Development of Hematopoietic Stem Cell Based Gene Therapy for HIV-1 Infection: Considerations for Proof of Concept Studies and Translation to Standard Medical Practice


Abstract
:1. Introduction
2. Results and Discussion
2.1. Initial Trial Using Lentiviral Vectors and Ablative Conditioning



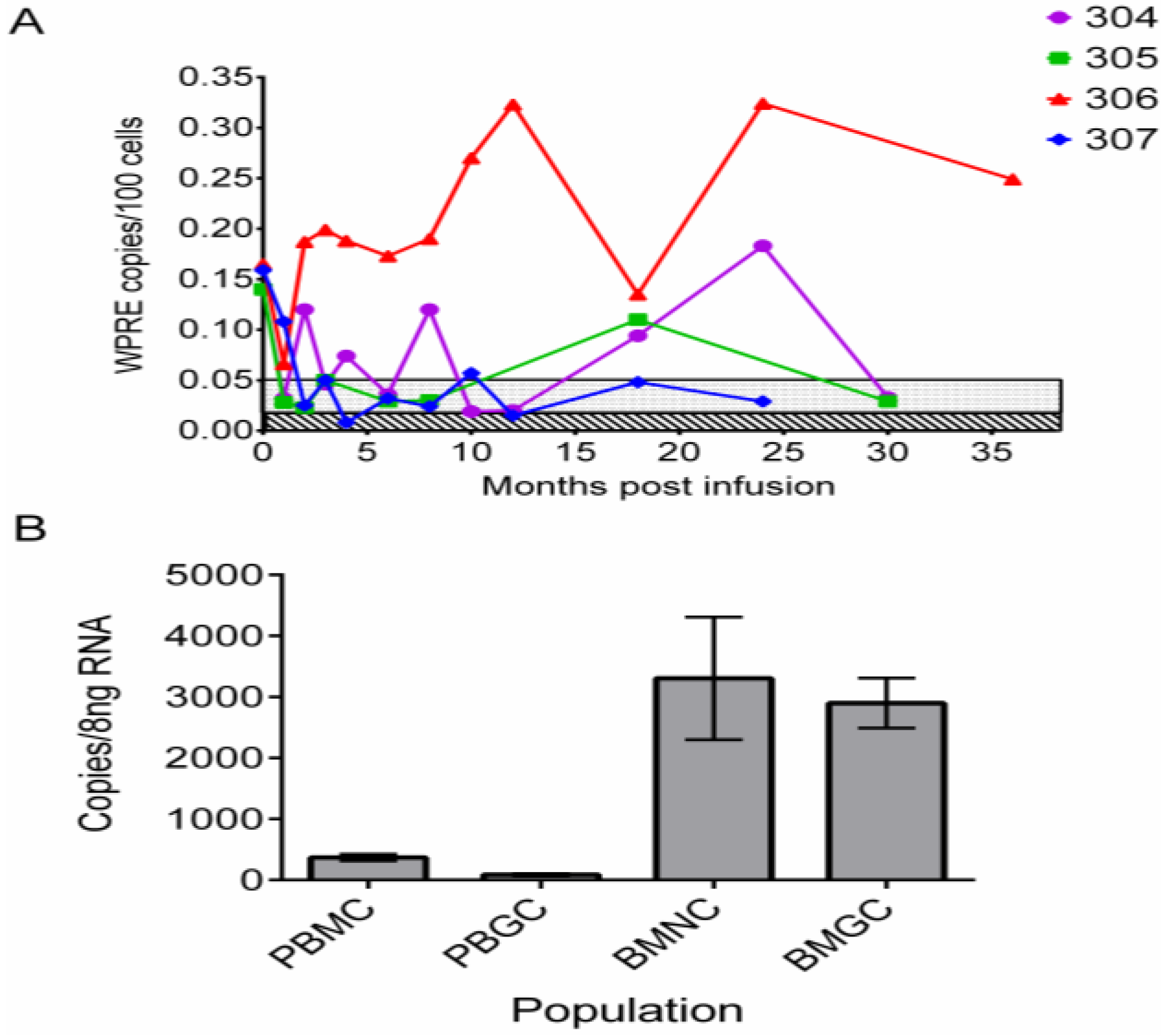{kind=link}
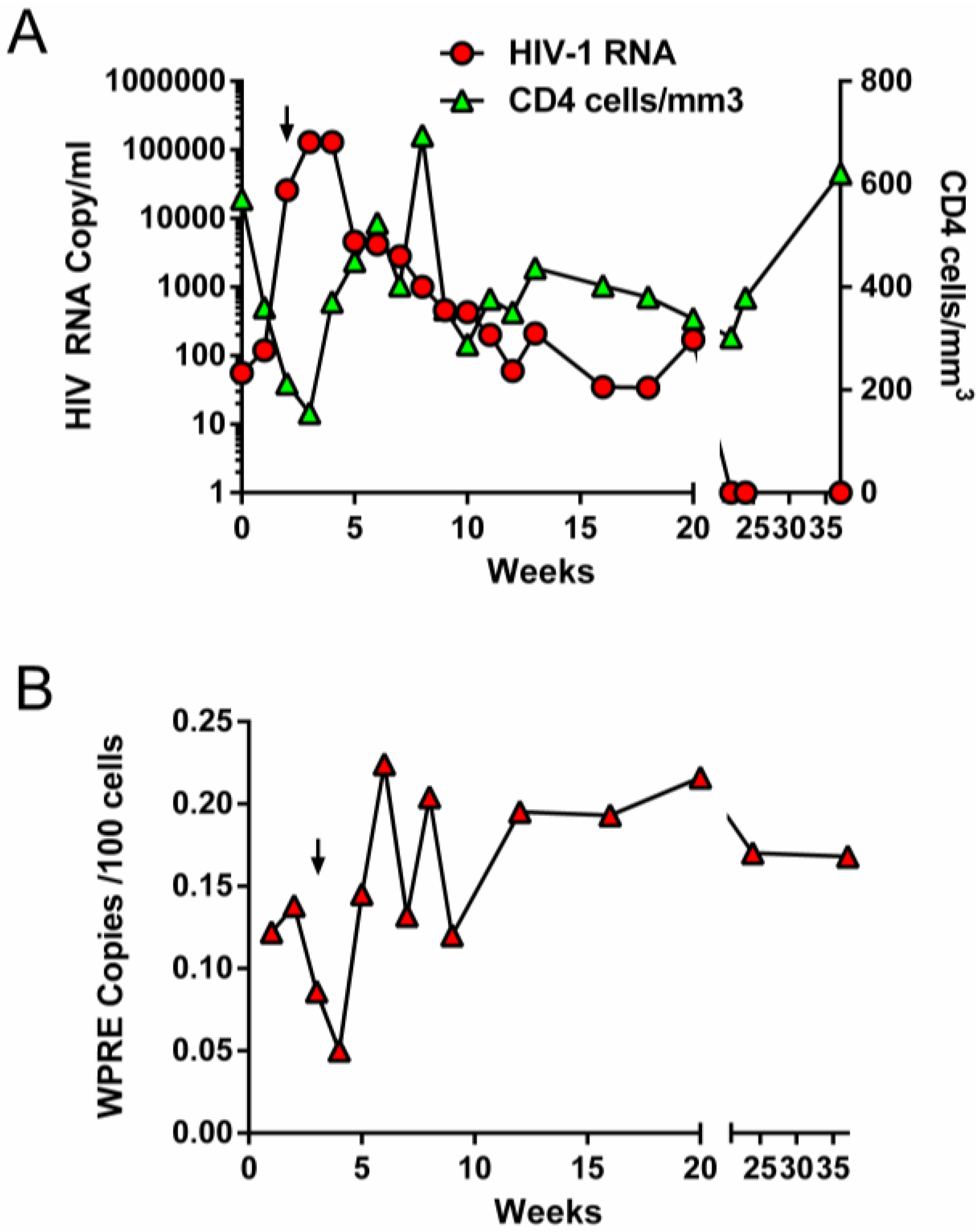{kind=link}
| Chromosome | Start | End | # of Sequence Reads | Gene | Relative Location |
|---|---|---|---|---|---|
| chr19 | 18,384,645 | 18,384,747 | 1,690,418 | KIAA1683 | intron |
| chr19 | 9,442,260 | 9,442,322 | 1,049,697 | ZNF559-ZNF177 | intron |
| chr21 | 15,383,973 | 15,384,065 | 211 | ANKRD20A11P | intergenic (3’) |
| chr19 | 324,134 | 324,282 | 84 | MIER2 | intron |
| chr2 | 20,368,493 | 20,368,580 | 60 | SDC1 | intergenic (5’) |
| chr1 | 121,485,117 | 121,485,410 | 56 | EMBP1 | intergenic (5’) |
| chr19 | 323,958 | 324,045 | 48 | MIER2 | intron |
| chr19 | 324,378 | 324,465 | 43 | MIER2 | intron |
| chr19 | 18,384,488 | 18,384,578 | 34 | KIAA1683 | intron |
| chr19 | 18,384,347 | 18,384,404 | 21 | KIAA1683 | intron |
2.2. Second Generation Anti-HIV-1 Constructs
2.2.1. Improved RNA Expression
2.2.2. Selective Enrichment of Gene-Modified Cells
2.3. Patient Selection
2.3.1. Risk:Benefit Considerations
2.4. Stem Cell Number and Quality
2.4.1. HSPC Collection
| No. | HIV/AIDS Subpopulation | Current Rx Options for HIV-1 infection | Aspects of SOC Rx for HIV-1 infection | Potential Benefit of Research Rx | Real or Potential Risks of Research Rx | Risk:Benefit Analysis |
|---|---|---|---|---|---|---|
| 1 | HIV/AIDS pts on cART (controlled viremia and CD4 counts >500/µL) | cART | <10% treatment failure Outcome expectations excellent | Minimal to no potential benefit since virus control and CD4 counts are adequate | Transient myeloid dysfunction Unknown effects of genetic modification & HSPC mobilization | Unfavorable; first in human trial cannot be justified in this group |
| 2 | AIDS pts off cART (side effects to cART or cART “fatigue”) | Symptomatic Rx if cART not tolerable | Heightened potential for AIDS progression | Improved control of HIV-1 | Transient myeloid dysfunction Unknown effects of genetic modification & HSPC mobilization | Favorable but conditioning adds unnecessary risk in these patients who are already drug adverse |
| 3 | AIDS pts on cART, with incomplete immune recovery with suboptimal CD4 levels | cART Treatment as indicated for infections | Poor expected outcome | Expansion of CD4 count Potential for improved control of HIV-1 | Transient myeloid dysfunction Unknown effects of genetic modification & HSPC mobilization | Favorable |
| 4 | AIDS pts who do not respond to cART | Research therapy with new antivirals | Poor expected outcome | Improved control of HIV-1 | Transient myeloid dysfunction Unknown effects of genetic modification & HSPC mobilization | Favorable but limitation of subject availability |
