [CrossRef]
- Smith, J.A.; White, E.A.; Sowa, M.E.; Powell, M.L.; Ottinger, M.; Harper, J.W.; Howley, P.M. Genome-wide siRNA screen identifies SMCX, EP400, and Brd4 as E2-dependent regulators of human papillomavirus oncogene expression. Proc. Natl. Acad. Sci. USA 2010, 107, 3752–3757. [Google Scholar]
- Schweiger, M.R.; Ottinger, M.; You, J.; Howley, P.M. Brd4 independent transcriptional repression function of the papillomavirus E2 proteins. J. Virol. 2007, 81, 9612–9622. [Google Scholar] [CrossRef]
- Wu, S.Y.; Lee, A.Y.; Hou, S.Y.; Kemper, J.K.; Erdjument-Bromage, H.; Tempst, P.; Chiang, C.M. Brd4 links chromatin targeting to HPV transcriptional silencing. Genes Dev. 2006, 20, 2383–2396. [Google Scholar] [CrossRef]
- Thierry, F.; Howley, P.M. Functional analysis of E2-mediated repression of the HPV18 P105 promoter. New Biol. 1991, 3, 90–100. [Google Scholar]
- Hwang, E.S.; Riese, D.J.D.; Settleman, J.; Nilson, L.A.; Honig, J.; Flynn, S.; DiMaio, D. Inhibition of cervical carcinoma cell line proliferation by the introduction of a bovine papillomavirus regulatory gene. J. Virol. 1993, 67, 3720–3729. [Google Scholar]
- Dowhanick, J.J.; McBride, A.A.; Howley, P.M. Suppression of cellular proliferation by the papillomavirus E2 protein. J. Virol. 1995, 69, 7791–7799. [Google Scholar]
- Desaintes, C.; Demeret, C.; Goyat, S.; Yaniv, M.; Thierry, F. Expression of the papillomavirus E2 protein in HeLa cells leads to apoptosis. EMBO J. 1997, 16, 504–514. [Google Scholar] [CrossRef]
- Stubenrauch, F.; Zobel, T.; Iftner, T. The E8 domain confers a novel long-distance transcriptional repression activity on the E8E2C protein of high-risk human papillomavirus type 31. J. Virol. 2001, 75, 4139–4149. [Google Scholar] [CrossRef]
- Chiang, C.M.; Broker, T.R.; Chow, L.T. An E1M^E2C fusion protein encoded by human papillomavirus type 11 is a sequence-specific transcription repressor. J. Virol. 1991, 65, 3317–3329. [Google Scholar]
- Powell, M.L.; Smith, J.A.; Sowa, M.E.; Harper, J.W.; Iftner, T.; Stubenrauch, F.; Howley, P.M. NCoR1 mediates papillomavirus E8;E2C transcriptional repression. J. Virol. 2010, 84, 4451–4460. [Google Scholar] [CrossRef]
- Pett, M.; Coleman, N. Integration of high-risk human papillomavirus: A key event in cervical carcinogenesis? J. Pathol. 2007, 212, 356–367. [Google Scholar] [CrossRef]
- Francis, D.A.; Schmid, S.I.; Howley, P.M. Repression of the integrated papillomavirus E6/E7 promoter is required for growth suppression of cervical cancer cells. J. Virol. 2000, 74, 2679–2686. [Google Scholar] [CrossRef]
- Jeon, S.; Allen-Hoffmann, B.L.; Lambert, P.F. Integration of human papillomavirus type 16 into the human genome correlates with a selective growth advantage of cells. J. Virol. 1995, 69, 2989–2997. [Google Scholar]
- Mohr, I.J.; Clark, R.; Sun, S.; Androphy, E.J.; MacPherson, P.; Botchan, M.R. Targeting the E1 replication protein to the papillomavirus origin of replication by complex formation with the E2 transactivator. Science 1990, 250, 1694–1699. [Google Scholar]
- Ustav, M.; Stenlund, A. Transient replication of BPV-1 requires two viral polypeptides encoded by the E1 and E2 open reading frames. EMBO J. 1991, 10, 449–457. [Google Scholar]
- Ustav, M.; Ustav, E.; Szymanski, P.; Stenlund, A. Identification of the origin of replication of bovine papillomavirus and characterization of the viral origin recognition factor E1. EMBO J. 1991, 10, 4321–4329. [Google Scholar]
- McBride, A.A. Replication and partitioning of papillomavirus genomes. Adv. Virus Res. 2008, 72, 155–205. [Google Scholar] [CrossRef]
- Sanders, C.M.; Stenlund, A. Recruitment and loading of the E1 initiator protein: An ATP-dependent process catalysed by a transcription factor. EMBO J. 1998, 17, 7044–7055. [Google Scholar] [CrossRef]
- Piirsoo, M.; Ustav, E.; Mandel, T.; Stenlund, A.; Ustav, M. Cis and trans requirements for stable episomal maintenance of the BPV-1 replicator. EMBO J. 1996, 15, 1–11. [Google Scholar]
