Molecular Characterization of Major Structural Protein Genes of Avian Coronavirus Infectious Bronchitis Virus Isolates in Southern China

{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Alignment Analysis of Nucleotide and the Deduced Amino Acid Sequences
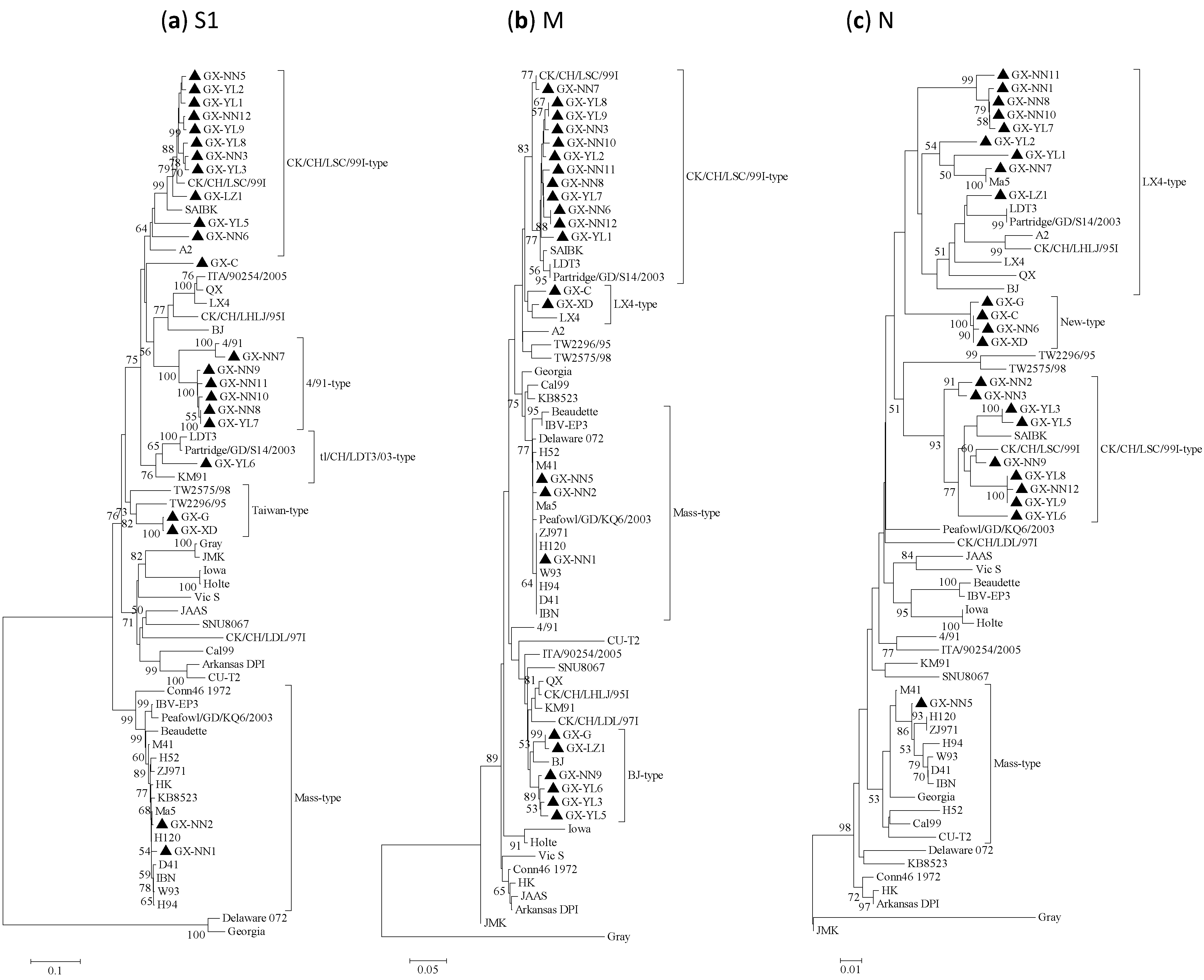
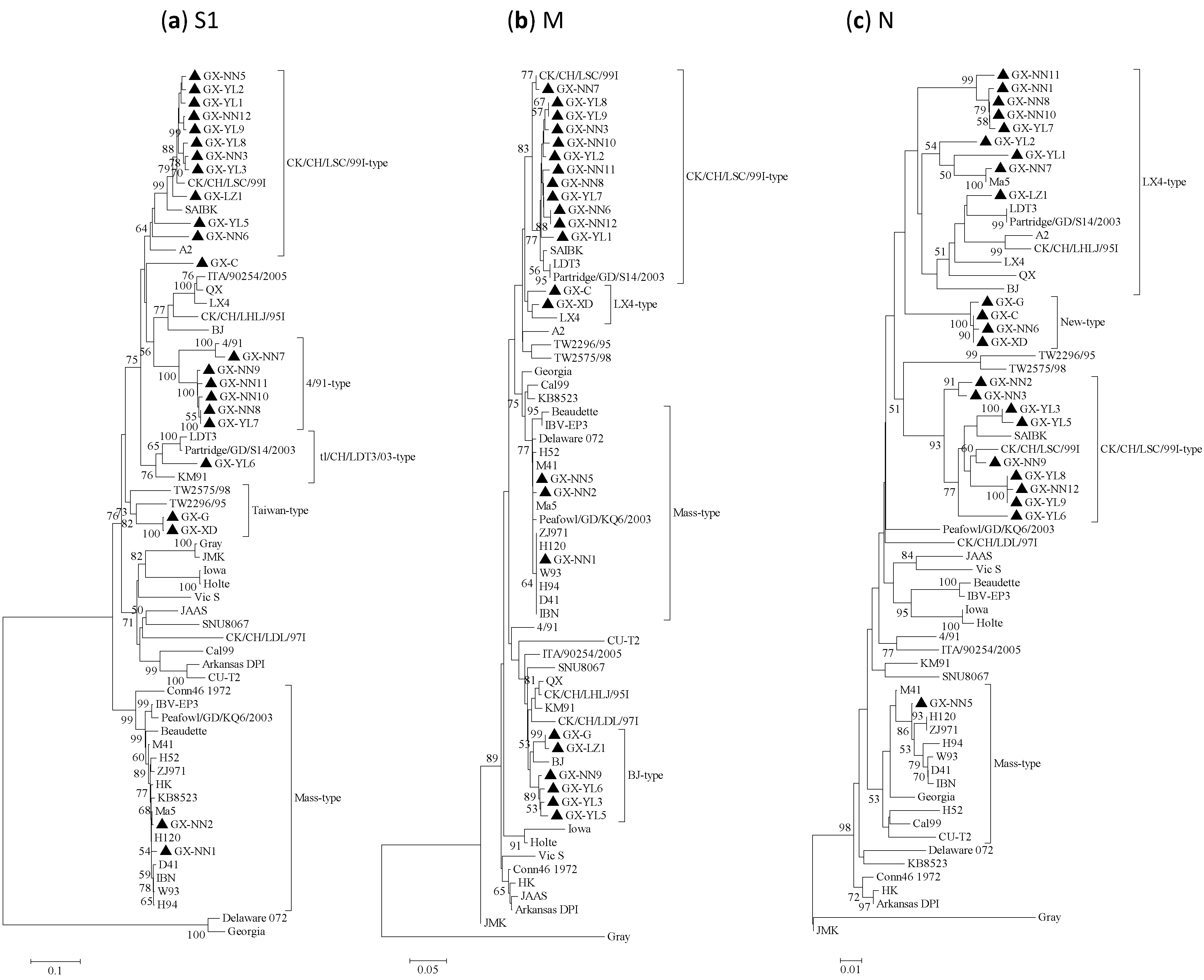
2.2. Phylogenetic Analysis
2.3. Positive Selection on the S1, M and N Proteins of IBVs
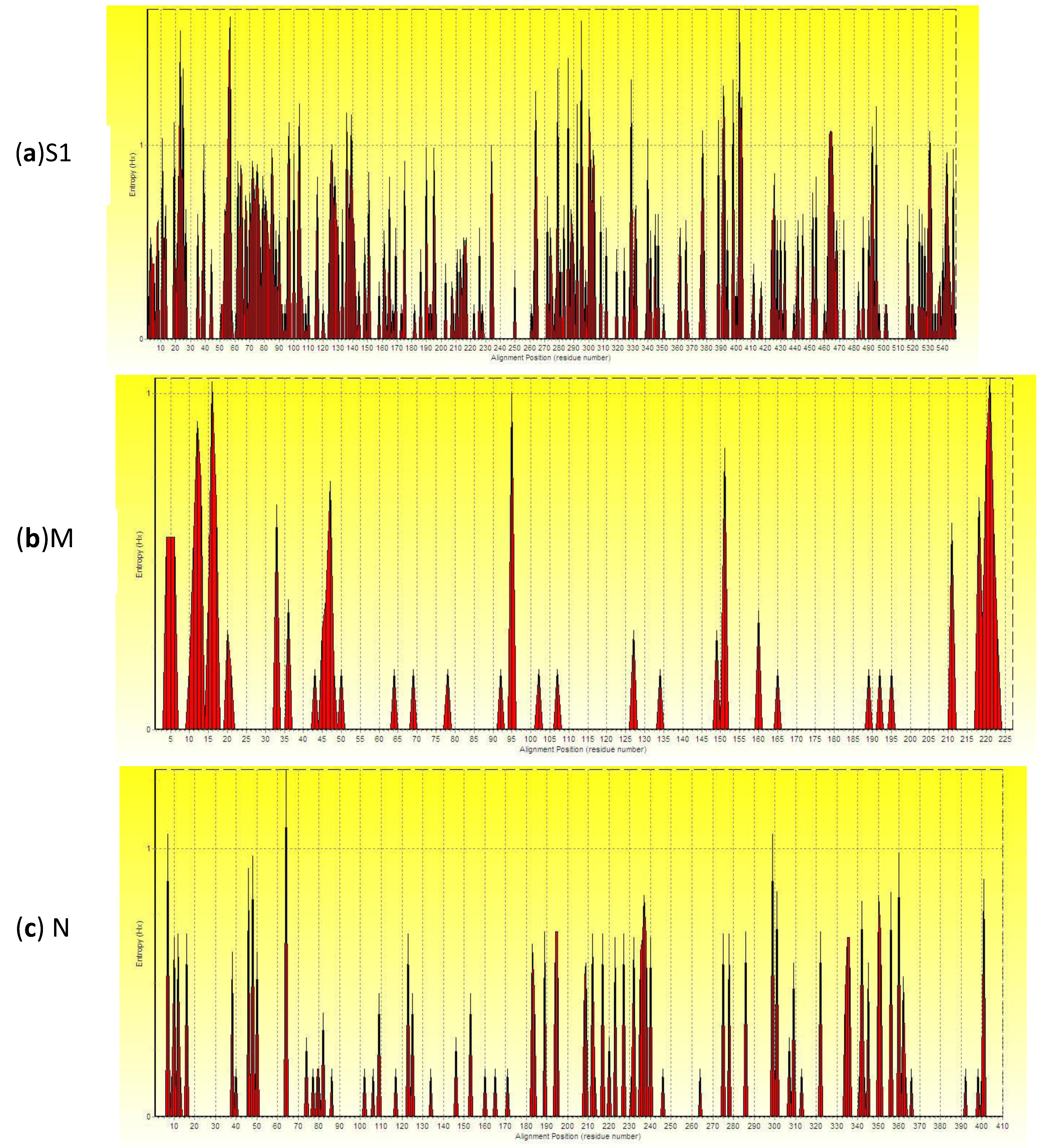
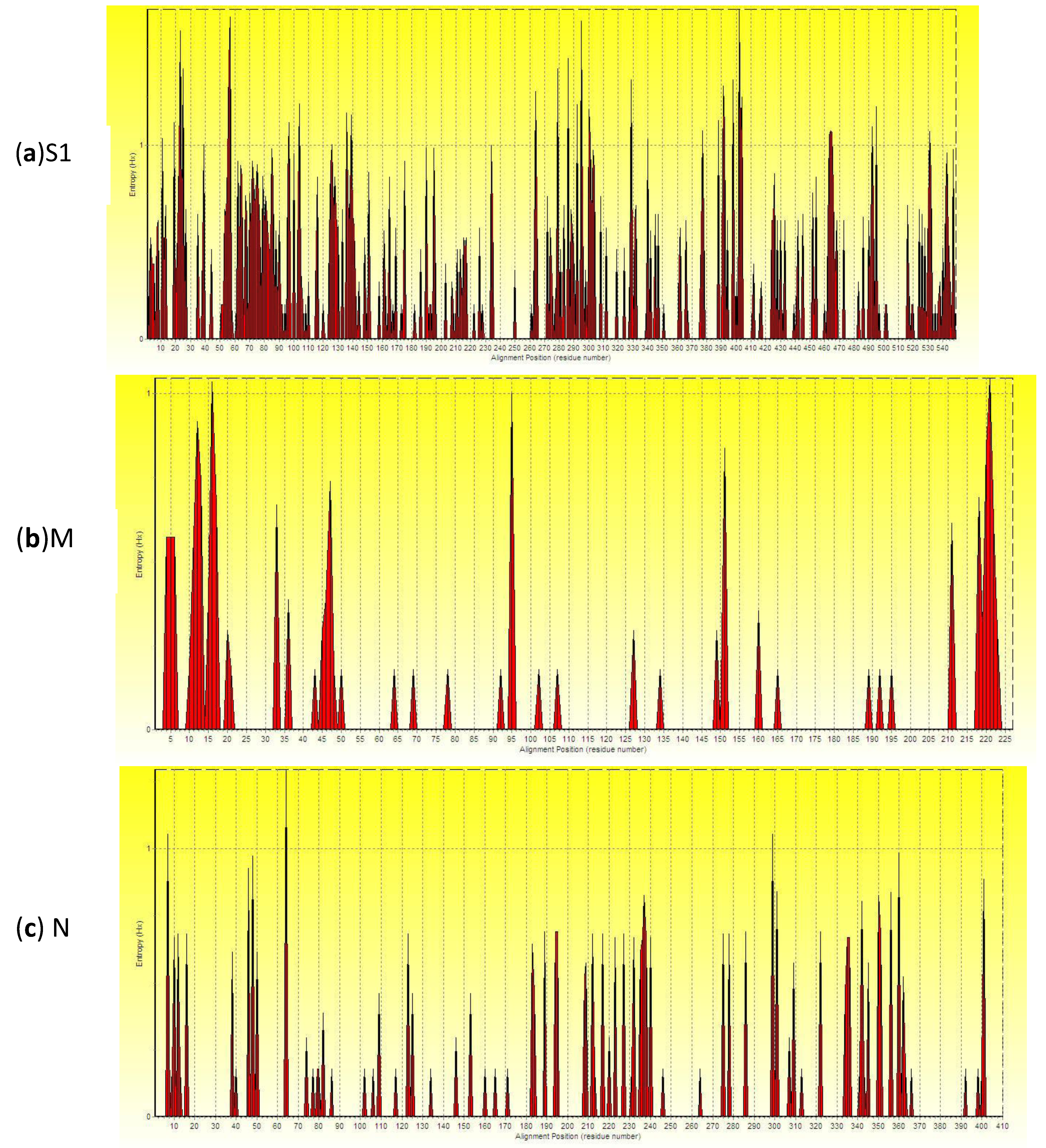
2.4. Analysis of Entropy of Amino Acid Sequences
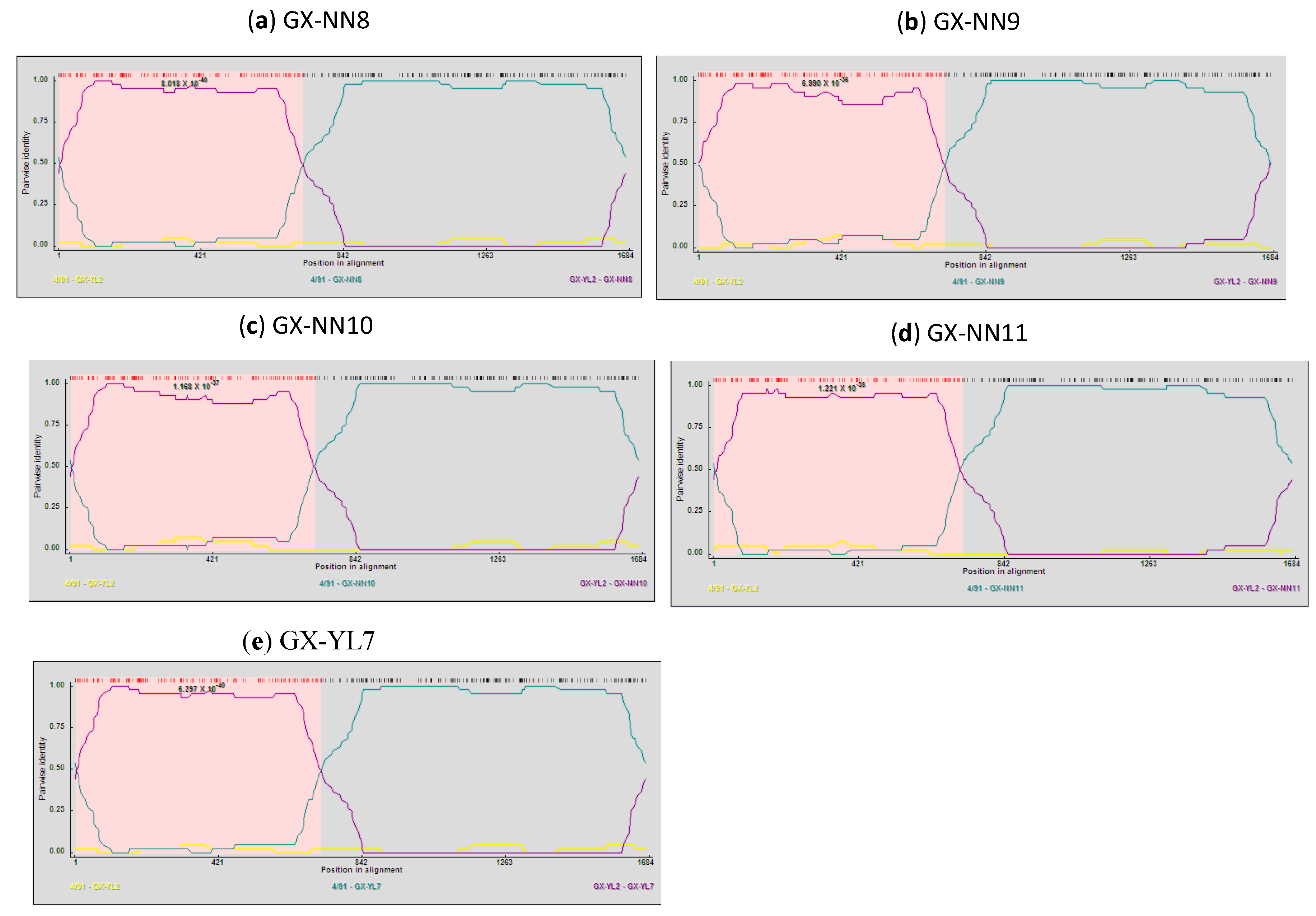
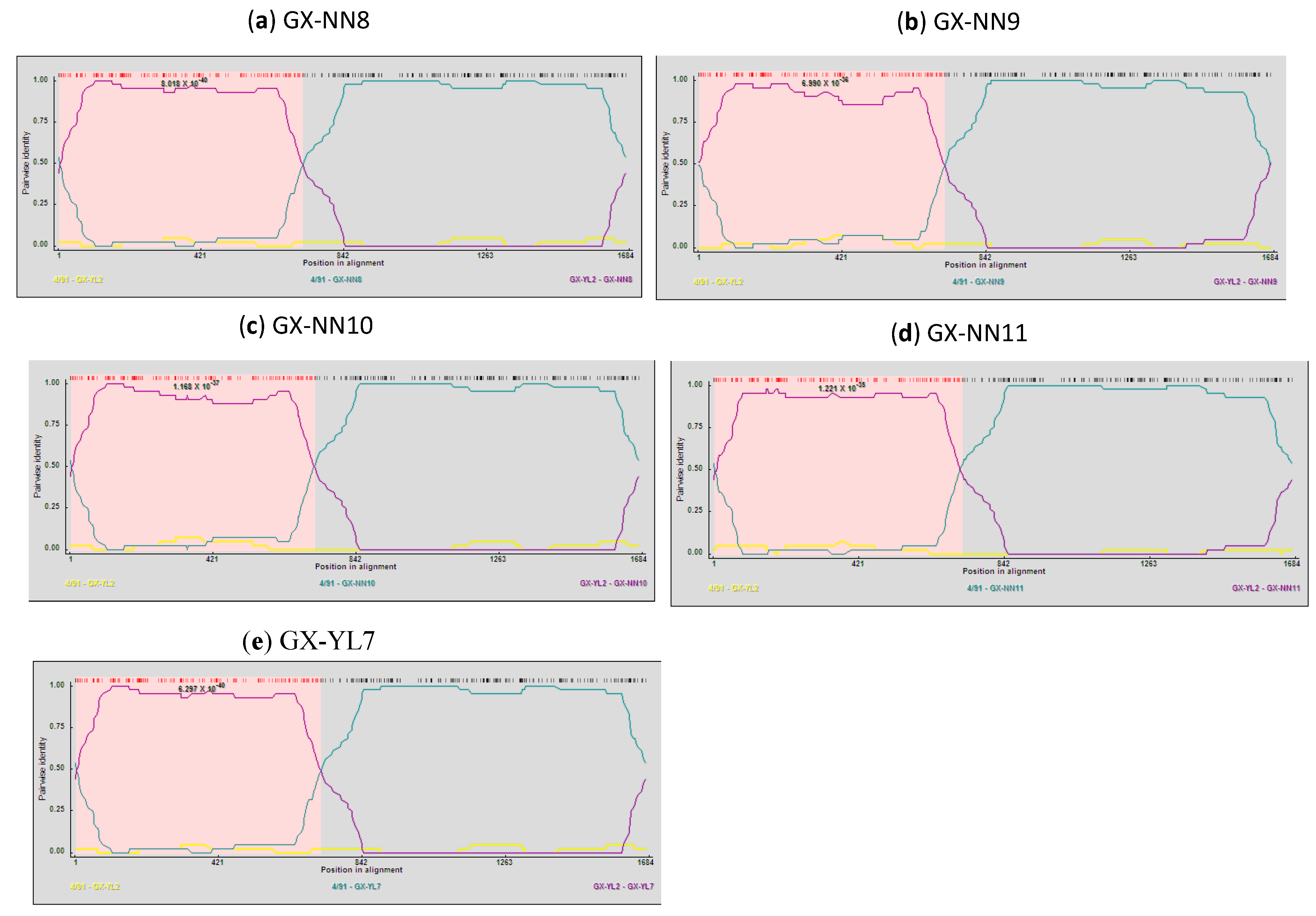
2.5. Analysis of Recombinants
3. Discussion
4. Experimental Section
4.1. Virus Isolation and Propagation
4.2. Primers for S1, M and N genes Amplification
4.3. RNA Extraction and Amplification of S1, N and M Genes
4.4. Gene Sequencing, Alignments and Phylogenetic Analysis
4.5. Analysis of Entropy of Amino Acid Sequences and Positive Selection
5. Conclusions
Acknowledgments
Conflicts of Interest
References
- Carstens, E. Ratification vote on taxonomic proposals to the International Committee on Taxonomy of Viruses (2009). Arch. Virol. 2010, 155, 133–146. [Google Scholar] [CrossRef]
- Cavanagh, D. Severe acute respiratory syndrome vaccine development: Experiences of vaccination against avian infectious bronchitis coronavirus. Avian. Pathol. 2003, 32, 567–582. [Google Scholar] [CrossRef]