| 5 | ARL pts in remission following frontline Rx | cART Treatment as indicated for infections | Remission stable Outcome expectations very good; concern for risk of myelodysplasia | Minimal to no potential benefit IF virus control and CD4 counts are adequate | Transient myeloid dysfunction Unknown effects of genetic modification | Less favorable due to potential for myelo-dysplasia post-chemotherapy and conditioning |
| 6 | ARL pts on salvage therapy (transplant) | cART Treatment as indicated for infections | Outcome expectations good; concern for myelodysplasia risk | Minimal to no potential benefit IF virus control and CD4 counts are adequate | Transient myeloid dysfunction Unknown effects of genetic modification | Less favorable due to 10%–20% potential for myelodysplasia post-transplant and conditioning |
2.4.2. HSPC and HIV-1 Infection
2.5. Preparation for Transplantation
2.6. Getting to Market
3. Experimental Section
4. Conclusions
Acknowledgments
Conflicts of Interest
References
- World Health Organization. Global HIV/AIDS response - epidemic update and health sector progress towards universal access: Progress report 2011. Available online: http://whqlibdoc.who.int/publications/2011/9789241502986_eng.pdf (accessed on 20 November 2013).
- Chun, T.W.; Stuyver, L.; Mizell, S.B.; Ehler, L.A.; Mican, J.A.; Baseler, M.; Lloyd, A.L.; Nowak, M.A.; Fauci, A.S. Presence of an inducible HIV-1 latent reservoir during highly active antiretroviral therapy. Proc. Natl. Acad. Sci. U S A 1997, 94, 13193–13197. [Google Scholar] [CrossRef]
- Finzi, D.; Blankson, J.; Siliciano, J.D.; Margolick, J.B.; Chadwick, K.; Pierson, T.; Smith, K.; Lisziewicz, J.; Lori, F.; Flexner, C.; et al. Latent infection of CD4+ T cells provides a mechanism for lifelong persistence of HIV-1, even in patients on effective combination therapy. Nat. Med. 1999, 5, 512–517. [Google Scholar] [CrossRef]
- Finzi, D.; Hermankova, M.; Pierson, T.; Carruth, L.M.; Buck, C.; Chaisson, R.E.; Quinn, T.C.; Chadwick, K.; Margolick, J.; Brookmeyer, R.; et al. Identification of a reservoir for HIV-1 in patients on highly active antiretroviral therapy. Science 1997, 278, 1295–1300. [Google Scholar] [CrossRef]
- Wong, J.K.; Hezareh, M.; Gunthard, H.F.; Havlir, D.V.; Ignacio, C.C.; Spina, C.A.; Richman, D.D. Recovery of replication-competent HIV despite prolonged suppression of plasma viremia. Science 1997, 278, 1291–1295. [Google Scholar] [CrossRef]
- Calza, L. Renal toxicity associated with antiretroviral therapy. HIV Clin. Trials 2012, 13, 189–211. [Google Scholar] [CrossRef]
- Domingo, P.; Estrada, V.; Lopez-Aldeguer, J.; Villaroya, F.; Martinez, E. Fat redistribution syndromes associated with HIV-1 infection and combination antiretroviral therapy. AIDS Rev. 2012, 14, 112–123. [Google Scholar]
- Hester, E.K. HIV medications: An update and review of metabolic complications. Nutr. Clin. Pract. 2012, 27, 51–64. [Google Scholar] [CrossRef]
- Jones, M.; Nunez, M. Liver toxicity of antiretroviral drugs. Semin. Liver Dis. 2012, 32, 167–176. [Google Scholar] [CrossRef]