- Skiadopoulos, M.H.; McBride, A.A. Bovine papillomavirus type 1 genomes and the E2 transactivator protein are closely associated with mitotic chromatin. J. Virol. 1998, 72, 2079–2088. [Google Scholar]
- Ilves, I.; Kivi, S.; Ustav, M. Long-term episomal maintenance of bovine papillomavirus type 1 plasmids is determined by attachment to host chromosomes, which is mediated by the viral E2 protein and its binding sites. J. Virol. 1999, 73, 4404–4412. [Google Scholar]
- Bastien, N.; McBride, A.A. Interaction of the papillomavirus E2 protein with mitotic chromosomes. Virology 2000, 270, 124–134. [Google Scholar] [CrossRef]
- Kim, K.; Lambert, P.F. E1 protein of bovine papillomavirus 1 is not required for the maintenance of viral plasmid DNA replication. Virology 2002, 293, 10–14. [Google Scholar] [CrossRef]
- Egawa, N.; Nakahara, T.; Ohno, S.; Narisawa-Saito, M.; Yugawa, T.; Fujita, M.; Yamato, K.; Natori, Y.; Kiyono, T. The E1 protein of human papillomavirus type 16 is dispensable for maintenance replication of the viral genome. J. Virol. 2012, 86, 3276–3283. [Google Scholar] [CrossRef]
- Xue, Y.; Bellanger, S.; Zhang, W.; Lim, D.; Low, J.; Lunny, D.; Thierry, F. HPV16 E2 is an immediate early marker of viral infection, preceding E7 expression in precursor structures of cervical carcinoma. Canc. Res. 2010, 70, 5316–5325. [Google Scholar] [CrossRef]
- Penrose, K.J.; McBride, A.A. Proteasome-mediated degradation of the papillomavirus E2-TA protein is regulated by phosphorylation and can modulate viral genome copy number. J. Virol. 2000, 74, 6031–6038. [Google Scholar] [CrossRef]
- Flores, E.R.; Lambert, P.F. Evidence for a switch in the mode of human papillomavirus type 16 DNA replication during the viral life cycle. J. Virol. 1997, 71, 7167–7179. [Google Scholar]
- Sakakibara, N.; Chen, D.; McBride, A.A. Papillomaviruses use Recombination Dependent Replication to Vegetatively Amplify their Genomes in Differentiated Cells. PLoS Pathog. 2013, in press. [Google Scholar]
- Gillespie, K.A.; Mehta, K.P.; Laimins, L.A.; Moody, C.A. Human papillomaviruses recruit cellular DNA repair and homologous recombination factors to viral replication centers. J. Virol 2012, 86, 9520–9526. [Google Scholar] [CrossRef]
- Moody, C.A.; Laimins, L.A. Human papillomaviruses activate the ATM DNA damage pathway for viral genome amplification upon differentiation. PLoS Pathog. 2009, 5, e1000605. [Google Scholar] [CrossRef]
- Fradet-Turcotte, A.; Bergeron-Labrecque, F.; Moody, C.A.; Lehoux, M.; Laimins, L.A.; Archambault, J. Nuclear accumulation of the papillomavirus E1 helicase blocks S-phase progression and triggers an ATM-dependent DNA damage response. J. Virol. 2011, 85, 8996–9012. [Google Scholar] [CrossRef]
- Sakakibara, N.; Mitra, R.; McBride, A.A. The papillomavirus E1 helicase activates a cellular DNA damage response in viral replication foci. J. Virol. 2011, 85, 8981–8995. [Google Scholar] [CrossRef]
- Reinson, T.; Toots, M.; Kadaja, M.; Pipitch, R.; Allik, M.; Ustav, E.; Ustav, M. Engagement of the ATR-dependent DNA damage response at the human papillomavirus 18 replication centers during the initial amplification. J. Virol. 2013, 87, 951–964. [Google Scholar] [CrossRef]
- Swindle, C.S.; Zou, N.; Van Tine, B.A.; Shaw, G.M.; Engler, J.A.; Chow, L.T. Human papillomavirus DNA replication compartments in a transient DNA replication system. J. Virol. 1999, 73, 1001–1009. [Google Scholar]
- Van Doorslaer, K.; Tan, Q.; Xirasagar, S.; Bandaru, S.; Gopalan, V.; Mohamoud, Y.; Huyen, Y.; McBride, A.A. The Papillomavirus Episteme: A central resource for papillomavirus sequence data and analysis. Nucleic Acids Res. 2013, 41, D571–D578. [Google Scholar] [CrossRef]
- Li, R.; Knight, J.; Bream, G.; Stenlund, A.; Botchan, M. Specific recognition nucleotides and their DNA context determine the affinity of E2 protein for 17 binding sites in the BPV-1 genome. Genes Dev. 1989, 3, 510–526. [Google Scholar] [CrossRef]
