Approaches for Identification of HIV-1 Entry Inhibitors Targeting gp41 Pocket




Abstract
:1. Introduction
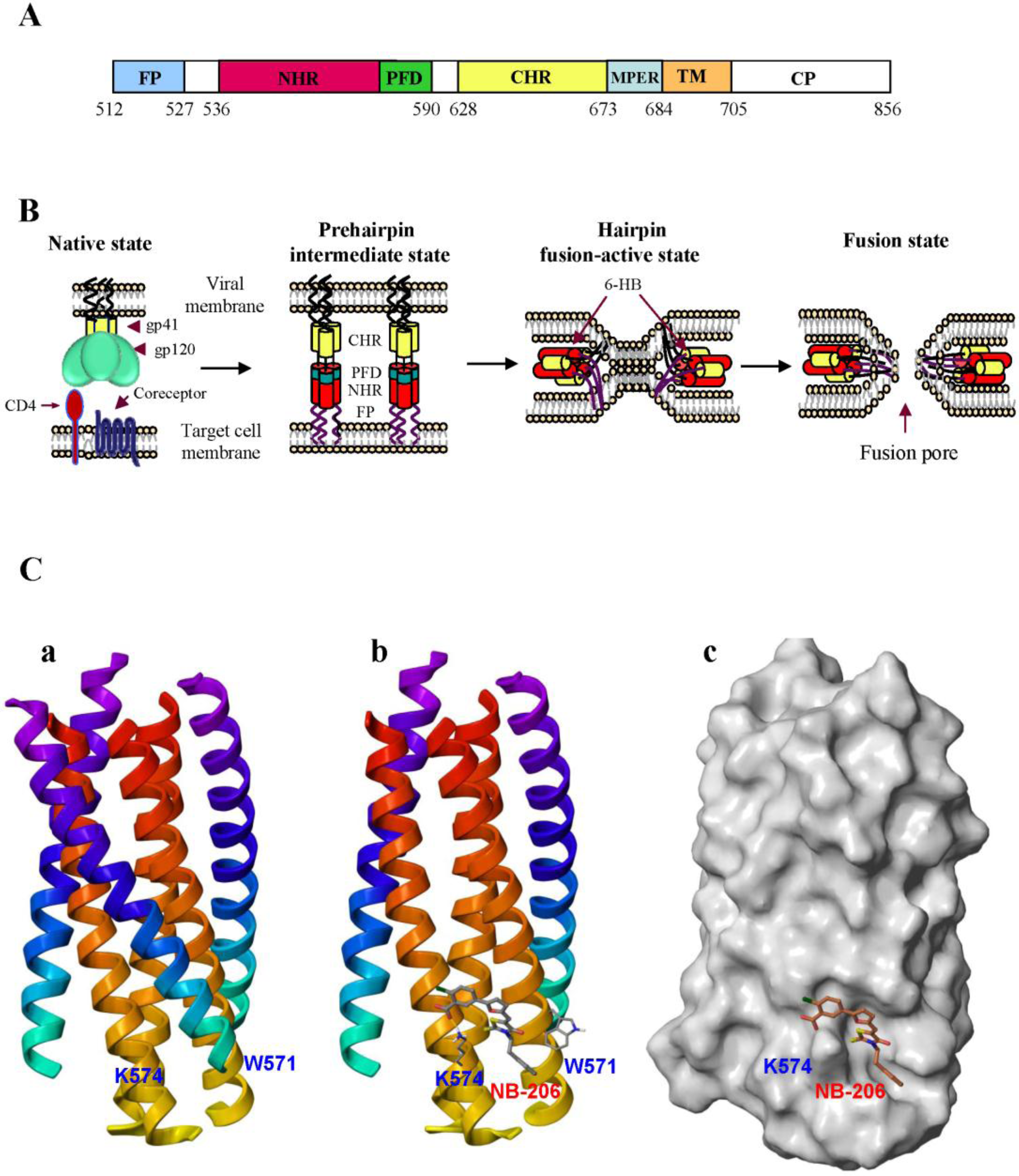
2. Development of HIV Entry Inhibitor Peptides Targeting to gp41
2.1. Development of Biophysical Methods for Identification of Inhibitors Against gp41 6-HB Formation
2.2. Peptides Targeting gp41 NHR-Trimer and Its Pocket
2.3. Rational Design of Peptides Targeting gp41 CHR-Helices
3. Computer Modeling-Based Virtual Screening of HIV-1 Fusion Inhibitors Targeting gp41 Pocket
3.1. Introduction of Computer-Aided Molecular Docking Techniques
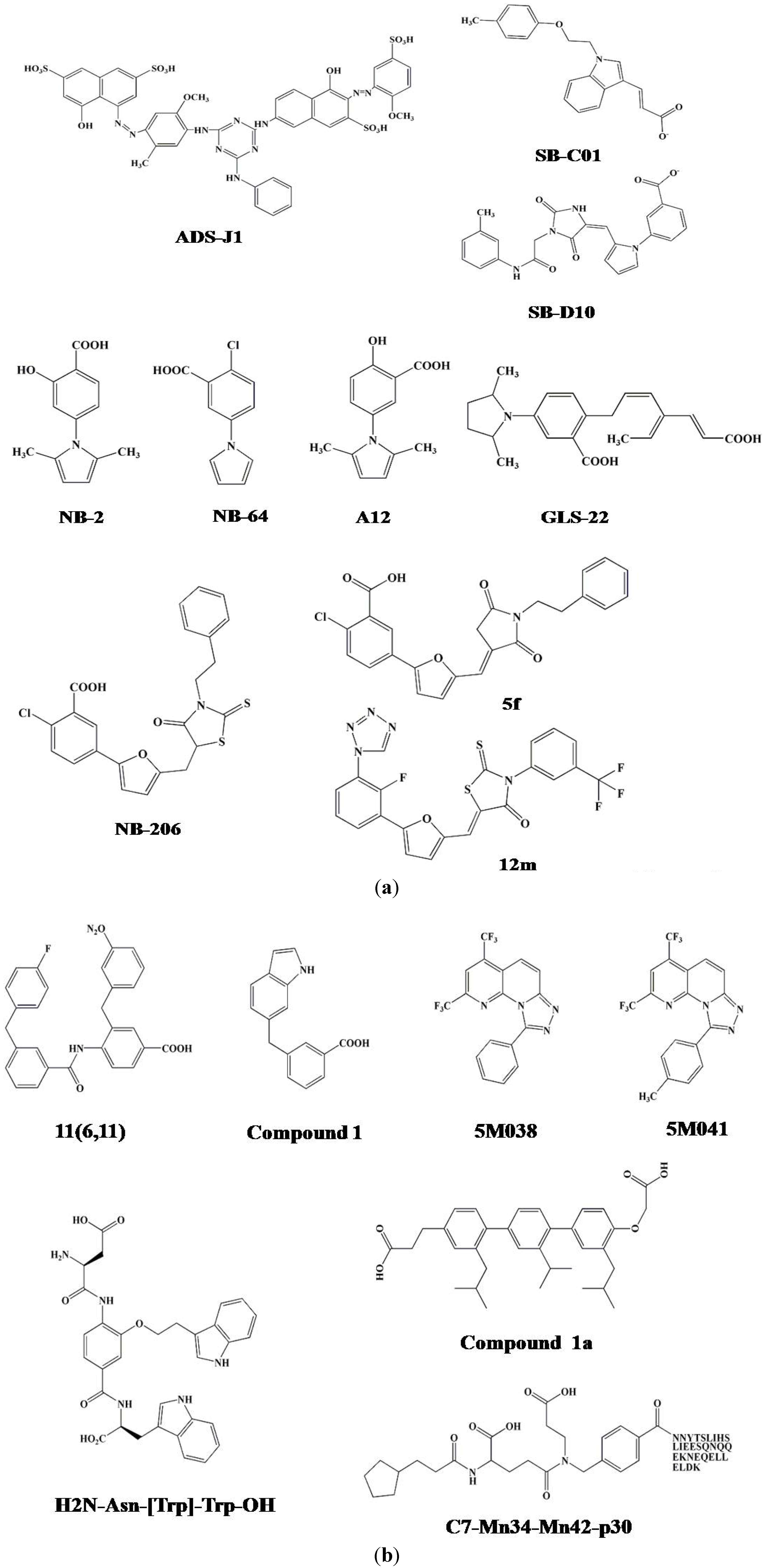
3.2. Identification of ADS-J1 and Derivatives from ComGenex Database Using the Virtual Screening Program DOCK3.5
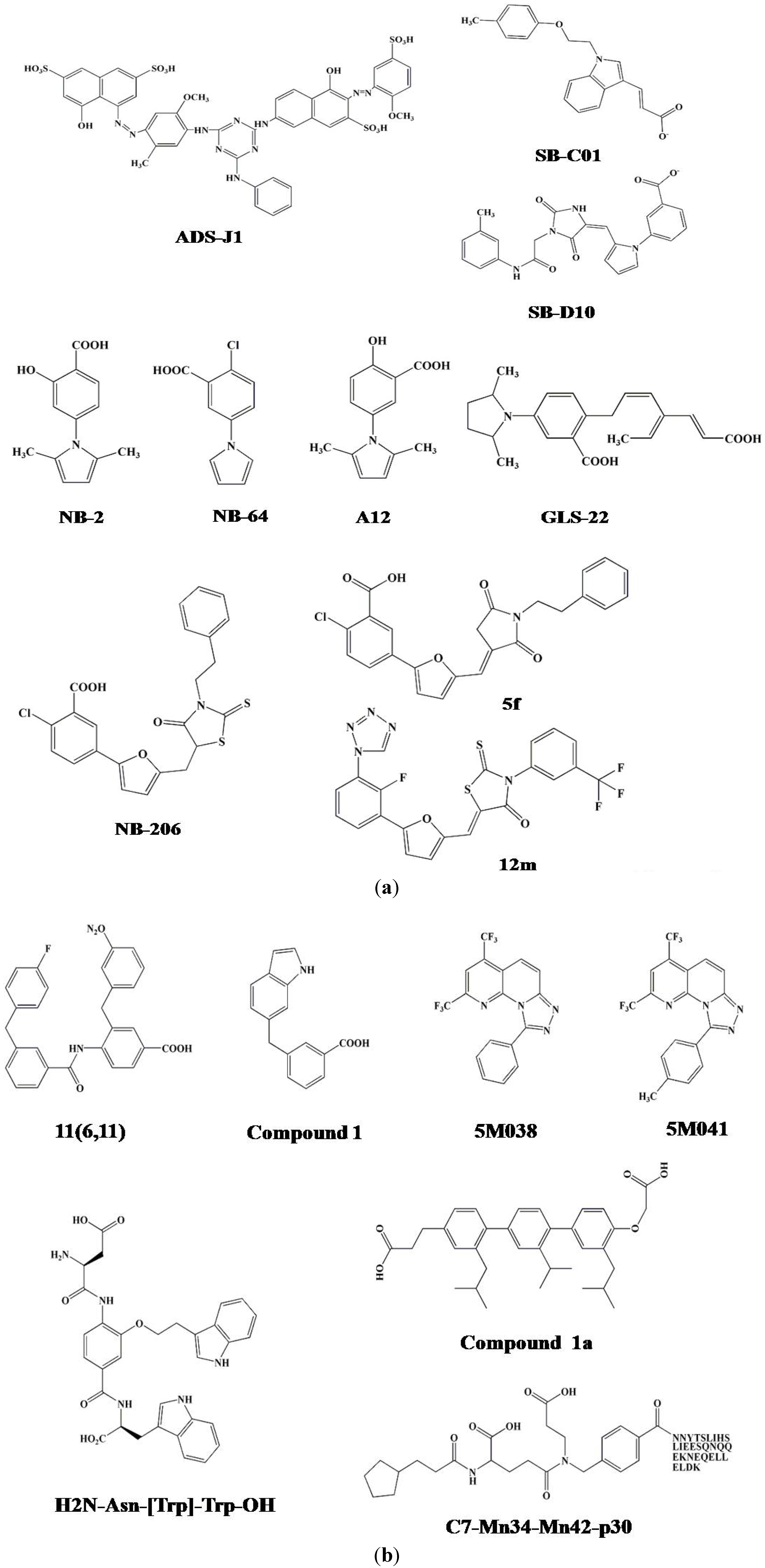
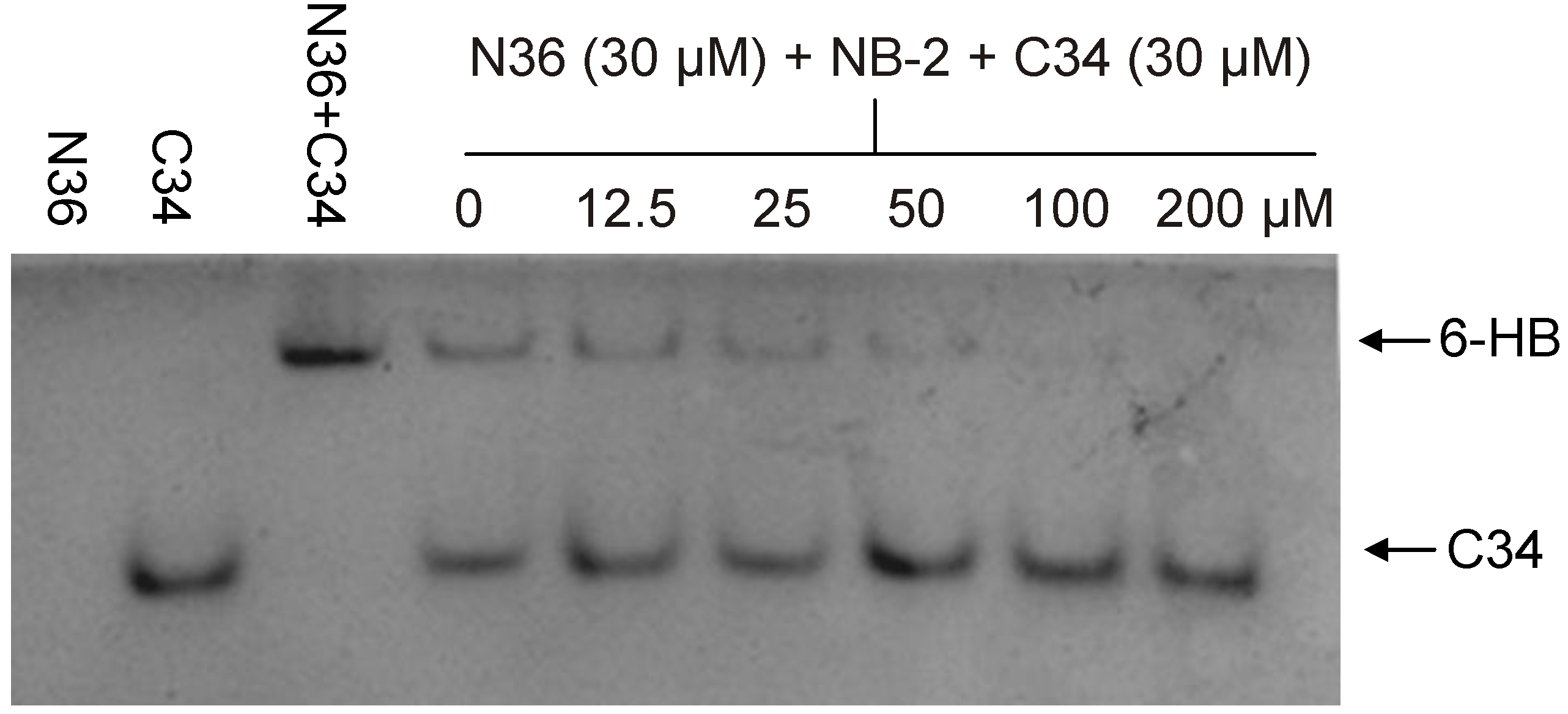
3.3. Identification of SB-D10, SB-C01 and their Derivatives from the ZINC Database Using the Virtual Screening Program DOCK6.5
4. ELISA- and FLISA-Based High-Throughput Screening (HTS) Assays for Identification of HIV-1 Fusion Inhibitors Targeting gp41 Pocket
4.1. Sandwich ELISA, Direct ELISA, and Fluorescence-Linked Immunosorbent Assay (FLISA)
5. Metal-Ion-Based NHR-Trimer as the Target for Screening of HIV-1 Fusion Inhibitors Targeting gp41 Pocket
6. Five-Helix-Based HTS Assay to Screen HIV-1 Fusion Inhibitors Targeting gp41 Pocket
7. Alpha-Helix Mimicry-Based Design or HTS Assay for Identification of HIV-1 Fusion Inhibitors Targeting gp41 Pocket

{kind=link}
{kind=link}
{kind=link}
| Inhibitors | IC50 (μM) | CC50 | SI | Ref. | ||
|---|---|---|---|---|---|---|
| Inhibiting 6-HB formation | Inhibiting p24 production | Inhibiting Cell-cell fusion | ||||
| Computer modeling-based virtual screening | ||||||
| ADS-J1 | 0.73 | 8.29 | 4.95 | 292.16 | 35.24 | [51] |
| ADS-J2 | 3.18 | 30.76 | 21.85 | 289.94 | 9.43 | [51] |
| SB-D10 | 160 | >100 | [57] | |||
| SC-01 | 43 | >50 | [57] | |||
| ELISA- or FLISA-based high-throughput screening (HTS) assays | ||||||
| NB-2 | 1.04 | 6.74 | 2755 | [14] | ||
| NB-64 | 58.74 | 2.21 | 29.92 | >4000 | [14] | |
| A12 | 29.39-37.36 | 0.69-28.19 | 43.24 | 133.5-333.8 | [66] | |
| GLS-22 | 20.73 | 4.91 | 3.60 | 255.28 | [67] | |
| GLS-23 | 21.75 | 8.30 | 8.30 | 227.27 | [67] | |
| NB-293(11a) | 0.098 | 19.35 | 440 | [68] | ||
| 11b | 0.031 | 42.14 | 426 | [68] | ||
| NB-206 (11d) | 0.017 | 16.82 | 330 | [68] | ||
| 12l | 0.018-0.32 | 66.34 | 3686 | [70] | ||
| 12m | 0.014-0.99 | 27.85 | 1989 | [70] | ||
| 5f | 15.55 | 6.90 | 43.55 | 118.45 | [69] | |
| 5h | 12.89 | 16.79 | 45.92 | >320 | [69] | |
| Fluorescence resonance energy transfer (FRET) using metal ion | ||||||
| 11(6,11) | 8 | [73] | ||||
| Compound 1 | 3.2 | >500 | [80] | |||
| Fluorescence polarization assay | ||||||
| 5M038 | 19 | 38 | [82] | |||
| 5M041 | 18 | 18 | [82] | |||
| alpha-helical mimicry | ||||||
| Compound 1a | 13.18 | 15.70 | [83] | |||
| H2N-Asn-[Trp]-Trp-OH | 6 | [85] | ||||
| H2N-Trp-[Trp]-Leu-OH | 5 | [85] | ||||
| H2N-TyrMe-[Trp]-Trp-OH | 8 | [85] | ||||
| Structure-based combinatorial approach | ||||||
| C7-Mn34-Mn42-p30 | 0.3 | [86] | ||||
8. Synergistic Combinations of Multiple HIV Entry Inhibitors
9. Conclusions
References
- Cohen, M.S.; Hellmann, N.; Levy, J.A.; DeCock, K.; Lange, J. The spread, treatment, and prevention of HIV-1: Evolution of a global pandemic. J. Clin. Invest. 2008, 118, 1244–1254. [Google Scholar] [CrossRef]