- Thor, S.W.; Hilt, D.A.; Kissinger, J.C.; Paterson, A.H.; Jackwood, M.W. Recombination in avian gamma-coronavirus infectious bronchitis virus. Viruses 2011, 3, 1777–1799. [Google Scholar] [CrossRef]
- Cavanagh, D.; Davis, P.J.; Cook, J.K.; Li, D.; Kant, A.; Koch, G. Location of the amino acid differences in the S1 spike glycoprotein subunit of closely related serotypes of infectious bronchitis virus. Avian Pathol. 1992, 21, 33–43. [Google Scholar] [CrossRef]
- Koch, G.; Hartog, L.; Kant, A.; van Roozelaar, D.J. Antigenic domains on the peplomer protein of avian infectious bronchitis virus: Correlation with biological functions. J. Gen. Virol. 1990, 71, 1929–1935. [Google Scholar] [CrossRef]
- Ignjatovic, J.; Galli, U. Immune responses to structural proteins of avian infectious bronchitis virus. Avian Pathol. 1995, 24, 313–332. [Google Scholar] [CrossRef]
- Zwaagstra, K.A.; van der Zeijst, B.A.; Kusters, J.G. Rapid detection and identification of avian infectious bronchitis virus. J. Clin. Microbiol. 1992, 30, 79–84. [Google Scholar]
- Lee, C.W.; Hilt, D.A.; Jackwood, M.W. Typing of field isolates of infectious bronchitis virus based on the sequence of the hypervariable region in the S1 gene. J. Vet. Diagn. Invest. 2003, 15, 344–348. [Google Scholar] [CrossRef]
- Corse, E.; Machamer, C.E. The cytoplasmic tails of infectious bronchitis virus E and M proteins mediate their interaction. Virology 2003, 312, 25–34. [Google Scholar] [CrossRef]