- Stubenrauch, F.; Lim, H.B.; Laimins, L.A. Differential requirements for conserved E2 binding sites in the life cycle of oncogenic human papillomavirus type 31. J. Virol. 1998, 72, 1071–1077. [Google Scholar]
- Van Doorslaer, K.; Khan, J.; Chapman, S.; McBride, A.A. Three E2 binding sites are sufficient for stable episomal maintenance of HPV18. National Institutes of Health: Bethesda, MD, USA, 2013; To be submitted for publication. [Google Scholar]
- Dey, A.; Ellenberg, J.; Farina, A.; Coleman, A.E.; Maruyama, T.; Sciortino, S.; Lippincott-Schwartz, J.; Ozato, K. A bromodomain protein, MCAP, associates with mitotic chromosomes and affects G(2)-to-M transition. Mol. Cell Biol. 2000, 20, 6537–6549. [Google Scholar] [CrossRef]
- Florence, B.; Faller, D.V. You bet-cha: A novel family of transcriptional regulators. Front. Biosci. 2001, 6, D1008–D1018. [Google Scholar] [CrossRef]
- Houzelstein, D.; Bullock, S.L.; Lynch, D.E.; Grigorieva, E.F.; Wilson, V.A.; Beddington, R.S. Growth and early postimplantation defects in mice deficient for the bromodomain-containing protein Brd4. Mol. Cell Biol. 2002, 22, 3794–3802. [Google Scholar] [CrossRef]
- Dey, A.; Chitsaz, F.; Abbasi, A.; Misteli, T.; Ozato, K. The double bromodomain protein Brd4 binds to acetylated chromatin during interphase and mitosis. Proc. Natl. Acad. Sci. USA 2003, 100, 8758–8763. [Google Scholar] [CrossRef]
- Dey, A.; Nishiyama, A.; Karpova, T.; McNally, J.; Ozato, K. Brd4 marks select genes on mitotic chromatin and directs postmitotic transcription. Mol. Biol. Cell 2009, 20, 4899–4909. [Google Scholar] [CrossRef]
- Mochizuki, K.; Nishiyama, A.; Jang, M.K.; Dey, A.; Ghosh, A.; Tamura, T.; Natsume, H.; Yao, H.J.; Ozato, K. The bromodomain protein Brd4 stimulates G(1) gene transcription and promotes progression to S phase. J. Biol. Chem. 2008, 283, 9040–9048. [Google Scholar] [CrossRef]
- Zhao, R.; Nakamura, T.; Fu, Y.; Lazar, Z.; Spector, D.L. Gene bookmarking accelerates the kinetics of post-mitotic transcriptional re-activation. Nat. Cell Biol. 2011, 13, 1295–1304. [Google Scholar] [CrossRef]
- Jang, M.K.; Mochizuki, K.; Zhou, M.; Jeong, H.S.; Brady, J.N.; Ozato, K. The bromodomain protein Brd4 is a positive regulatory component of P-TEFb and stimulates RNA polymerase II-dependent transcription. Mol. Cell 2005, 19, 523–534. [Google Scholar] [CrossRef]
- Yang, Z.; Yik, J.H.; Chen, R.; He, N.; Jang, M.K.; Ozato, K.; Zhou, Q. Recruitment of P-TEFb for stimulation of transcriptional elongation by the bromodomain protein Brd4. Mol. Cell 2005, 19, 535–545. [Google Scholar] [CrossRef]
- Devaiah, B.N.; Singer, D.S. Two faces of brd4: Mitotic bookmark and transcriptional lynchpin. Transcription 2013, 4, 13–17. [Google Scholar] [CrossRef]
- Filippakopoulos, P.; Picaud, S.; Mangos, M.; Keates, T.; Lambert, J.P.; Barsyte-Lovejoy, D.; Felletar, I.; Volkmer, R.; Muller, S.; Pawson, T.; et al. Histone recognition and large-scale structural analysis of the human bromodomain family. Cell 2012, 149, 214–231. [Google Scholar] [CrossRef]
- Vollmuth, F.; Blankenfeldt, W.; Geyer, M. Structures of the dual bromodomains of the P-TEFb-activating protein Brd4 at atomic resolution. J. Biol. Chem. 2009, 284, 36547–36556. [Google Scholar] [CrossRef]
- Schröder, S.; Cho, S.; Zeng, L.; Zhang, Q.; Kaehlcke, K.; Mak, L.; Lau, J.; Bisgrove, D.; Schnölzer, M.; Verdin, E. Two-pronged binding with bromodomain-containing protein 4 liberates positive transcription elongation factor b from inactive ribonucleoprotein complexes. J. Biol. Chem. 2012, 287, 1090–1099. [Google Scholar] [CrossRef]
- Huang, B.; Yang, X.D.; Zhou, M.M.; Ozato, K.; Chen, L.F. Brd4 coactivates transcriptional activation of NF-kappaB via specific binding to acetylated RelA. Mol. Cell Biol. 2009, 29, 1375–1387. [Google Scholar] [CrossRef]
