Understanding Bacteriophage Specificity in Natural Microbial Communities

Abstract
:1. Introduction
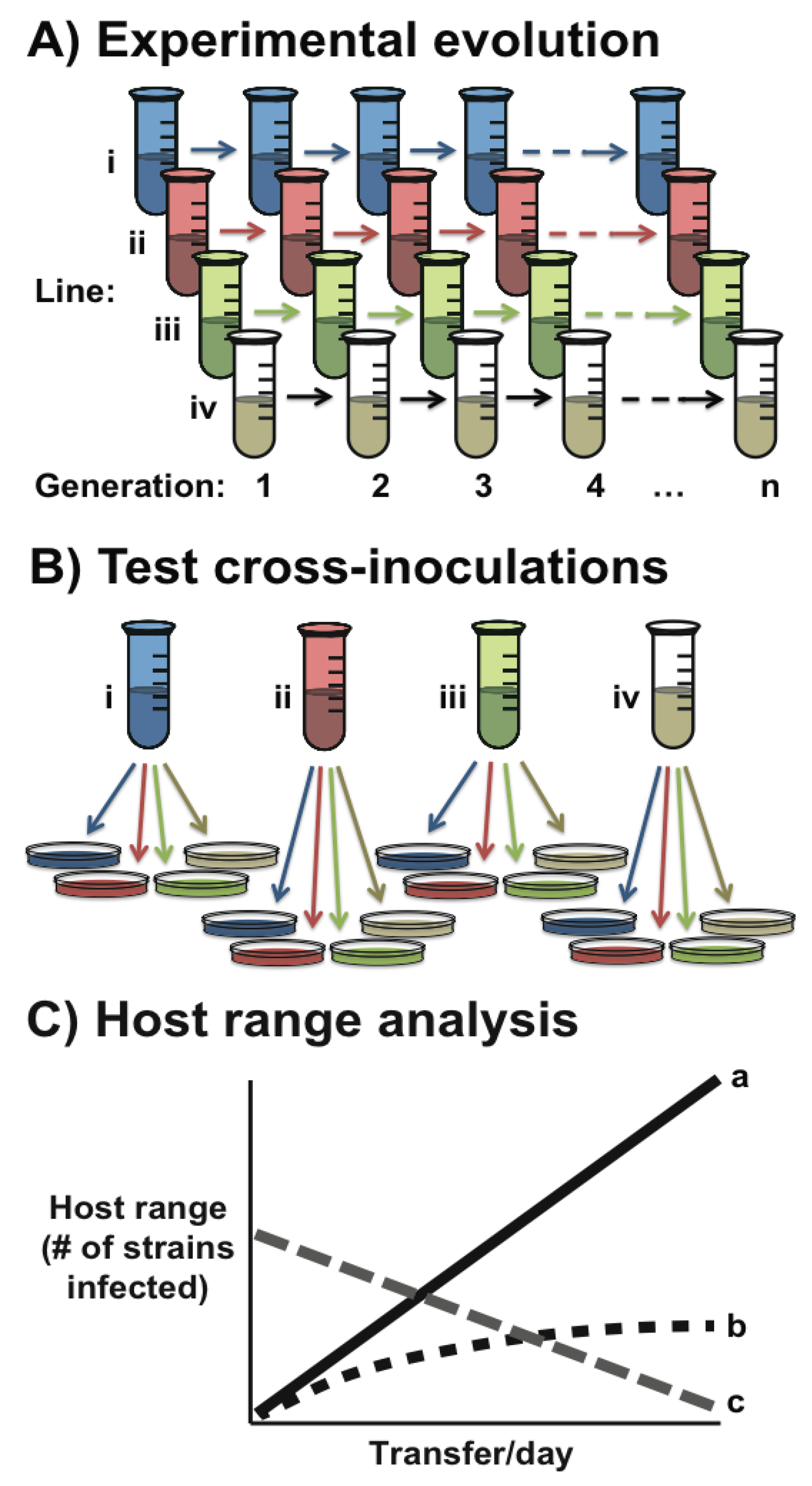
Box 1. Experimental evolution of phage specificity.
