Performance of VIDISCA-454 in Feces-Suspensions and Serum

{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
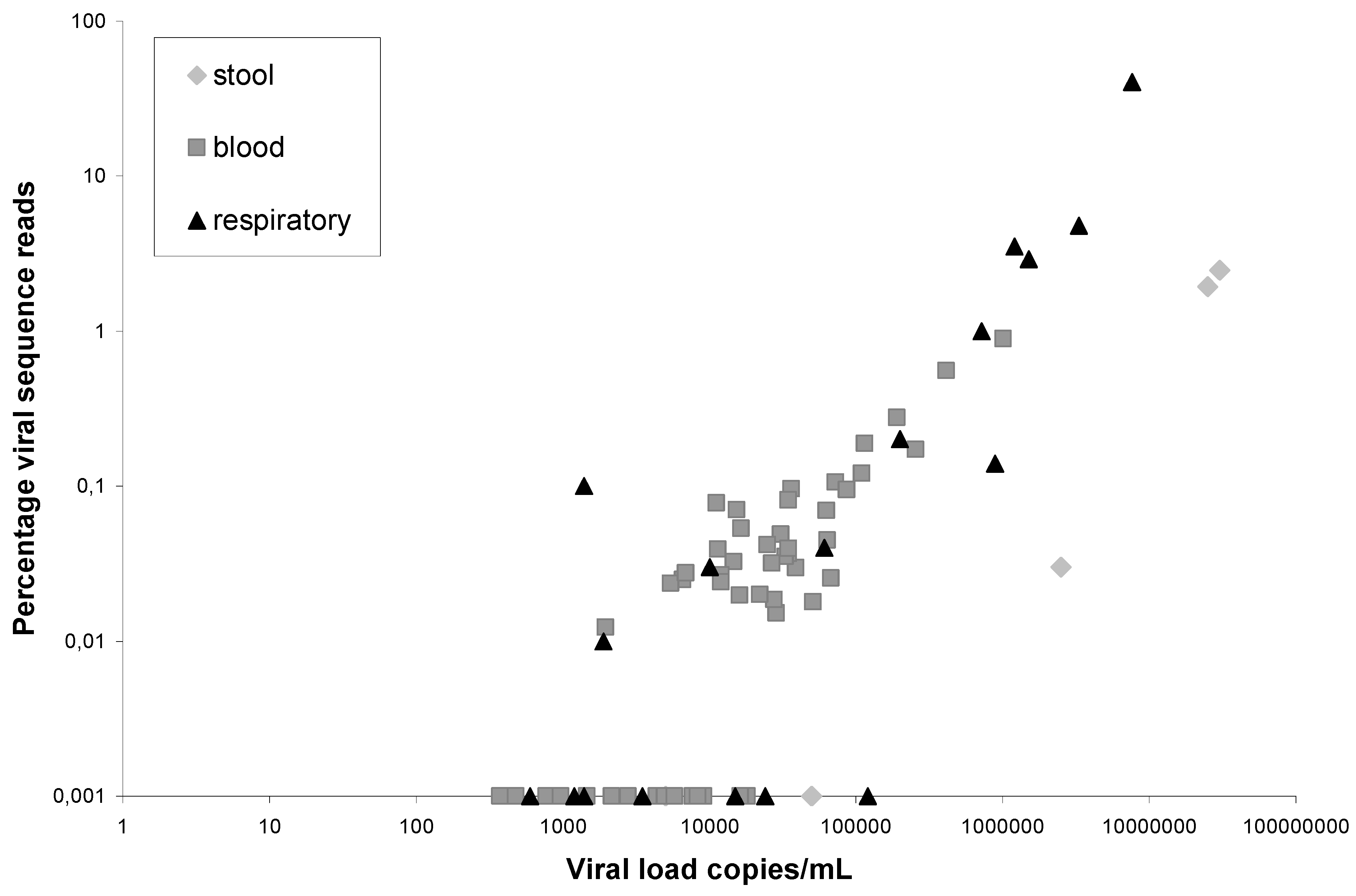
 ), and respiratory copan-collected swabs (▲). On the X-axis the viral load in a clinical sample is indicated, on the Y-axis the percentage of viral sequence reads: HIV-1 (blood), norovirus (stool), coronaviruses, adenoviruses, picornaviruses and influenzaviruses (respiratory material). The viruses and percentages in the respiratory samples have been published previously [6].