- Lin, Y.J.; Umehara, T.; Inoue, M.; Saito, K.; Kigawa, T.; Jang, M.K.; Ozato, K.; Yokoyama, S.; Padmanabhan, B.; Guntert, P. Solution structure of the extraterminal domain of the bromodomain-containing protein BRD4. Protein Sci. 2008, 17, 2174–2179. [Google Scholar] [CrossRef]
- Rahman, S.; Sowa, M.E.; Ottinger, M.; Smith, J.A.; Shi, Y.; Harper, J.W.; Howley, P.M. The Brd4 extraterminal domain confers transcription activation independent of pTEFb by recruiting multiple proteins, including NSD3. Mol. Cell Biol. 2011, 31, 2641–2652. [Google Scholar] [CrossRef]
- You, J.; Croyle, J.L.; Nishimura, A.; Ozato, K.; Howley, P.M. Interaction of the bovine papillomavirus E2 protein with Brd4 tethers the viral DNA to host mitotic chromosomes. Cell 2004, 117, 349–360. [Google Scholar] [CrossRef]
- Bisgrove, D.A.; Mahmoudi, T.; Henklein, P.; Verdin, E. Conserved P-TEFb-interacting domain of BRD4 inhibits HIV transcription. Proc. Natl. Acad. Sci. USA 2007, 104, 13690–13695. [Google Scholar]
- Wu, S.Y.; Lee, A.Y.; Lai, H.T.; Zhang, H.; Chiang, C.M. Phospho switch triggers brd4 chromatin binding and activator recruitment for gene-specific targeting. Mol. Cell 2013, 49, 843–857. [Google Scholar] [CrossRef]
- French, C.A.; Miyoshi, I.; Aster, J.C.; Kubonishi, I.; Kroll, T.G.; Dal Cin, P.; Vargas, S.O.; Perez-Atayde, A.R.; Fletcher, J.A. BRD4 bromodomain gene rearrangement in aggressive carcinoma with translocation t(15;19). Am. J. Pathol. 2001, 159, 1987–1992. [Google Scholar] [CrossRef]
- Reynoird, N.; Schwartz, B.E.; Delvecchio, M.; Sadoul, K.; Meyers, D.; Mukherjee, C.; Caron, C.; Kimura, H.; Rousseaux, S.; Cole, P.A.; et al. Oncogenesis by sequestration of CBP/p300 in transcriptionally inactive hyperacetylated chromatin domains. EMBO J. 2010, 29, 2943–2952. [Google Scholar] [CrossRef]
- Filippakopoulos, P.; Qi, J.; Picaud, S.; Shen, Y.; Smith, W.B.; Fedorov, O.; Morse, E.M.; Keates, T.; Hickman, T.T.; Felletar, I.; et al. Selective inhibition of BET bromodomains. Nature 2010, 468, 1067–1073. [Google Scholar] [CrossRef]
- Schwartz, B.E.; Hofer, M.D.; Lemieux, M.E.; Bauer, D.E.; Cameron, M.J.; West, N.H.; Agoston, E.S.; Reynoird, N.; Khochbin, S.; Ince, T.A.; et al. Differentiation of NUT midline carcinoma by epigenomic reprogramming. Canc. Res. 2011, 71, 2686–2696. [Google Scholar] [CrossRef]
- Yan, J.; Diaz, J.; Jiao, J.; Wang, R.; You, J. Perturbation of BRD4 protein function by BRD4-NUT protein abrogates cellular differentiation in NUT midline carcinoma. J. Biol. Chem. 2011, 286, 27663–27675. [Google Scholar] [CrossRef]
- Crawford, N.P.S.; Alsarraj, J.; Lukes, L.; Walker, R.C.; Officewala, J.S.; Yang, H.H.; Lee, M.P.; Ozato, K.; Hunter, K.W. Bromodomain 4 activation predicts breast cancer survival. Proc. Natl. Acad. Sci. USA 2008, 105, 6380–6385. [Google Scholar] [CrossRef]
- Lockwood, W.W.; Zejnullahu, K.; Bradner, J.E.; Varmus, H. Sensitivity of human lung adenocarcinoma cell lines to targeted inhibition of BET epigenetic signaling proteins. Proc. Natl. Acad. Sci. USA 2012, 109, 19408–19413. [Google Scholar]
- Ott, C.J.; Kopp, N.; Bird, L.; Paranal, R.M.; Qi, J.; Bowman, T.; Rodig, S.J.; Kung, A.L.; Bradner, J.E.; Weinstock, D.M. BET bromodomain inhibition targets both c-Myc and IL7R in high-risk acute lymphoblastic leukemia. Blood 2012, 120, 2843–2852. [Google Scholar] [CrossRef]
- Dawson, M.A.; Prinjha, R.K.; Dittmann, A.; Giotopoulos, G.; Bantscheff, M.; Chan, W.I.; Robson, S.C.; Chung, C.W.; Hopf, C.; Savitski, M.M.; et al. Inhibition of BET recruitment to chromatin as an effective treatment for MLL-fusion leukaemia. Nature 2011, 478, 529–533. [Google Scholar] [CrossRef]
- Delmore, J.E.; Issa, G.C.; Lemieux, M.E.; Rahl, P.B.; Shi, J.; Jacobs, H.M.; Kastritis, E.; Gilpatrick, T.; Paranal, R.M.; Qi, J.; et al. BET bromodomain inhibition as a therapeutic strategy to target c-Myc. Cell 2011, 146, 904–917. [Google Scholar] [CrossRef]