1.1. The Structure of Bacteria-Phage Interaction Networks
1.2. The Evolutionary Implications of Phage Host Range
1.3. The Applied Implications of Phage Host Range
2. Results and Discussion
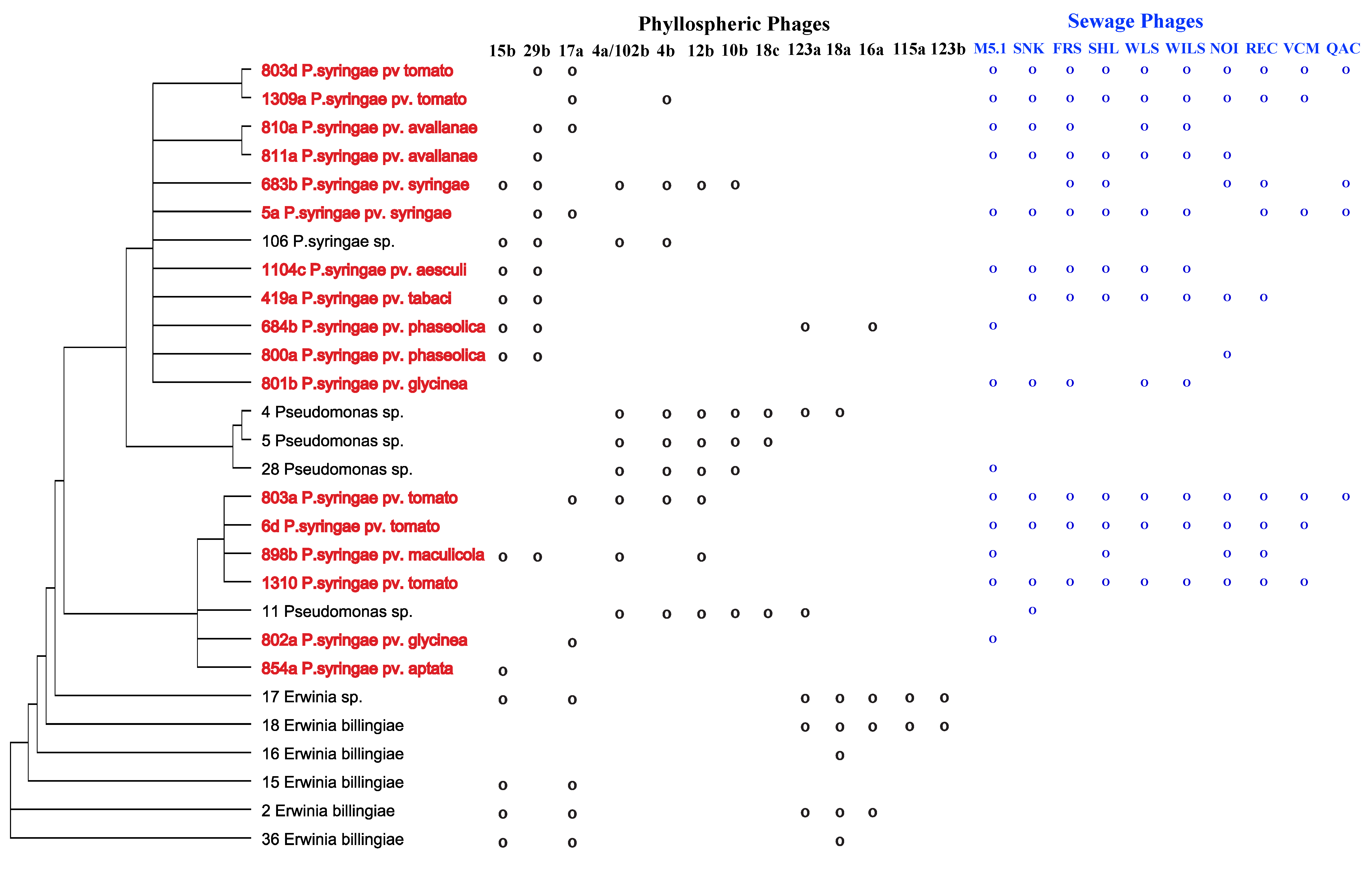
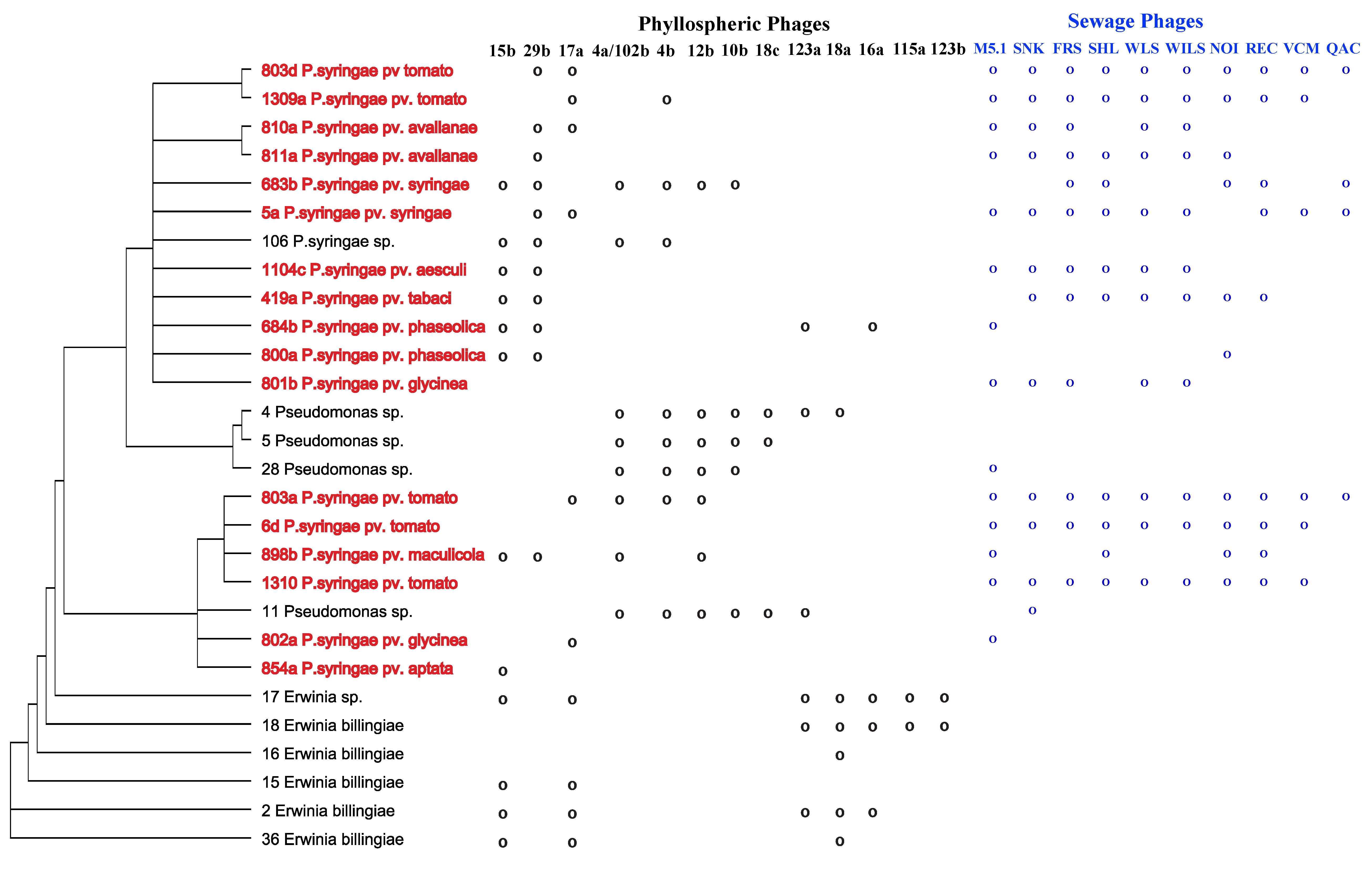
2.1. Specificity within a Natural Phyllosphere Environment

{kind=link}
{kind=link}
{kind=link}
| Host range | ||||||
|---|---|---|---|---|---|---|
| Habitat | Host | # Phages tested | Multi-species | Multi-genus | Within-species specificity | Reference |
| Rhizosphere | Pseudomonas | 5 | 4 | 0 | n/a | Campbell et al. 1995 [61] |
| Sewage | Multiple hosts | 11 | n/a | 11 | n/a | Jensen et al. 1998 [62] |
| Industrial | Leuconostoc | 6 | 0 | 0 | Yes | Barrangou et al. 2002 [63] |
| Marine | Vibrio | 13 | 10 | n/a | Yes | Comeau et al. 2005 [64] |
| Soil | Burkholderia | 6 | 6 | n/a | Yes | Seed and Dennis 2005 [65] |
| Effluent | Salmonella | 66 | n/a | 0 | Yes | McLaughlin et al. 2006 [66] |
| Marine | Cellulophaga | 46 | 0 | 0 | Yes | Holmfeldt et al. 2007 [67] |
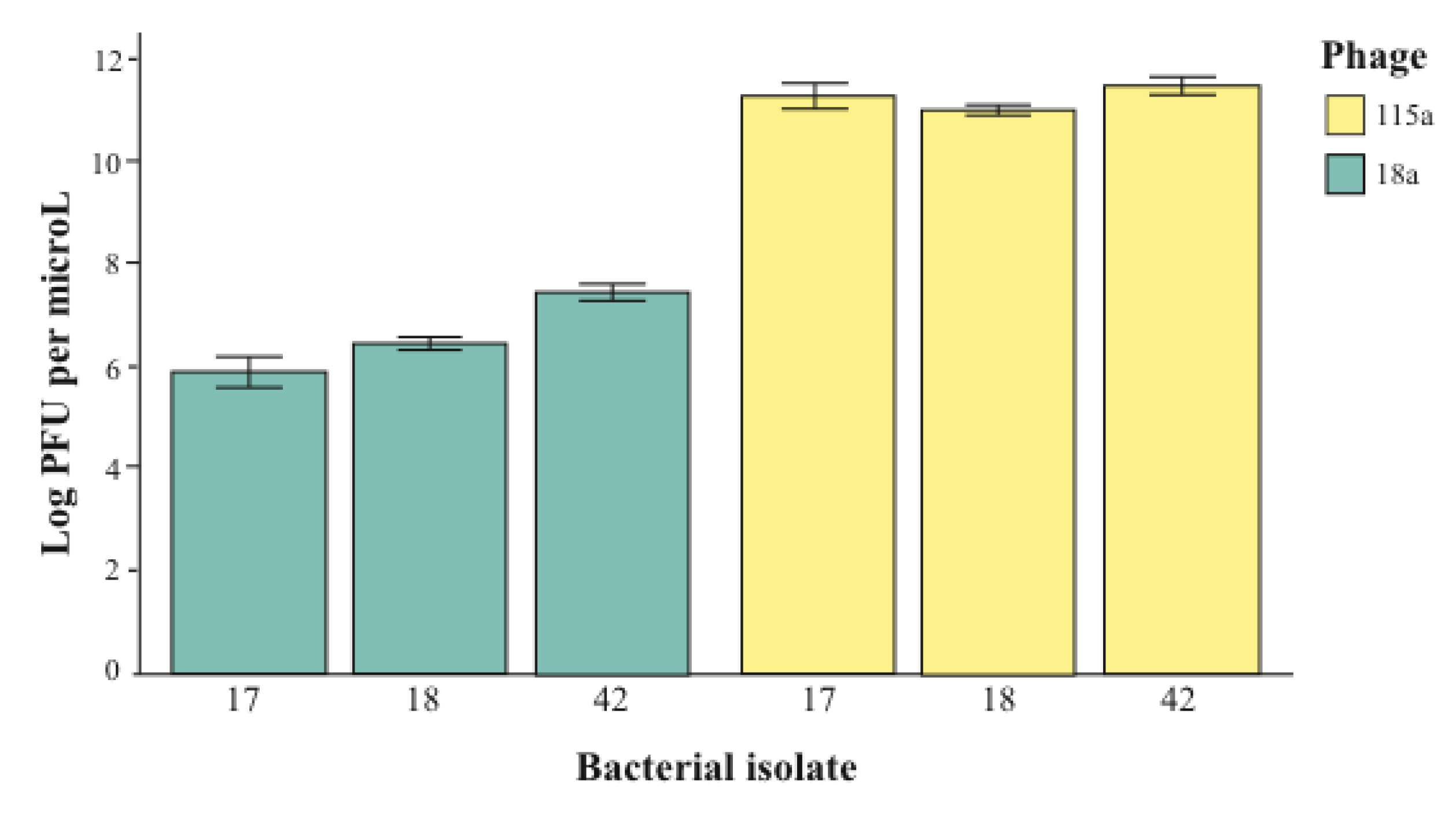
2.2. The Importance of Dose in Measuring Specificity
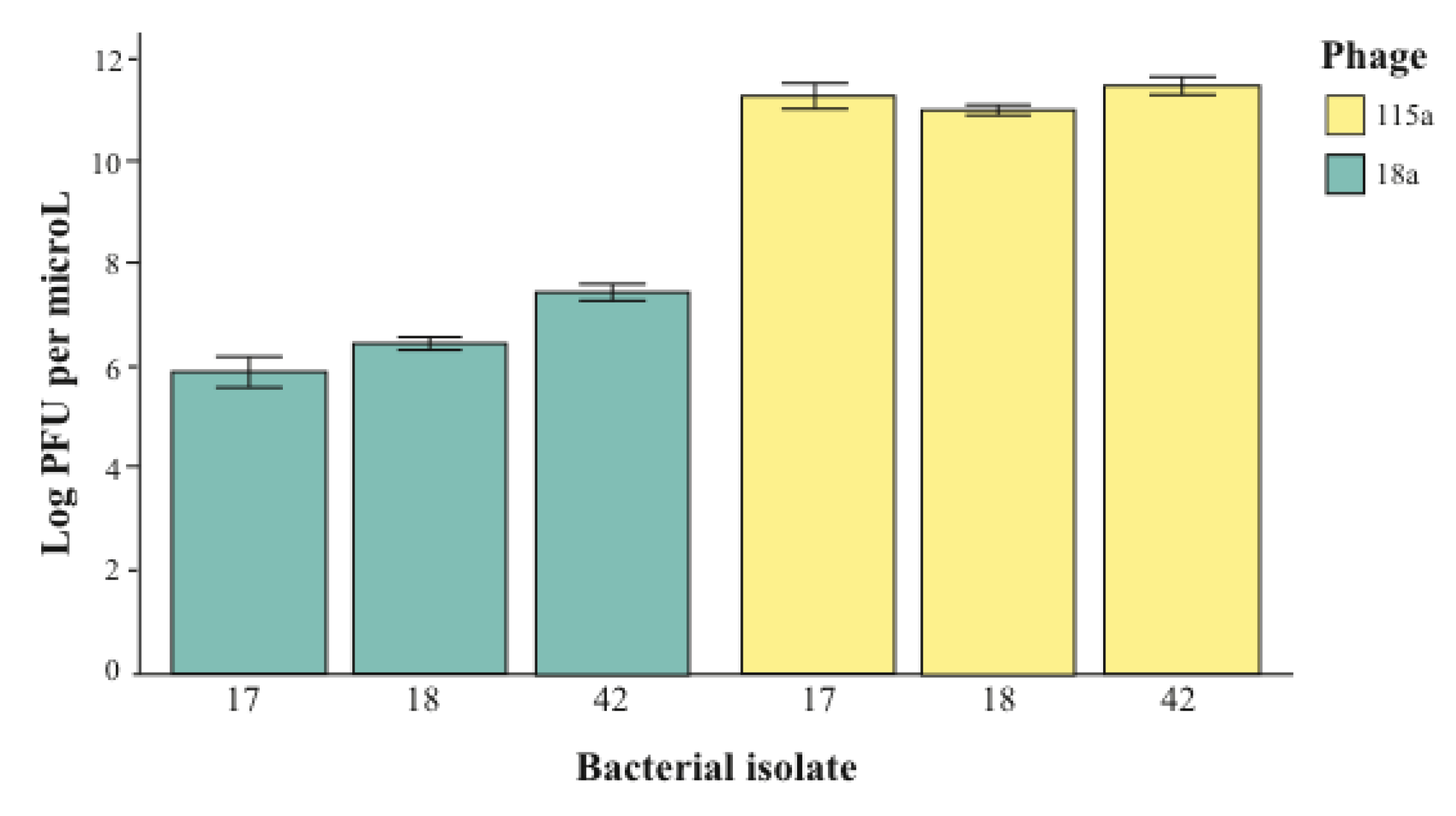
3. Conclusions
Acknowledgments
Conflict of Interest
References and Notes
- Gómez, P.; Buckling, A. Bacteria-phage antagonistic coevolution in soil. Science 2011, 332, 106–109. [Google Scholar] [CrossRef]
- Griffiths, R.I.; Thomson, B.C.; James, P.; Bell, T.; Bailey, M.; Whiteley, A.S. The bacterial biogeography of British soils. Environ. Microb. 2011, 13, 1642–1654. [Google Scholar] [CrossRef]
- Koskella, B.; Thompson, J.N.; Preston, G.M.; Buckling, A. Local biotic environment shapes the spatial scale of bacteriophage adaptation to bacteria. Am. Nat. 2011, 177, 440–451. [Google Scholar] [CrossRef]
- Lindow, S.E.; Brandl, M.T. Microbiology of the Phyllosphere. Appl. Environ. Microbiol. 2003, 69, 1875–1883. [Google Scholar] [CrossRef]
- Marston, M.F.; Pierciey, F.J.; Shepard, A.; Gearin, G.; Qi, J.; Yandava, C.; Schuster, S.C.; Henn, M.R.; Martiny, J.B.H. Rapid diversification of coevolving marine Synechococcus and a virus. PNAS 2012, 109, 4544–4549. [Google Scholar] [CrossRef]
- Pommier, T.; Douzery, E.J.P.; Mouillot, D. Environment drives high phylogenetic turnover among oceanic bacterial communities. Biol. Lett. 2012, 8, 562–566. [Google Scholar] [CrossRef]