), and respiratory copan-collected swabs (▲). On the X-axis the viral load in a clinical sample is indicated, on the Y-axis the percentage of viral sequence reads: HIV-1 (blood), norovirus (stool), coronaviruses, adenoviruses, picornaviruses and influenzaviruses (respiratory material). The viruses and percentages in the respiratory samples have been published previously [6].
), and respiratory copan-collected swabs (▲). On the X-axis the viral load in a clinical sample is indicated, on the Y-axis the percentage of viral sequence reads: HIV-1 (blood), norovirus (stool), coronaviruses, adenoviruses, picornaviruses and influenzaviruses (respiratory material). The viruses and percentages in the respiratory samples have been published previously [6].
), and respiratory copan-collected swabs (▲). On the X-axis the viral load in a clinical sample is indicated, on the Y-axis the percentage of viral sequence reads: HIV-1 (blood), norovirus (stool), coronaviruses, adenoviruses, picornaviruses and influenzaviruses (respiratory material). The viruses and percentages in the respiratory samples have been published previously [6]. 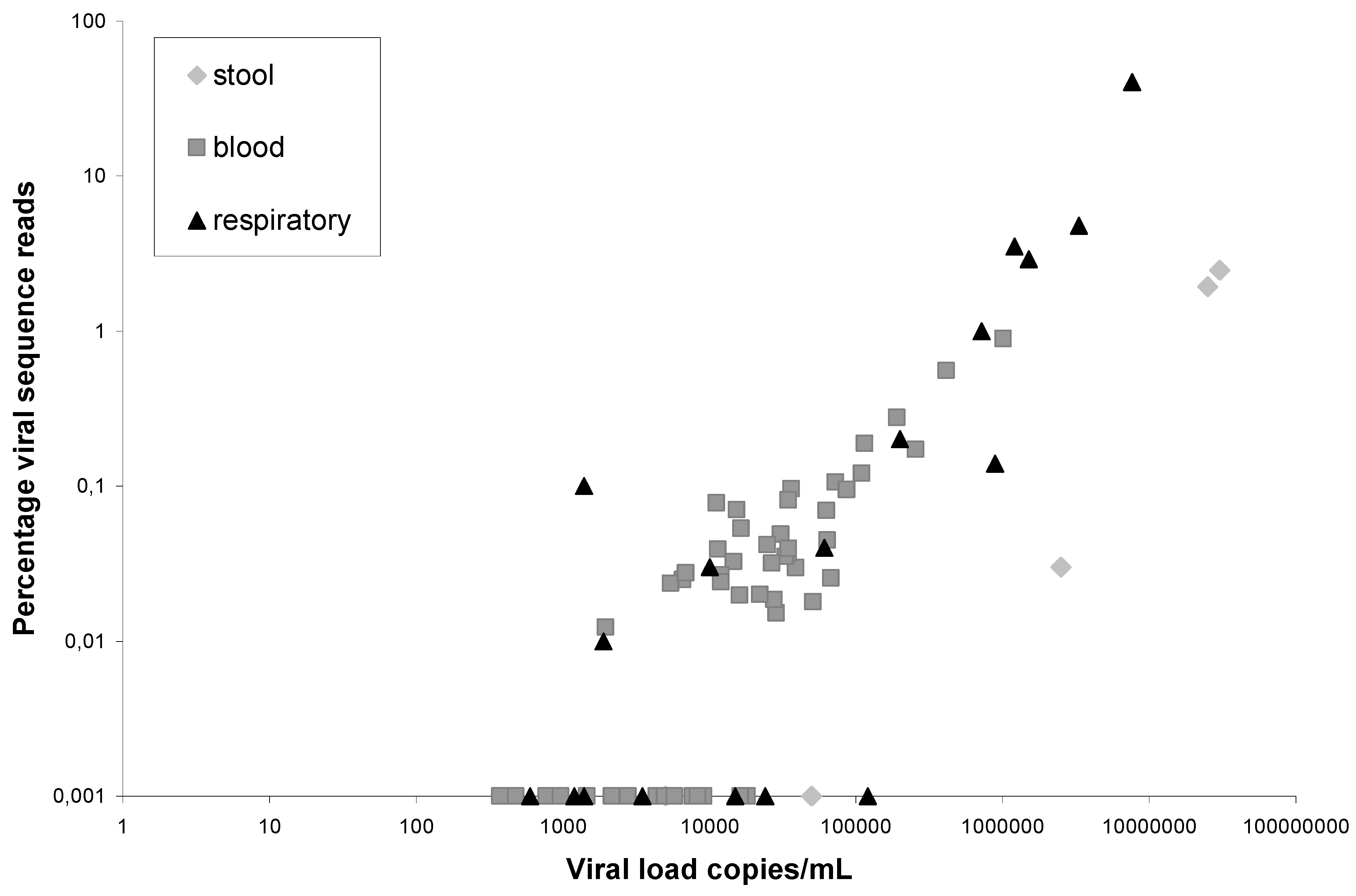
3. Experimental Section
3.1. Clinical Samples
3.2. Real Time HIV-1-RNA and Norovirus-RNA RT-PCR
3.3. VIDISCA-454
4. Conclusions
Acknowledgments
Conflict of Interest
References adn Notes
- Allander, T.; Emerson, S.U.; Engle, R.E.; Purcell, R.H.; Bukh, J. A virus discovery method incorporating DNase treatment and its application to the identification of two bovine parvovirus species. Proc. Natl. Acad. Sci. USA 2001, 98, 11609–11614. [Google Scholar]
- Kapoor, A.; Slikas, E.; Simmonds, P.; Chieochansin, T.; Naeem, A.; Shaukat, S.; Alam, M.M.; Sharif, S.; Angez, M.; Zaidi, S.; et al. A newly identified bocavirus species in human stool. J. Infect. Dis. 2009, 199, 196–200. [Google Scholar] [CrossRef]
- Woo, P.C.; Lau, S.K.; Huang, Y.; Tsoi, H.W.; Chan, K.H.; Yuen, K.Y. Phylogenetic and recombination analysis of coronavirus HKU1, a novel coronavirus from patients with pneumonia. Arch. Virol. 2005, 150, 2299–2311. [Google Scholar] [CrossRef]