- Mertz, J.A.; Conery, A.R.; Bryant, B.M.; Sandy, P.; Balasubramanian, S.; Mele, D.A.; Bergeron, L.; Sims, R.J., 3rd. Targeting MYC dependence in cancer by inhibiting BET bromodomains. Proc. Natl. Acad. Sci. USA 2011, 108, 16669–16674. [Google Scholar] [CrossRef]
- Olejnik-Schmidt, A.K.; Schmidt, M.T.; Kedzia, W.; Gozdzicka-Jozefiak, A. Search for cellular partners of human papillomavirus type 16 E2 protein. Arch. Virol. 2008, 153, 983–990. [Google Scholar] [CrossRef]
- Baxter, M.K.; McPhillips, M.G.; Ozato, K.; McBride, A.A. The mitotic chromosome binding activity of the papillomavirus E2 protein correlates with interaction with the cellular chromosomal protein, Brd4. J. Virol. 2005, 79, 4806–4818. [Google Scholar] [CrossRef]
- Platt, G.M.; Simpson, G.R.; Mittnacht, S.; Schulz, T.F. Latent nuclear antigen of Kaposi's sarcoma-associated herpesvirus interacts with RING3, a homolog of the Drosophila female sterile homeotic (fsh) gene. J. Virol. 1999, 73, 9789–9795. [Google Scholar]
- McPhillips, M.G.; Ozato, K.; McBride, A.A. Interaction of bovine papillomavirus E2 protein with Brd4 stabilizes its association with chromatin. J. Virol. 2005, 79, 8920–8932. [Google Scholar] [CrossRef]
- Lehman, C.W.; Botchan, M.R. Segregation of viral plasmids depends on tethering to chromosomes and is regulated by phosphorylation. Proc. Natl. Acad. Sci. USA 1998, 95, 4338–4343. [Google Scholar] [CrossRef]
- Abbate, E.A.; Voitenleitner, C.; Botchan, M.R. Structure of the papillomavirus DNA-tethering complex E2:Brd4 and a peptide that ablates HPV chromosomal association. Mol. Cell 2006, 24, 877–889. [Google Scholar] [CrossRef]
- McPhillips, M.G.; Oliveira, J.G.; Spindler, J.E.; Mitra, R.; McBride, A.A. Brd4 is required for e2-mediated transcriptional activation but not genome partitioning of all papillomaviruses. J. Virol. 2006, 80, 9530–9543. [Google Scholar] [CrossRef]
- Ottinger, M.; Christalla, T.; Nathan, K.; Brinkmann, M.M.; Viejo-Borbolla, A.; Schulz, T.F. The Kaposi’s Sarcoma-Associated Herpesvirus LANA-1 interacts with the short variant of BRD4 and releases cells from a BRD4- and BRD2/RING3-induced G1 cell cycle arrest. J. Virol. 2006, 80, 10772–10786. [Google Scholar] [CrossRef]
- You, J.; Srinivasan, V.; Denis, G.V.; Harrington, W.J., Jr.; Ballestas, M.E.; Kaye, K.M.; Howley, P.M. Kaposi’s sarcoma-associated herpesvirus latency-associated nuclear antigen interacts with bromodomain protein Brd4 on host mitotic chromosomes. J. Virol. 2006, 80, 8909–8919. [Google Scholar] [CrossRef]
- Lee, A.Y.; Chiang, C.M. Chromatin adaptor Brd4 modulates E2 transcription activity and protein stability. J. Biol. Chem. 2009, 284, 2778–2786. [Google Scholar]
- Gagnon, D.; Joubert, S.; Senechal, H.; Fradet-Turcotte, A.; Torre, S.; Archambault, J. Proteasomal degradation of the papillomavirus E2 protein is inhibited by overexpression of bromodomain-containing protein 4. J. Virol. 2009, 83, 4127–4139. [Google Scholar] [CrossRef]
- Zheng, G.; Schweiger, M.R.; Martinez-Noel, G.; Zheng, L.; Smith, J.A.; Harper, J.W.; Howley, P.M. Brd4 regulation of papillomavirus protein E2 stability. J. Virol. 2009, 83, 8683–8692. [Google Scholar] [CrossRef]
- Winokur, P.L.; McBride, A.A. Separation of the transcriptional activation and replication functions of the bovine papillomavirus-1 E2 protein. EMBO J. 1992, 11, 4111–4118. [Google Scholar]
- Sakai, H.; Yasugi, T.; Benson, J.D.; Dowhanick, J.J.; Howley, P.M. Targeted mutagenesis of the human papillomavirus type 16 E2 transactivation domain reveals separable transcriptional activation and DNA replication functions. J. Virol. 1996, 70, 1602–1611. [Google Scholar]
- Abroi, A.; Kurg, R.; Ustav, M. Transcriptional and replicational activation functions in the BPV1 E2 protein are encoded by different structural determinants. J. Virol. 1996, 70, 6169–6179. [Google Scholar]
- Breiding, D.E.; Grossel, M.J.; Androphy, E.J. Genetic analysis of the bovine papillomavirus E2 transcriptional activation domain. Virology 1996, 221, 34–43. [Google Scholar] [CrossRef]