- Lipshultz, S.E.; Mas, C.M.; Henkel, J.M.; Franco, V.I.; Fisher, S.D.; Miller, T.L. HAART to heart: Highly active antiretroviral therapy and the risk of cardiovascular disease in HIV-infected or exposed children and adults. Expert Rev. Anti-Infect. Ther. 2012, 10, 661–674. [Google Scholar] [CrossRef]
- Hutter, G.; Nowak, D.; Mossner, M.; Ganepola, S.; Mussig, A.; Allers, K.; Schneider, T.; Hofmann, J.; Kucherer, C.; Blau, O.; et al. Long-term control of HIV by CCR5 delta32/delta32 stem-cell transplantation. N. Engl. J. Med. 2009, 360, 692–698. [Google Scholar] [CrossRef]
- Allers, K.; Hutter, G.; Hofmann, J.; Loddenkemper, C.; Rieger, K.; Thiel, E.; Schneider, T. Evidence for the cure of HIV infection by CCR5delta32/delta32 stem cell transplantation. Blood 2011, 117, 2791–2799. [Google Scholar] [CrossRef]
- Yukl, S.A.; Boritz, E.; Busch, M.; Bentsen, C.; Chun, T.W.; Douek, D.; Eisele, E.; Haase, A.; Ho, Y.C.; Hutter, G.; et al. Challenges in detecting HIV persistence during potentially curative interventions: A study of the Berlin patient. PLoS Pathog. 2013, 9, e1003347. [Google Scholar] [CrossRef]
- Henrich, T.J.; Hu, Z.; Li, J.Z.; Sciaranghella, G.; Busch, M.P.; Keating, S.M.; Gallien, S.; Lin, N.H.; Giguel, F.F.; Lavoie, L.; et al. Long-term reduction in peripheral blood HIV-1 reservoirs following reduced-intensity conditioning allogeneic stem cell transplantation. J. Infect. Dis. 2013, 207, 1694–1702. [Google Scholar] [CrossRef]
- Hutter, G.; Zaia, J.A. Allogeneic haematopoietic stem cell transplantation in patients with human immunodeficiency virus: The experiences of more than 25 years. Clin. Exp. Immunol. 2011, 163, 284–295. [Google Scholar] [CrossRef]
- Burnett, J.C.; Zaia, J.A.; Rossi, J.J. Creating genetic resistance to HIV. Curr. Opin. Immunol. 2012, 24, 625–632. [Google Scholar] [CrossRef]
- Holt, N.; Wang, J.; Kim, K.; Friedman, G.; Wang, X.; Taupin, V.; Crooks, G.M.; Kohn, D.B.; Gregory, P.D.; Holmes, M.C.; et al. Human hematopoietic stem/progenitor cells modified by zinc-finger nucleases targeted to CCR5 control HIV-1 in vivo. Nat. Biotechnol. 2010, 28, 839–847. [Google Scholar] [CrossRef]
- Hur, E.M.; Patel, S.N.; Shimizu, S.; Rao, D.S.; Gnanapragasam, P.N.; An, D.S.; Yang, L.; Baltimore, D. Inhibitory effect of HIV-specific neutralizing IgA on mucosal transmission of HIV in humanized mice. Blood 2012, 120, 4571–4582. [Google Scholar] [CrossRef]
- Joseph, A.; Zheng, J.H.; Chen, K.; Dutta, M.; Chen, C.; Stiegler, G.; Kunert, R.; Follenzi, A.; Goldstein, H. Inhibition of in vivo HIV infection in humanized mice by gene therapy of human hematopoietic stem cells with a lentiviral vector encoding a broadly neutralizing anti-HIV antibody. J. Virol. 2010, 84, 6645–6653. [Google Scholar] [CrossRef]
- Ringpis, G.-E.E.; Shimizu, S.; Arokium, H.; Camba-Colón, J.; Carroll, M.V.; Cortado, R.; Xie, Y.; Kim, P.Y.; Sahakyan, A.; Lowe, E.L.; et al. Engineering HIV-1-resistant T-cells from short-hairpin RNA-expressing hematopoietic stem/progenitor cells in humanized BLT mice. PLoS One 2012, 7, e53492. [Google Scholar]