- Kilby, J.M.; Hopkins, S.; Venetta, T.M.; DiMassimo, B.; Cloud, G.A.; Lee, J.Y.; Alldredge, L.; Hunter, E.; Lambert, D.; Bolognesi, D.; et al. Potent suppression of HIV-1 replication in humans by T-20, a peptide inhibitor of gp41-mediated virus entry. Nat. Med. 1998, 4, 1302–1307. [Google Scholar]
- Jiang, S.; Zhao, Q.; Debnath, A.K. Peptide and non-peptide HIV fusion inhibitors. Curr. Pharm. Des. 2002, 8, 563–580. [Google Scholar] [CrossRef]
- Allan, J.S.; Coligan, J.E.; Barin, F.; McLane, M.F.; Sodroski, J.G.; Rosen, C.A.; Haseltine, W.A.; Lee, T.H.; Essex, M. Major glycoprotein antigens that induce antibodies in AIDS patients are encoded by HTLV-III. Science 1985, 228, 1091–1094. [Google Scholar]
- Moore, J.P.; Jameson, B.A.; Weiss, R.A.; Sattentau, Q.J. The HIV-Cell Fusion Reaction. In Viral Fusion Mechanisms; Bentz, J., Ed.; CRC Press: Boca Raton, FL, USA, 1993; pp. 233–289. [Google Scholar]
- Berger, E.A. HIV entry and tropism: the chemokine receptor connection. AIDS 1997, 11 Suppl. A, S3–S16. [Google Scholar]
- Chan, D.C.; Kim, P.S. HIV entry and its inhibition. Cell 1998, 93, 681–684. [Google Scholar] [CrossRef]
- Chan, D.C.; Chutkowski, C.T.; Kim, P.S. Evidence that a prominent cavity in the coiled coil of HIV type 1 gp41 is an attractive drug target. Proc. Natl. Acad. Sci. USA 1998, 95, 15613–15617. [Google Scholar]
- Ji, H.; Shu, W.; Burling, T.; Jiang, S.; Lu, M. Inhibition of HIV-1 infectivity by the gp41 core: Role of a conserved hydrophobic cavity in membrane fusion. J. Virol. 1999, 73, 8578–8586. [Google Scholar]
- Lu, M.; Blacklow, S.C.; Kim, P.S. A trimeric structural domain of the HIV-1 transmembrane glycoprotein. Nat. Struct. Biol. 1995, 2, 1075–1082. [Google Scholar] [CrossRef]
- Lu, M.; Kim, P.S. A trimeric structural subdomain of the HIV-1 transmembrane glycoprotein. J. Biomol. Struct. Dyn. 1997, 15, 465–471. [Google Scholar] [CrossRef]
- Liu, S.; Zhao, Q.; Jiang, S. Determination of the HIV-1 gp41 fusogenic core conformation modeled by synthetic peptides: applicable for identification of HIV-1 fusion inhibitors. Peptides 2003, 24, 1303–1313. [Google Scholar] [CrossRef]
- Liu, S.; Lu, H.; Niu, J.; Xu, Y.; Wu, S.; Jiang, S. Different from the HIV fusion inhibitor C34, the anti-HIV drug Fuzeon (T-20) inhibits HIV-1 entry by targeting multiple sites in gp41 and gp120. J. Biol. Chem. 2005, 280, 11259–11273. [Google Scholar]
- Jiang, S.; Lu, H.; Liu, S.; Zhao, Q.; He, Y.; Debnath, A.K. N-substituted pyrrole derivatives as novel human immunodeficiency virus type 1 entry inhibitors that interfere with the gp41 six-helix bundle formation and block virus fusion. Antimicrob. Agents Chemother. 2004, 48, 4349–4359. [Google Scholar] [CrossRef]
- Wang, H.; Qi, Z.; Guo, A.; Mao, Q.; Lu, H.; An, X.; Xia, C. ADS-J1 inhibits human immunodeficiency virus type 1 entry by interacting with the gp41 pocket region and blocking fusion-active gp41 core formation. Antimicrob. Agents Chemother. 2009, 53, 4987–4998. [Google Scholar] [CrossRef]
- Jiang, S.; Lin, K.; Strick, N.; Neurath, A.R. HIV-1 inhibition by a peptide. Nature 1993, 365, 113. [Google Scholar]
- Jiang, S.; Lin, K.; Strick, N.; Neurath, A.R. Inhibition of HIV-1 infection by a fusion domain binding peptide from HIV-1 envelope glycoprotein gp41. Biochem. Biophys. Res. Commun. 1993, 195, 533–538. [Google Scholar] [CrossRef]