- Smillie, C.S.; Smith, M.B.; Friedman, J.; Cordero, O.X.; David, L.A.; Alm, E.J. Ecology drives a global network of gene exchange connecting the human microbiome. Nature 2011, 480, 241–244. [Google Scholar]
- Hooper, L.V.; Littman, D.R.; Macpherson, A.J. Interactions Between the Microbiota and the Immune System. Science 2012, 336, 1268–1273. [Google Scholar] [CrossRef]
- De Wit, R.; Bouvier, T. “Everything is everywhere, but, the environment selects”; what did Baas Becking and Beijerinck really say? Environ. Microb. 2006, 8, 755–758. [Google Scholar] [CrossRef]
- O’Malley, M.A. “Everything is everywhere: but the environment selects”: Ubiquitous distribution and ecological determinism in microbial biogeography. Stud. Hist. Phil. Biol. Biomed. Sci. 2008, 39, 314–325. [Google Scholar]
- Lin, W.; Wang, Y.; Li, B.; Pan, Y. A biogeographic distribution of magnetotactic bacteria influenced by salinity. ISME J. 2011, 6, 475–479. [Google Scholar]
- Chu, H.; Fierer, N.; Lauber, C.L.; Caporaso, J.G.; Knight, R.; Grogan, P. Soil bacterial diversity in the Arctic is not fundamentally different from that found in other biomes. Environ. Microb. 2010, 12, 2998–3006. [Google Scholar] [CrossRef]
- Buckling, A.; Rainey, P.B. The role of parasites in sympatric and allopatric host diversification. Nature 2002, 420, 496–499. [Google Scholar] [CrossRef]
- Childs, L.M.; Held, N.L.; Young, M.J.; Whitaker, R.J.; Weitz, J.S. Multiscale model of CRISPR-induced coevolutionary dynamics: Diversification at the interface of Larmarck and Darwin. Evolution 2012, 66, 2015–2029. [Google Scholar] [CrossRef]
- Suttle, C.A. Viruses in the sea. Nature 2005, 437, 356–361. [Google Scholar] [CrossRef]
- Rodriguez-Valera, F.; Martin-Cuadrado, A.-B.; Rodriguez-Brito, B.; Pasic, L.; Thingstad, T.F.; Rohwer, F.; Mira, A. Explaining microbial population genomics through phage predation. Nat. Rev. Micro. 2009, 7, 828–836. [Google Scholar] [CrossRef]
- Clokie, M.R.J.; Millard, A.D.; Letarov, A.V.; Heaphy, S. Phages in nature. Bacteriophage 2011, 1, 31–45. [Google Scholar] [CrossRef]
- Flores, C.O.; Meyer, J.R.; Valverde, S.; Farr, L.; Weitz, J.S. Statistical structure of host–phage interactions. PNAS. 2011, 108, 288–297. [Google Scholar]
- Frisch, A.W.; Levine, P. Specificity of the Multiplication of Bacteriophage. J. Immunol. 1936, 30, 89–108. [Google Scholar]
- Fierer, N.; Lennon, J.T. The generation and maintenance of diversity in microbial communities. Am. J. Bot. 2011, 98, 439–448. [Google Scholar] [CrossRef]