- Vatakis, D.N.; Bristol, G.C.; Kim, S.G.; Levin, B.; Liu, W.; Radu, C.G.; Kitchen, S.G.; Zack, J.A. Using the BLT humanized mouse as a stem cell based gene therapy tumor model. J. Vis. Exp. 2012, ((70)), e4181. [Google Scholar] [CrossRef]
- Kohn, D.B.; Bauer, G.; Rice, C.R.; Rothschild, J.C.; Carbonaro, D.A.; Valdez, P.; Hao, Q.; Zhou, C.; Bahner, I.; Kearns, K.; et al. A clinical trial of retroviral-mediated transfer of a rev-responsive element decoy gene into CD34(+) cells from the bone marrow of human immunodeficiency virus-1-infected children. Blood 1999, 94, 368–371. [Google Scholar]
- Podsakoff, G.M.; Engel, B.C.; Carbonaro, D.A.; Choi, C.; Smogorzewska, E.M.; Bauer, G.; Selander, D.; Csik, S.; Wilson, K.; Betts, M.R.; et al. Selective survival of peripheral blood lymphocytes in children with HIV-1 following delivery of an anti-HIV gene to bone marrow CD34(+) cells. Mol. Ther. 2005, 12, 77–86. [Google Scholar] [CrossRef]
- Amado, R.G.; Mitsuyasu, R.T.; Rosenblatt, J.D.; Ngok, F.K.; Bakker, A.; Cole, S.; Chorn, N.; Lin, L.S.; Bristol, G.; Boyd, M.P.; et al. Anti-human immunodeficiency virus hematopoietic progenitor cell-delivered ribozyme in a phase I study: Myeloid and lymphoid reconstitution in human immunodeficiency virus type-1-infected patients. Hum. Gene Ther. 2004, 15, 251–262. [Google Scholar]
- Mitsuyasu, R.T.; Merigan, T.C.; Carr, A.; Zack, J.A.; Winters, M.A.; Workman, C.; Bloch, M.; Lalezari, J.; Becker, S.; Thornton, L.; et al. Phase 2 gene therapy trial of an anti-HIV ribozyme in autologous CD34+ cells. Nat. Med. 2009, 15, 285–292. [Google Scholar] [CrossRef]
- DiGiusto, D.L.; Krishnan, A.; Li, L.; Li, H.; Li, S.; Rao, A.; Mi, S.; Yam, P.; Stinson, S.; Kalos, M.; et al. RNA-based gene therapy for HIV with lentiviral vector-modified CD34(+) cells in patients undergoing transplantation for AIDS-related lymphoma. Sci. Transl. Med. 2010, 2, 1–8. [Google Scholar]
- Li, M.J.; Kim, J.; Li, S.; Zaia, J.; Yee, J.K.; Anderson, J.; Akkina, R.; Rossi, J.J. Long-term inhibition of HIV-1 infection in primary hematopoietic cells by lentiviral vector delivery of a triple combination of anti-HIV shRNA, anti-CCR5 ribozyme, and a nucleolar-localizing TAR decoy. Mol. Ther. 2005, 12, 900–909. [Google Scholar] [CrossRef]
- Ustek, D.; Sirma, S.; Gumus, E.; Arikan, M.; Cakiris, A.; Abaci, N.; Mathew, J.; Emrence, Z.; Azakli, H.; Cosan, F.; et al. A genome-wide analysis of lentivector integration sites using targeted sequence capture and next generation sequencing technology. Infect. Genet. Evol. 2012, 12, 1349–1354. [Google Scholar] [CrossRef]
- An, D.S.; Qin, F.X.; Auyeung, V.C.; Mao, S.H.; Kung, S.K.; Baltimore, D.; Chen, I.S. Optimization and functional effects of stable short hairpin RNA expression in primary human lymphocytes via lentiviral vectors. Mol. Ther. 2006, 14, 494–504. [Google Scholar] [CrossRef]