- Dolz, R.; Pujols, J.; Ordonez, G.; Porta, R.; Majo, N. Molecular epidemiology and evolution of avian infectious bronchitis virus in Spain over a fourteen-year period. Virology 2008, 374, 50–59. [Google Scholar] [CrossRef]
- McKinley, E.T.; Jackwood, M.W.; Hilt, D.A.; Kissinger, J.C.; Robertson, J.S.; Lemke, C.; Paterson, A.H. Attenuated live vaccine usage affects accurate measures of virus diversity and mutation rates in avian coronavirus infectious bronchitis virus. Virus Res. 2011, 158, 225–234. [Google Scholar]
- Ammayappan, A.; Upadhyay, C.; Gelb, J., Jr.; Vakharia, V.N. Identification of sequence changes responsible for the attenuation of avian infectious bronchitis virus strain Arkansas DP. Arch. Virol. 2009, 154, 495–499. [Google Scholar] [CrossRef]
- Park, J.Y.; Park, S.I.; Sung, H.W.; Kim, J.H.; Song, C.S.; Lee, C.W.; Kwon, H.M. Variations in the nucleocapsid protein gene of infectious bronchitis viruses isolated in Korea. Virus Genes 2005, 31, 153–162. [Google Scholar] [CrossRef]
- Ren, X.; Yin, J.; Ma, D.; Li, G. Characterization and membrane gene-based phylogenetic analysis of avian infectious bronchitis virus Chinese strain HH06. Virus Genes 2009, 38, 39–45. [Google Scholar] [CrossRef]
- Li, M.; Wang, X.Y.; Wei, P.; Chen, Q.Y.; Wei, Z.J.; Mo, M.L. Serotype and genotype diversity of infectious bronchitis viruses isolated during 1985–2008 in Guangxi, China. Arch. Virol. 2012, 157, 467–474. [Google Scholar] [CrossRef]