- Ferguson, M.F.; Botchan, M.R. Genetic analysis of the activation domain of bovine papillomavirus protein E2:its role in transcription and replication. J. Virol. 1996, 70, 4193–4199. [Google Scholar]
- Cooper, C.S.; Upmeyer, S.N.; Winokur, P.L. Identification of single amino acids in the human papillomavirus 11 E2 protein critical for the transactivation or replication functions. Virology 1998, 241, 312–322. [Google Scholar] [CrossRef]
- Antson, A.A.; Burns, J.E.; Moroz, O.V.; Scott, D.J.; Sanders, C.M.; Bronstein, I.B.; Dodson, G.G.; Wilson, K.S.; Maitland, N.J. Structure of the intact transactivation domain of the human papillomavirus E2 protein. Nature 2000, 403, 805–809. [Google Scholar] [CrossRef]
- Senechal, H.; Poirier, G.G.; Coulombe, B.; Laimins, L.A.; Archambault, J. Amino acid substitutions that specifically impair the transcriptional activity of papillomavirus E2 affect binding to the long isoform of Brd4. Virology 2007, 358, 10–17. [Google Scholar] [CrossRef]
- Schweiger, M.R.; You, J.; Howley, P.M. Bromodomain protein 4 mediates the papillomavirus e2 transcriptional activation function. J. Virol. 2006, 80, 4276–4285. [Google Scholar] [CrossRef]
- Jha, S.; Vande, P.S.; Banerjee, N.S.; Dutta, A.B.; Chow, L.T.; Dutta, A. Destabilization of TIP60 by human papillomavirus E6 results in attenuation of TIP60-dependent transcriptional regulation and apoptotic pathway. Mol. Cell 2010, 38, 700–711. [Google Scholar] [CrossRef]
- Kimura, A.; Horikoshi, M. Tip60 acetylates six lysines of a specific class in core histones in vitro. Gene. Cell. 1998, 3, 789–800. [Google Scholar] [CrossRef]
- Yan, J.; Li, Q.; Lievens, S.; Tavernier, J.; You, J. Abrogation of the Brd4-positive transcription elongation factor B complex by papillomavirus E2 protein contributes to viral oncogene repression. J. Virol. 2010, 84, 76–87. [Google Scholar] [CrossRef]
- Bechtold, V.; Beard, P.; Raj, K. Human papillomavirus type 16 E2 protein has no effect on transcription from episomal viral DNA. J. Virol. 2003, 77, 2021–2028. [Google Scholar] [CrossRef]
- Baxter, M.K.; McBride, A.A. An acidic amphipathic helix in the Bovine Papillomavirus E2 protein is critical for DNA replication and interaction with the E1 protein. Virology 2005, 332, 78–88. [Google Scholar] [CrossRef]
- Brokaw, J.L.; Blanco, M.; McBride, A.A. Amino acids critical for the functions of the bovine papillomavirus type 1 E2 transactivator. J. Virol. 1996, 70, 23–29. [Google Scholar]
- Ilves, I.; Maemets, K.; Silla, T.; Janikson, K.; Ustav, M. Brd4 is involved in multiple processes of the bovine papillomavirus type 1 life cycle. J. Virol. 2006, 80, 3660–3665. [Google Scholar] [CrossRef]
- Wang, X.; Helfer, C.M.; Pancholi, N.; Bradner, J.E.; You, J. Recruitment of brd4 to the human papillomavirus type 16 DNA replication complex is essential for replication of viral DNA. J. Virol. 2013, 87, 3871–3884. [Google Scholar] [CrossRef]
- Sakakibara, N.; Chen, D.; Jang, M.K.; McBride, A.A. The Brd4 Chromatin Adaptor Protein is displaced from nuclear foci as the HPV E2 protein switches from Transcriptional to Replicational Modes. National Institutes of Health: Bethesda, MD, USA, 2013; Submitted for publication. [Google Scholar]
- Stubenrauch, F.; Colbert, A.M.; Laimins, L.A. Transactivation by the E2 protein of oncogenic human papillomavirus type 31 is not essential for early and late viral functions. J. Virol. 1998, 72, 8115–8123. [Google Scholar]
- Maruyama, T.; Farina, A.; Dey, A.; Cheong, J.; Bermudez, V.P.; Tamura, T.; Sciortino, S.; Shuman, J.; Hurwitz, J.; Ozato, K. A Mammalian bromodomain protein, Brd4, interacts with replication factor C and inhibits progression to S phase. Mol. Cell Biol. 2002, 22, 6509–6520. [Google Scholar] [CrossRef]
- Jang, M.K.; McBride, A.A. National Institutes of Health: Bethesda, MD, USA, 2013; Unpublished work.