- Chang, Y.; Cesarman, E.; Pessin, M.S.; Lee, F.; Culpepper, J.; Knowles, D.M.; Moore, P.S. Identification of herpesvirus-like DNA sequences in AIDS-associated Kaposi’s sarcoma. Science 1994, 266, 1865–1869. [Google Scholar]
- Van der Hoek, L.; Pyrc, K.; Jebbink, M.F.; Vermeulen-Oost, W.; Berkhout, R.J.; Wolthers, K.C.; Wertheim-van Dillen, P.M.; Kaandorp, J.; Spaargaren, J.; Berkhout, B. Identification of a new human coronavirus. Nat. Med. 2004, 10, 368–373. [Google Scholar] [CrossRef]
- De Vries, M.; Deijs, M.; Canuti, M.; van Schaik, B.D.; Faria, N.R.; van de Garde, M.D.; Jachimowski, L.C.; Jebbink, M.F.; Jakobs, M.; Luyf, A.C.; et al. A sensitive assay for virus discovery in respiratory clinical samples. PLoS One 2011, 6, e16118. [Google Scholar]
- Canuti, M.; Eis-Huebinger, A.M.; Deijs, M.; de Vries, M.; Drexler, J.F.; Oppong, S.K.; Muller, M.A.; Klose, S.M.; Wellinghausen, N.; Cottontail, V.M.; et al. Two novel parvoviruses in frugivorous New and Old World bats. PLoS One 2011, 6, e29140. [Google Scholar]
- Van der Hoek, L.; Sol, C.J.; Maas, J.; Lukashov, V.V.; Kuiken, C.L.; Goudsmit, J. Genetic differences between human immunodeficiency virus type 1 subpopulations in faeces and serum. J. Gen. Virol. 1998, 79, 259–267. [Google Scholar]
- Jansen, R.R.; Schinkel, J.; Koekkoek, S.; Pajkrt, D.; Beld, M.; de Jong, M.D.; Molenkamp, R. Development and evaluation of a four-tube real time multiplex PCR assay covering fourteen respiratory viruses, and comparison to its corresponding single target counterparts. J. Clin. Virol. 2011, 51, 179–185. [Google Scholar] [CrossRef]
- Snijders, F.; Kuijper, E.J.; de Wever, B.; van der Hoek, L.; Danner, S.A.; Dankert, J. Prevalence of Campylobacter-associated diarrhea among patients infected with human immunodeficiency virus. Clin. Infect. Dis. 1997, 24, 1107–1113. [Google Scholar]
- Boom, R.; Sol, C.J.; Salimans, M.M.; Jansen, C.L.; Wertheim-van Dillen, P.M.; van der Noordaa, J. Rapid and simple method for purification of nucleic acids. J. Clin. Microbiol. 1990, 28, 495–503. [Google Scholar]