- Wild, C.; Dubay, J.W.; Greenwell, T.; Baird, T., Jr.; Oas, T.G.; McDanal, C.; Hunter, E.; Matthews, T. Propensity for a leucine zipper-like domain of human immunodeficiency virus type 1 gp41 to form oligomers correlates with a role in virus-induced fusion rather than assembly of the glycoprotein complex. Proc. Natl. Acad. Sci. USA 1994, 91, 12676–12680. [Google Scholar]
- Lalezari, J.P.; Henry, K.; O'Hearn, M.; Montaner, J.S.; Piliero, P.J.; Trottier, B.; Walmsley, S.;Cohen, C; Kuritzkes, D.R.; Eron, J.J.; et al. Enfuvirtide, an HIV-1 fusion inhibitor, for drug-resistant HIV infection in north and south America. N. Engl. J. Med. 2003, 348, 2175–2185. [Google Scholar] [CrossRef]
- Lazzarin, A.; Clotet, B.; Cooper, D.; Reynes, J.; Arasteh, K.; Nelson, M.; Katlama, C.; Stellbrink, H.J.; Delfraissy, J.F.; Lange, J.; et al. Efficacy of enfuvirtide in patients infected with drug-resistant HIV-1 in Europe and Australia. N. Engl. J. Med. 2003, 348, 2186–2195. [Google Scholar] [CrossRef]
- Walmsley, S.; Henry, K.; Katlama, C.; Nelson, M.; Castagna, A.; Reynes, J.; Clotet, B.; Hui, J.; Salgo, M.; Demasi, R.; et al. Enfuvirtide (T-20) cross-reactive glycoprotein 41 antibody does not impair the efficacy or safety of enfuvirtide. J. Infect Dis. 2003, 188, 1827–1833. [Google Scholar] [CrossRef]
- Hardy, H.; Skolnik, P.R. Enfuvirtide, a new fusion inhibitor for therapy of human immunodeficiency virus infection. Pharmacotherapy 2004, 24, 198–211. [Google Scholar] [CrossRef]
- Lu, J.; Deeks, S.G.; Hoh, R.; Beatty, G.; Kuritzkes, B.A.; Martin, J.N.; Kuritzkes, D.R. Rapid emergence of enfuvirtide resistance in HIV-1-infected patients: Results of a clonal analysis. J. Acquir. Immune. Defic. Syndr. 2006, 43, 60–64. [Google Scholar] [CrossRef]
- Rimsky, L.T.; Shugars, D.C.; Matthews, T.J. Determinants of human immunodeficiency virus type 1 resistance to gp41-derived inhibitory peptides. J. Virol. 1998, 72, 986–993. [Google Scholar]
- Poveda, E.; Briz, V.; Soriano, V. Enfuvirtide, the first fusion inhibitor to treat HIV infection. AIDS Rev. 2005, 7, 139–147. [Google Scholar]
- Lalezari, J.P.; Bellos, N.C.; Sathasivam, K.; Richmond, G.J.; Cohen, C.J.; Myers, R.A., Jr.; Henry, D.H.; Raskino, C.; Melby, T.; Murchison, H.; et al. T-1249 retains potent antiretroviral activity in patients who had experienced virological failure while on an enfuvirtide-containing treatment regimen. J. Infect. Dis. 2005, 191, 1155–1163. [Google Scholar] [CrossRef]
- Martin-Carbonero, L. Discontinuation of the clinical development of fusion inhibitor T-1249. AIDS Rev. 2004, 6, 61. [Google Scholar]
- Dwyer, J.J.; Wilson, K.L.; Davison, D.K.; Freel, S.A.; Seedorff, J.E.; Wring, S.A.; Tvermoes, N.A.; Matthews, T.J.; Greenberg, M.L.; Delmedico, M.K. Design of helical, oligomeric HIV-1 fusion inhibitor peptides with potent activity against enfuvirtide-resistant virus. Proc. Natl. Acad. Sci. USA 2007, 104, 12772–12777. [Google Scholar]
- Sista, P.R.; Melby, T.; Davison, D.; Jin, L.; Mosier, S.; Mink, M.; Nelson, E.L.; Demasi, R.; Cammack, N.; Salgo, M.P.; et al. Characterization of determinants of genotypic and phenotypic resistance to enfuvirtide in baseline and on-treatment HIV-1 isolates. AIDS 2004, 18, 1787–1794. [Google Scholar]