- Abroi, A.; Ilves, I.; Kivi, S.; Ustav, M. Analysis of chromatin attachment and partitioning functions of bovine papillomavirus type 1 E2 protein. J. Virol. 2004, 78, 2100–2113. [Google Scholar] [CrossRef]
- Cardenas-Mora, J.; Spindler, J.E.; Jang, M.K.; McBride, A.A. Dimerization of the papillomavirus E2 protein is required for efficient mitotic chromosome association and Brd4 binding. J. Virol. 2008, 82, 7298–7305. [Google Scholar] [CrossRef]
- You, J.; Schweiger, M.R.; Howley, P.M. Inhibition of E2 binding to Brd4 enhances viral genome loss and phenotypic reversion of bovine papillomavirus-transformed cells. J. Virol. 2005, 79, 14956–14961. [Google Scholar] [CrossRef]
- Brannon, A.R.; Maresca, J.A.; Boeke, J.D.; Basrai, M.A.; McBride, A.A. Reconstitution of papillomavirus E2-mediated plasmid maintenance in Saccharomyces cerevisiae by the Brd4 bromodomain protein. Proc. Natl. Acad. Sci. USA 2005, 102, 2998–3003. [Google Scholar] [CrossRef]
- Oliveira, J.G.; Colf, L.A.; McBride, A.A. Variations in the association of papillomavirus E2 proteins with mitotic chromosomes. Proc. Natl. Acad. Sci. USA 2006, 103, 1047–1052. [Google Scholar] [CrossRef]
- Poddar, A.; Reed, S.C.; McPhillips, M.G.; Spindler, J.E.; McBride, A.A. The human papillomavirus type 8 E2 tethering protein targets the ribosomal DNA loci of host mitotic chromosomes. J. Virol. 2009, 83, 640–650. [Google Scholar] [CrossRef]
- Sekhar, V.; Reed, S.C.; McBride, A.A. Interaction of the betapapillomavirus E2 tethering protein with mitotic chromosomes. J. Virol. 2010, 84, 543–557. [Google Scholar] [CrossRef]
- Sekhar, V.; McBride, A.A. Phosphorylation regulates binding of the human papillomavirus type 8 E2 protein to host chromosomes. J. Virol. 2012, 86, 10047–10058. [Google Scholar] [CrossRef]
- Woodard, C.; Shamay, M.; Liao, G.; Zhu, J.; Ng, A.N.; Li, R.; Newman, R.; Rho, H.S.; Hu, J.; Wan, J.; et al. Phosphorylation of the chromatin binding domain of KSHV LANA. PLoS Pathog. 2012, 8, e1002972. [Google Scholar] [CrossRef]
- McBride, A.A.; van Doorslaer, K. National Institutes of Health: Bethesda, MD, USA, 2013; Unpublished work.