- Mahmood, Z.H.; Sleman, R.R.; Uthman, A.U. Isolation and molecular characterization of Sul/01/09 avian infectious bronchitis virus, indicates the emergence of a new genotype in the Middle East. Vet. Microbiol. 2011, 150, 21–27. [Google Scholar] [CrossRef]
- Mo, M.L.; Huang, B.C.; Wei, P.; Wei, T.C.; Chen, Q.Y.; Wang, X.Y.; Li, M.; Fan, W.S. Complete genome sequences of two chinese virulent avian coronavirus infectious bronchitis virus variants. J. Virol. 2012, 86, 10903–10904. [Google Scholar]
- Mo, M.L.; Hong, S.M.; Kwon, H.J.; Kim, I.H.; Song, C.S.; Kim, J.H. Genetic diversity of spike, 3a, 3b and e genes of infectious bronchitis viruses and emergence of new recombinants in Korea. Viruses 2013, 5, 550–567. [Google Scholar] [CrossRef]
- Callison, S.A.; Jackwood, M.W.; Hilt, D.A. Molecular characterization of infectious bronchitis virus isolates foreign to the United States and comparison with United States isolates. Avian Dis. 2001, 45, 492–499. [Google Scholar] [CrossRef]
- Han, Z.; Sun, C.; Yan, B.; Zhang, X.; Wang, Y.; Li, C.; Zhang, Q.; Ma, Y.; Shao, Y.; Liu, Q.; et al. A 15-year analysis of molecular epidemiology of avian infectious bronchitis coronavirus in China. Infect. Genet. Evol. 2011, 11, 190–200. [Google Scholar] [CrossRef]