- Eggink, D.; Baldwin, C.E.; Deng, Y.; Langedijk, J.P.; Lu, M.; Sanders, R.W.; Berkhout, B. Selection of T1249-resistant human immunodeficiency virus type 1 variants. J. Virol. 2008, 82, 6678–6688. [Google Scholar] [CrossRef]
- He, Y.; Cheng, J.; Li, J.; Qi, Z.; Lu, H.; Dong, M.; Jiang, S.; Dai, Q. Identification of a critical motif for the human immunodeficiency virus type 1 (HIV-1) gp41 core structure: implications for designing novel anti-HIV fusion inhibitors. J. Virol. 2008, 82, 6349–6358. [Google Scholar]
- He, Y.; Cheng, J.; Lu, H.; Li, J.; Hu, J.; Qi, Z.; Liu, Z.; Jiang, S.; Dai, Q. Potent HIV fusion inhibitors against Enfuvirtide-resistant HIV-1 strains. Proc. Natl. Acad. Sci. USA 2008, 105, 16332–16337. [Google Scholar]
- Yu, X.; Lu, L.; Cai, L.; Tong, P.; Tan, S.; Zou, P.; Meng, F.; Chen, Y.H.; Jiang, S. Mutations of Gln64 in the HIV-1 gp41 N-terminal heptad repeat render viruses resistant to peptide HIV fusion inhibitors targeting the gp41 pocket. J. Virol. 2012, 86, 589–593. [Google Scholar] [CrossRef]
- Wang, R.R.; Yang, L.M.; Wang, Y.H.; Pang, W.; Tam, S.C.; Tien, P.; Zheng, Y.T. Sifuvirtide, a potent HIV fusion inhibitor peptide. Biochem. Biophys. Res. Commun. 2009, 382, 540–544. [Google Scholar] [CrossRef]
- Dai, S.J.; Dou, G.F.; Qiang, X.H.; Song, H.F.; Tang, Z.M.; Liu, D.S.; Liu, X.W.; Yang, L.M.; Zheng, Y.T.; Liang, Q. Pharmacokinetics of sifuvirtide, a novel anti-HIV-1 peptide, in monkeys and its inhibitory concentration in vitro. Acta. Pharmacol. Sin. 2005, 26, 1274–1280. [Google Scholar] [CrossRef]
- He, Y.; Xiao, Y.; Song, H.; Liang, Q.; Ju, D.; Chen, X.; Lu, H.; Jing, W.; Jiang, S.; Zhang, L. Design and evaluation of sifuvirtide, a novel HIV-1 fusion inhibitor. J. Biol. Chem. 2008, 283, 11126–11134. [Google Scholar]
- Wild, C.; Oas, T.; McDanal, C.; Bolognesi, D.; Matthews, T. A synthetic peptide inhibitor of human immunodeficiency virus replication: correlation between solution structure and viral inhibition. Proc. Natl. Acad. Sci. USA 1992, 89, 10537–10541. [Google Scholar] [CrossRef]
- Liu, S.; Wu, S.; Jiang, S. HIV entry inhibitors targeting gp41: From polypeptides to small-molecule compounds. Curr. Pharm. Des. 2007, 13, 143–162. [Google Scholar] [CrossRef]
- Cai, L.; Jiang, S. Development of peptide and small-molecule HIV-1 fusion inhibitors that target gp41. ChemMedChem. 2010, 5, 1813–1824. [Google Scholar] [CrossRef]
- Root, M.J.; Kay, M.S.; Kim, P.S. Protein design of an HIV-1 entry inhibitor. Science 2001, 291, 884–888. [Google Scholar] [CrossRef]
- Bewley, C.A.; Louis, J.M.; Ghirlando, R.; Clore, G.M. Design of a novel peptide inhibitor of HIV fusion that disrupts the internal trimeric coiled-coil of gp41. J. Biol. Chem. 2002, 277, 14238–14245. [Google Scholar]
- Sousa, S.F.; Fernandes, P.A.; Ramos, M.J. Protein-ligand docking: current status and future challenges. Proteins 2006, 65, 15–26. [Google Scholar] [CrossRef]
- Friesner, R.A.; Banks, J.L.; Murphy, R.B.; Halgren, T.A.; Klicic, J.J.; Mainz, D.T.; Repasky, M.P.; Knoll, E.H.; Shelley, M.; Perry, J.K. Glide: A new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J. Med. Chem. 2004, 47, 1739–1749. [Google Scholar] [CrossRef]
- Jones, G.; Willett, P.; Glen, R.C. Molecular recognition of receptor sites using a genetic algorithm with a description of desolvation. J. Mol. Biol. 1995, 245, 43–53. [Google Scholar] [CrossRef]