- Hyman, P.; Abedon, S.T. Bacteriophage host range and bacterial resistance. Adv. Appl. Microbiol. 2010, 70, 217–248. [Google Scholar] [CrossRef]
- Duplessis, M.; Moineau, S. Identification of a genetic determinant responsible for host specificity in Streptococcus thermophilus bacteriophages. Mol. Microbiol. 2001, 41, 325–336. [Google Scholar] [CrossRef]
- Miklič, A.; Rogelj, I. Characterization of lactococcal bacteriophages isolated from Slovenian dairies. Int. J. Food Sci. Tech. 2003, 38, 305–311. [Google Scholar] [CrossRef]
- Vos, M.; Birkett, P.J.; Birch, E.; Griffiths, R.I.; Buckling, A. Local adaptation of bacteriophages to their bacterial hosts in soil. Science 2009, 325, 833. [Google Scholar] [CrossRef]
- Hall, A.R.; Scanlan, P.D.; Morgan, A.D.; Buckling, A. Host–parasite coevolutionary arms races give way to fluctuating selection. Ecol. Lett. 2011, 14, 635–642. [Google Scholar] [CrossRef]
- Riede, I.; Degen, M.; Henning, U. The receptor specificity of bacteriophages can be determined by a tail fiber modifying protein. EMBO J. 1985, 4, 2343. [Google Scholar]
- Rakhuba, D.; Kolomiets, E.; Szwajcer Dey, E.; Novik, G. Bacteriophage receptors, mechanisms of phage adsorption and penetration into host cell. Pol. J. Microbiol. 2010, 59, 145–155. [Google Scholar]
- Chatterjee, S.; Rothenberg, E. Interaction of Bacteriophage l with Its E. coli Receptor, LamB. Viruses 2012, 4, 3162–3178. [Google Scholar] [CrossRef]
- Mahony, J.; van Sinderen, D. Structural Aspects of the Interaction of Dairy Phages with Their Host Bacteria. Viruses 2012, 4, 1410–1424. [Google Scholar] [CrossRef]
- Richter, C.; Chang, J.T.; Fineran, P.C. Function and Regulation of Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR)/CRISPR Associated (Cas) Systems. Viruses 2012, 4, 2291–2311. [Google Scholar] [CrossRef]
- de Vos, W.M.; Underwood, H.M.; Lyndon Davies, F. Plasmid encoded bacteriophage resistance in Streptococcus cremoris SK11. FEMS Microbiol. Lett. 1984, 23, 175–178. [Google Scholar]
- Deng, Y.-M.; Harvey, M.L.; Liu, C.-Q.; Dunn, N.W. A novel plasmid-encoded phage abortive infection system from Lactococcus lactis biovar. diacetylactis. FEMS Microbiol. Lett. 1997, 146, 149–154. [Google Scholar] [CrossRef]