- Liu, S.; Zhang, Q.; Chen, J.; Han, Z.; Shao, Y.; Kong, X.; Tong, G. Identification of the avian infectious bronchitis coronaviruses with mutations in gene 3. Gene 2008, 412, 12–25. [Google Scholar] [CrossRef]
- Ma, H.; Shao, Y.; Sun, C.; Han, Z.; Liu, X.; Guo, H.; Kong, X.; Liu, S. Genetic diversity of avian infectious bronchitis coronavirus in recent years in China. Avian Dis. 2012, 56, 15–28. [Google Scholar] [CrossRef]
- Teng, L.Q.; Wei, P.; Song, Z.B.; He, J.J.; Cui, Z.Z. Molecular epidemiological investigation of Marek’s disease virus from Guangxi, China. Arch. Virol. 2011, 156, 203–206. [Google Scholar] [CrossRef]
- Wang, C.H.; Huang, Y.C. Relationship between serotypes and genotypes based on the hypervariable region of the S1 gene of infectious bronchitis virus. Arch. Virol. 2000, 145, 291–300. [Google Scholar] [CrossRef]
- Schikora, B.M.; Shih, L.M.; Hietala, S.K. Genetic diversity of avian infectious bronchitis virus California variants isolated between 1988 and 2001 based on the S1 subunit of the spike glycoprotein. Arch. Virol. 2003, 148, 115–136. [Google Scholar] [CrossRef]
- Hughes, A.L. Recombinational histories of avian infectious bronchitis virus and turkey coronavirus. Arch. Virol. 2011, 156, 1823–1829. [Google Scholar] [CrossRef]
- Shieh, H.K.; Shien, J.H.; Chou, H.Y.; Shimizu, Y.; Chen, J.N.; Chang, P.C. Complete nucleotide sequences of S1 and N genes of infectious bronchitis virus isolated in Japan and Taiwan. J. Vet. Med. Sci. 2004, 66, 555–558. [Google Scholar] [CrossRef]