- Endoh, D.; Mizutani, T.; Kirisawa, R.; Maki, Y.; Saito, H.; Kon, Y.; Morikawa, S.; Hayashi, M. Species-independent detection of RNA virus by representational difference analysis using non-ribosomal hexanucleotides for reverse transcription. Nucleic Acids Res. 2005, 33. [Google Scholar]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar]
- Luyf, A.C.; van Schaik, B.D.; de Vries, M.; Baas, F.; van Kampen, A.H.; Olabarriaga, S.D. Initial steps towards a production platform for DNA sequence analysis on the grid. BMC Bioinformatics 2010, 11. [Google Scholar]
- Benson, D.A.; Karsch-Mizrachi, I.; Clark, K.; Lipman, D.J.; Ostell, J.; Sayers, E.W. GenBank. Nucleic Acids Res. 2012, 40, D48–D53. [Google Scholar]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
De Vries, M.; Oude Munnink, B.B.; Deijs, M.; Canuti, M.; Koekkoek, S.M.; Molenkamp, R.; Bakker, M.; Jurriaans, S.; Van Schaik, B.D.C.; Luyf, A.C.; et al. Performance of VIDISCA-454 in Feces-Suspensions and Serum. Viruses 2012, 4, 1328-1334. https://doi.org/10.3390/v4081328
De Vries M, Oude Munnink BB, Deijs M, Canuti M, Koekkoek SM, Molenkamp R, Bakker M, Jurriaans S, Van Schaik BDC, Luyf AC, et al. Performance of VIDISCA-454 in Feces-Suspensions and Serum. Viruses. 2012; 4(8):1328-1334. https://doi.org/10.3390/v4081328
Chicago/Turabian StyleDe Vries, Michel, Bas B. Oude Munnink, Martin Deijs, Marta Canuti, Sylvie M. Koekkoek, Richard Molenkamp, Margreet Bakker, Suzanne Jurriaans, Barbera D. C. Van Schaik, Angela C. Luyf, and et al. 2012. "Performance of VIDISCA-454 in Feces-Suspensions and Serum" Viruses 4, no. 8: 1328-1334. https://doi.org/10.3390/v4081328
APA StyleDe Vries, M., Oude Munnink, B. B., Deijs, M., Canuti, M., Koekkoek, S. M., Molenkamp, R., Bakker, M., Jurriaans, S., Van Schaik, B. D. C., Luyf, A. C., Olabarriaga, S. D., Van Kampen, A. H. C., & Van der Hoek, L. (2012). Performance of VIDISCA-454 in Feces-Suspensions and Serum. Viruses, 4(8), 1328-1334. https://doi.org/10.3390/v4081328