- Chung, J.; Zhang, J.; Li, H.; Ouellet, D.L.; DiGiusto, D.L.; Rossi, J.J. Endogenous MCM7 microRNA cluster as a novel platform to multiplex small interfering and nucleolar RNAs for combinational HIV-1 gene therapy. Hum. Gene Ther. 2012, 23, 1200–1208. [Google Scholar] [CrossRef]
- Davis, B.M.; Roth, J.C.; Liu, L.; Xu-Welliver, M.; Pegg, A.E.; Gerson, S.L. Characterization of the P140K, PVP(138-140)MLK, and G156A O6-methylguanine-DNA methyltransferase mutants: Implications for drug resistance gene therapy. Hum. Gene Ther. 1999, 10, 2769–2778. [Google Scholar] [CrossRef]
- Beard, B.C.; Trobridge, G.D.; Ironside, C.; McCune, J.S.; Adair, J.E.; Kiem, H.P. Efficient and stable MGMT-mediated selection of long-term repopulating stem cells in nonhuman primates. J. Clin. Invest. 2010, 120, 2345–2354. [Google Scholar] [CrossRef]
- Chinnasamy, D.; Fairbairn, L.J.; Neuenfeldt, J.; Treisman, J.S.; Hanson, J.P., Jr.; Margison, G.P.; Chinnasamy, N. Lentivirus-mediated expression of mutant MGMTP140K protects human CD34+ cells against the combined toxicity of O6-benzylguanine and 1,3-bis(2-chloroethyl)-nitrosourea or temozolomide. Hum. Gene Ther. 2004, 15, 758–769. [Google Scholar] [CrossRef]
- Gori, J.L.; Beard, B.C.; Ironside, C.; Karponi, G.; Kiem, H.P. In vivo selection of autologous mgmt gene-modified cells following reduced-intensity conditioning with BCNU and temozolomide in the dog model. Cancer Gene Ther. 2012, 19, 523–529. [Google Scholar] [CrossRef]
- Jansen, M.; Sorg, U.R.; Ragg, S.; Flasshove, M.; Seeber, S.; Williams, D.A.; Moritz, T. Hematoprotection and enrichment of transduced cells in vivo after gene transfer of MGMT(P140K) into hematopoietic stem cells. Cancer Gene Ther. 2002, 9, 737–746. [Google Scholar] [CrossRef]
- Neff, T.; Beard, B.C.; Peterson, L.J.; Anandakumar, P.; Thompson, J.; Kiem, H.P. Polyclonal chemoprotection against temozolomide in a large-animal model of drug resistance gene therapy. Blood 2005, 105, 997–1002. [Google Scholar]
- Younan, P.M.; Polacino, P.; Kowalski, J.P.; Peterson, C.W.; Maurice, N.J.; Williams, N.P.; Ho, O.; Trobridge, G.D.; Von Laer, D.; Prlic, M.; et al. Positive selection of mC46-expressing CD4+ T cells and maintenance of virus specific immunity in a primate AIDS model. Blood 2013, 122, 179–187. [Google Scholar] [CrossRef]
- Food and Drug Administration. Structured approach to benefit-risk assessment in drug regulatory decision-making. Draft PDUFA V Implementation Plan - February 2013; Fiscal Years 2013-2017. Available online: http://www.fda.gov/downloads/ForIndustry/UserFees/PrescriptionDrugUserFee/UCM329758.pdf (accessed on 20 November 2013).
- Coplan, P.M.; Noel, R.A.; Levitan, B.S.; Ferguson, J.; Mussen, F. Development of a framework for enhancing the transparency, reproducibility and communication of the benefit-risk balance of medicines. Clin. Pharmacol. Ther. 2011, 89, 312–315. [Google Scholar] [CrossRef]
- Food and Drug Administration. Public meeting on HIV patient-focused drug development and HIV cure research. 2013. Available online: http://www.fda.gov/ForIndustry/UserFees/PrescriptionDrugUserFee/ucm348598.htm (accessed on 20 November 2013).