- Chen, H.W.; Huang, Y.P.; Wang, C.H. Identification of Taiwan and China-like recombinant avian infectious bronchitis viruses in Taiwan. Virus research 2009, 140, 121–129. [Google Scholar] [CrossRef]
- Tang, X.; Li, G.; Vasilakis, N.; Zhang, Y.; Shi, Z.; Zhong, Y.; Wang, L.F.; Zhang, S. Differential stepwise evolution of SARS coronavirus functional proteins in different host species. BMC Evol. Biol. 2009, 9, 52. [Google Scholar] [CrossRef]
- Chen, H.W.; Huang, Y.P.; Wang, C.H. Identification of intertypic recombinant infectious bronchitis viruses from slaughtered chickens. Poult. Sci. 2010, 89, 439–446. [Google Scholar] [CrossRef]
- Kuo, M.; Kao, H.W.; Hou, M.H.; Wang, C.H.; Lin, S.H.; Su, H.L. Evolution of infectious bronchitis virus in Taiwan: Positively selected sites in the nucleocapsid protein and their effects on RNA-binding activity. Vet. Microbiol. 2013, 162, 408–418. [Google Scholar] [CrossRef]
- Pan, K.; Deem, M.W. Quantifying selection and diversity in viruses by entropy methods, with application to the haemagglutinin of H3N2 influenza. J.R. Soc. Interface. 2011, 8, 1644–1653. [Google Scholar] [CrossRef]
- Wang, K.; Samudrala, R. Incorporating background frequency improves entropy-based residue conservation measures. BMC Bioinforma. 2006, 7, 385. [Google Scholar] [CrossRef]