- Antonovics, J.; Boots, M.; Ebert, D.; Koskella, B.; Poss, M.; Sadd, B.M. The origins of specificity by means of natural selection: Evolved and nonhost resistance in host-pathogen interactions. Evolution 2012, 6, 1–9. [Google Scholar]
- Straub, C.S.; Ives, A.R.; Gratton, C. Evidence for a trade-off between host-range breadth and host-use efficiency in aphid parasitoids. Am. Nat. 2011, 177, 389–395. [Google Scholar] [CrossRef]
- Poullain, V.; Gandon, S.; Brockhurst, M.A.; Buckling, A.; Hochberg, M.E. The evolution of specificity in evolving and coevolving antagonistic interactions between a bacteria and its phage. Evolution 2008, 62, 1–11. [Google Scholar]
- Duffy, S.; Turner, P.E.; Burch, C.L. Pleiotropic costs of niche expansion in the RNA bacteriophage Φ6. Genetics 2006, 172, 751–757. [Google Scholar] [CrossRef]
- Avrani, S.; Schwartz, D.A.; Lindell, D. Virus-host swinging party in the oceans: Incorporating biological complexity into paradigms of antagonistic coexistence. Mob. Gen. Elem. 2012, 2, 88–95. [Google Scholar] [CrossRef]
- Koskella, B.; Lin, D.M.; Buckling, A.; Thompson, J.N. The costs of evolving resistance in heterogeneous parasite environments. Proc. R. So. B: Biol. Sci. 2012, 279, 1896–1903. [Google Scholar] [CrossRef]
- Gaba, S.; Ebert, D. Time-shift experiments as a tool to study antagonistic coevolution. TREE 2009, 24, 226–232. [Google Scholar]
- Hall, A.R.; Scanlan, P.D.; Buckling, A. Bacteria—Phage Coevolution and the Emergence of Generalist Pathogens. Am. Nat. 2011, 177, 44–53. [Google Scholar]
- Buckling, A.; Rainey, P.B. Antagonistic coevolution between a bacterium and a bacteriophage. Proc. R. So. B: Biol. Sci. 2002, 269, 931–936. [Google Scholar] [CrossRef]
- Bono, L.M.; Gensel, C.L.; Pfennig, D.W.; Burch, C.L. Competition and the origins of novelty: Experimental evolution of niche-width expansion in a virus. Biol. Let. 2013, 9. [Google Scholar]
- Dennehy, J.J.; Friedenberg, N.A.; Holt, R.D.; Turner, P.E. Viral ecology and the maintenance of novel host use. Am. Nat. 2006, 167, 429–439. [Google Scholar]
- Duffy, S.; Burch, C.L.; Turner, P.E. Evolution of host specificity drives reproductive isolation among RNA viruses. Evolution 2007, 61, 2614–2622. [Google Scholar] [CrossRef]
- Scanlan, P.D.; Hall, A.R.; Lopez-Pascua, L.D.C.; Buckling, A. Genetic basis of infectivity evolution in a bacteriophage. Molec. Ecol. 2011, 20, 981–989. [Google Scholar] [CrossRef]