- National Institutes of Health. AIDSinfo. Federally approved HIV/AIDS medical practice guidelines. 2013. Available online: http://aidsinfo.nih.gov/guidelines (accessed on 20 November 2013).
- Lewden, C.; Chene, G.; Morlat, P.; Raffi, F.; Dupon, M.; Dellamonica, P.; Pellegrin, J.L.; Katlama, C.; Dabis, F.; Leport, C.; et al. HIV-infected adults with a CD4 cell count greater than 500 cells/mm3 on long-term combination antiretroviral therapy reach same mortality rates as the general population. J. Acquir. Immune Defic. Syndr. 2007, 46, 72–77. [Google Scholar] [CrossRef]
- Engsig, F.N.; Gerstoft, J.; Kronborg, G.; Larsen, C.S.; Pedersen, G.; Roge, B.; Jensen, J.; Nielsen, L.N.; Obel, N. Long-term mortality in HIV patients virally suppressed for more than three years with incomplete CD4 recovery: A cohort study. BMC Infect. Dis. 2010, 10, 318. [Google Scholar] [CrossRef] [Green Version]
- Gaardbo, J.C.; Hartling, H.J.; Gerstoft, J.; Nielsen, S.D. Incomplete immune recovery in HIV infection: Mechanisms, relevance for clinical care, and possible solutions. Clin. Dev. Immunol. 2012, 2012, 670957. [Google Scholar]
- Piketty, C.; Weiss, L.; Thomas, F.; Mohamed, A.S.; Belec, L.; Kazatchkine, M.D. Long-term clinical outcome of human immunodeficiency virus-infected patients with discordant immunologic and virologic responses to a protease inhibitor-containing regimen. J. Infect. Dis. 2001, 183, 1328–1335. [Google Scholar] [CrossRef]
- Re, A.; Cattaneo, C.; Skert, C.; Balsalobre, P.; Michieli, M.; Bower, M.; Ferreri, A.J.; Hentrich, M.; Ribera, J.M.; Allione, B.; et al. Stem cell mobilization in HIV seropositive patients with lymphoma. Haematologica 2013, 98, 1762–1768. [Google Scholar] [CrossRef]
- Krishnan, A.; Bhatia, S.; Slovak, M.L.; Arber, D.A.; Niland, J.C.; Nademanee, A.; Fung, H.; Bhatia, R.; Kashyap, A.; Molina, A.; et al. Predictors of therapy-related leukemia and myelodysplasia following autologous transplantation for lymphoma: An assessment of risk factors. Blood 2000, 95, 1588–1593. [Google Scholar]
- Agarwal, D.; Chakravarty, J.; Chaube, L.; Rai, M.; Agrawal, N.R.; Sundar, S. High incidence of zidovudine induced anaemia in HIV infected patients in Eastern India. Indian J. Med. Res. 2010, 132, 386–389. [Google Scholar]
- Costantini, A.; Giuliodoro, S.; Mancini, S.; Butini, L.; Regnery, C.M.; Silvestri, G.; Greco, F.; Leoni, P.; Montroni, M. Impaired in-vitro growth of megakaryocytic colonies derived from CD34 cells of HIV-1-infected patients with active viral replication. AIDS 2006, 20, 1713–1720. [Google Scholar] [CrossRef]
- Isgro, A.; Leti, W.; De Santis, W.; Marziali, M.; Esposito, A.; Fimiani, C.; Luzi, G.; Pinti, M.; Cossarizza, A.; Aiuti, F.; et al. Altered clonogenic capability and stromal cell function characterize bone marrow of HIV-infected subjects with low CD4+ T cell counts despite viral suppression during HAART. Clin. Infect. Dis. 2008, 46, 1902–1910. [Google Scholar] [CrossRef]
- Thiebot, H.; Louache, F.; Vaslin, B.; de Revel, T.; Neildez, O.; Larghero, J.; Vainchenker, W.; Dormont, D.; Le Grand, R. Early and persistent bone marrow hematopoiesis defect in simian/human immunodeficiency virus-infected macaques despite efficient reduction of viremia by highly active antiretroviral therapy during primary infection. J. Virol. 2001, 75, 11594–11602. [Google Scholar] [CrossRef]
- Costantini, A.; Giuliodoro, S.; Butini, L.; Silvestri, G.; Leoni, P.; Montroni, M. Abnormalities of erythropoiesis during HIV-1 disease: A longitudinal analysis. J. Acquir. Immune Defic. Syndr. 2009, 52, 70–74. [Google Scholar] [CrossRef]
- Costantini, A.; Giuliodoro, S.; Butini, L.; Silvestri, G.; Leoni, P.; Montroni, M. HIV-induced abnormalities in myelopoiesis and their recovery following antiretroviral therapy. Curr. HIV Res. 2010, 8, 336–339. [Google Scholar] [CrossRef]
- Sauce, D.; Larsen, M.; Fastenackels, S.; Pauchard, M.; Ait-Mohand, H.; Schneider, L.; Guihot, A.; Boufassa, F.; Zaunders, J.; Iguertsira, M.; et al. HIV disease progression despite suppression of viral replication is associated with exhaustion of lymphopoiesis. Blood 2011, 117, 5142–5151. [Google Scholar] [CrossRef]