- Ewing, T.J.A.; Kuntz, I.D. Critical evaluation of search algorithms for automated molecular docking and database screening. J. Comput. Chem. 1997, 18, 1175–1189. [Google Scholar] [CrossRef]
- Ewing, T.J.; Makino, S.; Skillman, A.G.; Kuntz, I.D. DOCK 4.0: Search strategies for automated molecular docking of flexible molecule databases. J. Comput. Aided Mol. Des. 2001, 15, 411–428. [Google Scholar] [CrossRef]
- Liu, S.; Jiang, S. High throughput screening and characterization of HIV-1 entry inhibitors targeting gp41: Theories and techniques. Curr. Pharm. Des. 2004, 10, 1827–1843. [Google Scholar] [CrossRef]
- Chan, D.C.; Fass, D.; Berger, J.M.; Kim, P.S. Core structure of gp41 from the HIV envelope glycoprotein. Cell 1997, 89, 263–273. [Google Scholar] [CrossRef]
- Weissenhorn, W.; Dessen, A.; Harrison, S.C.; Skehel, J.J.; Wiley, D.C. Atomic Structure of the Ectodomain from HIV-1 gp41. Nature 1997, 387, 426–428. [Google Scholar] [CrossRef]
- Tan, K.; Liu, J.; Wang, J.; Shen, S.; Liu, M. Atomic structure of a thermostable subdomain of HIV-1 gp41. Proc. Natl. Acad. Sci. USA 1997, 94, 12303–12308. [Google Scholar] [CrossRef]
- Debnath, A.K.; Radigan, L.; Jiang, S. Structure-based identification of small molecule antiviral compounds targeted to the gp41 core structure of the human immunodecifiency virus type 1. J. Med. Chem. 1999, 42, 3203–3209. [Google Scholar] [CrossRef]
- Jiang, S.; Lin, K.; Zhang, L.; Debnath, A.K. A screening assay for antiviral compounds targeted to the HIV-1 gp41 core structure using a conformation-specific monoclonal antibody. J. Virol. Methods 1999, 80, 85–96. [Google Scholar] [CrossRef]
- Liu, S.; Jiang, S. High throughput screening and characterization of HIV-1 entry inhibitors targeting gp41: Theories and techniques. Curr. Pharm. Des. 2004, 10, 1827–1843. [Google Scholar] [CrossRef]
- Eckert, D.M.; Malashkevich, V.N.; Hong, L.H.; Carr, P.A.; Kim, P.S. Inhibiting HIV-1 entry: Discovery of D-peptide inhibitors that target the gp41 coiled-coil pocket. Cell 1999, 99, 103–115. [Google Scholar] [CrossRef]
- Este, J.A.; Cabrera, C.; Schols, D.; Cherepanov, P.; Gutierrez, A.; Witvrouw, M.; Pannecouque, C.; Debyser, Z.; Rando, R.F.; Clotet, B. Human immunodeficiency virus glycoprotein gp120 as the primary target for the antiviral action of AR177 (Zintevir). Mol. Pharmacol. 1998, 53, 340–345. [Google Scholar]
- Armand-Ugon, M.; Clotet-Codina, I.; Tintori, C.; Manetti, F.; Clotet, B.; Botta, M.; Este, J.A. The anti-HIV activity of ADS-J1 targets the HIV-1 gp120. Virology 2005, 343, 141–149. [Google Scholar] [CrossRef]
- Holden, P.M.; Kaur, H.; Goyal, R.; Gochin, M.; Rizzo, R.C. Footprint-based identification of viral entry inhibitors targeting HIVgp41. Bioorg. Med. Chem. Lett. 2012, 22, 3011–3016. [Google Scholar]
- Irwin, J.J.; Shoichet, B.K. ZINC—a free database of commercially available compounds for virtual screening. J. Chem. Inf. Model. 2005, 45, 177–182. [Google Scholar] [CrossRef]
- Lang, P.T.; Brozell, S.R.; Mukherjee, S.; Pettersen, E.F.; Meng, E.C.; Thomas, V.; Rizzo, R.C.; Case, D.A.; James, T.L.; Kuntz, I.D. DOCK 6: combining techniques to model RNA-small molecule complexes. RNA. 2009, 15, 1219–1230. [Google Scholar] [CrossRef]