- He, X.; Zhou, J.; Bartlam, M.; Zhang, R.; Ma, J.; Lou, Z.; Li, X.; Li, J.; Joachimiak, A.; Zeng, Z.; et al. Crystal structure of the polymerase PA(C)-PB1(N) complex from an avian influenza H5N1 virus. Nature 2008, 454, 1123–1126. [Google Scholar] [CrossRef]
- Stewart, J.J.; Lee, C.Y.; Ibrahim, S.; Watts, P.; Shlomchik, M.; Weigert, M.; Litwin, S. A Shannon entropy analysis of immunoglobulin and Tcell receptor. Mol. Immunol. 1997, 34, 1067–1082. [Google Scholar] [CrossRef]
- Jackwood, M.W.; Boynton, T.O.; Hilt, D.A.; McKinley, E.T.; Kissinger, J.C.; Paterson, A.H.; Robertson, J.; Lemke, C.; McCall, A.W.; Williams, S.M.; et al. Emergence of a group 3 coronavirus through recombination. Virology 2010, 398, 98–108. [Google Scholar] [CrossRef]
- Ovchinnikova, E.V.; Bochkov, Y.A.; Shcherbakova, L.O.; Nikonova, Z.B.; Zinyakov, N.G.; Elatkin, N.P.; Mudrak, N.S.; Borisov, A.V.; Drygin, V.V. Molecular characterization of infectious bronchitis virus isolates from Russia and neighbouring countries: Identification of intertypic recombination in the S1 gene. Avian Pathol. 2011, 40, 507–514. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, H.N.; Wang, T.; Fan, W.Q.; Zhang, A.Y.; Wei, K.; Tian, G.B.; Yang, X. Complete genome sequence and recombination analysis of infectious bronchitis virus attenuated vaccine strain H120. Virus Genes 2010, 41, 377–388. [Google Scholar] [CrossRef]