- Krishnan, A. HIV-infected patients. Biol. Blood Marrow Transplant 2009, 15, 142–145. [Google Scholar] [CrossRef]
- Krishnan, A.; Molina, A.; Zaia, J.; Smith, D.; Vasquez, D.; Kogut, N.; Falk, P.M.; Rosenthal, J.; Alvarnas, J.; Forman, S.J. Durable remissions with autologous stem cell transplantation for high-risk HIV-associated lymphomas. Blood 2005, 105, 874–878. [Google Scholar] [CrossRef]
- Schooley, R.T.; Mladenovic, J.; Sevin, A.; Chiu, S.; Miles, S.A.; Pomerantz, R.J.; Campbell, T.B.; Bell, D.; Ambruso, D.; Wong, R.; et al. Reduced mobilization of CD34+ stem cells in advanced human immunodeficiency virus type 1 disease. J. Infect. Dis. 2000, 181, 148–157. [Google Scholar] [CrossRef]
- Keating, G.M. Plerixafor: A review of its use in stem-cell mobilization in patients with lymphoma or multiple myeloma. Drugs 2011, 71, 1623–1647. [Google Scholar] [CrossRef]
- McNamara, L.A.; Ganesh, J.A.; Collins, K.L. Latent HIV-1 infection occurs in multiple subsets of hematopoietic progenitor cells and is reversed by NF-kappaB activation. J. Virol. 2012, 86, 9337–9350. [Google Scholar] [CrossRef]
- Nixon, C.C.; Vatakis, D.N.; Reichelderfer, S.N.; Dixit, D.; Kim, S.G.; Uittenbogaart, C.H.; Zack, J.A. HIV-1 infection of hematopoietic progenitor cells in vivo in humanized mice. Blood 2013, 122, 2195–2204. [Google Scholar] [CrossRef]
- Onafuwa-Nuga, A.; McNamara, L.A.; Collins, K.L. Towards a cure for HIV: The identification and characterization of HIV reservoirs in optimally treated people. Cell Res. 2010, 20, 1185–1187. [Google Scholar] [CrossRef]
- Eisele, E.; Siliciano, R.F. Redefining the viral reservoirs that prevent HIV-1 eradication. Immunity 2012, 37, 377–388. [Google Scholar] [CrossRef]
- McNamara, L.A.; Onafuwa-Nuga, A.; Sebastian, N.T.; Riddell, J.T.; Bixby, D.; Collins, K.L. CD133+ hematopoietic progenitor cells harbor HIV genomes in a subset of optimally treated people with long-term viral suppression. J. Infect. Dis. 2013, 207, 1807–1816. [Google Scholar] [CrossRef]
- Sachdeva, M.; Fischl, M.A.; Pahwa, R.; Sachdeva, N.; Pahwa, S. Immune exhaustion occurs concomitantly with immune activation and decrease in regulatory T cells in viremic chronically HIV-1-infected patients. J. Acquir. Immune. Defic. Syndr. 2010, 54, 447–454. [Google Scholar] [CrossRef]
- Biffi, A.; Montini, E.; Lorioli, L.; Cesani, M.; Fumagalli, F.; Plati, T.; Baldoli, C.; Martino, S.; Calabria, A.; Canale, S.; et al. Lentiviral hematopoietic stem cell gene therapy benefits metachromatic leukodystrophy. Science 2013, 341, 1233158. [Google Scholar] [CrossRef]
- Aiuti, A.; Biasco, L.; Scaramuzza, S.; Ferrua, F.; Cicalese, M.P.; Baricordi, C.; Dionisio, F.; Calabria, A.; Giannelli, S.; Castiello, M.C.; et al. Lentiviral hematopoietic stem cell gene therapy in patients with Wiskott-Aldrich syndrome. Science 2013, 341, 1233151. [Google Scholar] [CrossRef]
- Candotti, F.; Shaw, K.L.; Muul, L.; Carbonaro, D.; Sokolic, R.; Choi, C.; Schurman, S.H.; Garabedian, E.; Kesserwan, C.; Jagadeesh, G.J.; et al. Gene therapy for adenosine deaminase-deficient severe combined immune deficiency: Clinical comparison of retroviral vectors and treatment plans. Blood 2012, 120, 3635–3646. [Google Scholar] [CrossRef]
- Gutierrez-Aguirre, C.H.; Gomez-Almaguer, D.; Cantu-Rodriguez, O.G.; Gonzalez-Llano, O.; Jaime-Perez, J.C.; Herena-Perez, S.; Manzano, C.A.; Estrada-Gomez, R.; Gonzalez-Carrillo, M.L.; Ruiz-Arguelles, G.J. Non-myeloablative stem cell transplantation in patients with relapsed acute lymphoblastic leukemia: Results of a multicenter study. Bone Marrow Transplant. 2007, 40, 535–539. [Google Scholar]
- Slavin, S.; Nagler, A.; Naparstek, E.; Kapelushnik, Y.; Aker, M.; Cividalli, G.; Varadi, G.; Kirschbaum, M.; Ackerstein, A.; Samuel, S.; et al. Nonmyeloablative stem cell transplantation and cell therapy as an alternative to conventional bone marrow transplantation with lethal cytoreduction for the treatment of malignant and nonmalignant hematologic diseases. Blood 1998, 91, 756–763. [Google Scholar]