- Moustakas, D.T.; Lang, P.T.; Pegg, S.; Pettersen, E.; Kuntz, I.D.; Brooijmans, N.; Rizzo, R.C. Development and validation of a modular, extensible docking program: DOCK 5. J. Comput. Aided Mol. Des. 2006, 20, 601–619. [Google Scholar] [CrossRef]
- Katritzky, A.R.; Tala, S.R.; Lu, H.; Vakulenko, A.V.; Chen, Q.Y.; Sivapackiam, J.; Pandya, K.; Jiang, S; Debnath, A.K. Design, synthesis, and structure-activity relationship of a novel series of 2-aryl 5-(4-oxo-3-phenethyl-2-thioxothiazolidinylidenemethyl)furans as HIV-1 entry inhibitors. J. Med. Chem. 2009, 52, 7631–7639. [Google Scholar] [CrossRef]
- Zhou, G.; Wu, D.; Snyder, B.; Ptak, R.G.; Kaur, H.; Gochin, M. Development of indole compounds as small molecule fusion inhibitors targeting HIV-1 glycoprotein-41. J. Med. Chem. 2011, 54, 7220–7231. [Google Scholar] [CrossRef]
- Jiang, S.; Lin, K.; Zhang, L.; Debnath, A.K. A screening assay for antiviral compounds targeted to the HIV-1 gp41 core structure using a conformation-specific monoclonal antibody. J. Virol. Methods 1999, 80, 85–96. [Google Scholar] [CrossRef]
- Jiang, S.; Lin, K.; Lu, M. A conformation-specific monoclonal antibody reacting with fusion-active gp41 from the human immunodeficiency virus type 1 envelope glycoprotein. J. Virol. 1998, 72, 10213–10217. [Google Scholar]
- Liu, S.; Boyer-Chatenet, L.; Lu, H.; Jiang, S. Rapid and automated fluorescence-linked immunosorbent assay for high-throughput screening of HIV-1 fusion inhibitors targeting gp41. J. Biomol. Screen. 2003, 8, 685–693. [Google Scholar] [CrossRef]
- Liu, K.; Lu, H.; Hou, L.; Qi, Z.; Teixeira, C.; Barbault, F.; Fan, B.T.; Liu, S.; Jiang, S.; Xie, L. Design, synthesis, and biological evaluation of N-carboxyphenylpyrrole derivatives as potent HIV fusion inhibitors targeting gp41. J. Med. Chem. 2008, 51, 7843–7854. [Google Scholar]
- Wang, Y.; Lu, H.; Zhu, Q.; Jiang, S.; Liao, Y. Structure-based design, synthesis and biological evaluation of new N-carboxyphenylpyrrole derivatives as HIV fusion inhibitors targeting gp41. Bioorg. Med. Chem. Lett. 2010, 20, 189–192. [Google Scholar]
- Katritzky, A.R.; Tala, S.R.; Lu, H.; Vakulenko, A.V.; Chen, Q.Y.; Sivapackiam, J.; Pandya, K.; Jiang, S.; Debnath, A.K. Design, synthesis, and structure-activity relationship of a novel series of 2-Aryl 5-(4-oxo-3-phenethyl-2-thioxothiazolidin-ylidenemethyl)furans as HIV-1 entry inhibitors. J. Med. Chem. 2009, 52, 7631–7639. [Google Scholar]
- Jiang, S.; Tala, S.R.; Lu, H.; Zou, P.; Avan, I.; Ibrahim, T.S.; bo-Dya, N.E.; Abdelmajeid, A.; Debnath, A.K.; Katritzky, A.R. Design, synthesis, and biological activity of a novel series of 2,5-disubstituted furans/pyrroles as HIV-1 fusion inhibitors targeting gp41. Bioorg. Med. Chem. Lett. 2011, 21, 6895–6898. [Google Scholar]
- Jiang, S.; Tala, S.R.; Lu, H.; bo-Dya, N.E.; Avan, I.; Gyanda, K.; Lu, L.; Katritzky, A.R.; Debnath, A.K. Design, synthesis, and biological activity of novel 5-((arylfuran/1H-pyrrol-2-yl)methylene)-2-thioxo-3-(3-(trifluoromethyl)phenyl)thi azolidin-4-ones as HIV-1 fusion inhibitors targeting gp41. J. Med. Chem. 2011, 54, 572–579. [Google Scholar] [CrossRef]
- Caffrey, M.; Cai, M.; Kaufman, J.; Stahl, S.J.; Wingfield, P.T.; Covell, D.G.; Gronenborn, A.M.; Clore, G.M. Three-dimensional solution structure of the 44 kDa ectodomain of SIV gp41. EMBO J. 1998, 17, 4572–4584. [Google Scholar]