- Donaldson, M.M.; Boner, W.; Morgan, I.M. TopBP1 regulates human papillomavirus type 16 E2 interaction with chromatin. J. Virol. 2007, 81, 4338–4342. [Google Scholar] [CrossRef]
- Dao, L.D.; Duffy, A.; van Tine, B.A.; Wu, S.Y.; Chiang, C.M.; Broker, T.R.; Chow, L.T. Dynamic localization of the human papillomavirus type 11 origin binding protein E2 through mitosis while in association with the spindle apparatus. J. Virol. 2006, 80, 4792–4800. [Google Scholar] [CrossRef]
- Yu, T.; Peng, Y.C.; Androphy, E.J. Mitotic kinesin-like protein 2 binds and colocalizes with papillomavirus E2 during mitosis. J. Virol. 2007, 81, 1736–1745. [Google Scholar] [CrossRef]
- Parish, J.L.; Bean, A.M.; Park, R.B.; Androphy, E.J. ChlR1 is required for loading papillomavirus E2 onto mitotic chromosomes and viral genome maintenance. Mol. Cell 2006, 24, 867–876. [Google Scholar] [CrossRef]
- Jang, M.K.; Sakakibara, N.; McBride, A.A. Papillomavirus Genomes associate with the Cellular Protein Brd4 to replicate at Fragile Sites in the Host Genome. National Institutes of Health: Bethesda, MD, USA, 2013; Submitted for publication. [Google Scholar]
- Silla, T.; Mannik, A.; Ustav, M. Effective formation of the segregation-competent complex determines successful partitioning of the bovine papillomavirus genome during cell division. J. Virol. 2010, 84, 11175–11188. [Google Scholar] [CrossRef]
- Jang, M.K.; Kwon, D.; McBride, A.A. Papillomavirus E2 proteins and the host BRD4 protein associate with transcriptionally active cellular chromatin. J. Virol. 2009, 83, 2592–2600. [Google Scholar] [CrossRef]
- Weidner-Glunde, M.; Ottinger, M.; Schulz, T.F. WHAT do viruses BET on? Front. Biosci. 2010, 15, 537–549. [Google Scholar] [CrossRef]
- Lin, A.; Wang, S.; Nguyen, T.; Shire, K.; Frappier, L. The EBNA1 protein of Epstein-Barr virus functionally interacts with Brd4. J. Virol. 2008, 82, 12009–12019. [Google Scholar] [CrossRef]
- Viejo-Borbolla, A.; Kati, E.; Sheldon, J.A.; Nathan, K.; Mattsson, K.; Szekely, L.; Schulz, T.F. A Domain in the C-terminal region of latency-associated nuclear antigen 1 of Kaposi’s sarcoma-associated Herpesvirus affects transcriptional activation and binding to nuclear heterochromatin. J. Virol. 2003, 77, 7093–7100. [Google Scholar]
- Wang, X.; Li, J.; Schowalter, R.M.; Jiao, J.; Buck, C.B.; You, J. Bromodomain protein Brd4 plays a key role in Merkel cell polyomavirus DNA replication. PLoS Pathog. 2012, 8, e1003021. [Google Scholar] [CrossRef]
- Li, Z.; Guo, J.; Wu, Y.; Zhou, Q. The BET bromodomain inhibitor JQ1 activates HIV latency through antagonizing Brd4 inhibition of Tat-transactivation. Nucleic Acids Res. 2013, 41, 277–287. [Google Scholar]
- Boehm, D.; Calvanese, V.; Dar, R.D.; Xing, S.; Schroeder, S.; Martins, L.; Aull, K.; Li, P.C.; Planelles, V.; Bradner, J.E.; et al. BET bromodomain-targeting compounds reactivate HIV from latency via a Tat-independent mechanism. Cell Cycle 2013, 12, 452–462. [Google Scholar] [CrossRef]
- Zhu, J.; Gaiha, G.D.; John, S.P.; Pertel, T.; Chin, C.R.; Gao, G.; Qu, H.; Walker, B.D.; Elledge, S.J.; Brass, A.L. Reactivation of latent HIV-1 by inhibition of BRD4. Cell Rep. 2012, 2, 807–816. [Google Scholar] [CrossRef]
- Banerjee, C.; Archin, N.; Michaels, D.; Belkina, A.C.; Denis, G.V.; Bradner, J.; Sebastiani, P.; Margolis, D.M.; Montano, M. BET bromodomain inhibition as a novel strategy for reactivation of HIV-1. J. Leukoc. Biol. 2012, 92, 1147–1154. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
McBride, A.A.; Jang, M.K. Current Understanding of the Role of the Brd4 Protein in the Papillomavirus Lifecycle. Viruses 2013, 5, 1374-1394. https://doi.org/10.3390/v5061374
McBride AA, Jang MK. Current Understanding of the Role of the Brd4 Protein in the Papillomavirus Lifecycle. Viruses. 2013; 5(6):1374-1394. https://doi.org/10.3390/v5061374
Chicago/Turabian StyleMcBride, Alison A., and Moon Kyoo Jang. 2013. "Current Understanding of the Role of the Brd4 Protein in the Papillomavirus Lifecycle" Viruses 5, no. 6: 1374-1394. https://doi.org/10.3390/v5061374
APA StyleMcBride, A. A., & Jang, M. K. (2013). Current Understanding of the Role of the Brd4 Protein in the Papillomavirus Lifecycle. Viruses, 5(6), 1374-1394. https://doi.org/10.3390/v5061374