- Meyer, J.R.; Dobias, D.T.; Weitz, J.S.; Barrick, J.E.; Quick, R.T.; Lenski, R.E. Repeatability and contingency in the evolution of a key innovation in phage lambda. Science 2012, 335, 428–432. [Google Scholar] [CrossRef]
- Held, N.; Whitaker, R.J. Viral biogeography revealed by signatures in Sulfolobus islandicus genomes. Environ. Microb. 2009, 11, 457–466. [Google Scholar] [CrossRef]
- Kunin, V.; He, S.; Warnecke, F.; Peterson, S.B.; Garcia Martin, H.; Haynes, M.; Ivanova, N.; Blackall, L.L.; Breitbart, M.; Rohwer, F.; et al. A bacterial metapopulation adapts locally to phage predation despite global dispersal. Gen. Res. 2008, 18, 293–297. [Google Scholar] [CrossRef]
- Fuhrman, J.A.; Schwalbach, M. Viral Influence on Aquatic Bacterial Communities. Biol. Bull. 2003, 204, 192–195. [Google Scholar] [CrossRef]
- Rodriguez-Brito, B.; Li, L.; Wegley, L.; Furlan, M.; Angly, F.; Breitbart, M.; Buchanan, J.; Desnues, C.; Dinsdale, E.; Edwards, R.; et al. Viral and microbial community dynamics in four aquatic environments. ISME J. 2010, 4, 739–751. [Google Scholar] [CrossRef]
- Shapiro, O.H.; Kushmaro, A.; Brenner, A. Bacteriophage predation regulates microbial abundance and diversity in a full-scale bioreactor treating industrial wastewater. ISME J. 2010, 4, 327–336. [Google Scholar] [CrossRef]
- Waterbury, J.B.; Valois, F.W. Resistance to co-occurring phages enables marine Synechococcus communities to coexist with cyanophages abundant in seawater. Appl. Environ. Microb. 1993, 59, 3393. [Google Scholar]
- Yu, X.; Lund, S.P.; Scott, R.A.; Greenwald, J.W.; Records, A.H.; Nettleton, D.; Lindow, S.E.; Gross, D.C.; Beattie, G.A. Transcriptional responses of Pseudomonas syringae to growth in epiphytic versus apoplastic leaf sites. PNAS. 2013, 110, 425–434. [Google Scholar] [CrossRef]
- Fineran, P.C.; Petty, N.K.; Salmond, G.P.C. Transduction: Host DNA Transfer by Bacteriophages. In The Encyclopedia of Microbiology; Schaechter, M., Ed.; Elsevier, 2009. [Google Scholar]
- Chen, J.; Novick, R.P. Phage-mediated intergeneric transfer of toxin genes. Science 2009, 323, 139–141. [Google Scholar] [CrossRef]
- Mazaheri Nezhad Fard, R.; Barton, M.; Heuzenroeder, M. Bacteriophage - mediated transduction of antibiotic resistance in enterococci. Lett. Appl. Microb. 2011, 52, 559–564. [Google Scholar] [CrossRef]
- Beumer, A.; Robinson, J.B. A Broad-Host-Range, Generalized Transducing Phage (SN-T) Acquires 16S rRNA Genes from Different Genera of Bacteria. Appl. Environ. Microb. 2005, 71, 8301–8304. [Google Scholar] [CrossRef]