- Gochin, M.; Kiplin, G.R.; Case, M.A. A metallopeptide assembly of the HIV-1 gp41 coiled coil is an ideal receptor in fluorescence detection of ligand binding. Angew. Chem. Int. Ed. Engl. 2003, 42, 5325–5328. [Google Scholar] [CrossRef]
- Cai, L.; Gochin, M. A novel fluorescence intensity screening assay identifies new low-molecular-weight inhibitors of the gp41 coiled-coil domain of human immunodeficiency virus type 1. Antimicrob. Agents Chemother. 2007, 51, 2388–2395. [Google Scholar] [CrossRef]
- Root, M.J.; Kay, M.S.; Kim, P.S. Protein design of an HIV-1 entry inhibitor. Science 2001, 291, 884–888. [Google Scholar]
- Weissenhorn, W.; Calder, L.J.; Dessen, A.; Laue, T.; Skehel, J.J.; Wiley, D.C. Assembly of a rod-shaped chimera of a trimeric GCN4 zipper and the HIV-1 gp41 ectodomain expressed in Escherichia coli. Proc. Natl. Acad. Sci. USA 1997, 94, 6065–6069. [Google Scholar]
- Chen, X.; Lu, L.; Qi, Z.; Lu, H.; Wang, J.; Yu, X.; Chen, Y.; Jiang, S. Novel recombinant engineered gp41 N-terminal heptad repeat trimers and their potential as anti-HIV-1 therapeutics or microbicides. J. Biol. Chem. 2010, 285, 25506–25515. [Google Scholar]
- Louis, J.M.; Nesheiwat, I.; Chang, L.; Clore, G.M.; Bewley, C.A. Covalent trimers of the internal N-terminal trimeric coiled-coil of gp41 and antibodies directed against them are potent inhibitors of HIV envelope-mediated cell fusion. J. Biol. Chem. 2003, 278, 20278–20285. [Google Scholar]
- Gochin, M.; Khorosheva, V.; Case, M.A. Structural characterization of a paramagnetic metal-ion-assembled three-stranded alpha-helical coiled coil. J. Am. Chem. Soc. 2002, 124, 11018–11028. [Google Scholar]
- Gochin, M.; Savage, R.; Hinckley, S.; Cai, L. A fluorescence assay for rapid detection of ligand binding affinity to HIV-1 gp41. Biol. Chem. 2006, 387, 477–483. [Google Scholar]
- Zhou, G.; Wu, D.; Hermel, E.; Balogh, E.; Gochin, M. Design, synthesis, and evaluation of indole compounds as novel inhibitors targeting Gp41. Bioorg. Med. Chem. Lett. 2010, 20, 1500–1503. [Google Scholar] [CrossRef]
- Root, M.J.; Steger, H.K. HIV-1 gp41 as a target for viral entry inhibition. Curr. Pharm. Des. 2004, 10, 1805–1825. [Google Scholar] [CrossRef]
- Frey, G.; Rits-Volloch, S.; Zhang, X.Q.; Schooley, R.T.; Chen, B.; Harrison, S.C. Small molecules that bind the inner core of gp41 and inhibit HIV envelope-mediated fusion. Proc. Natl. Acad. Sci. USA 2006, 103, 13938–13943. [Google Scholar]
- Ernst, J.T.; Kutzki, O.; Debnath, A.K.; Jiang, S.; Lu, H.; Hamilton, A.D. Design of a protein surface antagonist based on alpha-helix mimicry: inhibition of gp41 assembly and viral fusion. Angew. Chem. Int. Ed. Engl. 2002, 41, 278–281. [Google Scholar]
- Orner, B.P.; Ernst, J.T.; Hamilton, A.D. Toward proteomimetics: terphenyl derivatives as structural and functional mimics of extended regions of an alpha-helix. J. Am. Chem. Soc. 2001, 123, 5382–5383. [Google Scholar]
- Whitby, L.R.; Boyle, K.E.; Cai, L.; Yu, X.; Gochin, M.; Boger, D.L. Discovery of HIV fusion inhibitors targeting gp41 using a comprehensive alpha-helix mimetic library. Bioorg. Med. Chem. Lett. 2012, 22, 2861–2865. [Google Scholar] [CrossRef]
- Ferrer, M.; Kapoor, T.M.; Strassmaier, T.; Weissenhorn, W.; Skehel, J.J.; Oprian, D.; Schreiber, S.L.; Wiley, D.C.; Harrison, S.C. Selection of gp41-mediated HIV-1 cell entry inhibitors from biased combinatorial libraries of non-natural binding elements. Nat. Struct. Biol. 1999, 6, 953–960. [Google Scholar] [CrossRef]