- Liu, X.; Ma, H.; Xu, Q.; Sun, N.; Han, Z.; Sun, C.; Guo, H.; Shao, Y.; Kong, X.; Liu, S. Characterization of a recombinant coronavirus infectious bronchitis virus with distinct S1 subunits of spike and nucleocapsid genes and a 3' untranslated region. Vet. Microbiol. 2013, 162, 429–436. [Google Scholar] [CrossRef]
- Li, L.; Xue, C.; Chen, F.; Qin, J.; Xie, Q.; Bi, Y.; Cao, Y. Isolation and genetic analysis revealed no predominant new strains of avian infectious bronchitis virus circulating in South China during 2004–2008. Vet. Microbiol. 2010, 143, 145–154. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Mo, M.-L.; Li, M.; Huang, B.-C.; Fan, W.-S.; Wei, P.; Wei, T.-C.; Cheng, Q.-Y.; Wei, Z.-J.; Lang, Y.-H. Molecular Characterization of Major Structural Protein Genes of Avian Coronavirus Infectious Bronchitis Virus Isolates in Southern China. Viruses 2013, 5, 3007-3020. https://doi.org/10.3390/v5123007
Mo M-L, Li M, Huang B-C, Fan W-S, Wei P, Wei T-C, Cheng Q-Y, Wei Z-J, Lang Y-H. Molecular Characterization of Major Structural Protein Genes of Avian Coronavirus Infectious Bronchitis Virus Isolates in Southern China. Viruses. 2013; 5(12):3007-3020. https://doi.org/10.3390/v5123007
Chicago/Turabian StyleMo, Mei-Lan, Meng Li, Bai-Cheng Huang, Wen-Sheng Fan, Ping Wei, Tian-Chao Wei, Qiu-Ying Cheng, Zheng-Ji Wei, and Ya-Hui Lang. 2013. "Molecular Characterization of Major Structural Protein Genes of Avian Coronavirus Infectious Bronchitis Virus Isolates in Southern China" Viruses 5, no. 12: 3007-3020. https://doi.org/10.3390/v5123007