- Loc-Carrillo, C.; Abedon, S.T. Pros and cons of phage therapy. Bacteriophage 2011, 1, 111–114. [Google Scholar] [CrossRef]
- Vongkamjan, K.; Switt, A.M.; den Bakker, H.C.; Fortes, E.D.; Wiedmann, M. Silage Collected from Dairy Farms Harbors an Abundance of Listeriaphages with Considerable Host Range and Genome Size Diversity. Appl. Environ. Microb. 2012, 78, 8666–8675. [Google Scholar]
- Flores, C.O.; Meyer, J.R.; Valverde, S.; Farr, L.; Weitz, J.S. Statistical structure of host–phage interactions. PNAS. 2011, 108, 288–297. [Google Scholar]
- Campbell, J.I.A.; Albrechtsen, M.; Sørensen, J. Large Pseudomonas phages isolated from barley rhizosphere. FEMS Microb. Ecol. 1995, 18, 63–74. [Google Scholar]
- Jensen, E.C.; Schrader, H.S.; Rieland, B.; Thompson, T.L.; Lee, K.W.; Nickerson, K.W.; Kokjohn, T.A. Prevalence of Broad-Host-Range Lytic Bacteriophages of Sphaerotilus natans, Escherichia coli, andPseudomonas aeruginosa. Appl. Environ. Microb. 1998, 64, 575–580. [Google Scholar]
- Barrangou, R.; Yoon, S.-S.; Breidt, J.F.; Fleming, H.P.; Klaenhammer, T.R. Characterization of Six Leuconostoc fallax Bacteriophages Isolated from an Industrial Sauerkraut Fermentation. Appl. Environ. Microb. 2002, 68, 5452–5458. [Google Scholar] [CrossRef]
- Comeau, A.M.; Buenaventura, E.; Suttle, C.A. A Persistent, Productive, and Seasonally Dynamic Vibriophage Population within Pacific Oysters (Crassostrea gigas). Appl. Environ. Microb. 2005, 71, 5324–5331. [Google Scholar] [CrossRef]
- Seed, K.D.; Dennis, J.J. Isolation and characterization of bacteriophages of the Burkholderia cepacia complex. FEMS Microb. Lett. 2005, 251, 273–280. [Google Scholar] [CrossRef]
- McLaughlin, M.R.; Balaa, M.F.; Sims, J.; King, R. Isolation of Salmonella Bacteriophages from Swine Effluent Lagoons. Journal article number J-10632 of the Mississippi Agricultural and Forestry Experiment Station. J. Environ. Qual. 2006, 35, 522–528. [Google Scholar] [CrossRef]
- Holmfeldt, K.; Middelboe, M.; Nybroe, O.; Riemann, L. Large Variabilities in Host Strain Susceptibility and Phage Host Range Govern Interactions between Lytic Marine Phages and Their Flavobacterium Hosts. Appl. Environ. Microb. 2007, 73, 6730–6739. [Google Scholar] [CrossRef]