- Zhou, G.; Ferrer, M.; Chopra, R.; Kapoor, T.M.; Strassmaier, T.; Weissenhorn, W.; Skehel, J.J.; Oprian, D.; Schreiber, S.L.; Harrison, S.C.; et al. The structure of an HIV-1 specific cell entry inhibitor in complex with the HIV-1 gp41 trimeric core. Bioorg. Med. Chem. 2000, 8, 2219–2227. [Google Scholar] [CrossRef]
- Hammer, S.M.; Katzenstein, D.A.; Hughes, M.D.; Gundacker, H.; Schooley, R.T.; Haubrich, R.H.; Henry, W.K.; Lederman, M.M.; Phair, J.P.; Niu, M.; et al. A trial comparing nucleoside monotherapy with combination therapy in HIV-infected adults with CD4 cell counts from 200 to 500 per cubic millimeter. AIDS Clinical Trials Group Study 175 Study Team. N. Engl. J. Med. 1996, 335, 1081–1090. [Google Scholar]
- Nagashima, K.A.; Thompson, D.A.; Rosenfield, S.I.; Maddon, P.J.; Dragic, T.; Olson, W.C. Human immunodeficiency virus type 1 entry inhibitors PRO 542 and T-20 are potently synergistic in blocking virus-cell and cell-cell fusion. J. Infect. Dis. 2001, 183, 1121–1125. [Google Scholar] [CrossRef]
- Tremblay, C.L.; Kollmann, C.; Giguel, F.; Chou, T.C.; Hirsch, M.S. Strong in vitro synergy between the fusion inhibitor T-20 and the CXCR4 blocker AMD-3100. J. Acquir. Immune Defic. Syndr. 2000, 25, 99–102. [Google Scholar] [CrossRef]
- Tremblay, C.L.; Giguel, F.; Kollmann, C.; Guan, Y.; Chou, T.C.; Baroudy, B.M.; Hirsch, M.S. Anti-human immunodeficiency virus interactions of SCH-C (SCH 351125), a CCR5 antagonist, with other antiretroviral agents in vitro. Antimicrob. Agents Chemother. 2002, 46, 1336–1339. [Google Scholar] [CrossRef]
- Pan, C.; Lu, H.; Qi, Z.; Jiang, S. Synergistic efficacy of combination of enfuvirtide and sifuvirtide, the first- and next-generation HIV-fusion inhibitors. AIDS 2009, 23, 639–641. [Google Scholar] [CrossRef]
- Pan, C.; Cai, L.; Lu, H.; Qi, Z.; Jiang, S. Combinations of the first and next generations of human immunodeficiency virus (HIV) fusion inhibitors exhibit a highly potent synergistic effect against enfuvirtide- sensitive and -resistant HIV type 1 strains. J. Virol. 2009, 83, 7862–7872. [Google Scholar] [CrossRef]
- Pan, C.; Cai, L.; Lu, H.; Lu, L.; Jiang, S. A novel chimeric protein-based HIV-1 fusion inhibitor targeting gp41 glycoprotein with high potency and stability. J. Biol. Chem. 2011, 286, 28425–28434. [Google Scholar]
- Cai, L.; Pan, C.; Xu, L.; Shui, Y.; Liu, K.; Jiang, S. Interactions between different generation HIV-1 fusion inhibitors and the putative mechanism underlying the synergistic anti-HIV-1 effect resulting from their combination. FASEB J. 2012, 26, 1018–1026. [Google Scholar]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Yu, F.; Lu, L.; Du, L.; Zhu, X.; Debnath, A.K.; Jiang, S. Approaches for Identification of HIV-1 Entry Inhibitors Targeting gp41 Pocket. Viruses 2013, 5, 127-149. https://doi.org/10.3390/v5010127
Yu F, Lu L, Du L, Zhu X, Debnath AK, Jiang S. Approaches for Identification of HIV-1 Entry Inhibitors Targeting gp41 Pocket. Viruses. 2013; 5(1):127-149. https://doi.org/10.3390/v5010127
Chicago/Turabian StyleYu, Fei, Lu Lu, Lanying Du, Xiaojie Zhu, Asim K. Debnath, and Shibo Jiang. 2013. "Approaches for Identification of HIV-1 Entry Inhibitors Targeting gp41 Pocket" Viruses 5, no. 1: 127-149. https://doi.org/10.3390/v5010127