- Aiuti, A.; Cattaneo, F.; Galimberti, S.; Benninghoff, U.; Cassani, B.; Callegaro, L.; Scaramuzza, S.; Andolfi, G.; Mirolo, M.; Brigida, I.; et al. Gene therapy for immunodeficiency due to adenosine deaminase deficiency. N. Engl. J. Med. 2009, 360, 447–458. [Google Scholar] [CrossRef]
- Aiuti, A.; Slavin, S.; Aker, M.; Ficara, F.; Deola, S.; Mortellaro, A.; Morecki, S.; Andolfi, G.; Tabucchi, A.; Carlucci, F.; et al. Correction of ADA-SCID by stem cell gene therapy combined with nonmyeloablative conditioning. Science 2002, 296, 2410–2413. [Google Scholar] [CrossRef]
- Kang, E.M.; Choi, U.; Theobald, N.; Linton, G.; Long Priel, D.A.; Kuhns, D.; Malech, H.L. Retrovirus gene therapy for X-linked chronic granulomatous disease can achieve stable long-term correction of oxidase activity in peripheral blood neutrophils. Blood 2010, 115, 783–791. [Google Scholar]
- Kang, H.J.; Bartholomae, C.C.; Paruzynski, A.; Arens, A.; Kim, S.; Yu, S.S.; Hong, Y.; Joo, C.W.; Yoon, N.K.; Rhim, J.W.; et al. Retroviral gene therapy for X-linked chronic granulomatous disease: Results from phase I/II trial. Mol. Ther. 2011, 19, 2092–2101. [Google Scholar] [CrossRef]
- Ott, M.G.; Schmidt, M.; Schwarzwaelder, K.; Stein, S.; Siler, U.; Koehl, U.; Glimm, H.; Kuhlcke, K.; Schilz, A.; Kunkel, H.; et al. Correction of X-linked chronic granulomatous disease by gene therapy, augmented by insertional activation of MDS1-EVI1, PRDM16 or SETBP1. Nat. Med. 2006, 12, 401–409. [Google Scholar] [CrossRef]
- Lafeuillade, A. Potential strategies for an HIV infection cure. HIV Clin. Trials 2011, 12, 121–130. [Google Scholar] [CrossRef]
- Kiem, H.P.; Jerome, K.R.; Deeks, S.G.; McCune, J.M. Hematopoietic-stem-cell-based gene therapy for HIV disease. Cell Stem Cell 2012, 10, 137–147. [Google Scholar]
- Khera, N.; Zeliadt, S.B.; Lee, S.J. Economics of hematopoietic cell transplantation. Blood 2012, 120, 1545–1551. [Google Scholar] [CrossRef]
- Sloan, C.E.; Champenois, K.; Choisy, P.; Losina, E.; Walensky, R.P.; Schackman, B.R.; Ajana, F.; Melliez, H.; Paltiel, A.D.; Freedberg, K.A.; et al. Newer drugs and earlier treatment: Impact on lifetime cost of care for HIV-infected adults. AIDS 2012, 26, 45–56. [Google Scholar] [CrossRef]
- Walensky, R.P.; Sax, P.E.; Nakamura, Y.M.; Weinstein, M.C.; Pei, P.P.; Freedberg, K.A.; Paltiel, A.D.; Schackman, B.R. Economic savings versus health losses: The cost-effectiveness of generic antiretroviral therapy in the United States. Ann. Intern Med. 2013, 158, 84–92. [Google Scholar] [CrossRef]
- Gabriel, R.; Eckenberg, R.; Paruzynski, A.; Bartholomae, C.C.; Nowrouzi, A.; Arens, A.; Howe, S.J.; Recchia, A.; Cattoglio, C.; Wang, W.; et al. Comprehensive genomic access to vector integration in clinical gene therapy. Nat. Med. 2009, 15, 1431–1436. [Google Scholar] [CrossRef]
- Drmanac, R.; Drmanac, S.; Baier, J.; Chui, G.; Coleman, D.; Diaz, R.; Gietzen, D.; Hou, A.; Jin, H.; Ukrainczyk, T.; et al. DNA sequencing by hybridization with arrays of samples or probes. Methods Mol. Biol. 2001, 170, 173–179. [Google Scholar]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
DiGiusto, D.L.; Stan, R.; Krishnan, A.; Li, H.; Rossi, J.J.; Zaia, J.A. Development of Hematopoietic Stem Cell Based Gene Therapy for HIV-1 Infection: Considerations for Proof of Concept Studies and Translation to Standard Medical Practice. Viruses 2013, 5, 2898-2919. https://doi.org/10.3390/v5112898
DiGiusto DL, Stan R, Krishnan A, Li H, Rossi JJ, Zaia JA. Development of Hematopoietic Stem Cell Based Gene Therapy for HIV-1 Infection: Considerations for Proof of Concept Studies and Translation to Standard Medical Practice. Viruses. 2013; 5(11):2898-2919. https://doi.org/10.3390/v5112898
Chicago/Turabian StyleDiGiusto, David L., Rodica Stan, Amrita Krishnan, Haitang Li, John J. Rossi, and John A. Zaia. 2013. "Development of Hematopoietic Stem Cell Based Gene Therapy for HIV-1 Infection: Considerations for Proof of Concept Studies and Translation to Standard Medical Practice" Viruses 5, no. 11: 2898-2919. https://doi.org/10.3390/v5112898