- Turner, S.; Pryer, K.M.; Miao, V.P.W.; Palmer, J.D. Investigating Deep Phylogenetic Relationships among Cyanobacteria and Plastids by Small Subunit rRNA Sequence Analysis1. J. Euk. Microb. 1999, 46, 327–338. [Google Scholar] [CrossRef]
- Edgar, R.C. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucl. Acid. Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef]
- Weitz, J.S.; Poisot, T.; Meyer, J.R.; Flores, C.O.; Valverde, S.; Sullivan, M.B.; Hochberg, M.E. Phage—bacteria infection networks. Trends Microb. 2012, 21, 82–91. [Google Scholar]
- Green, S.; Laue, B.; Fossdal, C.G.; A'Hara, S.W.; Cottrell, J.E. Infection of horse chestnut (Aesculus hippocastanum) by Pseudomonas syringae pv. aesculi and its detection by quantitative real-time PCR. Plant Path. 2009, 58, 731–744. [Google Scholar] [CrossRef]
- Anderson, T.K.; Sukhdeo, M.V.K. Host Centrality in Food Web Networks Determines Parasite Diversity. PLoS One 2011, 6, e26798. [Google Scholar] [CrossRef]
- Poulin, R.; Moulillot, D. Parasite specialization from a phylogenetic perspective: a new index of host specificity. Parasitol. 2003, 126, 473–480. [Google Scholar] [CrossRef]
- Luijckx, P.; Ben-Ami, F.; Mouton, L.; Du Pasquier, L.; Ebert, D. Cloning of the unculturable parasite Pasteuria ramosa and its Daphnia host reveals extreme genotype–genotype interactions. Ecol. Lett. 2011, 14, 125–131. [Google Scholar] [CrossRef]
- Poisot, T.; Lepennetier, G.; Martinez, E.; Ramsayer, J.; Hochberg, M.E. Resource availability affects the structure of a natural bacteria–bacteriophage community. Biology Letters 2011, 7, 201–204. [Google Scholar] [CrossRef]
- Seeley, N.D.; Primrose, S.B. The Effect of Temperature on the Ecology of Aquatic Bacteriophages. J. Gen. Virol. 1980, 46, 87–95. [Google Scholar] [CrossRef]
- Lennon, J.; Khatana, S.; Marston, M.; Martiny, J. Is there a cost of virus resistance in marine cyanobacteria? ISME J. 2007, 1, 300. [Google Scholar]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Koskella, B.; Meaden, S. Understanding Bacteriophage Specificity in Natural Microbial Communities. Viruses 2013, 5, 806-823. https://doi.org/10.3390/v5030806
Koskella B, Meaden S. Understanding Bacteriophage Specificity in Natural Microbial Communities. Viruses. 2013; 5(3):806-823. https://doi.org/10.3390/v5030806
Chicago/Turabian StyleKoskella, Britt, and Sean Meaden. 2013. "Understanding Bacteriophage Specificity in Natural Microbial Communities" Viruses 5, no. 3: 806-823. https://doi.org/10.3390/v5030806